

Abstract
Background
The 90-kDa heat-shock protein (HSP90) is an abundant cytosolic chaperone and inhibition of HSP90 by 17-allylamino-17-demethoxygeldanamycin (17-AAG) compromises Transforming growth factor (TGF)-β-mediated transcriptional responses by enhancing TGF-β receptor I and II degradation, thus preventing Smad 2/3 activation. Here, we evaluated whether HSP90 regulates TGF-β signaling in the pathogenesis and treatment of keloids.
Methods
Keloid fibroblasts were treated with 17-AAG (10 μM), and mRNA levels of collagen type I and III were determined by real-time RT-PCR. Also, secreted TGF-β1 was assessed by Enzyme-linked immunosorbent assay (ELISA). The effect of 17-AAG on protein levels of Smad 2/3 complex was determined by Western blot. Additionally, in 17-AAG-treated keloid spheroids, the collagen deposition and expression of major extracellular matrix proteins were investigated by Masson's trichrome staining and immunohistochemistry.
Results
We found that HSP90 is overexpressed in human keloid tissue compared to adjacent normal tissue, and 17-AAG decreased mRNA levels of type I collagen, secreted TGF-β1, Smad 2/3 complex protein expression in keloid fibroblasts. Masson's trichrome staining revealed that collagen deposition was decreased in 17-AAG-treated keloid spheroids, and immunohistochemical analysis showed that expression of collagen I and III, elastin, and fibronectin were markedly decreased in 17-AAG-treated keloid spheroids.
Conclusion
These results suggest that the antifibrotic action of HSP90 inhibitors such as 17-AAG may have therapeutic effects on keloids.
INTRODUCTION
A Keloid is the result of abnormal fibroblast proliferations of the dermal layer of the skin, resulting in excessive deposition of extracellular matrix (ECM) components. Although many strategies are available for keloid scars, none are completely effective1–3. Abnormal fibroblasts also cause keloids and organ fibrosis. During abnormal dermal fibrosis, activated fibroblasts acquire a myofibroblast-like phenotype characterized by increased proliferation and excessive ECM synthesis4,5. Therefore, suppression of keloid fibroblasts (KFs) proliferation and activation has been proposed as a therapeutic strategy for the treatment and prevention of keloids. Transforming growth factor (TGF)-β is a key regulatory growth factor of ECM assembly and remodeling, and TGF-β/Smad signaling plays a central role in keloid pathogenesis1,6,7. Therefore, modulation of TGF-β synthesis or activity represents a potential approach to treat hypertrophic scar and keloids.
The 90-kDa heat-shock protein (HSP90) is an abundant cytosolic protein, which is induced in response to a wide variety of physiological and environmental stress8 and is involved in intracellular signaling pathways that promote cell proliferation and/or cell survival. HSP90 facilitates protein folding and stabilization, and HSP90 forms complexes with many client proteins, which are important for cell growth, survival, and differentiation8–11. The small-molecule 17-allylamino-17-demethoxygeldanamycin (17-AAG) is a geldanamycin analog that specifically inhibits the ATPase activity of HSP909,10,12. Inhibition of HSP90 alters TGF-β–dependent transcriptional responses by increasing TGF-β receptor ubiquitination and degradation in a Smurf2 ubiquitin E3 ligase-dependent manner, thus preventing Smad 2/3 activation13–15. TGF- β receptor I and II directly interact with HSP90 and are clients of this cellular chaperone13. However, the clinical significance of HSP90 inhibitors such as 17-AAG in disease characterized by aberrant TGF- β responses (e.g., keloid and hypertrophic scarring) remains unclear.
Here, we hypothesized whether HSP90 regulate TGF-β signaling in the pathogenesis and in the treatment of keloids, and investigated the expression of HSP90 in keloid tissue and normal tissue by immunohistochemistry (IHC). Based on this findings, we treated HSP90 inhibitor like 17-AAG on KFs to examine the therapeutic potential of 17-AAG for treating keloid and hypertrophic scar. Additionally, the expression levels of ECM such as type I and III collagen, fibronectin, and elastin were investigated by IHC in keloid spheroids16 treated with 17-AAG.
MATERIALS AND METHOD
Human dermal fibroblast and keloid-derived fibroblast cells
Human dermal fibroblasts (HDFs) and keloid fibroblasts (KFs) were obtained from the American Type Culture Collection (Manassas, VA). Cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; GIBCO, Grand Island, NY) supplemented with 10% heat-inactivated fetal bovine serum (FBS), penicillin (100 UmL−1), streptomycin (100 μgmL−1). The culture medium was changed every 2 to 3 days. KFs cells was treated with 17-allylamino-17-demethoxygeldanamycin (17-AAG) for 48 h, incubated in CO2 incubator at 37 °C.
Real time RT-PCR analysis of collagen I and III expression
HDFs and KFs (5×105 cells) were treated for 48 h with 10 μM 17-AAG (Sigma, Saint Louis, Mo). After 2 days, the cells were harvested, and total RNA was prepared with TRIzol® reagent (Gibco BRL, Grand Island, NY). Complementary DNA was prepared from 0.5 μg total RNA by random priming using a first-strand cDNA synthesis kit (Promega Corp., Madison, WI). The following amplification conditions were used: 95°C for 5 min, 37°C for 2 h, and 75°C for 15 min. Taqman® primer/probe kits [assay ID: Hs00164004_m1 (collagen type I) and Hs00164103_m1 (collagen type III)] were used to analyze mRNA levels with an ABI Prism® 7500 HT Sequence Detection System (primer kits and instrument from Applied Biosystems, Foster City, CA). The mRNA levels of target genes were compared with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as an internal control (assay ID: Hs99999905_m1, Applied Biosystems). For cDNA amplification, AmpliTaqGold® DNA polymerase (Applied Biosystems) was activated by 10-min incubation at 95°C; this was followed by 40 cycles of 15 sec at 95°C and 1 min at 60°C for each cycle. To determine cDNA levels, the threshold cycle, at which fluorescence was first detected above baseline, was determined, and a standard curve was drawn between starting nucleic acid concentrations and the threshold cycle. Target mRNA expression levels were normalized to GAPDH levels, and relative quantization was expressed as fold-induction compared with control conditions in each cell type.
Enzyme-linked immunosorbent assay (ELISA) for secreted TGF-β1 expression
KFs (2×105 cells) in 6-cm culture dishes were treated with 10 μM 17-AAG. At 2 days post-infection, supernatants were collected by centrifugation at 15,000×g for 10 min at 4°C, and secreted TGF-β1 protein was assessed using an ELISA kit (R&D Systems, Minneapolis, MN).
Western blotting analysis for Smad 2/3 complex
Keloid fibroblasts were grown to 70% confluence in 100×20 mm cell culture dishes. Cultured keloid fibroblasts were exposed to 17-AAG (5 μM) for 48 h. Cells were lysed in 50 mM Tris-HCl (pH 7.6), 1% Nonidet P-40 (NP-40), 150 mM NaCl, and 0.1 mM zinc acetate in the presence of protease inhibitors. Protein concentration was determined by the Lowry method (Bio-Rad, Hercules, CA), and 20 µg of each sample was separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The gels were then electrophoretically transferred onto a PVDF membrane (Millipore, Billerica, MA). The membrane was blocked with blocking buffer for 1 h and then incubated overnight at 4°C with primary antibodies against Smad 2/3 complex (Cell Signaling Technology, Beverly, MA) and actin (mouse monoclonal; Sigma). After a 2 h incubation at room temperature with the secondary antibodies horseradish peroxidase (HRP)-conjugated rabbit antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) and HRP-conjugated mouse antibody (Santa Cruz Biotechnology), the protein bands were visualized using electrochemiluminescence detection reagents (Amersham Pharmacia Biotech Inc., Piscataway, NJ) according to the manufacturer’s instructions. Protein expression was analyzed using Image J software (National Institutes of Health).
keloid spheroid preparation and 17-AAG treatment
Keloid tissues were obtained from active-stage keloid patients (n=4), after obtaining informed consent. All experiments involving humans were performed in adherence to the Declaration of Helsinki tenets, and the study protocol was approved by the Yonsei University College of Medicine institutional review board. Keloid spheroids were prepared by dissecting tissue from the central of the keloids into 2-mm diameter pieces with sterile 21-gauge needles. The explants were plated individually in HydroCell® 12-well plates (designed to prevent cell attachment; Nunc, Rochester, NY) and cultured in DMEM supplemented with 10% FBS. The keloid spheroids treated with 17-AAG (10 μM) for 3 days and then fixed with 10% formalin, paraffin-embedded, and cut into 5-μm thick sections.
Immunohistochemistry (IHC)
Formaldehyde-fixed tissues were transferred to a paraffin-embedded block, sectioned at 4-μm thicknesses. After tissue deparaffinization and rehydration, endogenous peroxidase activity was blocked by a 10-min incubation at room temperature with absolute methanol containing 1% hydrogen peroxide. The tissue sections were incubated with a primary antibody against HSP90 (1:250, Rabbit polyclonal, Abcam Inc., Cambridge, MA) at 4°C overnight. After incubation with the secondary antibody (Super Sensitive™ Polymer-HRP IHC, BioGenex) for 1 h at room temperature, the bound complexes were visualized by incubating tissue sections with 0.05% diaminobenzidine and 0.003% hydrogen peroxide. The sections were counterstained with Harris hematoxylin and then dehydrated and mounted. HSP90 protein levels were semiquantitatively analyzed using MetaMorph® image analysis software (Universal Image Corp., Buckinghamshire, UK). Results are expressed as mean optical density of six different digital images per sample.
To evaluate expression of ECM proteins, keloid spheroid sections were incubated at 4°C overnight with the following primary antibodies: mouse, anti-collagen type I (ab6308; Abcam, Ltd., Cambridge, UK), mouse anti-collagen type III (C7805; Sigma), mouse anti-elastin (E4013; Sigma), or mouse anti-fibronectin (sc-52331; Santa Cruz Biotechnology). The sections were incubated at room temperature for 20 min with the secondary antibody (EnVision™ Kit; Dako, Glostrup, Denmark). The intensity of fibrosis was examined using Masson’s trichrome staining, and the expression of type I and III collagen, elastin, and fibronectin were semiquantitatively analyzed using MetaMorph® image analysis software (Universal Image Corp., Buckinghamshire, UK). Results are expressed as mean optical density of six different digital images.
Statistics
Results are expressed as the mean ± standard error of the mean (SEM). Data were analyzed by a repeated-measures one-way ANOVA. Two sets of independent sample data were compared using a paired t-test; p-values<0.05 was considered significant.
RESULTS
HSP 90 expression was increased in keloid tissues compared with adjacent normal tissues
Keloid tissue stained with hematoxylin and eosin showed dense and excessive collagen deposition that extended beyond the clinical keloid margin (Fig. 1, above left). HSP90 expression in keloid tissues of four patients was assessed by immunohistochemistry (Fig. 1, above right). The results showed that HSP90 protein levels were markedly higher in the central and peripheral regions of the keloids (optical density; 20,629 ± 2235) (Fig. 1, below left) than in normal tissue (optical density; 4303 ± 488.1) (Fig. 1, below center). Protein levels of HSP90 in keloid tissue were 4.8 times that of normal tissue (p<0.01) (Fig. 1, below right).
Figure 1.
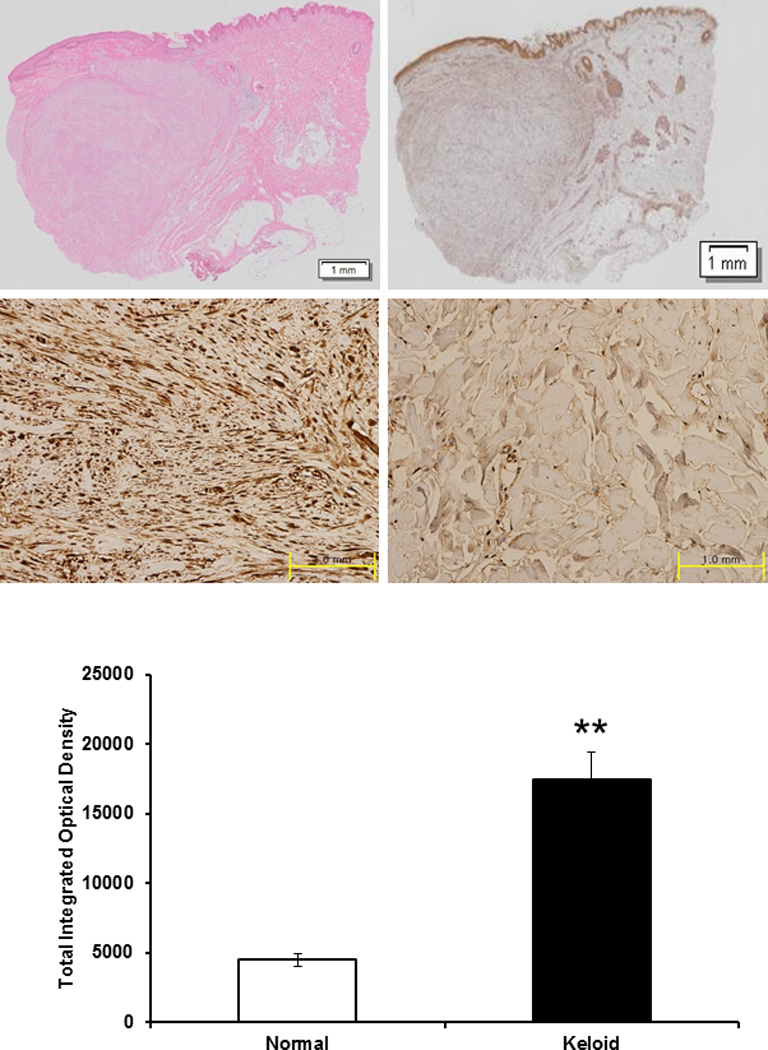
Histologic analysis of keloid tissue. (above left) H&E staing. Under the light microscope (×12), keloid tissue had a dense and excessive deposition of collagen. Results are representative of four different keloid tissue specimens. (above right) Immunohistochemical staining of keloids and adjacent normal dermal tissues. The expression of HSP90 in keloid tissue (center left) was increased than that in adjacent normal tissue (center right) using immunohistochemistry. (below) On the semi-quantitative analysis using Metamorph image analysis software, the expression of HSP90 increased by 4.8 times than normal tissue and this difference was statistically significant (**p<0.01).
17-AAG down-regulates mRNA expression of type I collagen in KFs
Type I and III collagen mRNA levels were examined in HDFs and KFs by real time RT–PCR. Type I collagen mRNA in HDFs was not changed after 17-AAG treatment (10 μM) (Fig. 2, above). However, TGF-β1 (10 ng/ml) treated HDFs and KFs was significantly decreased with 17-AAG treatment (10 μM) by 68% and 53%, respectively, compared with untreated controls (*p<0.05, **p<0.01) (Fig. 2, above and center). Also, the type I/III collagen mRNA ratio in KFs was significantly decreased by 77% after 17-AAG treatment (10 μM) in comparison to untreated control (**p<0.01; Fig. 2, below). However, mRNA expression of type III collagen was not reduced (datas not shown).
Figure 2.
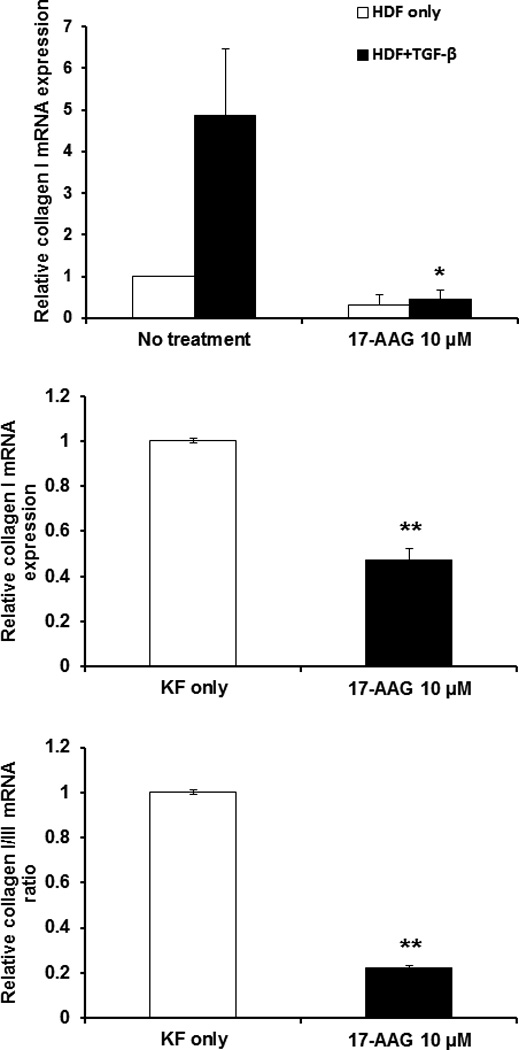
17-AAG down-regulates mRNA expression of type I collagen in KFs. (above) qRT–PCR analysis indicated that type I collagen mRNA levels in HDFs treated with 17-AAG treatment (10 μM) were not changed. However, type I collagen mRNA levels in TGF-β1 (10 ng/ml) treated HDFs were significantly decreased with 17-AAG treatment (10 μM) (*p<0.05) versus 17-AAG untreated HDFs. (center) Real time RT–PCR analysis indicated that type I collagen mRNA levels in KFs treated with 17-AAG treatment (10 μM) was significantly decreased (**p<0.01) compared with non-treated KFs. (below) Also, the type I/III collagen mRNA ratio was significantly decreased by 77% (**p<0.01) in KFs treated with 17-AAG treatment (10 μM). Each experiment was performed at least four times, Standard error bars are shown.
To examine the mechanism by which the 17-AAG suppressed type I collagen mRNA expression, secreted TGF-β1 and expression of Smad 2/3 complex protein were next investigated using ELISA and Western blot analysis, respectively. As shown in Figure 3 (above), decreased secreted TGF-β1 in KFs compared with untreated cells (*p<0.05). In addition, protein levels of Smad 2/3 complex was decreased by 37% in 17-AAG-treated KFs compared with untreated cells (**p<0.01) (Fig. 3 center and below). These results revealed that 17-AAG decreased collagen type I mRNA expression by inhibiting TGF-β1 expression and TGF-β-mediated transcriptional responses, thus blocking Smad 2/3 activation.
Figure 3.
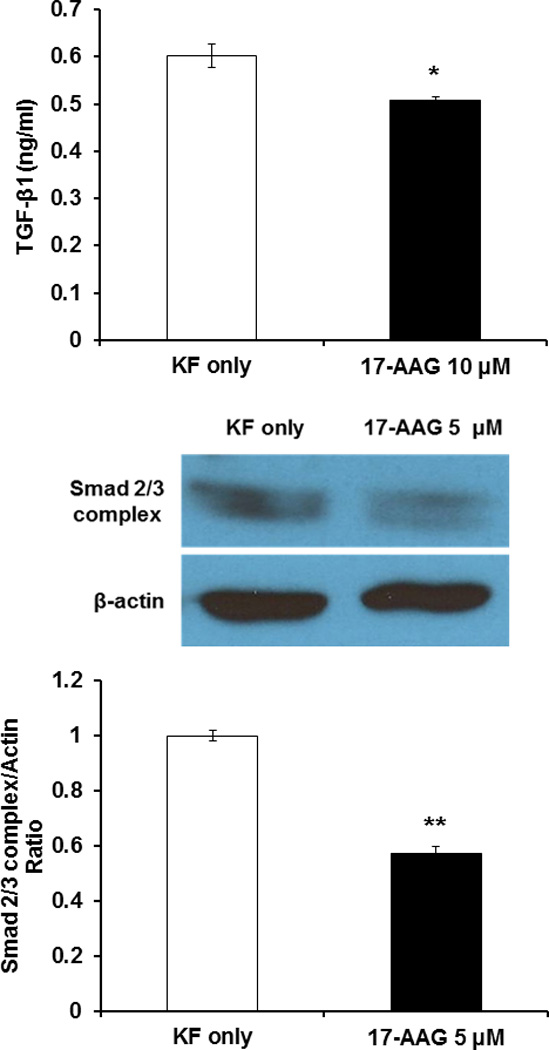
Effect of 17-AAG on TGF-β1 secretion and Smad 2/3 complex expression. (above) Secreted TGF-β1 protein measured by ELISA. 17-AAG treatments (10 μM) decreased secreted TGF-β1 protein level in KFs compared with untreated cells (*p<0.05). (center and below) Immunoblot analysis of Smad 2/3 complex protein in 17-AAG-treated KFs (5 µM) was significantly reduced compared to untreated KFs (**p<0.01). Results are representative of four independent experiments.
Collagen deposition and intensity were decreased in keloid spheroids treated with 17-AAG
We have previously developed a 3-dimensional organotypic multicellular spheroid (3-D OMS) model to mimic the microenvironment of human keloid tissues16. Keloid spheroids cultivated ex vivo retained the major characteristics of keloids, such as high levels of collagen I and TGF-β expression for up to 7 days. In order to verify the antifibrotic effect of 17-AAG on keloid tissues, we have cultured keloid spheroids derived from active-stage keloid patients (n=4) for 3 days with 17-AAG (10 μM). Masson’s trichrome staining revealed excessive deposition of collagen arranged in thick, irregular bundles in control untreated keloid spheroid tissue. However, collagen deposition and intensity was decreased in keloid spheroids treated with 17-AAG (10 μM) compared with untreated controls. In addition, thick and coarse collagen bundles were replaced by thin, shallow collagen bundles in 17-AAG–treated keloid spheroids (Fig. 4, left). On the semi-quantitative analysis using Metamorph image analysis software, significantly reduced collagen deposition in 17-AAG (10 μM) treated keloid spheroids by 81% versus non-treated keloid spheroids (**p<0.01)(Fig. 4, right).
Figure 4.
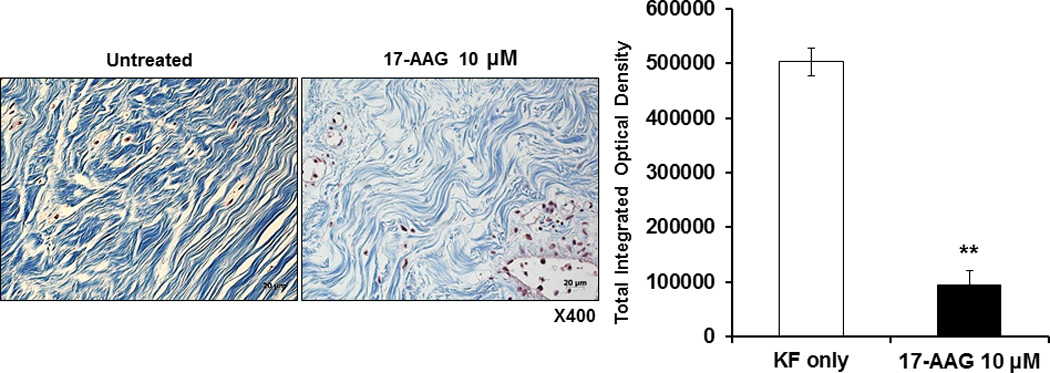
Masson’s trichrome staining of the keloid spheroid. (left) The keloid tissue had more dense and excessive deposition of collagen compared with adjacent normal dermal tissue. Also, irregular bundle-shaped collagen arrangement was showed on the keloid spheroid tissues. After treatment with 17-AAG (10 μM), collagen deposition and intensity were decreased. Also, dense and coarse collagen bundles were replaced by thin and shallow collagen bundles. (right) On the semi-quantitative analysis using Metamorph image analysis software, significantly reduced collagen deposition in 17-AAG (10 μM) treated keloid spheroids by 81% versus non-treated keloid spheroids (**p<0.01).
17-AAG decreases expression of collagen I and III, elastin, and fibronectin in keloid spheroids
Keloid spheroids derived from active-stage keloid patients (n=4) were cultured for 3 days with treatment of 17-AAG (10 μM) (Fig. 5, above). Immunohistochemical staining revealed significantly reduced type I collagen, type III collagen, elastin, and fibronection in 17-AAG (10 μM) treated keloid spheroids, by 11%, 32%, 8%, and 16%, respectively, versus non-treated keloid spheroids (*p<0.05, **p<0.01; Fig. 5, below). Taken together, these data strongly suggest that expression of major ECM components was decreased by HSP90 inhibitor like 17-AAG, which has a prominent role in remodeling ECM components.
Figure 5.
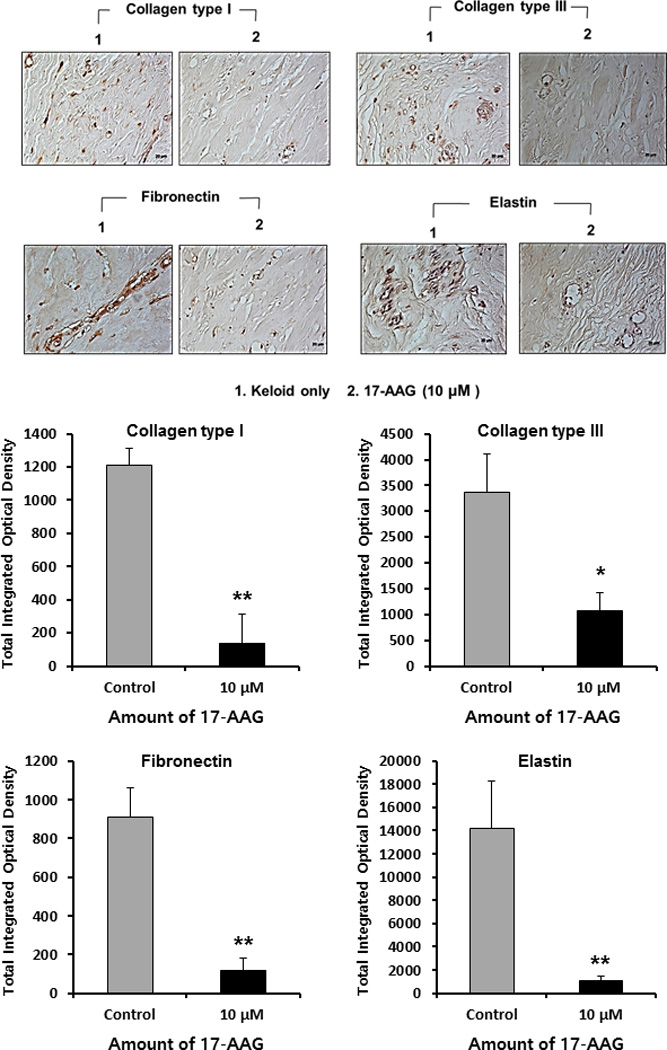
Immunohistochemical staining of keloid spheroid sections for type I, III collagen, fibronectin and elastin from 17-AAG-treated keloid tissues. (above) Representative light micrographs of collagen I, III, elastin and fibronectin immunohistochemistry of spheroid tissues cultured with 17-AAG (10 μM, ×400). (below) Semi-quantitative analysis of panel showed the expression of collagen I, III, fibronectin and elastin protein was significantly decreased in keloid spheroid with 17-AAG treatment (10 μM) ( *p<0.05, **p<0.01).
DISCUSSION
In this study we demonstrated that HSP90 is overexpressed in human keloid tissue compared with adjacent normal tissue, and the HSP90 inhibitor 17-AAG decreases expression of collagen type I and Smad 2/3 complex in keloid fibroblasts. Results of Masson’s trichrome staining revealed that thick, coarse collagen bundles were replaced by thin, shallow collagen bundles in keloid spheroid sections treated with 17-AAG. In addition, results of immunohistochemistry demonstrated that expression of extracellular matrix proteins collagen I and III, fibronectin, and elastin were markedly decreased in keloid spheroids treated with 17-AAG.
Hypertrophic scars and keloids are caused by excessive extracellular matrix accumulation resulting from an aberrant extracellular matrix protein synthesis and degradation. Potential explanations for keloid formation include altered growth factor regulation, immune dysfunction, aberrant collagen turnover, sebum or sebocytes as self-antigens, altered mechanics, and altered apoptotic signaling in keloid fibroblasts3,17. Abnormal increases in cytokines and growth factors are also involved in the pathogenesis of keloids. In particular, TGF-β appears to play a critical role in keloid formation1,6,7; therefore, inhibiting TGF-β1-dependent signaling by TGF-βI/TGF-βII receptor-neutralizing antibodies, truncated receptors, antisense oligonucleotides, or Smad 2-/Smad 3-specific siRNAs can decrease procollagen gene expression and inhibit fibrosis progression 3,7,18–21.
During remodeling, the predominance of type III collagen in ECM gradually become converted type I collagen, which strengthens the scar7,17. The ratio of type I/III collagen has been shown to be significantly elevated in keloids compared with normal scars22,23. In our study we found that type I collagen mRNA level and the type I/III collagen ratio were decreased in 17-AAG–treated KFs, suggesting that 17-AAG can change the ratio of type I/III collagen in keloids to resemble that of normal scars.
The growth factor TGF-β increases tropoelastin mRNA abundance and elastin formation24,25. In addition, elastin mRNA is stabilized by TGF-β signaling26. Moreover, TGF-β1 inhibits elastin degradation by decreasing the expression and activity of matrix metalloproteinases 2 and 927. Thus, inhibiting TGF-β with 17-AAG reduces elastin accumulation in scar tissue. Fibronectin is also induced by TGF-β28, and the induction of connective tissue growth factor and fibronectin increase the profibrotic effects of TGF-β29. In other words, these proteins enhance the cellular response to TGF-β, prolonging the wound healing and fibrotic response29. Treatment with 17-AAG can attenuate these phenomena by reducing fibronectin formation.
In this study we have demonstrated that 17-AAG reduces ECM accumulation via TGF-β /Smad-dependent pathways. Keloid formation involves various intracellular signal molecules, including extracellular signal-regulated protein kinase 1/2 (ERK1/2), p38 mitogen-activated protein kinase, Sma- and Mad-related proteins (Smads), signal transducer and activator of transcription-3 (STAT3), and phosphatidylinositol-3-kinase (PI3K)/Akt. Thus, many researchers have suggested the modulation of these signaling mediators to treat keloids. Previous studies have reported that suppression of Smad3, PI3K, ERK, and STAT3 pathways is sufficient to inhibit ECM production in keloids30–34. Future studies investigating the effect of 17-AAG on Smad-independent pathways will be helpful to determine its mechanism of action.
Fibrotic diseases affect various organs, including the liver, kidney, lung, and skin, and HSP90 inhibitors have shown promising results in fibrotic disease models12,14. In this study we demonstrated that an HSP90 inhibitor may be useful in the treatment of keloid scars by decreasing the accumulation of ECM components. However, future studies are needed to determine the effect of HSP90 on proliferation, migration, and apoptosis of keloid fibroblasts.
CONCLUSION
The HSP90 inhibitor, 17-AAG, decreases collagen synthesis by keloid fibroblasts and attenuates accumulation of the extracellular matrix components type I and III collagen, elastin, and fibronectin in a keloid spheroid model. The effects of 17-AAG are mediated through its inhibitory action on TGF-β expression and the Smad pathway. Therefore, antifibrotic effects of HSP90 inhibitors such as 17-AAG may have therapeutic potential for the treatment of keloids.
Acknowledement
This work was supported by grants from the National Research Foundation of Korea (2010-0029220, 2013K1A1A2A02050188, 2013M3A9D3045879), and the National Institutes of Health, USA (CA 107070). This work was partially supported by the Brain Korea 21 plus Future Biopharmaceutical Human Resources Training and Research Team. This study was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (No. 2014051295, WJ Lee) and a faculty research grant of Yonsei University College of Medicine for 2014 (6-2011-0173).
Footnotes
Financial Disclosure: The authors have no proprietary or commercial interest in any materials discussed in this article.
Conflict of interest: No authors have conflicts of interest that could potentially influence the described research.
REFERENCES
- 1.Burd A, Huang L. Hypertrophic response and keloid diathesis: two very different forms of scar. Plast Reconstr Surg. 2005;116:150e–157e. doi: 10.1097/01.prs.0000191977.51206.43. [DOI] [PubMed] [Google Scholar]
- 2.Atiyeh BS, Costagliola M, Hayek SN. Keloid or hypertrophic scar: the controversy: review of the literature. Ann Plast Surg. 2005;54:676–680. doi: 10.1097/01.sap.0000164538.72375.93. [DOI] [PubMed] [Google Scholar]
- 3.Al-Attar A, Mess S, Thomassen JM, Kauffman CL, Davison SP. Keloid pathogenesis and treatment. Plast Reconstr Surg. 2006;117:286–300. doi: 10.1097/01.prs.0000195073.73580.46. [DOI] [PubMed] [Google Scholar]
- 4.Piera-Velazquez S, Jimenez SA. Molecular mechanisms of endothelial to mesenchymal cell transition (EndoMT) in experimentally induced fibrotic diseases. Fibrogenesis Tissue Repair. 2012;5(Suppl 1):S7. doi: 10.1186/1755-1536-5-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kis K, Liu X, Hagood JS. Myofibroblast differentiation and survival in fibrotic disease. Expert Rev Mol Med. 2011;13:e27. doi: 10.1017/S1462399411001967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Russell SB, Russell JD, Trupin KM, et al. Epigenetically altered wound healing in keloid fibroblasts. J Invest Dermatol. 2010;130:2489–2496. doi: 10.1038/jid.2010.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bran GM, Goessler UR, Hormann K, Riedel F, Sadick H. Keloids: current concepts of pathogenesis (review) Int J Mol Med. 2009;24:283–293. doi: 10.3892/ijmm_00000231. [DOI] [PubMed] [Google Scholar]
- 8.Joly AL, Wettstein G, Mignot G, Ghiringhelli F, Garrido C. Dual role of heat shock proteins as regulators of apoptosis and innate immunity. J Innate Immun. 2010;2:238–247. doi: 10.1159/000296508. [DOI] [PubMed] [Google Scholar]
- 9.Vaughan CK, Neckers L, Piper PW. Understanding of the Hsp90 molecular chaperone reaches new heights. Nat Struct Mol Biol. 2010;17:1400–1404. doi: 10.1038/nsmb1210-1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neckers L. Hsp90 inhibitors as novel cancer chemotherapeutic agents. Trends Mol Med. 2002;8:S55–S61. doi: 10.1016/s1471-4914(02)02316-x. [DOI] [PubMed] [Google Scholar]
- 11.Citri A, Harari D, Shohat G, et al. Hsp90 recognizes a common surface on client kinases. J Biol Chem. 2006;281:14361–14369. doi: 10.1074/jbc.M512613200. [DOI] [PubMed] [Google Scholar]
- 12.Myung SJ, Yoon JH, Kim BH, et al. Heat shock protein 90 inhibitor induces apoptosis and attenuates activation of hepatic stellate cells. J Pharmacol Exp Ther. 2009;330:276–282. doi: 10.1124/jpet.109.151860. [DOI] [PubMed] [Google Scholar]
- 13.Wrighton KH, Lin X, Feng XH. Critical regulation of TGFbeta signaling by Hsp90. Proc Natl Acad Sci U S A. 2008;105:9244–9249. doi: 10.1073/pnas.0800163105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noh H, Kim HJ, Yu MR, et al. Heat shock protein 90 inhibitor attenuates renal fibrosis through degradation of transforming growth factor-beta type II receptor. Lab Invest. 2012;92:1583–1596. doi: 10.1038/labinvest.2012.127. [DOI] [PubMed] [Google Scholar]
- 15.Zhang K, Lu Y, Yang P, et al. HILI inhibits TGF-beta signaling by interacting with Hsp90 and promoting TbetaR degradation. PLoS One. 2012;7:e41973. doi: 10.1371/journal.pone.0041973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee WJ, Choi IK, Lee JH, Kim YO, Yun CO. A novel three-dimensional model system for keloid study: organotypic multicellular scar model. Wound Repair Regen. 2013;21:155–165. doi: 10.1111/j.1524-475X.2012.00869.x. [DOI] [PubMed] [Google Scholar]
- 17.Shih B, Garside E, McGrouther DA, Bayat A. Molecular dissection of abnormal wound healing processes resulting in keloid disease. Wound Repair Regen. 2010;18:139–153. doi: 10.1111/j.1524-475X.2009.00553.x. [DOI] [PubMed] [Google Scholar]
- 18.Shah M, Foreman DM, Ferguson MW. Neutralisation of TGF-beta 1 and TGF-beta 2 or exogenous addition of TGF-beta 3 to cutaneous rat wounds reduces scarring. J Cell Sci. 1995;108(Pt 3):985–1002. doi: 10.1242/jcs.108.3.985. [DOI] [PubMed] [Google Scholar]
- 19.Gao Z, Wang Z, Shi Y, et al. Modulation of collagen synthesis in keloid fibroblasts by silencing Smad2 with siRNA. Plast Reconstr Surg. 2006;118:1328–1337. doi: 10.1097/01.prs.0000239537.77870.2c. [DOI] [PubMed] [Google Scholar]
- 20.Bran GM, Goessler UR, Schardt C, et al. Effect of the abrogation of TGF-beta1 by antisense oligonucleotides on the expression of TGF-beta-isoforms and their receptors I and II in isolated fibroblasts from keloid scars. Int J Mol Med. 2010;25:915–921. doi: 10.3892/ijmm_00000422. [DOI] [PubMed] [Google Scholar]
- 21.Wang Z, Gao Z, Shi Y, et al. Inhibition of Smad3 expression decreases collagen synthesis in keloid disease fibroblasts. J Plast Reconstr Aesthet Surg. 2007;60:1193–1199. doi: 10.1016/j.bjps.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 22.Zhang GY, Gao WY, Li X, et al. Effect of camptothecin on collagen synthesis in fibroblasts from patients with keloid. Ann Plast Surg. 2009;63:94–99. doi: 10.1097/SAP.0b013e3181872775. [DOI] [PubMed] [Google Scholar]
- 23.Friedman DW, Boyd CD, Mackenzie JW, et al. Regulation of collagen gene expression in keloids and hypertrophic scars. J Surg Res. 1993;55:214–222. doi: 10.1006/jsre.1993.1132. [DOI] [PubMed] [Google Scholar]
- 24.McGowan SE, McNamer R. Transforming growth factor-beta increases elastin production by neonatal rat lung fibroblasts. Am J Respir Cell Mol Biol. 1990;3:369–376. doi: 10.1165/ajrcmb/3.4.369. [DOI] [PubMed] [Google Scholar]
- 25.Katsuta Y, Ogura Y, Iriyama S, et al. Fibulin-5 accelerates elastic fibre assembly in human skin fibroblasts. Exp Dermatol. 2008;17:837–842. doi: 10.1111/j.1600-0625.2008.00709.x. [DOI] [PubMed] [Google Scholar]
- 26.Kucich U, Rosenbloom JC, Abrams WR, Rosenbloom J. Transforming growth factor-beta stabilizes elastin mRNA by a pathway requiring active Smads, protein kinase C-delta, and p38. Am J Respir Cell Mol Biol. 2002;26:183–188. doi: 10.1165/ajrcmb.26.2.4666. [DOI] [PubMed] [Google Scholar]
- 27.Dai J, Losy F, Guinault AM, et al. Overexpression of transforming growth factor-beta1 stabilizes already-formed aortic aneurysms: a first approach to induction of functional healing by endovascular gene therapy. Circulation. 2005;112:1008–1015. doi: 10.1161/CIRCULATIONAHA.104.523357. [DOI] [PubMed] [Google Scholar]
- 28.Hocevar BA, Brown TL, Howe PH. TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J. 1999;18:1345–1356. doi: 10.1093/emboj/18.5.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.LEASK A, ABRAHAM DJ. TGF-β signaling and the fibrotic response. The FASEB Journal. 2004;18:816–827. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- 30.Lim CP, Phan TT, Lim IJ, Cao X. Stat3 contributes to keloid pathogenesis via promoting collagen production, cell proliferation and migration. Oncogene. 2006;25:5416–5425. doi: 10.1038/sj.onc.1209531. [DOI] [PubMed] [Google Scholar]
- 31.Lim IJ, Phan TT, Tan EK, et al. Synchronous activation of ERK and phosphatidylinositol 3-kinase pathways is required for collagen and extracellular matrix production in keloids. J Biol Chem. 2003;278:40851–40858. doi: 10.1074/jbc.M305759200. [DOI] [PubMed] [Google Scholar]
- 32.Park G, Yoon BS, Moon JH, et al. Green tea polyphenol epigallocatechin-3-gallate suppresses collagen production and proliferation in keloid fibroblasts via inhibition of the STAT3-signaling pathway. J Invest Dermatol. 2008;128:2429–2441. doi: 10.1038/jid.2008.103. [DOI] [PubMed] [Google Scholar]
- 33.Song J, Xu H, Lu Q, et al. Madecassoside suppresses migration of fibroblasts from keloids: involvement of p38 kinase and PI3K signaling pathways. Burns. 2012;38:677–684. doi: 10.1016/j.burns.2011.12.017. [DOI] [PubMed] [Google Scholar]
- 34.Xia W, Longaker MT, Yang GP. P38 MAP kinase mediates transforming growth factor-beta2 transcription in human keloid fibroblasts. Am J Physiol Regul Integr Comp Physiol. 2006;290:R501–R508. doi: 10.1152/ajpregu.00472.2005. [DOI] [PubMed] [Google Scholar]