

Abstract
Purpose
Autosomal dominant nanophthalmos is an inherited eye disorder characterized by a structurally normal but smaller eye. Patients with nanophthalmos have high hyperopia (far-sightedness), a greater incidence of angle-closure glaucoma, and increased risk of surgical complications. In this study, the clinical features and the genetic basis of nanophthalmos were investigated in two large autosomal dominant nanophthalmos pedigrees.
Methods
Fourteen members of a Caucasian pedigree from the United States and 15 members of a pedigree from the Mariana Islands enrolled in a genetic study of nanophthalmos and contributed DNA samples. Twenty of 29 family members underwent eye examinations that included measurement of axial eye length and/or refractive error. The genetic basis of nanophthalmos in the pedigrees was studied with linkage analysis, whole exome sequencing, and candidate gene (i.e., TMEM98) sequencing to identify the nanophthalmos-causing gene.
Results
Nine members of the pedigree from the United States and 11 members of the pedigree from the Mariana Islands were diagnosed with nanophthalmos that is transmitted as an autosomal dominant trait. The patients with nanophthalmos had abnormally short axial eye lengths, which ranged from 15.9 to 18.4 mm. Linkage analysis of the nanophthalmos pedigree from the United States identified nine large regions of the genome (greater than 10 Mbp) that were coinherited with disease in this family. Genes within these “linked regions” were examined for disease-causing mutations using exome sequencing, and a His196Pro mutation was detected in the TMEM98 gene, which was recently reported to be a nanophthalmos gene. Sanger sequencing subsequently showed that all other members of this pedigree with nanophthalmos also carry the His196Pro TMEM98 mutation. Testing the Mariana Islands pedigree for TMEM98 mutations identified a 34 bp heterozygous deletion that spans the 3′ end of exon 4 in all affected family members. Neither TMEM98 mutation was detected in public exome sequence databases.
Conclusions
A recent report identified a single TMEM98 missense mutation in a nanophthalmos pedigree. Our discovery of two additional TMEM98 mutations confirms the important role of the gene in the pathogenesis of autosomal dominant nanophthalmos.
Introduction
A spectrum of eye disorders ranges from small eyes that are otherwise normal (nanophthalmos or simple microphthalmia) to small eyes that have a grossly disorganized and dysfunctional structure (microphthalmia). Clinical features of nanophthalmos include a short axial eye length (less than 20 mm), high hyperopia (spherical refraction greater than +6 diopters), and small corneal diameter [1-4]. These anatomic features of nanophthalmos result in a crowded anterior chamber and a high incidence of angle closure glaucoma. Nanophthalmic eyes also have thick and impermeable sclera, which has been associated with a high risk of developing choroidal effusions after any intraocular surgery [5]. Nanophthalmos may occur in isolation or as part of an ocular or systemic syndrome.
Nanophthalmos has a strong genetic basis, and autosomal dominant [3,4] and autosomal recessive [6] forms of inheritance have been reported. Mutations in the membrane-type frizzled-related protein (MFRP, OMIM 606227) have been detected in a large recessive nanophthalmos pedigree [7]. Similarly, a large autosomal dominant nanophthalmos pedigree was recently studied with linkage analysis, and a disease-causing gene was mapped to chromosome 17p12-q12 [8]. Subsequently, a mutation in the transmembrane protein 98 (TMEM98, OMIM 615949) gene was detected that is coinherited with nanophthalmos in this pedigree [4].
In this study, we describe two additional families with autosomal dominant inherited nanophthalmos, a Caucasian family of German heritage living in the United States and a Pacific Islander family of Micronesian heritage living in the Mariana Islands. We confirm the identification of TMEM98 as a gene for autosomal dominant nanophthalmos with the identification of novel disease-causing mutations in the coding sequence of the TMEM98 gene.
Methods
Patient resources
Research was approved by local Internal Review Boards and all patients provided written consent in advance of the study. The study design met the tenets of the Declaration of Helsinki and Association for Research in Vision and Ophthalmology guidelines. Participants were examined by an ophthalmologist and underwent an eye examination that included measurement of visual acuity, refractive error, and intraocular pressure as well as a slit-lamp examination of the anterior chamber and fundus examination. Axial eye length was measured in some patients with A-scan ultrasound (Alcon, Fort Worth, TX). Patients were diagnosed with nanophthalmos when short axial eye length (<20 mm) or significant hyperopia (spherical equivalent > +6.00) were observed in the absence of other ocular abnormalities. The control subjects showed no evidence of nanophthalmos. Some members of both pedigrees contributed DNA samples but were unavailable for examination, and medical records could not be obtained. These subjects are indicated on the pedigree drawing with stars (Figure 1), and their affection status was determined according to the family history report.
Figure 1.

Nanophthalmos pedigree 991-F from the United States. Subjects with nanophthalmos are indicated with black symbols, while healthy subjects are indicated with white symbols. Gray symbols indicate unknown affection status. Diagnosis was made with a clinical exam or a review of the medical records for all available family members. Diagnosis of family members indicated with stars was determined by family history. The subjects who contributed a DNA sample for the study are indicated with an “X.”
Genetic studies
Venous blood samples were obtained from the participants for the study. Genomic DNA was extracted from peripheral whole blood using the QIAamp Blood Maxi Kit (Qiagen, Valencia, CA). Pedigree 991-F was analyzed with a genome-wide linkage analysis using Illumina 6K human mapping microarrays (Illumina, Santa Clara, CA), which examine 6,020 single nucleotide polymorphisms (SNPs). Sample processing and labeling were performed following the manufacturer’s instructions. Genomic regions consistent with linkage were identified, and multipoint non-parametric linkage (NPL) scores were calculated using the MERLIN software package [9]. Penetrance and disease gene frequency were set to 99% and 0.001%, respectively.
Exome capture was performed on one member of pedigree 991-F (IV:3, Figure 1) with SureSelect Human All Exon v2 (Agilent, Santa Clara, CA) using the manufacturer’s protocol. Paired-end, 50 nucleotide reads were obtained using a HiSeq2000 (Illumina, San Diego, CA) that achieved average exon coverage of greater than 50X.
The entire coding region of TMEM98 (NM_15544.2) was amplified from patient and control DNA using overlapping primer pairs in PCR reactions containing 12.5 ng of each patient’s DNA, 1.25 μl of 10× buffer (100 mM Tris – HCL, pH 8.3, 500 mM KCl, 15 mM MgCl2), 300 μM of each dCTP, dATP, dGTP and dTTP, 1 pmol of each primer, and 0.25 U Biolase polymerase (Biolase) in a 8.35 μl of PCR solution. Primer sequences are listed in Appendix 1. Amplified DNA was scanned for mutations with bidirectional DNA sequencing with an Applied Biosystems model 3730 automated sequencer as previously described [10]. The mutations that result in amino acid substitutions were evaluated using the BLOSUM62 matrix [11], SIFT [12], and PolyPhen2 [13].
Results
Clinical features of nanophthalmos
Ophthalmic examinations were performed on 20 members of two large nanophthalmos pedigrees (pedigree 991-F from the United States, Figure 1; pedigree 951-N from the Marianas Islands, Figure 2). Both pedigrees exhibit an autosomal dominant inheritance pattern with high penetrance that spans at least four generations. Six of ten examined members of pedigree 991-F were diagnosed with nanophthalmos while an additional three members had nanophthalmos according to the family history report. Mean axial eye lengths were 17.1 mm±0.31 mm for the available patients. Affected family members also had a high degree of hyperopia with spherical equivalent refractions of +13.0±0.89 diopters (Table 1). Unaffected family members had much less hyperopia (+0.71±0.81 diopters).
Figure 2.
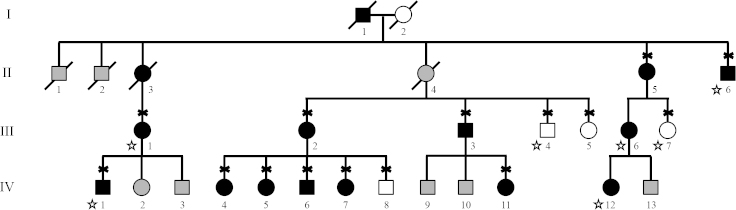
Nanophthalmos pedigree 951-N from the Marianas Islands. Subjects with nanophthalmos are indicated with black symbols, while healthy subjects are indicated with white symbols. Gray symbols indicate unknown affection status. Diagnosis was made with a clinical exam or a review of the medical records for all available family members. Diagnosis of family members indicated with stars was determined by family history. The subjects who contributed a DNA sample for the study are indicated with an “X.”
Table 1. Clinical features of Pedigree 991-F.
| Individual | Diagnosis | AEL OD (mm) | AEL OS (mm) | Spherical Equivalent OD (diopters) | Spherical Equivalent OS (diopters) | |
|---|---|---|---|---|---|---|
| III-3 |
Nanophthalmia |
NA |
NA |
12.75 |
13 |
|
| III-4 |
Nanophthalmia |
NA |
NA |
14.125 |
13.375 |
|
| IV-2 |
Nanophthalmia |
16.7 |
17.1 |
12.875 |
13.125 |
|
| IV-3 |
Nanophthalmia |
17.4 |
17.3 |
11.125 |
12.25 |
|
| V-1 |
Nanophthalmia |
NA |
NA |
13.75 |
14 |
|
| V-2 |
Nanophthalmia |
NA |
NA |
13.375 |
14.13 |
|
| Nanophthalmia mean |
17.1±0.31 mm |
+13.2±0.87 diopters |
||||
| III-1 |
Normal |
NA |
NA |
1.5 |
1.5 |
|
| III-2 |
Normal |
NA |
NA |
1 |
0.75 |
|
| III-5 |
Normal |
NA |
NA |
0.5 |
0 |
|
| III-6 |
Normal |
NA |
NA |
−0.5 |
0 |
|
| Normal Mean | NA | +0.59±0.73 diopters | ||||
Family members are identified by their pedigree symbols from Figure 1. NA - Not available.
Eight of ten examined members of Pacific Islander pedigree 951-N had nanophthalmos and an average axial eye length of 17.4 mm±0.94 mm, while unaffected family members had more normal eye length (22.7±0.38 mm; Table 2). Along with short axial eye lengths, family members with nanophthalmos also had hyperopia with spherical equivalent refractions of +3.93±1.57 diopters. Three additional family members had nanophthalmos according to the family history report; however, they were not available for examinations, and no medical records could be obtained.
Table 2. Clinical features of Pedigree 951-N.
| Individual | Diagnosis | AEL OD (mm) | AEL OS (mm) | Spherical Equivalent OD (diopters) | Spherical Equivalent OS (diopters) |
|---|---|---|---|---|---|
| II-5 |
Nanophthalmia |
NA |
15.86 |
NA |
7.00 |
| III-2 |
Nanophthalmia |
18 |
18 |
6 |
NA |
| III-3 |
Nanophthalmia |
16.81 |
16.62 |
NA |
NA |
| IV-4 |
Nanophthalmia |
18.43 |
18.21 |
4 |
4 |
| IV-5 |
Nanophthalmia |
17.08 |
17.19 |
2.5 |
4 |
| IV-6 |
Nanophthalmia |
17.78 |
18.35 |
5 |
6 |
| IV-7 |
Nanophthalmia |
18.38 |
18.27 |
2 |
1.88 |
| IV-11 |
Nanophthalmia |
15.87 |
16.19 |
NA |
NA |
| Nanophthalmia Mean |
17.4±0.94 mm |
+3.93±1.57 diopters |
|||
| III-5 |
Normal |
22.36 |
22.35 |
0 |
0 |
| IV-8 |
Normal |
23.01 |
23.02 |
0 |
0 |
| Normal Mean | 22.7±0.38 mm | 0.00 diopters | |||
Family members are identified by their pedigree symbols from Figure 2. NA - Not available.
Genetic studies of nanophthalmos pedigrees
DNA from 20 affected family members of pedigrees 991-F and 951-N was genotyped at 6,020 SNPs. The genotypes were evaluated for coinheritance with nanophthalmos using linkage analysis. In pedigree 991-F, a 20.2 Mbp segment of chromosome 17p12-q12, bounded by SNPs rs530664 and rs16523, was transmitted with nanophthalmos in all members of the family. An overlapping 19.7 Mb section of chromosome 17, bounded by SNPs rs917541 and rs16523, was similarly coinherited with nanophthalmos in pedigree 951-N. These data suggest that the gene that causes autosomal dominant nanophthalmos in pedigrees 991-F and 951-N lies between SNPs rs917541 and rs16523 on chromosome 17 (Figure 3). This pericentric region of chromosome 17 has previously been linked to autosomal dominant nanophthalmos in a Chinese pedigree [8].
Figure 3.
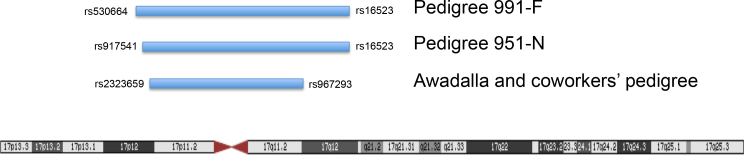
Diagram of the overlapping regions of chromosome 17 that are coinherited with nanophthalmos in the pedigrees (991-F and 951-N) and in the Awadalla and coworkers’ pedigree [4].
Exome sequencing was performed for patient IV:3 in pedigree 991-F (Figure 1) to identify the DNA sequence variants contained within the linked genomic regions. Within the linked region on chromosome 17, four non-synonymous DNA sequence variations were detected in four genes, ZNF286A, ZNF287, SLC47A1 (OMIM 609832), and TMEM98 (Figure 3). Each variant was evaluated as a potential nanophthalmos-causing mutation by testing the rest of pedigree 991-F for coinheritance with disease and evaluating the pathogenicity of the mutation using sequence analysis programs, including SIFT, PolyPhen, and BLOSUM62. Of the four variants, only the His196Pro mutation in TMEM98 was coinherited with nanophthalmos, absent from healthy family members, and had pathogenic scores on all three sequence analysis systems (Table 3). The TMEM98 missense mutation in pedigree 991-F, His196Pro, produced a BLOSUM62 score of −2, a PolyPhen score of 0.998 (probably damaging), and a SIFT score of 0 (damaging). All indicate that this mutation is likely pathogenic (Table 3). As data from this experiment were being collected, another research group reported the discovery of an autosomal dominant nanophthalmos gene, trans membrane 98 gene (TMEM98), that was located within the same chromosome 17 locus linked with disease in our pedigree [4] (Figure 3). Consequently, the TMEM98 gene has become the top candidate gene for causing glaucoma in our pedigrees.
Table 3. Non-synonymous mutations detected in nanophthalmos patient IV-3 of Pedigree 991-F.
| Gene | Mutation | Co-inheritance with disease in pedigree | SIFT | Polyphen | Blosum62 |
|---|---|---|---|---|---|
| ZNF286A |
Pro87Ser |
No |
0.01 (damaging) |
0.001 (benign) |
−1 |
| ZNF287 |
Cys48Arg |
No |
0.07 (tolerated) |
0.993 (probably damaging) |
−3 |
| SLC47A1 |
Ile414Thr |
Yes |
0 (damaging) |
0.45 (benign) |
−1 |
| TMEM98 | His196Pro | Yes | 0 (damaging) | 0.998 (probably damaging) | −2 |
Exome sequencing identified four non-synonymous mutations in the chromosome 17 critical region. Mutations were evaluated for pathogenicity based on co-inheritance with nanophthalmos in pedigree 991-F and with three mutation analysis software packages.
Pedigree 951-N was subsequently analyzed for TMEM98 mutations. Eleven members with nanophthalmos and four healthy family members were tested for disease-causing mutations using Sanger DNA sequencing. A novel 34 bp heterozygous deletion was detected in all affected family members that spans the last 28 bp of exon 4 and the first 6 bp of intron 4 (c.694_721delAGAATGAAGACTGGATCGAAGATGCCTCgtaagg). This TMEM98 deletion eliminates a splice site and results in a premature termination of translation. The mutation is coinherited with nanophthalmos in pedigree 951-F and is absent from all healthy family members.
Sequence analysis of both TMEM98 mutations provided additional support for their pathogenicity. Neither the His196Pro mutation nor the 34 bp deletion was detected in public DNA sequence databases including the Exome Variation Server and the 1000 Genomes Project [14]. Both mutations span highly conserved sequences based on the UCSC Genome Browser and BlastP. The His196Pro mutation detected in pedigree 991-F alters a histidine amino acid that is highly conserved across many disparate species (Figure 4), which provides additional support for the mutation’s potential pathogenicity.
Figure 4.

TMEM98 sequence analysis. Alignment of protein sequences encoded by human TMEM98 with orthologous genes spanning the His196Pro mutation detected in pedigree 991-F. The histidine amino acid at codon 196 indicated in blue letters is conserved across many species; the flanking amino acids that are not conserved are indicated in red.
Discussion
Some forms of nanophthalmos are inherited as a Mendelian trait and are caused primarily by a single gene. Several cases of recessive nanophthalmos have been attributed to mutations in the MFRP gene [7]. Recently, Awadalla and coworkers identified TMEM98 as the first autosomal dominant nanophthalmos gene. A missense mutation in the TMEM98 gene (A193P) was associated with autosomal dominant nanophthalmos in one large pedigree [4].
TMEM98 encodes a transmembrane protein that has ubiquitous expression in humans [4]. Furthermore, Awadalla and coworkers demonstrated that TMEM98 is also produced within ocular tissues including the sclera, choroid, retinal pigment epithelium, and iris that may have central roles in the pathogenesis of nanophthalmos [4,15]. Little is known about the function of the protein encoded by TMEM98; however, its expression in the sclera suggest that mutations may contribute to its pathologic thickening in nanophthalmos. Moreover, TMEM98 expression in tissues of the outflow pathway makes it plausible that TMEM98 mutations might contribute to the development of secondary glaucoma in nanophthalmos. The cause of the abnormally small eyes in nanophthalmos is unknown. However, identification and future studies of TMEM98 may provide new insights into the developmental mechanisms that determine eye size, scleral thickness, and risk for glaucoma.
In this study, we confirmed the association between TMEM98 mutations and nanophthalmos by reporting two novel TMEM98 mutations (His196Pro and c.694_721delAGAATGAAGACTGGATCGAAGATGCCTCgtaagg) in autosomal dominant pedigrees. Both TMEM98 mutations identified in our nanophthalmos pedigrees are coinherited with disease, absent from sequencing databases, alter conserved DNA sequences, and are judged likely to be pathogenic based on their destructive effects on the encoded TMEM98. Our contribution of two additional large nanophthalmos pedigrees with TMEM98 variants that are coinherited with disease greatly strengthens Awadalla and coworkers’ discovery and assertion that mutations in TMEM98 cause nanophthalmos [4,16].
Acknowledgments
Funding was provided in part by The Polakoff Foundation, Research to Prevent Blindness, and the NIH (NEI R01EY023512). The data in this manuscript was previously presented at the annual meeting of the Association for Research in Vision and Ophthalmology in 2015.
Appendix 1: Oligo Sequences for amplification of TMEM98.
To access the data, click or select the words “Appendix 1.”
References
- 1.Brockhurst RJ. Nanophthalmos with uveal effusion. A new clinical entity. Arch Ophthalmol. 1975;93:1989–99. doi: 10.1001/archopht.1975.01010020923001. [DOI] [PubMed] [Google Scholar]
- 2.Singh OS, Simmons RJ, Brockhurst RJ, Trempe CL. Nanophthalmos: a perspective on identification and therapy. Ophthalmology. 1982;89:1006–12. [PubMed] [Google Scholar]
- 3.Vingolo EM, Steindl K, Forte R, Zompatori L, Iannaccone A, Sciarra A, Del Porto G, Pannarale MR. Autosomal dominant simple microphthalmos. J Med Genet. 1994;31:721–5. doi: 10.1136/jmg.31.9.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Awadalla MS, Burdon KP, Souzeau E, Landers J, Hewitt AW, Sharma S, Craig JE. Mutation in TMEM98 in a large white kindred with autosomal dominant nanophthalmos linked to 17p12-q12. JAMA Ophthalmol. 2014;132:970–7. doi: 10.1001/jamaophthalmol.2014.946. [DOI] [PubMed] [Google Scholar]
- 5.Calhoun FP. The management of glaucoma in nanophthalmos. Trans Am Ophthalmol Soc. 1975;73:97–122. [PMC free article] [PubMed] [Google Scholar]
- 6.Martorina M. Familial nanophthalmos. J Fr Ophtalmol. 1988;11:357–61. [PubMed] [Google Scholar]
- 7.Sundin OH, Leppert GS, Silva ED, Yang JM, Dharmaraj S, Maumenee IH, Santos LC, Parsa CF, Traboulsi EI, Broman KW, Dibernardo C, Sunness JS, Toy J, Weinberg EM. Extreme hyperopia is the result of null mutations in MFRP, which encodes a Frizzled-related protein. Proc Natl Acad Sci USA. 2005;102:9553–8. doi: 10.1073/pnas.0501451102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu Z, Yu C, Li J, Wang Y, Liu D, Xiang X, Su W, Pan Q, Xie L, Xia K. A novel locus for congenital simple microphthalmia family mapping to 17p12-q12. Invest Ophthalmol Vis Sci. 2011;52:3425–9. doi: 10.1167/iovs.10-6747. [DOI] [PubMed] [Google Scholar]
- 9.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin|[mdash]|rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 10.Fingert JH, Robin AL, Ben R.Roos, Davis LK, Scheetz TE, Wassink TH, et alCopy number variations on chromosome 12q14 in patients with normal tension glaucoma. Hum Mol Genet 2011202482–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eddy SR. Where did the BLOSUM62 alignment score matrix come from? Nat Biotechnol. 2004;22:1035–6. doi: 10.1038/nbt0804-1035. [DOI] [PubMed] [Google Scholar]
- 12.Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. Cold Spring Harbor Lab. 2001;11:863–74. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.1000 Genomes Project Consortium, Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wagner AH, Anand VN, Wang WH, Chatterton JE, Sun D, Shepard AR, Jacobson N, Pang IH, Deluca AP, Casavant TL, Scheetz TE, Mullins RF, Braun TA, Clark AF. Exon-level expression profiling of ocular tissues. Exp Eye Res. 2013;111:105–11. doi: 10.1016/j.exer.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Does the Association Between TMEM98 and Nanophthalmos Require Further Confirmation?-. Reply. JAMA Ophthalmol. 2015;133:359–60. doi: 10.1001/jamaophthalmol.2014.4919. [DOI] [PubMed] [Google Scholar]