

Abstract
Idiopathic pulmonary fibrosis (IPF) is a progressive disease with poor prognosis. The molecular mechanisms involved in the progression of IPF are not fully understood; however, the platelet-derived growth factor (PDGF)/PDGF receptor pathway is thought to play a critical role in fibrogenesis of the lungs. Other growth factors, including fibroblast growth factor and vascular endothelial growth factor, are also thought to contribute to the pathogenesis of pulmonary fibrosis. Nintedanib is an inhibitor of multiple tyrosine kinases, including receptors for PDGF, fibroblast growth factor, and vascular endothelial growth factor. In the Phase II TOMORROW trial, treatment with 150 mg of nintedanib twice daily showed a trend to slow the decline in lung function and significantly decrease acute exacerbations in patients with IPF, while showing an acceptable safety profile. The Phase III INPULSIS trials demonstrated a significant decrease in the annual rate of decline in forced vital capacity in IPF patients treated with 150 mg nintedanib twice daily. In the INPULSIS-2 trial, the time to the first acute exacerbation significantly increased in IPF patients who were treated with 150 mg of nintedanib twice daily. Pirfenidone, another antifibrotic drug, was shown to limit the decline in pulmonary function in patients with IPF in the ASCEND trial. Combination therapy with nintedanib and pirfenidone is anticipated, although further evaluation of its long-term safety is needed. There is limited evidence for the safety of the combination therapy although a Phase II trial conducted in Japan demonstrated that combination therapy with nintedanib and pirfenidone was tolerable for 1 month. Available antifibrotic agents (ie, pirfenidone and N-acetylcysteine) have limited efficacy as single therapies for IPF; therefore, further study of combination therapy with antifibrotic agents is needed.
Keywords: platelet-derived growth factor, fibroblast growth factor, vascular endothelial growth factor, pirfenidone, forced vital capacity
Core evidence clinical impact summary for the treatment of patients with IPF with 150 mg nintedanib
| Outcome measure | Evidence | Implications |
|---|---|---|
| Disease-oriented evidence | Clinical trials | Nintedanib treatment has been shown to significantly decrease the annual rate of decline in forced vital capacity and be effective in increasing the time to the first acute exacerbation. |
| Patient-oriented evidence | Clinical trials | Nintedanib treatment has been shown to reduce the numerical risk of all-cause mortality and improve health-related quality of life; however, the results are not statistically significant. The most frequent adverse event was diarrhea and that was almost always mild or moderate. Nintedanib therapy has been shown to be well tolerated without serious adverse events. |
| Economic evidence | None currently | No formal cost-effectiveness analysis has been conducted. |
Introduction
Idiopathic pulmonary fibrosis (IPF) is the most common form of chronic fibrosing idiopathic interstitial pneumonia and is characterized by worsening dyspnea and progressive loss of lung function. The pathogenesis of IPF is thought to involve aberrant wound healing mediated by multiple signaling pathways.1 Activation of cell signaling pathways through profibrotic growth factors, including platelet-derived growth factor (PDGF), fibroblast growth factor (FGF), and vascular endothelial growth factor (VEGF), has been implicated in the pathogenesis of IPF.2–4 Nintedanib (formerly known as BIBF1120) is a potent intracellular inhibitor of tyrosine kinases, which targets PDGF receptors (PDGFRs), FGF receptors (FGFRs), and VEGF receptors (VEGFRs).5 Recently, two randomized trials of nintedanib showed significant reductions in the annual decline of forced vital capacity (FVC) in patients with IPF, demonstrating that nintedanib should become the standard treatment for patients with IPF.6,7 Pirfenidone has been licensed in Japan, Europe, and USA, and is currently a first-line treatment in European and UK guidelines; nintedanib would be a suitable addition to current practice.8–10
Molecular pathogenesis of IPF
The molecular mechanisms involved in the progression of IPF are not fully understood; however, alveolar epithelial type II cells (AECII) and fibroblasts are thought to be the main mediators of the process.11,12 The first event in the progression of IPF is AECII injury caused by endoplasmic reticulum stress, lysosomal stress, and mitochondrial and DNA damage.11,12 Patients with familial pulmonary fibrosis may have genetic predispositions, including mutations in surfactant protein C or A2, telomerase, or MUC5B, that lead to increased susceptibility of AECII to secondary stimuli such as cigarette smoking or viral infection. After exposure to the secondary stimuli, injured AECs show a tendency to undergo apoptosis. The link between injured AECII and fibrosis is not well understood, but it has been reported that AECII produces profibrotic mediators, among which transforming growth factor (TGF)-β and PDGF are thought to play a critical role in fibrogenesis of the lungs. Other growth factors, including FGF, VEGF, and insulin-like growth factor (IGF)-1, are also thought to contribute to the pathogenesis of pulmonary fibrosis.
Roles of growth factors in pulmonary fibrosis
Profibrotic growth factors, including PDGF, TGF-β, and FGF, have been studied to clarify their roles in pulmonary fibrosis, and their contributions are summarized in Table 1.
Table 1.
Producing cells and biological effects of growth factors related to pulmonary fibrosis
| Growth factor | Producing cells
|
Biological effects for fibroblasts
|
Profibrotic in vitro | |||||
|---|---|---|---|---|---|---|---|---|
| Epithelial cells | Fibroblasts | Fibrocytes | Macrophases | Proliferation | Migration | Collagen production | ||
| TGF-β | + | + | + | + | → | ↑ | ↑↑ | ↑ |
| PDGF | + | + | + | + | ↑↑ | ↑↑ | ↑ | ↑ |
| FGF-2 | − | + | + | + | ↑ | ↑ | ↑ | ? |
Notes: +, produce; −, not produce; The arrows show the level of stimulation: →, not stimulate; ↑, weak; ↑↑, strong; ?, not determined.
Abbreviations: TGF, transforming growth factor; PDGF, platelet-derived growth factor; FGF, fibroblast growth factor.
PDGF and PDGFRs
PDGF is a homo- or heterodimeric molecule with a molecular weight of 30 kDa.13 There are four PDGF genes, PDGF-A, PDGF-B, PDGF-C, and PDGF-D, which are located on chromosomes 7, 22, 4, and 11, respectively.14,15 There are two types of PDGFRs, α and β, which have a molecular weight of 170–180 kDa and are composed of homo- or heterodimers. In vitro studies have shown that the possible PDGF/PDGFR interactions are complex.15 However, in vivo studies have shown that PDGF-AA and PDGF-CC bind to PDGFR-α, while PDGF-BB binds to PDGFR-β.15
PDGFs are expressed in many types of cells, including fibroblasts, vascular endothelial cells, macrophages, and platelets/megakaryocytes.15 PDGFs are major mitogens for mesenchymal cells, and PDGF is the strongest identified proliferative stimulus for fibroblasts.13,15 In addition, binding of PDGF to PDGFRs induces Ca2+ influx and rearrangement of the cytoskeleton involving changes in the arrangement of actin stress fibers. Through these cytoskeletal responses, PDGF stimulates the migration of various cell types.
Knockout of the PDGF-A gene in mice is homozygous lethal with two different restriction points, one prenatal and one postnatal.16 Postnatally, PDGF-A-deficient mice develop lung emphysema due to loss of alveolar myofibroblasts expressing PDGFR-α. In contrast, PDGFR-α null mice showed cranial malformations and deficient myotome formation.17 Mice deficient in PDGF-B or PDGFR-β showed renal, cardiovascular, and hematological abnormalities, but their lungs were normal.18,19 These results show that the PDGF-A/PDGFR-α pathway plays a role in the secondary septation process, because PDGFR-α-expressing cells located in the alveolar entry ring have characteristics of myofibroblasts.20 PDGF is known to play a role in the pathogenesis of pulmonary fibrosis.13,15 Bleomycin (BLM)-induced pulmonary fibrosis in mice and rats has been used in the analyses of the molecular basis of pathogenesis of pulmonary fibrosis. Maeda et al reported increased expression of the PDGF-A gene in mice with BLM-induced pulmonary fibrosis.21 Walsh et al examined the bronchoalveolar lavage fluid (BAL) of rats treated with BLM and found that 38–40 kDa and 29 kDa peptides detected with anti-PDGF-BB and anti-PDGF-AA antibodies, respectively, showed growth-promoting activity in lung fibroblasts; however, anti-PDGF-BB and anti-PDGF-AA antibodies reduced this growth-promoting activity by 64% and 15%, respectively.22 In contrast, Zhuo et al showed that expression of PDGF-C, but not of PDGF-A, PDGF-B, or PDGF-D, was induced in the lungs of BLM-treated mice.23 Shimizu et al reported that PDGF-A and PDGF-B expression was elevated in the lungs of BLM-treated mice at the mRNA and protein levels.24 Adoptive transfer of an adenovirus expressing the PDGF-B gene into the lung induced severe fibrosis in mice.25 Based on these reports, expression of PDGF isoforms could be enhanced during fibrogenesis of the lungs, but further study is necessary to determine the precise mechanisms underlying this phenomenon.
Enhanced expression of PDGF in epithelial cells and alveolar macrophages in the lungs of patients with IPF has been reported.26,27 However, the mechanisms involved in enhanced PDGF expression and activity in the fibrotic lung are poorly understood. Recently, Gochuico et al examined growth factors in alveolar lining fluid from patients with rheumatoid arthritis complicated with pulmonary fibrosis and reported that PDGF-AB and PDGF-BB, but not TGF-β and PDGF-AA, were associated with the progressive stage of pulmonary fibrosis,28 indicating the importance of PDGF-B in fibrogenesis of the lungs.
The evidence described above suggests that targeting the PDGF/PDGFR signaling pathway may have therapeutic effects against pulmonary fibrosis. This hypothesis has been investigated using animal models of pulmonary fibrosis with specific PDGFR inhibitors. First, Rice et al reported that AG1296, a PDGFR inhibitor, prevented pulmonary fibrosis induced by vanadium pentoxide (V2O5) in rats.29 Next, imatinib mesylate (Gleevec in the United States and Glivec in Europe), a potent and specific inhibitor of tyrosine kinases c-abl, bcr-abl, and c-kit, was tested in animal models of pulmonary fibrosis because imatinib is also a specific inhibitor of PDGFR.30 Studies using various pulmonary fibrosis models have shown that imatinib strongly inhibits fibrogenesis in the lungs.31–33 In addition, Yoshida et al reported that in vivo gene transfer of an extracellular domain of PDGFR-β reduced BLM-induced pulmonary fibrosis.34 Recently, nilotinib, a compound with a profile similar to that of imatinib, was reported to inhibit fibrotic activity more effectively than imatinib.35 These observations suggest that PDGFR is a potential therapeutic target for pulmonary fibrosis.36
FGF and FGFRs
FGF and the FGFR are thought to be involved in fibrogenesis in the lungs. The FGF/FGFR family is composed of 18 FGF ligands and four FGFRs.37,38 Alternative splicing of domain III of FGFR1–3 yields two different isoforms: IIIb in epithelial tissue and IIIc in mesenchymal tissue. Heparan sulfate glycosaminoglycan binds to both FGF and the FGFR and stabilizes their binding by facilitating dimerization. The specificity of different FGFs for different receptor isoforms has been reported.39
The physiological functions of FGF are diverse and complex. In vitro studies with FGF proteins and in vivo studies with genetically modified mice have been performed.37–39 Because FGFs possess mitogenic, chemotactic, and angiogenic activities, they have been studied in cancer research. In addition, FGF knockout mice have demonstrated the role of FGFs in the embryonic development of some organs. However, the physiological roles of most FGFs and FGFRs are unclear. Fgf1 knockout (−/−) and Fgf2 −/− mice are viable, fertile, and show some abnormalities in the vascular system, and exogenous FGF2 induces proliferation of endothelial cells, smooth muscle cells, and fibroblasts in these mice.
The profibrogenic activity of FGF-2 (basic FGF) has been confirmed experimentally. Hetzel et al reported a proliferative effect of FGF-2 in lung fibroblasts.40 In comparison with normal fibroblasts, fibroblasts derived from IPF patients showed reduced response to FGF-2, IGF-1, and EGF, but not to PDGFs. Kanazawa et al reported that FGF-2 promoted migration of skin fibroblasts; however, FGF-2 did not stimulate the production of fibronectin in lung fibroblasts.41 No data exist regarding the production of FGF in the lungs of patients with IPF, but expression of FGF-2 mRNA was reported to be upregulated in the lungs of BLM-treated mice.42 Immunohistochemical staining for FGF-2 showed FGF-2 expression in inflammatory cells; however, the role of FGF-2/FGFR signaling in mouse models of lung fibrosis remains unclear. Ju et al reported that administration of a soluble FGFR2c ectodomain significantly reduced TGF-β-induced lung fibrosis in mice.43 On the other hand, Guzy et al recently reported that Fgf2 −/− mice showed increased mortality due to epithelial injury induced by BLM, without an associated change in lung fibrosis.42 These conflicting results make it impossible to conclude regarding the role of FGF-2 in pulmonary fibrosis in mice and human beings.
In contrast to FGF-2, FGF-1 has been reported to be an antifibrotic factor. FGF-1 reverses the profibrogenic effects of TGF-β such as alpha-smooth muscle actin (α-SMA) induction.44 In addition, FGF-1 reduced expression of collagen I and induced apoptosis in lung fibroblasts.45,46
It is difficult to determine the overall roles of FGF/FGFR in the pathogenesis of IPF from published reports. Further studies using specific FGFR inhibitors in pulmonary fibrosis models may help to clarify the roles of FGF/FGFR, although specific analysis of each isoform of FGF/FGFR will be difficult.
VEGF and VEGFR
VEGF and VEGFR play central roles in physiological and pathological angiogenesis.47 The VEGF family is composed of seven members: VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-E, PIGF, and svVEGF. The three main subtypes of VEGFR are VEGFR1, VEGFR2, and VEGFR3. VEGF-A has been reported to play a key role in angiogenesis. The splicing of the VEGF gene also controls angiogenesis and can produce the pro-angiogenic VEGF165 isoform, which is generated by proximal splice site selection in exon 8a, or the anti-angiogenic VEGF165b isoform, which is generated by distal splice site selection 66 bp further into exon 8 and generates mRNA isoforms that contain exon 8b.48 Ebina et al reported that the number of CD34-positive vessels in the lung increased in the early phase of IPF.49 However, the role of angiogenesis in the development of lung fibrogenesis remains unclear, and there is little evidence on the relationship between VEGF and lung fibrosis. Antoniou et al showed elevated VEGF levels in the BAL of IPF patients.50 Farkas et al reported direct effects of VEGF-A on lung fibroblasts and found that VEGF-A enhanced collagen I expression induced by TGF-β.51 Hamada et al demonstrated that gene therapy with a soluble form of the flt-1 gene reduced BLM-induced pulmonary fibrosis in mice.52 Ou et al reported that VEGFR-2 antagonist SU5416 attenuated BLM-induced lung fibrosis in mice.53 These reports suggest the possibility that VEGF-targeted therapy might ameliorate pulmonary fibrosis, although the identity of the best target is an unresolved question.
Development and pharmacological activities of nintedanib (BIBF1120)
The development of nintedanib began with the identification of a lead compound from a group of six-substituted indolinones via assessment of tumor angiogenesis inhibition, and this compound was shown to inhibit VEGFR-2.54 Derivatives of the lead compound were screened for triple angiokinase (eg, VEGFR-2, FGFR-1, and PDGFRα) inhibitory activity. Two compounds, BIBF1000 and BIBF1120, were developed, their antitumor activities were assessed, and BIBF1120 was selected for clinical trials (Figure 1).54 The kinase inhibition profile of BIBF1120 is provided in Table 2.4
Figure 1.
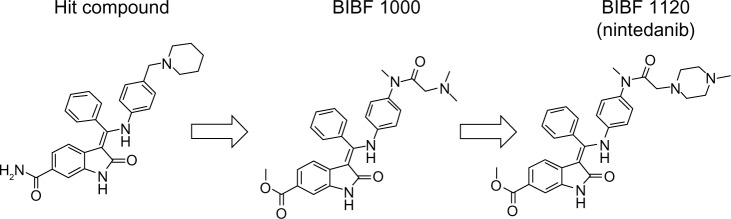
Chemical structure of nintedanib.
Notes: Indolinone lead compounds substituted at position 6 were selected by assessing inhibition of tumor angiogenesis and shown to inhibit VEGFR-2. Derivatives of the lead compound were further screened to enhance triple angiokinase (VEGFR-2, FGFR-1, and PDGFR-α) inhibitory activity. Two compounds, BIBF1000 and BIBF1120, were tested in assays of antitumor activity, and BIBF1120 was selected for clinical trials.
Abbreviations: VEGFR, vascular endothelial growth factor receptor; FGFR, fibroblast growth factor receptor; PDGFR, platelet-derived growth factor receptor.
Table 2.
Tyrosine kinase inhibition by nintedanib in a cellular BA/F3 assay
| Assay | IC50 (nM) |
|---|---|
| FGFR1 | 300–1,000 |
| FGFR2 | 257 |
| FGFR3 | 300–1,000 |
| FGFR4 | 300–1,000 |
| PDGFRα | 41 |
| PDGFRβ | 58 |
| VEGFR1 | 300–1,000 |
| VEGFR2 | 46 |
| VEGFR3 | 33 |
| LCK | 22 |
| LYN | 300–1,000 |
| SRC | 811 |
| FLT-3 | 17 |
Abbreviations: FGFR, fibroblast growth factor receptor; PDGFR, platelet-derived growth factor receptor; VEGFR, vascular endothelial growth factor receptor.
As mentioned in the section PDGF and PDGFRs, the PDGF/PDGFR pathway plays a critical role in the development of lung fibrosis. The antifibrotic effects of BIBF1000 – a compound similar to BIBF1120 – have been demonstrated in a BLM model in rats, and BIBF1000 weakly inhibited TGF-β activity and subsequent α-SMA expression in lung fibroblasts in vitro.2 However, the kinase inhibitory profile of nintedanib suggests that PDGFR, FGFR, and VEGFR play important roles in lung fibrosis. Interestingly, nintedanib had a similar IC50 for PDGFRs and VEGFRs, but PDGFRs seem to be more critical for the antifibrotic effects of nintedanib. In addition, Wollin et al reported sustained inhibition of PDGFRs in human lung fibroblasts by nintedanib, and they found that nintedanib inhibited PDGFR activity more effectively than imatinib (by 20–50 fold).55 Recently, the preventive and therapeutic effects of nintedanib were clearly demonstrated in a mouse model of pulmonary fibrosis induced by BLM and silica.2
TGF-β is thought to play a central role in lung fibrosis. However, Andrianifahanana et al showed that profibrotic TGF-β responses require the cooperative action of PDGF and ErbB receptor tyrosine kinases.56 Moreover, the action of TGF-β is partly mediated by the production of connective tissue growth factor (CTGF) and FGF-2.57,58 These reports demonstrate that the profibrotic effects of TGF-β are mediated by several growth factors, including PDGF, EGF, CTGF, and FGF-2, indicating that the role of TGF-β in lung fibrogenesis may have been overestimated in previous studies. The wide range of growth factors involved in pulmonary fibrosis that are inhibited by nintedanib is shown in Figure 2.
Figure 2.
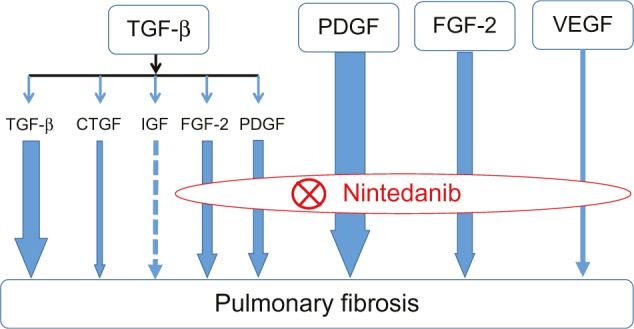
Mode of action of nintedanib in pulmonary fibrosis.
Notes: The profibrotic effects of TGF-β are mediated by several growth factors, including PDGF and FGF-2. Nintedanib improves pulmonary fibrosis by inhibiting the action of PDGF, FGF-2, and vascular endothelial growth factor, indicating indirect inhibitory effects on TGF-β signaling. The arrow size indicates the degree of contribution to pulmonary fibrosis suggested by the published data. The dotted line indicates that very little data was available.
Abbreviations: TGF, transforming growth factor; PDGF, platelet-derived growth factor; FGF, fibroblast growth factor; VEGF, vascular endothelial growth factor; CTGF, connective tissue growth factor.
Clinical trials
To evaluate the efficacy and safety of four different doses of nintedanib in patients with IPF, a randomized, double-blind, placebo-controlled, Phase II trial known as the TOMORROW trial was conducted.6 IPF patients (>40 years old) with FVC that was 50% or more of their predicted value and a diffusing capacity of the lung for carbon monoxide (DLCO) that was 30%–79% of their predicted value were recruited for the study. A total of 432 patients with IPF were randomly assigned to receive placebo or one of the four different nintedanib doses: 50 mg once per day, 50 mg twice per day, 100 mg twice per day, or 150 mg twice per day for 12 months. In the group that received 150 mg of nintedanib twice daily, the primary endpoint of the annual rate of decline in FVC was 60 mL, while that of the placebo group was 190 mL, corresponding to a reduction of 68.4% in the rate of FVC loss after nintedanib treatment. Despite the failure to achieve the primary endpoint, a number of clinically important and significant changes in secondary endpoints were found. Treatment with 150 mg nintedanib twice daily resulted in a significantly reduced incidence of acute exacerbations, which was an important result because acute exacerbation of IPF is associated with high morbidity and mortality, as well as a worsened prognosis.59 Although chronic therapy of IPF patients may reduce the risk of acute exacerbation,60 prevention of acute exacerbation is an important effect. Treatment with 150 mg nintedanib twice daily also resulted in a small decrease in the St George’s Respiratory Questionnaire score in comparison with placebo. Although there were no significant differences in the rate of death from any cause between any of the groups, non-significant trends for fewer deaths from respiratory causes were found in the groups receiving 100 mg and 150 mg nintedanib twice daily in comparison with the placebo group. The most frequent adverse event in the group receiving 150 mg nintedanib twice daily was diarrhea (55.3% vs 15.3% in the placebo group), followed by nausea (23.5%) and vomiting (12.9%). The adverse events most frequently leading to discontinuation were also diarrhea, nausea, and vomiting, and the proportion of patients who discontinued the study medication because of adverse events was 30.6% in the group receiving 150 mg nintedanib twice daily, compared with 25.9% in the placebo group. Clinically significant elevations in liver enzyme levels were observed in eight patients in the group receiving nintedanib; however, only two patients discontinued the study medication because of abnormal results in liver function tests.
The Phase II TOMORROW trial showed clinical benefits of nintedanib with an acceptable safety profile in IPF patients receiving 150 mg nintedanib twice daily; therefore, the Phase III INPULSIS™ trials were conducted to evaluate the efficacy and safety of 150 mg nintedanib twice daily in comparison with placebo.7 The INPULSIS™ trials were a series of two randomized, double-blind, 52-week, Phase III trials (INPUL-SIS-1 and INPULSIS-2). The eligibility criteria for IPF patients more than 40 years old were identical to those of the TOMORROW trial: FVC that was ≥50% of their predicted value and a DLCO that was 30%–79% of their predicted value. In the TOMORROW and INPULSIS trials, concomitant therapy with ≤15 mg of prednisone per day or the equivalent was permitted if the treatment dose had been stable for at least 8 weeks before screening. In the INPULSIS-1 and INPULSIS-2 trials, the annual rate of decline in FVC (the primary endpoint) in the nintedanib group was significantly lower than that of the placebo group: −114.7 mL per year in the nintedanib group in comparison with −239.9 mL per year in the placebo group for INPULSIS-1 and −113.6 mL per year in the nintedanib group in comparison with −207.3 mL per year in the placebo group for INPULSIS-2. A prespecified pooled analysis of data from the INPULSIS-1 and INPULSIS-2 trials showed that the proportion of patients with a decline in the percentage of predicted FVC that was not more than 5% or 10% in the nintedanib group was significantly greater than the corresponding proportion in the placebo group.
Although chronic treatment of IPF may reduce the risk of acute exacerbation,60 prevention of acute exacerbation is an important treatment effect. In the INPULSIS-2 trial and a prespecified sensitivity analysis of pooled data, the time to the first acute exacerbation (a secondary endpoint) was significantly increased in the nintedanib group in comparison with the placebo group, but not in the INPULSIS-1 trial. The difference between the results could have several causes. The diagnostic criteria of acute exacerbation were not standardized internationally, and different investigators and difference in the number of cases performed with chest computed tomography were responsible for the diagnosis of acute exacerbation. Furthermore, there was a difference in the incidence of acute exacerbation in placebo arms between the TOMORROW and the INPULSIS trials. The incidence of concomitant use of corticosteroids (CS) was different between TOMORROW (~49%) and INPULSIS (~20%) even though the eligibility criteria in both trials allowed concomitant use of <15 mg CS per day. The PANTHER trial reported the danger of CS in-between the two nintedanib trials, and this certainly influenced the different incidence.61 The incidence of concomitant use of CS might influence the difference of acute exacerbation in placebo arms because inappropriate dose reduction of CS and/or long-term use of CS could be a cause of acute exacerbation during management of IPF. There was no significant between-group difference in death in the prespecified pooled analysis of the INPULSIS-1 and INPULSIS-2 trials; however, a prespecified pooled analysis also revealed that patients treated with nintedanib had a numerical risk reduction in all-cause mortality by 30% in comparison with patients in the placebo group.
The most frequent adverse event in the nintedanib groups in the INPULSIS-1 and INPULSIS-2 trials was diarrhea, which was mostly mild or moderate. Treatment with nintedanib was associated with significant improvements in the primary endpoint and many secondary outcomes in the INPULSIS-1 and INPULSIS-2 trials, suggesting that this agent is a promising treatment for IPF. The lower incidence of acute exacerbation in the INPULSIS-2 trial may have contributed to the more stable quality of life observed in this trial. Moreover, this result indicates that treatment with nintedanib provided a benefit with respect to health-related quality of life.
To assess the impact of baseline FVC on the effect of nintedanib, a prespecified subgroup analysis of patients with baseline FVC >70% and ≤70% of the predicted value was conducted using pooled data from both INPULSIS trials.62 There was no difference in the effect of nintedanib between IPF patient subgroups defined by FVC% predicted at baseline. Treatment with pirfenidone was shown to have a better response in patients with IPF who have preserved lung function;63 however, this result indicated that nintedanib was effective in IPF patients despite severe lung function impairment at baseline. The incidence of acute exacerbation in patients with baseline FVC ≤70% of the predicted value was higher than that of patients with baseline FVC >70% of the predicted value. Moreover, in patients with baseline FVC ≤70% of the predicted value, the incidence of acute exacerbation in the nintedanib group was lower than that of the placebo group. The St George’s Respiratory Questionnaire score of the nintedanib group was also lower than that of the placebo group in patients with baseline FVC ≤70% of the predicted value. There are limitations to subgroup analysis, but the data indicate that nintedanib might even be effective in patients with moderate-to-severe lung function impairment.
In clinical trials in patients with IPF, change in FVC has been used as a primary endpoint because it is reproducible and provides a reasonable surrogate for long-term prognosis and survival.64,65 However, the choice of an endpoint in such studies remains controversial. Measurement of lung function (ie, FVC and DLCO) and exercise tolerance tends to be difficult in patients with greater lung function impairment, and the incidence of acute exacerbation could be low in patients with lesser lung function impairment.66,67 Therefore, in comparison with nontreated groups, it is suggested that measurement of lung function, a 6-minute walk test, and the GAP index (gender [G], age [A], and two lung physiology variables [P] [FVC and DLCO])68 should be assessed in mild-to-moderate IPF patients, while the incidence of acute exacerbation, overall survival, mortality, and health-related quality of life should be assessed in advanced IPF patients.
Mechanism and clinical evidence of pirfenidone
Pirfenidone is an oral antifibrotic therapeutic that has been evaluated for the treatment of IPF. Pirfenidone is a small molecule with combined anti-inflammatory, antioxidant, and antifibrotic effects in experimental models of pulmonary fibrosis.69–75 Pirfenidone suppresses BLM-induced infiltration of inflammatory cells into the BAL, and an effect on TGF-β and PDGF has been reported.76,77 In an experimental model, both pirfenidone and prednisolone suppressed lung inflammatory edema; however, pulmonary fibrosis was only significantly suppressed by pirfenidone. Pirfenidone prevented the BLM-induced decrease in lung interferon (IFN)-γ levels and suppressed elevation of lung bFGF and TGF-β1 levels, but prednisolone had no such effect. Pirfenidone exerts its antifibrotic effect through regulation of lung IFN-γ, bFGF, and TGF-β1 levels during the development of BLM-induced pulmonary fibrosis in mice.78 Nintedanib and pirfenidone have different mechanisms of action so combination therapy with these two drugs could be effective.
To date, several placebo-controlled randomized control trials have evaluated the efficacy of pirfenidone in IPF patients.63,79,80 A Japanese Phase III study demonstrated a significant effect on decline of FVC and progression-free survival.79 In the CAPACITY 004 and 006 studies, the primary end point was the change in the percentage of the predicted FVC from baseline to week 72, and this was met in study 004 but not in study 006; an additional trial to support the approval of pirfenidone was then requested.80 In patients with IPF in the Phase III ASCEND trial, treatment with pirfenidone significantly reduced the change in the percentage of the predicted FVC (the primary endpoint) from baseline in comparison with placebo.81 The mean decline from baseline in FVC was significantly reduced in the pirfenidone group (235 mL) compared with the placebo group (428 mL). Pirfenidone treatment also reduced the decline in distances on the 6-minute walk test. In a prespecified mortality analysis combining the 1-year results from both the ASCEND trial and the previously reported CAPACITY trials, there was evidence that pirfenidone reduced mortality in IPF patients.81
Concomitance with pirfenidone
Although treatment with nintedanib and pirfenidone reduced the rate of decline in FVC, the mean annual declines in FVC in the pirfenidone and placebo groups were −235 mL and −428 mL, respectively, in the ASCEND trial, while the mean annual declines in FVC in the nintedanib and placebo groups in the INPULSIS trials were −95 mL and −205 mL, respectively. The inclusion criteria in the ASCEND and INPULSIS trials were different, and this prohibits a simple comparison of these trials. However, these differences in the results of the ASCEND and INPULSIS trials could be due to the differences in the populations of patients with normal FVC values. Only the INPULSIS trial included patients with ≥90% of the predicted FVC value meaning that the IPF patients in the INPULSIS trial had a milder disease than the patients in the ASCEND trial. With respect to adverse events, there is some overlap in the adverse event profiles of pirfenidone and nintedanib in patients with IPF in terms of gastrointestinal events and increases in liver enzymes.6,7,80,82
Treatment with either nintedanib or pirfenidone significantly reduced the decline in FVC (primary endpoint); therefore, potential differences in secondary end points are discussed. Nintedanib improved the time to first acute exacerbation and health-related quality of life in INPULSIS-2; however, pirfenidone significantly slowed the reduction in the average distance walked in 6 minutes and improved progression-free survival. In a prespecified analysis combining results from the ASCEND and CAPACITY trials, all-cause death at 1 year was significantly reduced from 6.7% in the placebo group to 3.5% in the pirfenidone group. These findings suggest that patients with IPF should be treated with nintedanib and pirfenidone; however, there has been little evidence of the efficacy of combination therapy with these drugs or superiority of one drug over the other. Until further data are available, clinicians should consider cost, local availability, individual tolerance, and risk of disbenefit when deciding on a single drug with which to treat an IPF patient.83
A randomized, double-blind, Phase II, dose-escalation trial was conducted to assess the safety, tolerability, and pharmacokinetics of nintedanib in Japanese patients with IPF, both alone and when added to ongoing pirfenidone therapy.84 The results of this study show that, even when introduced to patients undergoing ongoing pirfenidone therapy, treatment with 150 mg nintedanib twice daily had an acceptable safety and tolerability profile in Japanese patients with IPF, and all adverse events reported in this trial were mild or moderate in intensity. Coadministration with nintedanib had no effect on the pharmacokinetics of pirfenidone; however, there was a trend toward reduced exposure to nintedanib when it was administered in combination with pirfenidone. Pirfenidone is metabolized by various cytochrome P450 enzymes, and the metabolism of nintedanib is predominantly characterized by ester cleavage yielding BIBF1202 ZW.84,85 Ogura et al reported that there are differences in the metabolic pathways of nintedanib and pirfenidone; therefore, the lower bioavailability of nintedanib coadministered with pirfenidone may reflect reduced absorption.84
There is little evidence regarding whether nintedanib and pirfenidone are beneficial in patients with more advanced IPF who were excluded from the pivotal trials; the drugs show long-term efficacy, and the drugs were effective in patients with pulmonary fibrosis associated with the presence of connective tissue disease or any other cause. Nintedanib and pirfenidone are the primary treatments for IPF, and future studies should assess the efficacy of combination therapy as well as identify additional effects of these drugs.
Conclusion
The pathogenesis of IPF is complex and involves the interaction of multiple signaling pathways, including PDGF, FGF, and VEGF. Therefore, treatment of patients with IPF requires targeting of a number of redundant pathways. Nintedanib has recently been shown to slow the progression of pulmonary fibrosis and to improve clinical outcomes. Questions remain regarding clinical treatment with nintedanib, and further evaluation of this agent is needed for better management of patients with IPF.
Footnotes
Disclosure
Arata Azuma was a Steering Committee Member and Advisory Board Member of Nintedanib in clinical trials for IPF in Boehringer Ingelheim. Minoru Inomata and Yasuhiko Nishioka report no conflicts of interest in this work.
References
- 1.Selman M, King TE, Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001;134:136–151. doi: 10.7326/0003-4819-134-2-200101160-00015. [DOI] [PubMed] [Google Scholar]
- 2.Chaudhary NI, Roth GJ, Hilberg F, et al. Inhibition of PDGF, VEGF and FGF signalling attenuates fibrosis. Eur Respir J. 2007;29:976–985. doi: 10.1183/09031936.00152106. [DOI] [PubMed] [Google Scholar]
- 3.Coward WR, Saini G, Jenkins G. The pathogenesis of idiopathic pulmonary fibrosis. Ther Adv Respir Dis. 2010;4:367–388. doi: 10.1177/1753465810379801. [DOI] [PubMed] [Google Scholar]
- 4.Wollin L, Maillet I, Quesniaux V, Holweg A, Ryffel B. Anti-fibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis. J Pharmacol Exp Ther. 2014;349:209–220. doi: 10.1124/jpet.113.208223. [DOI] [PubMed] [Google Scholar]
- 5.Hilberg F, Roth GJ, Krssak M, et al. BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008;68:4774–4782. doi: 10.1158/0008-5472.CAN-07-6307. [DOI] [PubMed] [Google Scholar]
- 6.Richeldi L, Costabel U, Selman M, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011;365(12):1079–1087. doi: 10.1056/NEJMoa1103690. [DOI] [PubMed] [Google Scholar]
- 7.Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071–2082. doi: 10.1056/NEJMoa1402584. [DOI] [PubMed] [Google Scholar]
- 8.National Clinical Guideline Centre (UK) Diagnosis and Management of Suspected Idiopathic Pulmonary Fibrosis: Idiopathic Pulmonary Fibrosis [Internet] London: Royal College of Physicians (UK); 2013. [PubMed] [Google Scholar]
- 9.Landells LJ, Naidoo B, Robertson J, Clark P. NICE guidance on pirfenidone for treating idiopathic pulmonary fibrosis. Lancet Respir Med. 2013;1(3):191–192. doi: 10.1016/S2213-2600(13)70065-1. [DOI] [PubMed] [Google Scholar]
- 10.Xaubet A, Behr J, Bendstrup E, et al. Review of IPF diagnosis and management recommendations in Europe. Sarcoidosis Vasc Diffuse Lung Dis. 2013;30(4):249–261. [PubMed] [Google Scholar]
- 11.Günther A, Korfei M, Mahavadi P, von der Beck D, Ruppert C, Markart P. Unravelling the progressive pathophysiology of idiopathic pulmonary fibrosis. Eur Respir Rev. 2012;21:152–160. doi: 10.1183/09059180.00001012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Noble PW, Barkauskas CE, Jiang D. Pulmonary fibrosis: patterns and perpetrators. J Clin Invest. 2012;122:2756–2762. doi: 10.1172/JCI60323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herdin CH, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev. 1999;79:1283–1316. doi: 10.1152/physrev.1999.79.4.1283. [DOI] [PubMed] [Google Scholar]
- 14.Li X, Eriksson U. Novel PDGF family members. PDGF-C and PDGF-D. Cytokine Growth Factor Rev. 2003;14:91–98. doi: 10.1016/s1359-6101(02)00090-4. [DOI] [PubMed] [Google Scholar]
- 15.Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008;22:1276–1312. doi: 10.1101/gad.1653708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boström H, Willetts K, Pekny M, et al. PDGF-A signaling is a critical event in lung alveolar myofibroblast development and alveogenesis. Cell. 1996;14:863–873. doi: 10.1016/s0092-8674(00)81270-2. [DOI] [PubMed] [Google Scholar]
- 17.Soriano P. The PDGFa receptor is required for neural crest cell development and for normal patterning of the somites. Development. 1997;124:2691–2700. doi: 10.1242/dev.124.14.2691. [DOI] [PubMed] [Google Scholar]
- 18.Levéen P, Pekny M, Gebre-Medhin S, Swolin B, Larsson E, Betsholtz C. Mice deficient for PDGF B show renal, cardiovascular, and hematological abnormalities. Genes Dev. 1994;8:1875–1887. doi: 10.1101/gad.8.16.1875. [DOI] [PubMed] [Google Scholar]
- 19.Soriano P. Abnormal kidney development and hematological disorders in PDGF beta-receptor mutant mice. Genes Dev. 1996;8:1888–1896. doi: 10.1101/gad.8.16.1888. [DOI] [PubMed] [Google Scholar]
- 20.McGowan SE, Grossmann RE, Kimani PW, Holmes AJ. Platelet-derived growth factor receptor-alpha-expressing cells localize to the alveolar entry ring and have characteristics of myofibroblasts during pulmonary alveolar septal formation. Anat Rec (Hoboken) 2008;291:1649–1661. doi: 10.1002/ar.20764. [DOI] [PubMed] [Google Scholar]
- 21.Maeda A, Hiyama K, Yamakido H, Ishioka S, Yamakido M. Increased expression of platelet-derived growth factor A and insulin-like growth factor-I in BAL cells during the development of bleomycin-induced pulmonary fibrosis in mice. Chest. 1996;109:780–786. doi: 10.1378/chest.109.3.780. [DOI] [PubMed] [Google Scholar]
- 22.Walsh J, Absher M, Kelley J. Variable expression of platelet-derived growth factor family proteins in acute lung injury. Am J Respir Cell Mol Biol. 1993;9:637–644. doi: 10.1165/ajrcmb/9.6.637. [DOI] [PubMed] [Google Scholar]
- 23.Zhuo Y, Zhang J, Laboy M, Lasky JA. Modulation of PDGF-C and PDGF-D expression during bleomycin-induced lung fibrosis. Am J Physiol Lung Cell Mol Physiol. 2004;286:L182–L188. doi: 10.1152/ajplung.00083.2003. [DOI] [PubMed] [Google Scholar]
- 24.Shimizu S, Gabazza EC, Taguchi O, et al. Activated protein C inhibits the expression of platelet-derived growth factor in the lung. Am J Respir Crit Care Med. 2003;167:1416–1426. doi: 10.1164/rccm.200206-515OC. [DOI] [PubMed] [Google Scholar]
- 25.Yoshida M, Sakuma J, Hayashi S, et al. A histologically distinctive interstitial pneumonia induced by overexpression of the interleukin 6, transforming growth factor beta 1, or platelet-derived growth factor B gene. Proc Natl Acad Sci U S A. 1995;92:9570–9574. doi: 10.1073/pnas.92.21.9570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Antoniades HN, Bravo MA, Avila RE, et al. Platelet-derived growth factor in idiopathic pulmonary fibrosis. J Clin Invest. 1990;86:1055–1064. doi: 10.1172/JCI114808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Homma S, Nagaoka I, Abe H, et al. Localization of platelet-derived growth factor and insulin-like growth factor I in the fibrotic lung. Am J Respir Crit Care Med. 1995;152:2084–2089. doi: 10.1164/ajrccm.152.6.8520779. [DOI] [PubMed] [Google Scholar]
- 28.Gochuico BR, Avila NA, Chow CK, et al. Progressive preclinical interstitial lung disease in rheumatoid arthritis. Arch Intern Med. 2008;168:159–166. doi: 10.1001/archinternmed.2007.59. [DOI] [PubMed] [Google Scholar]
- 29.Rice AB, Moomaw CR, Morgan DL, Bonner JC. Specific inhibitors of platelet-derived growth factor or epidermal growth factor receptor tyrosine kinase reduce pulmonary fibrosis in rats. Am J Pathol. 1999;55:213–221. doi: 10.1016/S0002-9440(10)65115-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Druker BJ, Tamura S, Buchdunger E, et al. Effects of s selective inhibitor of the abl tyrosine kinase on the growth of bcr-abl positive cells. Nat Med. 1996;2:561–566. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 31.Aono Y, Nishioka Y, Inayama M, et al. Imatinib as a novel antifibrotic agent in bleomycin-induced pulmonary fibrosis in mice. Am J Respir Crit Care Med. 2005;171:1279–1285. doi: 10.1164/rccm.200404-531OC. [DOI] [PubMed] [Google Scholar]
- 32.Daniels CE, Wilkes MC, Edens M, et al. Imatinib mesylate inhibits the profibrogenic activity of TGF-β and prevents bleomycin-mediated lung fibrosis. J Clin Invest. 2004;114:1308–1316. doi: 10.1172/JCI19603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abdollahi A, Li M, Ping G, et al. Inhibition of platelet-derived growth factor signaling attenuates pulmonary fibrosis. J Exp Med. 2005;201:925–935. doi: 10.1084/jem.20041393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoshida M, Sakuma-Mochizuki J, Abe K, et al. In vivo gene transfer of an extracellular domain of platelet-derived growth factor beta receptor by the HVJ-liposome method ameliorates bleomycin-induced pulmonary fibrosis. Biochem Biophys Res Commun. 1999;265:503–508. doi: 10.1006/bbrc.1999.1647. [DOI] [PubMed] [Google Scholar]
- 35.Rhee CK, Lee SH, Yoon HK, et al. Effect of nilotinib on bleomycin-induced acute lung injury and pulmonary fibrosis in mice. Respiration. 2011;82:273–287. doi: 10.1159/000327719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nishioka Y, Azuma M, Kishi M, Aono Y. Targeting platelet-derived growth factor as a therapeutic approach in pulmonary fibrosis. J Med Invest. 2013;60(3,4):175–183. doi: 10.2152/jmi.60.175. [DOI] [PubMed] [Google Scholar]
- 37.Turner N, Grose R. Fibroblast growth factor signaling: from development to cancer. Nat Rev. 2010;10:116–120. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- 38.Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov. 2009;8:235–253. doi: 10.1038/nrd2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Powers CJ, McLeskey SW, Wellstein A. Fibroblast growth factors, their receptors and signaling. Endocr Relat Cancer. 2000;7:165–197. doi: 10.1677/erc.0.0070165. [DOI] [PubMed] [Google Scholar]
- 40.Hetzel M, Bachem M, Anders D, Trischler G, Faehling M. Different effects of growth factors on proliferation and matrix production of normal and fibrotic human lung fibroblasts. Lung. 2005;183:225–237. doi: 10.1007/s00408-004-2534-z. [DOI] [PubMed] [Google Scholar]
- 41.Kanazawa S, Fujiwara T, Matsuzaki S, et al. bFGF regulates PI3-kinase-Rac1-JNK pathway and promotes fibroblast migration in wound healing. PLoS One. 2010;5:e12228. doi: 10.1371/journal.pone.0012228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guzy RD, Stoilov I, Elton TJ, Mecham RP, Ornitz DM. Fibroblast growth factor 2 is required for epithelial recovery, but not for pulmonary fibrosis, in response to bleomycin. Am J Respir Cell Mol Biol. 2015;52:116–128. doi: 10.1165/rcmb.2014-0184OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ju W, Zhihong Y, Zhiyou Z, et al. Inhibition of α-SMA by the ectodomain of FGFR2c attenuates lung fibrosis. Mol Med. 2012;18:992–1002. doi: 10.2119/molmed.2011.00425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ramos C, Becerril C, Montaño M, et al. FGF-1 reverts epithelial-mesenchymal transition induced by TGF-b1 through MAPK/ERK kinase pathway. Am J Physiol Lung Cell Mol Physiol. 2010;299:L222–L231. doi: 10.1152/ajplung.00070.2010. [DOI] [PubMed] [Google Scholar]
- 45.Ramos C, Montaño M, Becerril C, et al. Acidic fibroblast growth factor decreases a-smooth muscle actin expression and induces apoptosis in human normal lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2006;291:L871–L879. doi: 10.1152/ajplung.00019.2006. [DOI] [PubMed] [Google Scholar]
- 46.Becerril C, Pardo A, Montaño M, Ramos C, Ramírez R, Selman M. Acidic fibroblast growth factor induces an antifibrogenic phenotype in human lung fibroblasts. Am J Respir Cell Mol Biol. 1999;20:1020–1027. doi: 10.1165/ajrcmb.20.5.3288. [DOI] [PubMed] [Google Scholar]
- 47.Shibuya M. Vascular endothelial growth factor receptor-1 (VEGFR-1/Flt-1): a dual regulator for angiogenesis. Angiogenesis. 2006;9:225–230. doi: 10.1007/s10456-006-9055-8. [DOI] [PubMed] [Google Scholar]
- 48.Nowak DG, Amin EM, Rennel ES, et al. Regulation of vascular endothelial growth factor (VEGF) splicing from pro-angiogenic to anti-angiogenic isoforms. a novel therapeutic strategy for angiogenesis. J Biol Chem. 2010;285(8):5532–5540. doi: 10.1074/jbc.M109.074930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ebina M, Shimizukawa M, Shibata N, et al. Heterogeneous increase in CD34-positive alveolar capillaries in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2004;169(11):1203–1208. doi: 10.1164/rccm.200308-1111OC. [DOI] [PubMed] [Google Scholar]
- 50.Antoniou KM, Soufla G, Proklou A, et al. Different activity of the biological axis VEGF-Flt-1 (fms-like tyrosine kinase 1) and CXC chemokines between pulmonary sarcoidosis and idiopathic pulmonary fibrosis: a bronchoalveolar lavage study. Clin Dev Immunol. 2009;2009:537929. doi: 10.1155/2009/537929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Farkas L, Farkas D, Ask K, et al. VEGF ameliorates pulmonary hypertension through inhibition of endothelial apoptosis in experimental lung fibrosis in rats. J Clin Invest. 2009;119:1298–1311. doi: 10.1172/JCI36136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hamada N, Kuwano K, Yamada M, et al. Anti-vascular endothelial growth factor gene therapy attenuates lung injury and fibrosis in mice. J Immunol. 2005;175:1224–1231. doi: 10.4049/jimmunol.175.2.1224. [DOI] [PubMed] [Google Scholar]
- 53.Ou XM, Li WC, Liu DS, et al. VEGFR-2 antagonist SU5416 attenuates bleomycin-induced pulmonary fibrosis. Int Immunopharmacol. 2009;9:70–79. doi: 10.1016/j.intimp.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 54.Roth GJ, Heckel A, Colbatzky F, et al. Design, synthesis, and evaluation of indolinones as triple angiokinase inhibitors and the discovery of a highly specific 6-methoxycarbonyl-substituted indolinone (BIBF 1120) J Med Chem. 2009;52:4466–4480. doi: 10.1021/jm900431g. [DOI] [PubMed] [Google Scholar]
- 55.Wollin L, Neugebauer J, Ostermann A, et al. Sustained inactivation of human lung fibroblasts by nintedanib. Am J Respir Crit Care Med. 2013;188:A3378. [Google Scholar]
- 56.Andrianifahanana M, Wilkes MC, Gupta SK, et al. Profibrotic TGFb responses require the cooperative action of PDGF and ErbB receptor tyrosine kinases. FASEB J. 2013;27:4444–4454. doi: 10.1096/fj.12-224907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li CM, Khosla J, Pagan I, Hoyle P, Sannes PL. TGF-β1 and fibroblast growth factor-1 modify fibroblast growth factor-2 production in type II cells. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1038–L1046. doi: 10.1152/ajplung.2000.279.6.L1038. [DOI] [PubMed] [Google Scholar]
- 58.Kucich U, Rosenbloom JC, Herrick DJ, et al. Signaling events required for transforming growth factor-beta stimulation of connective tissue growth factor expression by cultured human lung fibroblasts. Arch Biochem Biophys. 2001;395(1):103–112. doi: 10.1006/abbi.2001.2571. [DOI] [PubMed] [Google Scholar]
- 59.Collard HR, Moore BB, Flaherty KR, et al. Idiopathic Pulmonary Fibrosis Clinical Research Network Investigators Acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2007;176(7):636–643. doi: 10.1164/rccm.200703-463PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim DS. Acute exacerbations in patients with idiopathic pulmonary fibrosis. Respir Res. 2013;14:86. doi: 10.1186/1465-9921-14-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Idiopathic Pulmonary Fibrosis Clinical Research Network. Raghu G, Anstrom KJ, King TE, Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. 2012;366(21):1968–1977. doi: 10.1056/NEJMoa1113354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Costabel U, Inoue Y, Richeldi L, et al. Effect of baseline FVC on decline in lung function with nintedanib: results from the INPULSIS™ trials. Eur Respir J. 2014;44(Suppl 58):1907. [Google Scholar]
- 63.Azuma A, Nukiwa T, Tsuboi E, et al. Double-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2005;171:1040–1047. doi: 10.1164/rccm.200404-571OC. [DOI] [PubMed] [Google Scholar]
- 64.Vancheri C, Failla M, Crimi N, Raghu G. Idiopathic pulmonary fibrosis: a disease with similarities and links to cancer biology. Eur Respir J. 2010;35(3):496–504. doi: 10.1183/09031936.00077309. [DOI] [PubMed] [Google Scholar]
- 65.du Bois RM, Nathan SD, Richeldi L, Schwarz MI, Noble PW. Idiopathic pulmonary fibrosis: lung function is a clinically meaningful endpoint for phase III trials. Am J Respir Crit Care Med. 2012;186(8):712–715. doi: 10.1164/rccm.201206-1010PP. [DOI] [PubMed] [Google Scholar]
- 66.Collard HR, Yow E, Richeldi L, Anstrom KJ, Glazer C, IPFnet investigators Suspected acute exacerbation of idiopathic pulmonary fibrosis as an outcome measure in clinical trials. Respir Res. 2013;14:73. doi: 10.1186/1465-9921-14-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Song JW, Hong SB, Lim CM, Koh Y, Kim DS. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J. 2011;37:356–363. doi: 10.1183/09031936.00159709. [DOI] [PubMed] [Google Scholar]
- 68.Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med. 2012;156:684–691. doi: 10.7326/0003-4819-156-10-201205150-00004. [DOI] [PubMed] [Google Scholar]
- 69.Margolin S, Margolin B, Margolin D. Removal of interstitial pulmonary fibrosis (asbestos-induced) by oral chemotherapy with pirfenidone. Fed Proc. 1982;41:1550. [Google Scholar]
- 70.Margolin SB, Lefkowitz S. Pirfenidone: a novel pharmacologic agent for prevention and resolution of lung fibrosis. FASEB J. 1994;8:A382. [Google Scholar]
- 71.Nakazato H, Oku H, Yamane S, Tsuruta Y, Suzuki R. A novel anti-fibrotic agent pirfenidone suppresses tumor necrosis factor-α at the translational level. Eur J Pharmacol. 2002;446:177–185. doi: 10.1016/s0014-2999(02)01758-2. [DOI] [PubMed] [Google Scholar]
- 72.Oku H, Nakazato H, Horikawa T, Tsuruta Y, Suzuki R. Pirfenidone suppresses tumor necrosis factor-α, enhances interleukin-10 and protects mice from endotoxic shock. Eur J Pharmacol. 2002;446:167–176. doi: 10.1016/s0014-2999(02)01757-0. [DOI] [PubMed] [Google Scholar]
- 73.Iyer SN, Gurujeyalakshmi G, Giri SN. Effects of pirfenidone on procollagen gene expression at the transcriptional level in bleomycin hamster model of lung fibrosis. J Pharmacol Exp Ther. 1999;289:211–218. [PubMed] [Google Scholar]
- 74.Misra HP, Rabideau C. Pirfenidone inhibits NADPH-dependent microsomal lipid peroxidation and scavenges hydroxyl radicals. Mol Cell Biochem. 2000;204:119–126. doi: 10.1023/a:1007023532508. [DOI] [PubMed] [Google Scholar]
- 75.Iyer SN, Wild JS, Schiedt MJ, Hyde DM, Margolin SB, Giri SN. Dietary intake of pirfenidone ameliorates bleomycin-induced lung fibrosis in hamsters. J Lab Clin Med. 1995;125:779–785. [PubMed] [Google Scholar]
- 76.Iyer SN, Gurujeyalakshmi G, Giri SN. Effects of pirfenidone on transforming growth factor-beta gene expression at the transcriptional level in bleomycin hamster model of lung fibrosis. J Pharmacol Exp Ther. 1999;291(1):367–373. [PubMed] [Google Scholar]
- 77.Gurujeyalakshmi G, Hollinger MA, Giri SN. Pirfenidone inhibits PDGF isoforms in bleomycin hamster model of lung fibrosis at the translational level. Am J Physiol. 1999;276(2 pt 1):L311–L318. doi: 10.1152/ajplung.1999.276.2.L311. [DOI] [PubMed] [Google Scholar]
- 78.Oku H, Shimizu T, Kawabata T, et al. Antifibrotic action of pirfenidone and prednisolone: different effects on pulmonary cytokines and growth factors in bleomycin-induced murine pulmonary fibrosis. Eur J Phar-macol. 2008;590(1–3):400–408. doi: 10.1016/j.ejphar.2008.06.046. [DOI] [PubMed] [Google Scholar]
- 79.Taniguchi H, Ebina M, Kondoh Y, et al. Pirfenidone Clinical Study Group in Japan Pirfenidone Clinical Study Group in Japan. Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J. 2010;35(4):821–829. doi: 10.1183/09031936.00005209. [DOI] [PubMed] [Google Scholar]
- 80.Noble PW, Albera C, Bradford WZ, et al. CAPACITY Study Group Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 2011;377:1760–1769. doi: 10.1016/S0140-6736(11)60405-4. [DOI] [PubMed] [Google Scholar]
- 81.King TE, Jr, Bradford WZ, Castro-Bernardini S, et al. ASCEND Study Group ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083–2092. doi: 10.1056/NEJMoa1402582. [DOI] [PubMed] [Google Scholar]
- 82.Hunninghake GM. A new hope for idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2142–2143. doi: 10.1056/NEJMe1403448. [DOI] [PubMed] [Google Scholar]
- 83.Lake F. Pirfenidone reduced disease progression in idiopathic pulmonary fibrosis. Ann Intern Med. 2014;161:JC4–JC5. doi: 10.7326/0003-4819-161-8-201410210-02004. [DOI] [PubMed] [Google Scholar]
- 84.Ogura T, Taniguchi H, Azuma A, et al. Safety and pharmacokinetics of nintedanib and pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J. 2015;45(5):1382–1392. doi: 10.1183/09031936.00198013. [DOI] [PubMed] [Google Scholar]
- 85.European Medicines Agency CHMP Assessment Report: Esbriet; 2013 [cited December 16, 2010] [Accessed September 4, 2013]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002154/WC500103073.pdf.