

Abstract
Background
Cranberries (Vaccinium macrocarpon Ait.), renowned for their excellent health benefits, are an important berry crop. Here, we performed transcriptome sequencing of one cranberry cultivar, from fruits at two different developmental stages, on the Illumina HiSeq 2000 platform. Our main goals were to identify putative genes for major metabolic pathways of bioactive compounds and compare the expression patterns between white fruit (W) and red fruit (R) in cranberry.
Results
In this study, two cDNA libraries of W and R were constructed. Approximately 119 million raw sequencing reads were generated and assembled de novo, yielding 57,331 high quality unigenes with an average length of 739 bp. Using BLASTx, 38,460 unigenes were identified as putative homologs of annotated sequences in public protein databases, including NCBI NR, NT, Swiss-Prot, KEGG, COG and GO. Of these, 21,898 unigenes mapped to 128 KEGG pathways, with the metabolic pathways, secondary metabolites, glycerophospholipid metabolism, ether lipid metabolism, starch and sucrose metabolism, purine metabolism, and pyrimidine metabolism being well represented. Among them, many candidate genes were involved in flavonoid biosynthesis, transport and regulation. Furthermore, digital gene expression (DEG) analysis identified 3,257 unigenes that were differentially expressed between the two fruit developmental stages. In addition, 14,473 simple sequence repeats (SSRs) were detected.
Conclusions
Our results present comprehensive gene expression information about the cranberry fruit transcriptome that could facilitate our understanding of the molecular mechanisms of fruit development in cranberries. Although it will be necessary to validate the functions carried out by these genes, these results could be used to improve the quality of breeding programs for the cranberry and related species.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1842-4) contains supplementary material, which is available to authorized users.
Keywords: Cranberry, Illumina sequencing, Fruit transcriptome, Flavonoid
Background
The American, or large cranberries (Vaccinium macrocarpon Ait), renowned for their health benefits, are an important berry crop [1]. Today, cranberries are commercially cultivated in the USA, Canada, Chile, Europe and China. In 2012, 504,030 tonnes were produced worldwide [2]. Most commercial cranberries produce round and red fruits, which are useful for fresh and frozen products, juice, wine, fruit beverages, jelly and jam. Cranberries are a popular fruit because of their attractive color, special flavor and nutritional values [3].
Their nutritional quality is mainly determined by the production and accumulation of bioactive compounds. The bioactive compounds of V. macrocarpon have been analyzed previously in fresh fruit and cranberry juice by high-performance liquid chromatography (HPLC), ultraviolet–visible (UV/Vis) and mass spectrometer (MS) detection [4–6]. There have been 10,038 phytochemicals detected in cranberry metabolomics profiles [6]. The major bioactive compounds in cranberries are flavonoids, including anthocyanins, proanthocyanidins (PAs), flavonols, flavan-3-ols (catechins), and a series of phenolic acid derivatives [7]. These phytochemicals have high antioxidant potential and beneficial health properties, including the prevention and treatment of cardiovascular diseases, various cancers, obesity, and infections involving the urinary tract, dental health, and Helicobacter pylori-induced stomach ulcers and cancers [8–17].
The cranberry fruit is an accessory fruit that develops from the development of the inferior ovary, which consists of the ovary wall and the floral tube [18]. Fruit ripening has an impact on the levels of various phytochemicals, such as vitamins, flavonoids and the phenolic acid derivatives. Hence, the study of the growth and ripening of cranberry fruits is an important field of research, as it influences the quality of the fruit, affecting pigment accumulation, flavor, nutritional value, and functionality. In particular, pigment accumulation during fruit ripening takes place from a bright green to white, then pink, and finally red, conferring the natural pigmentation to mature fruits [19, 20]. Given the distribution of these phytochemicals vary differently among different plants, even within different populations of the same species and different organs of plants, for this reason the molecular mechanisms of their biosynthesis, transport and regulation might be diverse and complex. Therefore, it is essential to use modern genetic tools for dissect out the complexities involve with phytochemicals in cranberry.
There has been some previous research into the cranberry, including simple sequence repeats (SSRs) mining and validation, genetic map construction, and quantitative trait loci (QTL) analysis [21–23]. Recently, a sequencing of one cultivar of the cranberry was published [24]. However, information about the development of the fruit of the cranberry is still scarce. The application of next-generation sequencing (NGS) technologies such as deep-sequencing dependent RNA sequencing (RNA-Seq), in particular de novo sequencing, provides a cost-effective means for sequencing the transcriptome of an organism [19–23]. Among the new-generation sequencing methods, 454 pyrosequencing techniques and Illumina sequencing are widely used to analyze transcriptomes [25–29]. Compared with 454 pyrosequencing, Illumina HiSeq 2000 costs less and has a much greater output. This makes HiSeq 2000 an enabling approach for high-throughput RNA-Seq. HiSeq 2000 sequencing technology has become a valuable tool for the study of fruit trees, such as the black raspberry [30], Lycium chinense [31], and the longan [32].
In this work, we present a de novo assembly of the fruit transcriptome of V. macrocarpon using high-throughput Illumina HiSeq 2000 sequencing. Furthermore, differential gene expression between white and red fruits was investigated to reveal differential regulation of key pathways. This study provides an important genetic resource for understanding molecular mechanisms on the cranberry fruit ripening, and the results may be helpful for further gene expression and functional genomic studies, and molecular breeding of the cranberry.
Results and discussion
cDNA sequence generation, de novo assembly and mapping to the cranberry genome
To obtain a complete profile of the cranberry transcriptome during fruit development, two cDNA libraries were built for two fruit developmental stages: white fruit (W) and red fruit (R). 59,986,374 and 59,690,570 raw reads were generated from the W and R libraries, respectively. A summary of these sequencing results are presented in Table 1. After removing low quality short sequences, 52,413,112 and 53,352,808 clean reads for W and R, respectively, remained and were used for assembly. The Q20 percentages (sequencing error rate <1 %) and GC percentages obtained from the W and R libraries were 97.86 % and 46.93 %, and 97.93 % and 47.24 %, respectively. These results suggest that the sequencing data have sufficient quantity and quality to ensure accurate sequence assembly and adequate transcriptome coverage. Cleaned reads from each library were assembled independently using Trinity tool. Inchworm assembly of reads, the first step of Trinity, resulted in 105,377 and 114,265 contigs with mean sizes of 359 bp and 367 bp, and N50s of 739 bp and 783 bp, for W and R, respectively. After clustering with the TGICL software [33], the contigs were assembled into 69,540 unigenes for W with a mean length of 510 bp and an N50 of 763 bp, and 66,917 unigenes for R with a mean length of 597 bp and an N50 of 1,020 bp. At last, these two sets of unigenes were merged with TGICL resulting in a final assembly of 57,331 All-unigenes (with a total length of ~42 Mb), with a mean size of 739 bp and an N50 of 1,209 bp (Additional file 1). The N50 value is one of the most popular metrics to assess assembly quality, which reflects a continuous and complete assembly [34]. The N50 value of the cranberry fruit transcriptome is longer than that reported in previous studies on blueberry fruit transcriptomes (1,100 bp) [35]. The unigene size distribution showed the following: 76.1 % (43,643) of the unigenes were between 300 and 1,000 bp in length; 22.2 % (12,720) of the unigenes were between 1,000 and 3,000 bp; and 1.7 % (969) were more than 3,000 bp long (Fig. 1). The assembled unigenes were also mapped to the cranberry genome to examine the accuracy of the transcriptomes. 47,316 (82.5 %) unigenes were mapped to the cranberry genome sequence using the NCBI Mega BLAST program [36]. The remaining unigenes that did not map to the cranberry genome may be owing to differences between cultivars, gaps in the genome sequence or too short exons.
Table 1.
Overview of the sequencing and assembly
| W | R | All | |
|---|---|---|---|
| Total raw reads | 59,986,374 | 59,690,570 | |
| Total clean reads | 52,413,112 | 53,352,808 | |
| Total clean nucleotides (bp) | 4,717,180,080 | 4,801,752,720 | |
| Q20 percentage | 97.86 % | 97.93 % | |
| GC percentage | 46.93 % | 47.24 % | |
| Step-wise assembly | |||
| Contig | |||
| Total number of contigs | 105,377 | 114,265 | |
| Length of all contig (bp) | 37,852,028 | 41,902,139 | |
| Average length of contigs (bp) | 359 | 367 | |
| Contig N50 (bp) | 739 | 783 | |
| Unigene | |||
| Total number of unigenes | 69,540 | 66,917 | 57,331 |
| Length of all unigene (bp) | 35,488,277 | 39,943,235 | 42,391,141 |
| Average length of unigenes (bp) | 510 | 597 | 739 |
| Unigene N50 (bp) | 763 | 1,020 | 1,209 |
Fig. 1.
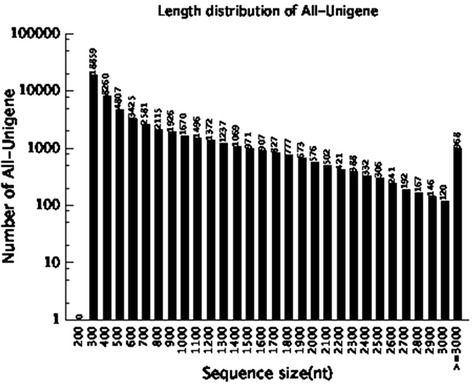
Length distribution of All-unigenes in cranberry fruit transcriptome
Functional annotation by searching similarity
For validation and annotation of the assembled unigenes, all unigenes were aligned against the NCBI non-redundant protein (NR), Swiss-Prot protein, Kyoto Encyclopedia of Genes and Genomes (KEGG), Clusters of Orthologous Group (COG) and Gene Ontology (GO) databases using BLASTx, and the nucleotide database (NT) by BlastN with an E-value threshold of 1e-5. A total of 38,460 unigenes (67.08 %) could be matched to known genes in the public databases. This suggests that many genes of unknown function play important roles during cranberry fruit development, and that this process is unexpectedly complex (Table 2). The identity distribution and species distribution were analyzed (Fig. 2). According to the E-value distribution of the top hits in the NR databases, 52.3 % of the matched sequences showed strong homology (<1e-45), while 47.8 % of the matched sequences showed moderate homology (between 1e-5 and 1e-45) (Fig. 2a). For the similarity distribution of the predicted proteins, 67.1 % of the sequences had a similarity higher than 60 %, whereas 32.8 % showed similarity between 18 % and 60 % (Fig. 2b). The species distribution of the top BLAST hits against the NR database for the cranberry fruit transcriptome showed that these unigenes had the greatest number of matches with genes of Vitis vinifera (37.5 %), followed by other species including Prunus persica (9.5 %), Solanum lycopersicum (9.3 %), Ricinus communis (7.1 %), Populus trichocarpa (6.9 %), Fragaria vesca subsp. vesca (4.0 %) and Glycine max (2.8 %) (Fig. 2c). The other 22.9 % unigenes had first hits with other species. This indicates that the transcriptome of V. macrocarpon is more closely related to that of V. vinifera than to other plant genomes that are present in current public databases. In summary, the annotation results suggest that the HiSeq 2000 sequencing project in this study generated a substantial number of assembled transcripts of cranberry fruits.
Table 2.
Statistics of annotation results against the public databases
| Public databases | Number of unigenes | Percentage of unigenes |
|---|---|---|
| NR | 36,411 | 63.51 % |
| NT | 29,884 | 52.13 % |
| Swiss-Prot | 24,033 | 41.92 % |
| KEGG | 21,898 | 56.94 % |
| COG | 14,758 | 38.20 % |
| GO | 27,286 | 47.59 % |
| All | 38,460 | 67.08 % |
Fig. 2.
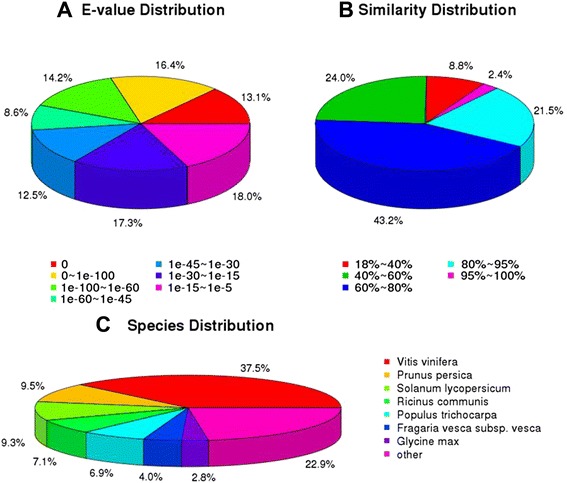
Distribution of the homology search of unigenes against the non redundant (NR) protein database. a E-value distribution of the top Blastx hits against the NR database for each unigene; (b) Similarly distribution of the top Blastx hits against the NR database for each unigene; (c) Species distribution of unigenes matching the top seven species using Blastx in the NR database
Functional classification by GO and COG
GO is a useful program for annotating and analyzing the functional categorization of annotated genes. To facilitate the organization of the cranberry transcripts into putative functional groups, GO terms were assigned using Blast2GO [37]. A total of 27,286 unigenes (47.59 %) were assigned into GO ontologies based on their similarity to sequences with previously known functions, including 31,474 sequences assigned to the molecular function category, 104,552 to the biological process category and 83,221 to the cellular component category (Table 2 and Fig. 3). The assigned sequences were divided into 55 functional subcategories. Because 86 % of the unigenes were assigned to more than one GO term, the total number of GO terms was larger than the total number of the unigenes with GO assignments. “Cellular process”, “cell” and “catalytic activity” were the dominant subcategories in the biological process, cellular component and molecular function categories, respectively. Moreover, “metabolic process”, “single-organism process”, “cell part”, “organelle” and “binding” were also well represented, which suggests that many novel genes involved in extensive metabolic activities could be playing important roles during the growth and development stages of the fruit. However, few genes were assigned to the categories “locomotion”, “extracellular matrix”, “extracellular matrix part”, “extracellular region part”, “virion”, “virion part”, “channel regulator activity”, “metallochaperone activity”, “nutrient reservoir activity”, “protein tag”, and “translation regulator activity”. Overall, these GO terms account for almost half of the overall unigenes in the cranberry fruit transcriptomic dataset, suggesting that a large number of cranberry unigenes may be conserved across different plant species.
Fig. 3.
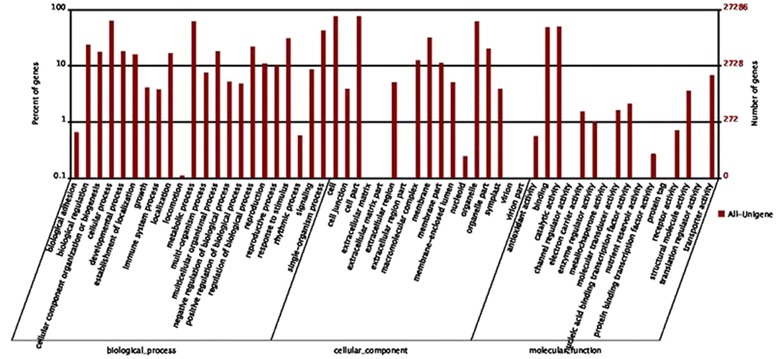
Histogram of GO analysis of All-Unigenes
The COG database can phylogenetically predict and classify the completeness of the unigene sequences. Based on sequence homology, 14,758 unigenes (25.7 % of all the unigenes) were assigned into 25 COG categories (Fig. 4). “General function prediction only” represented the largest group (4,511, 15.5 %), followed by “transcription” (2,690, 9.24 %), “translation, ribosomal structure and biogenesis” (2,317, 7.96 %) and “posttranslational modification, protein turnover, chaperones” (2,292, 7.87 %). “RNA processing and modification” (174, 0.60 %), “extracellular structures” (11, 0.04 %) and “nuclear structure” (4, 0.01 %) were the smallest groups. It is noteworthy that 816 unigenes (5.5 %) were classified into the group of “secondary metabolites biosynthesis, transport and catabolism”, suggesting that those secondary metabolite processes play important roles in cranberry fruit development.
Fig. 4.
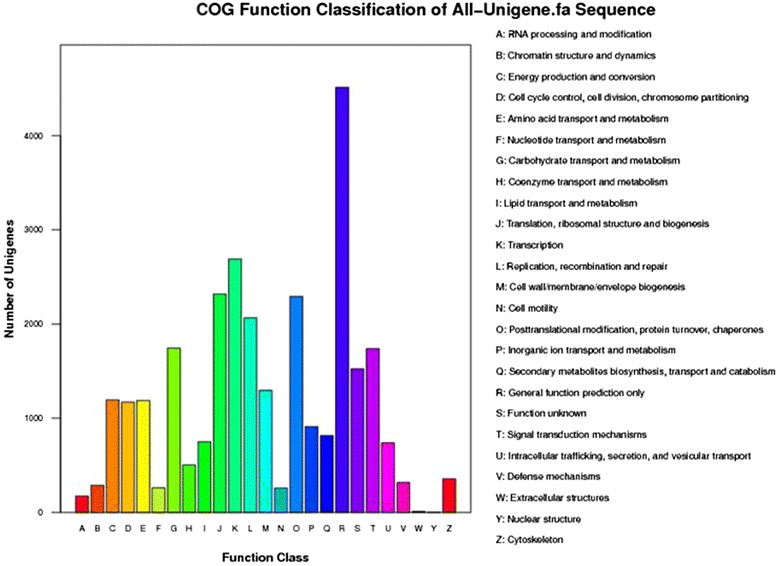
Histogram of COG classification of All-Unigenes
KEGG pathway mapping
KEGG analysis can help us further understand the specific processes, gene functions and gene interactions at a transcriptome level. In our study, 21,898 (38.2 %) all-unigenes were mapped to 128 predicted metabolic pathways through the KEGG database (Table 2 and Additional file 2). The largest category was metabolic pathways (5,522) which included biosynthesis of secondary metabolites (2,603), glycerophospholipid metabolism (973), ether lipid metabolism (824), starch and sucrose metabolism (578), purine metabolism (574), pyrimidine metabolism (501) and other subcategories. In the secondary metabolism category, 31 subcategories comprised 2,603 unigenes, the most represented of which were phenylpropanoid biosynthesis (313), flavonoid biosynthesis (228), limonene and pinene degradation (192), stilbenoid, diarylheptanoid and gingerol biosynthesis (169), terpenoid backbone biosynthesis (150), zeatin biosynthesis (136), carotenoid biosynthesis (130), and flavone and flavonol biosynthesis (108). Moreover, isoflavonoid biosynthesis (53), folate biosynthesis (34), anthocyanin biosynthesis (26), and betalain biosynthesis (6) were also classified. These results suggest that the secondary metabolic processes are active pathways in cranberry fruit development. In addition to metabolism pathways, genes involved in genetic information processing (6,781) and cellular processes (1,718) were highly represented categories. Endocytosis, ribosome, RNA transport, spliceosome, protein processing in endoplasmic reticulum, ribosome biogenesis in eukaryotes, RNA degradation, mRNA surveillance pathway, ubiquitin mediated proteolysis, RNA polymerase, phagosome and peroxisome were included in these categories.
Candidate genes involved in flavonoid biosynthesis
Given that cranberries are rich in flavonoids, we focus on identifying the candidate genes involved in flavonoid biosynthesis. In plants, flavonoids are a group of polyphenolic secondary metabolites, which play many diverse physiological functions [38]. The flavonoid biosynthetic pathway has largely been characterized in Arabidopsis thaliana, Zea mays, and V. vinifera [39–41]. However, the overall molecular mechanism of flavonoid biosynthesis and accumulation in the cranberry is not fully understood.
Multiple transcripts encoding almost all known enzymes involved in flavonoid biosynthesis were identified in the annotated cranberry fruit transcriptome. A brief schematic is shown in Fig. 5, which is modified version from Jaakola’s figure [42]. The biosynthesis of anthocyanins, PAs and flavonols shares the upstream phenylpropanoid pathway activated by a cytosolic multienzyme complex [43, 44]. In particular, some of these enzymes belong to the cytochrome-P450 family [45, 46]. First, phenylalanines are converted to chalcones via the phenylpropanoid pathway by the enzymes phenylalanine ammonia lyase (PAL, 11 unigenes), cinnamate 4-hydroxylase (C4H, 4 unigenes), 4-coumarate CoA ligase (4CL, 13 unigenes) and chalcone synthase (CHS, 2 unigenes). Subsequently, chalcone isomerase (CHI, 3 unigenes) catalyzes the stereo-specific cyclization of chalcones into naringenins or flavanoes; flavanone 3-hydroxylase (F3H, 6 unigenes) catalyzes the hydroxylation of flavanones to form the dihydrokaempferols that are continually converted to dihydroquercetins by flavonoid 3′-hydroxylase (F3′H, 6 unigenes), or converted to dihydromyricetins by flavonoid 3′,5′-hydroxylase (F3′5′H, 7 unigenes). Finally, flavonol synthase (FLS, 3 unigenes) catalyzes the conversion of dihydrokaempferols, dihydroquercetins and dihydromyricetins to flavonols.
Fig. 5.
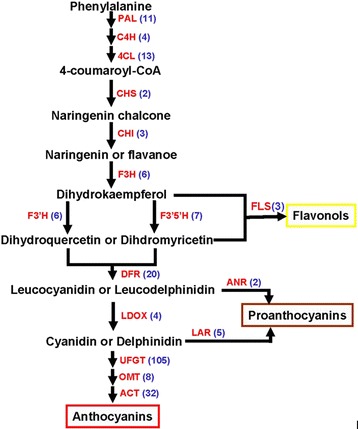
A schematic representation of the anthocyanin biosynthetic pathway emphasizing the anthocyanins, proanthocyanidins and flavonols found in blueberry. Enzyme abbreviations: PAL, phenylalanine ammonia lyase; C4H, cinnamate 4-hydroxylase; 4CL, 4-coumarate CoA ligase; CHS, chalcone synthase; CHI, chalcone isomerase; F3H, flavonoid 3-hydroxylase; F3′H, flavonoid 3′-hydroxylase; F3′5′H, flavonoid 3′,5′-hydroxylase; DFR, dihydroflavonol 4-reductase; LDOX, leucoanthocyanidin dioxygenase; FLS, flavonol synthase; LAR, leucoanthocyanidin reductase; ANR, anthocyanidin reductase; UFGT, UDP-glucose flavonoid 3-O-glucosyl transferase; OMT, O-methyltransferase; ACT, anthocyanin acyltransferase
In the anthocyanin branch, the dihydroquercetins and dihydromyricetins are converted to leucocyanidins or leucodelphinidins by dihydroflavonol 4-reductase (DFR, 20 unigenes). Leucoanthocyanidin dioxygenase (LDOX, 4 unigenes), known as anthocyanidin synthase (ANS), converts leucocyanidins or leucodelphinidins to cyanidins or delphinidins, that are the first colored compounds in the anthocyanin pathway. The final modification steps in the anthocyanin pathway are glycosylation by UDP-glucose flavonoid 3-O-glucosyl transferase (UFGT, 105 unigenes), methylation by O-methyltransferase (OMT, 8 unigenes) and acylation by anthocyanin acyltransferase (ACT, 32 unigenes). The synthesis of PAs branches off the anthocyanin pathway after the reduction of leucoanthocyanins (or anthocyanidins) to catechins (or epicatechins). PAs, also known as condensed tannins, are oligomeric or polymeric phenolics that result from the polymerization of flavan-3-ol units. Leucoanthocyanidin reductase (LAR) and anthocyanidin reductase (ANR) are both key enzymes of PA biosynthesis. For the PA pathway, the formation of flavan-3-ols (2,3-cis-(−)-flavan-3-ols and 2,3-trans-(+)-flavan-3-ols) is achieved by LAR (5 unigenes) and ANR (2 unigenes).
When comparing the cranberry transcriptome to the blueberry fruit transcriptome [35], we found that the cranberry fruit transcriptome data show more abundant flavonoid biosynthesis enzymes (Table 3), especially the number of UFGT enzymes. This suggests that the cranberry contains diverse flavonoid compounds, which have different chemical, size, three-dimensional shape, and physical and biochemical properties. In a previous study that carried out cranberry genome assembly and generated a leaf transcriptome dataset, it was suggested that the cranberry might be lacking two enzymes, F3′5′H and LAR [24]. However, we found seven unigenes encoding F3′5′H and five unigenes encoding LAR in the cranberry fruit transcriptome. Given that flavonoid biosynthesis has temporal and spatial properties, RNA-Seq data of special organs or tissues from different growth and developmental stages could dissect out the complexities involved in flavonoid biosynthesis. Therefore, our cranberry fruit transcriptome undoubtedly provides a powerful supplement to the previous cranberry genome and leaf transcriptome dataset. Furthermore, the metabolomics results showed that the American cranberry contains small amounts of Malvidin, Pelargonidin, Delphinidin, Petunidin, (+)-catechin and (epi)gallocatechins [6, 7]. Therefore, it is not surprising that F3′5′H and LAR are expressed in cranberry fruit.
Table 3.
The numbers of candidate genes involved in flavonoid transport in the cranberry fruit transcriptome
| Family | Number of annotated sequences |
|---|---|
| ATP-binding cassette (ABC) transporter | 341 |
| H+-ATPases | 174 |
| glutathione S-transferase (GST) | 78 |
| multidrug and toxic compound extrusion (MATE) | 75 |
| H+-PPases | 41 |
| soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNARE) | 30 |
| vacuolar sorting receptor (VSR) | 6 |
Candidate genes involved in flavonoid transport
According to previous research performed in plant species including Arabidopsis and grape, two mechanisms have been proposed to explain both flavonoid transport from the ER to the vacuole, and the reverse transport from storage sites to other cell targets [47]. Flavonoids could be accumulated into vacuoles or cell walls by the membrane transporter-mediated transport (MTT) system. The proton gradient between the cytosol and the vacuole (or cell wall) by H+-ATPases (and H+-PPases in the tonoplast) has been proposed to be the main driving force for the transport of some flavonoids [48]. ATP-binding cassette (ABC) transporters have also been claimed to play a role in sequestration of flavonoids into the vacuole [49–51]. In particular, multidrug and toxic compound extrusion protein (MATE) transporters have been described as participating in flavonoid vacuolar sequestration in the tomato, Arabidopsis and grapes [48, 52, 53]. Besides the MTT system, the membrane vesicle-mediated transport (MVT) mechanism could also be involved in flavonoid accumulation. The MVT mechanism involves flavonoid-containing vesicles releasing their content into the accumulation targets by fusion [54]. These vesicles require vacuolar sorting receptor (VSR) proteins and soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNARE) proteins to be addressed to the correct compartment and fuse to the membrane target [47]. Additionally, glutathione S-transferase (GST) gene has also been demonstrated to catalyze the conjugation of the tripeptide glutathione (c-Glu-Cys-Gly, GSH) as flavonoid binding proteins, and also be responsible for vesicle uploading or vacuolar transport [55, 56]. In the cranberry fruit transcriptome, 341 unigenes encoding ABC, 75 unigenes encoding MATE, 174 unigenes encoding H+-ATPases and 41 unigenes encoding H+-PPases were identified. Moreover, 78 unigenes encoding GST, 6 unigenes encoding VSR and 30 unigenes encoding SNARE were found. These results imply that the two distinct transport mechanisms (MTT and MVT) could both be present, and transport may be a multifactorial process in the cranberry, involving different strategies and the contribution of several enzymes. Unlike similar studies in the carnation and the grape vine, we did not find bilitranslocase (BTL), which is a putative flavonoid that is localized in the liver and gastric mucosa in mammals [57, 58]. These findings provide a powerful genomic tool to research the mechanisms of transport and accumulation of flavonoids in the cranberry.
Candidate transcription factors involved in flavonoid biosynthesis and transport
The main structural genes encoding enzymes involved in the flavonoid biosynthetic pathway have been studied in many species, including Arabidopsis, maize, the petunia, the snapdragon, apples and grapes [59]. In particular, the expression of structural genes involved in flavonoid synthesis is largely controlled by basic helix–loop–helix (bHLH) transcription factors (TFs), MYB proteins and WD-repeat-containing proteins [60, 61]. bHLH TFs belong to multigenic families and are structurally organized into basic helix-loop-helix DNA-binding conserved motifs [62, 63]. MYB TFs are a large gene family that is thought to be one of the most important plant regulatory gene families [64, 65]. In plants, MYB TFs have been demonstrated to be involved in control of phenylpropanoid secondary metabolism, including the biosynthesis of flavonoids and anthocyanins [66–69]. WD-repeat-containing proteins consist of four or more copies of the WD (tryptophan-aspartate) repeat, which is a sequence motif approximately 31 amino acids long that encodes a structural repeat [70]. It has been reported that certain MYB TFs interact with bHLH and WD40 proteins to form a MYB-bHLH-WD40 complex in the regulation of the flavonoid biosynthetic pathway [71, 72]. Moreover, TFs also control the regulation of flavonoid transport. For example, Arabidopsis R2R3-MYB TF (AtTT2) regulates the expression of the MATE transporter gene TT12 to control the flavonoids in developing siliques; and the maize ABCC transporter protein ZmMRP3 is regulated by the R (bHLH family) and C1 (R2R3-MYB) TFs to control anthocyanin transport [73, 74]. Based on our results, a total of 669 unigenes were identified as putative TFs or regulators, with 641 of the unigenes belonging to 65 subclasses (Table 4). The unigenes encoding WD40 (202), bHLH (53), MYB (41) and WRKY (41) are the most abundant in the cranberry fruit transcriptome. By comparison to the number of WD40 unigenes detected in the blueberry fruit transcriptome, the cranberry shows more than twice the number of WD40 unigenes. These results imply that the WD40 proteins may be playing an important role in the regulation of flavonoid biosynthesis and transport. Because all major TF subclasses known to be involved in regulating anthocyanin biosynthesis are highly expressed in the cranberry fruit transcriptome, we conclude that flavonoid biosynthesis and transport are regulated by TFs in the cranberry, such as WD40, bHLH, MYB and WRKY. The control mechanism of flavonoid biosynthesis and transport is complex in the cranberry, and may be independently regulated by a single TF, or controlled by combinations of TF complexes.
Table 4.
The numbers of transcription factors involved in flavonoid biosynthetic and transport in the cranberry fruit transcriptome
| Family | Number of annotated sequences | Family | Number of annotated sequences |
|---|---|---|---|
| WD40 | 202 | RNA polymerase II | 3 |
| bHLH | 53 | UNE | 3 |
| MYB | 41 | CRF | 2 |
| WRKY | 41 | global | 2 |
| GATA | 28 | HD | 2 |
| TCP | 19 | ICE | 2 |
| bZIP | 18 | jumonji domain-containing | 2 |
| ERF | 18 | MYC | 2 |
| nuclear TF | 18 | RAP | 2 |
| GTE | 17 | RAV | 2 |
| GRAS | 16 | RAX | 2 |
| AP2 | 15 | sfc | 2 |
| HSF | 10 | WER | 2 |
| trihelix | 10 | WRI | 2 |
| BTF | 8 | AS | 1 |
| ccaat-binding | 7 | COL | 1 |
| PIF | 7 | CPP | 1 |
| TGA | 7 | DOF | 1 |
| ILR | 6 | DPB | 1 |
| NAC | 5 | DRE | 1 |
| RF | 5 | yellow 2 | 1 |
| ASG | 4 | HY5 | 1 |
| BIM | 4 | IIIB | 1 |
| general | 4 | KNOTTED | 1 |
| HBP | 4 | m50 | 1 |
| KAN | 4 | NAP | 1 |
| scarecrow | 4 | Pur-alpha | 1 |
| APETALA | 3 | RWP-RK | 1 |
| B3 | 3 | SPATULA | 1 |
| CPC | 3 | UPBEAT | 1 |
| E2F | 3 | VIP | 1 |
| MADS-box | 3 | YABBY | 1 |
| ORG | 3 | others | 28 |
SSR motif discovery
SSRs have been widely used in the study of genetic identification and fingerprint mapping, due to their high polymorphic information content, simple technology, and high reproducibility. The transcriptome is also an important resource for rapid and cost-effective development of genetic markers. To further evaluate the quality of assembly and develop new molecular markers, the cranberry unigenes generated in this study were used to identify SSRs. The distribution of mono-, di-, tri-, quad-, penta- and hexa-nucleotide SSRs in these unigenes are shown (Table 5). A total of 14,473 SSRs were identified in 11,980 unigenes. Of the11,980 unigenes, 2,215 and 851 unigenes contained more than one SSR and SSRs in compound formation, respectively. This indicates that nearly 21 % of the cranberry unigenes contained SSRs, which is higher than the SSR ratio (8.6 %) reported in the cranberry leaf transcriptome, but lower than the SSR ratio (37.8 %) in the cranberry genome [24]. The largest fraction of SSRs identified was di-nucleotide repeats (62.4 %), followed by tri- (17.4 %) and mono- repeats (17.3 %). The di-nucleotide repeats were the most abundant type, accounting for 62.4 %, which is higher than the findings in the cranberry genome (44 %) and the leaf transcriptome (35 %) [24]. SSR motifs were further analyzed for the number of repeating units, and the most represented repeats of potential SSRs was nine (Additional file 3). The most frequent mono-, di-, tri-, quad-, and penta-nucleotide motifs were A, GA, GAA, AAAG and AAAAG, accounting for 8.7 %, 18 %, 1.4 %, 0.3 % and 0.8 %, respectively. These results show that the SSRs identified in the cranberry fruit transcriptome were GA rich, which is similar to the earlier reports on the cranberry genome (16.5 %) and the leaf transcriptome (15 %). The SSRs identified in the present study provide an abundant resource for molecular marker studies in the cranberry.
Table 5.
Summary of SSR identified in cranberry fruit transcriptome
| Searching item | Numbers |
|---|---|
| Total number of sequences examined | 57,331 |
| Total size of examined sequences (bp) | 42,391,141 |
| Total number of identified SSRs | 14,473 |
| Number of SSR containing sequences | 11,980 |
| Number of sequences containing more than 1 SSR | 2,115 |
| Number of SSRs present in compound formation | 851 |
SSR marker validation
We designed primer pairs for 12 unigenes from the GO term “response to stimulus/chemicals”. The sequences of the primers are in Additional file 4. Silver stained polyacrylamide gels results showed that 11 pairs (91.7 %) successfully amplified bands. Among the 11 primer pairs, four primer pairs that showed polymorphic bands and seven that showed monomorphic bands. The four polymorphic SSR loci are shown in Fig. 6. The primers for CL2296.Contig2 amplified bigger band (~200 bp) than expected size (Fig. 6a), that may be due to the regions between the two primers contain introns. The primers for CL284.Contig2 amplified expected band (Fig. 6b), the primers for CL2216.Contig1 and CL2216.Contig4 both amplified two alleles (Fig. 6c, d). Thus, these results indicate that the novel SSR primers show good transferability to cultivars of cranberry. However, the polymorphic rate is low owing to the conserved nature of expressed sequence tag SSRs (EST-SSRs) [75]. Therefore, the novel primers developed may represent highly-conserved genes involved in the response to stimuli/chemicals, which would be useful for the breeding of resistant cranberry cultivars and other Vaccinium plants in future studies.
Fig. 6.
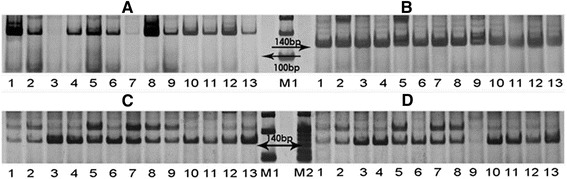
Silver stained polyacrylamide gels of four SSRs on CL2296.Contig1 a, CL284.Contig2 (b), CL2216.Contig1 (c) and CL2216.Contig4 (d); PCR products from line1 to line13 are Pilgrim, Stankawich, Howes, Bergmen, WAU108, Brewer, Bain Fav. No.1, Bain11, Le Muryon, Hollister Red, Mathewes, Bain6 and Washington, respectively. (M1: Marker DL2000; M2: Marker DL100)
Difference in gene expression between white and red fruit
To obtain a digital expression profile of the differentially expressed genes between the two fruit development stages in the cranberry, we used the FPKM (Fragments per kb per million fragments) method to perform gene expression analysis between the W and R libraries [76]. The transcripts with at least a two-fold difference between white and red fruits are shown in Fig. 7. A total of 3,257 unigenes were identified as differentially expressed genes (DEGs) between the two developmental stages of cranberry fruit (Fig. 8). Among them, 2,125 and 1,132 unigenes were highly expressed in the red and white fruits, respectively. More genes were highly expressed in red fruits, suggesting that more genes were involved in complex metabolites during the full ripening stage of the fruit. Moreover, 3,010 and 1,328 unigenes were only expressed in red and white fruits, respectively. This indicates that the unigenes may be specifically expressed in the different development stages of cranberry fruits.
Fig. 7.
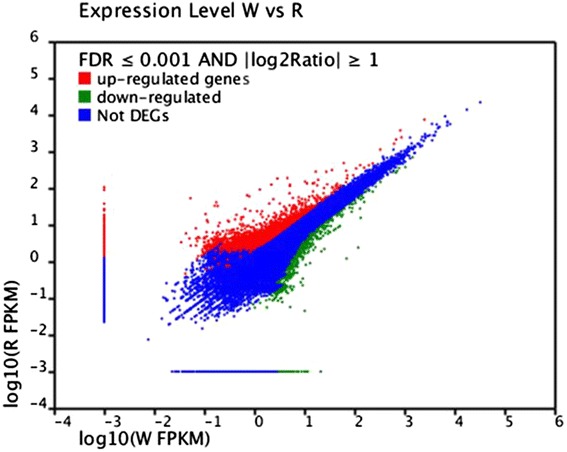
Gene transcription profile between W and R libraries. For comparing gene expression level between the two libraries, each library was normalized to 1 million tags. Red dots represent transcripts more prevalent in R library, green dots show those present at a lower frequency in W library and blue dots indicate transcripts that did not change significantly
Fig. 8.
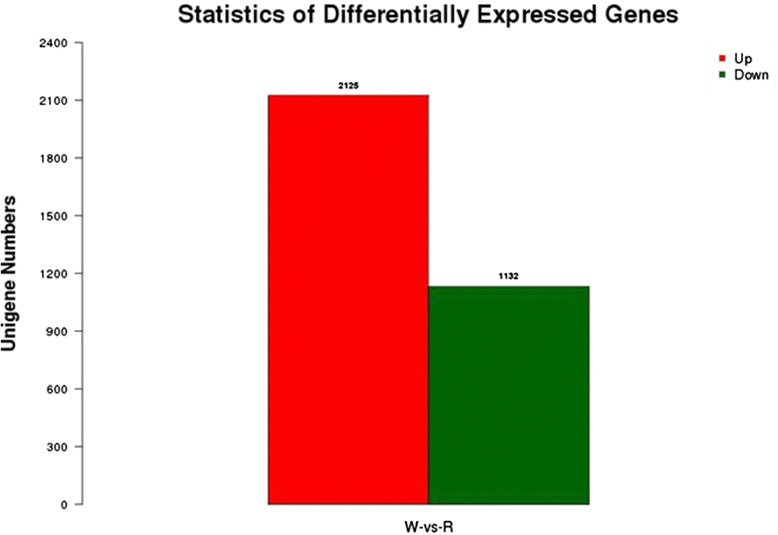
The number of up-regulated and down-regulated genes between W and R libraries
We identified 1,246 DEGs that were assigned to 49 significantly enriched GO terms. The most abundant GO terms were “metabolic process”, “cell and cell part” and “catalytic activity” in the biological process, cellular component and molecular function categories, respectively (Fig. 9). Moreover, there are some interesting biological processes which are associated with fruit development, such as “developmental process”, “growth”, “reproduction”, “reproductive process”, “antioxidant activity” and “transporter activity”.
Fig. 9.
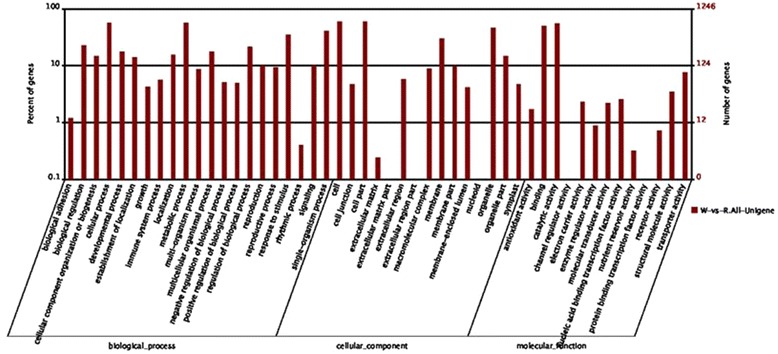
Histogram of GO analysis of DEGs
We also found that a total of 113 pathways were affected by 1,134 DEGs (Additional file 5). Notably, the greatest number of DEGs were mapped to “metabolic process” (384, 33.86 %) and “biosynthesis of secondary metabolites” (202, 17.81 %). In secondary metabolites biosynthesis, 39 (3.44 %) DEGs are mapped to “phenylpropanoid biosynthesis”, 28 (2.47 %) DEGs to flavonoid biosynthesis, 15 (1.32 %) DEGs to flavone and flavonol biosynthesis, 8 (0.71 %) DEGs to benzoxazinoid biosynthesis, 16 (1.41 %) DEGs to stilbenoid, diarylheptanoid and gingerol biosynthesis, 1 (0.09 %) DEG to betalain biosynthesis, 3 (0.26 %) DEGs to isoquinoline alkaloid biosynthesis, 3 (0.26 %) DEGs to tropane, piperidine and pyridine alkaloid biosynthesis, 1 (0.09 %) DEG to anthocyanin biosynthesis, and 2 (0.18 %) DEGs to isoflavonoid biosynthesis. This would reflect a higher specialization in partitioning the secondary metabolism intermediates towards different classes of end derivatives which accumulate in the same organ during the different stages of berry development.
To validate the data from our digital expression analysis, quantitative Real Time PCR (qRT-PCR) assays were performed on 10 DEGs involved in the phenylpropanoid biosynthesis, flavone and flavonol biosynthesis, and flavonoid biosynthetic pathways (Table 6). The qRT-PCR expression pattern of these unigenes is shown in Fig. 10. Except for the unigenes encoding UDP-glycosyltransferase (CL754.Contig1_All), cytochrome P450 (CL1200.Contig1_All, CL1200.Contig2_All, CL3597.Contig3_All, CL3597.Contig4_All, CL3597.Contig5_All and Unigene7973_All), flavonoid 3′ hydroxylase (Unigene20161_All), carboxylesterase (CL1068.Contig2_All), and UDP-glycosyltransferase (Unigene 24441_All), the unigenes were up-regulated in the red fruit stage. The results of the qRT-PCR are consistent with our digital expression data (Table 6). This also showed an association with flavonoid production, and it is likely that these genes are involved in the fruit development stage. Plant cytochrome P450s are a large superfamily of heme-containing monooxygenases, which are involved in a wide range of secondary metabolite biosynthetic reactions, such as terpenoids, phenylpropanoids, and nitrogen-containing compounds, including alkaloids, cyanogenic glucosides, and glucosinolates [77]. Therefore, the selected unigenes encoding cytochrome P450s may play important roles in flavonoid accumulation in cranberry fruit.
Table 6.
All-unigene selected for qRT-PCR in Illumina/Solexa sequencing library of cranberry fruit
| Gene ID | Gene Length | W_FPKM | R_FPKM | log2 Ratio | Up/Down | Nr-annotation |
|---|---|---|---|---|---|---|
| CL1200.Contig1_All | 2042 | 6.2174 | 34.4668 | 2.4708 | Up | cytochrome P450 |
| CL1200.Contig2_All | 930 | 1.421 | 23.6309 | 4.0557 | Up | cytochrome P450 |
| CL3597.Contig3_All | 343 | 7.7056 | 56.6377 | 2.8778 | Up | cytochrome P450 |
| CL3597.Contig4_All | 2327 | 1.1155 | 2.909 | 1.3828 | Up | cytochrome P450 |
| CL3597.Contig5_All | 2143 | 1.894 | 5.2357 | 1.4669 | Up | cytochrome P450 |
| Unigene7973_All | 1103 | 15.7038 | 55.2759 | 1.8155 | Up | cytochrome P450 |
| Unigene20161_All | 1637 | 4.0652 | 12.8019 | 1.655 | Up | flavonoid 3'-hydroxylase |
| CL1068.Contig2_All | 200 | 3.0678 | 9.9684 | 1.7002 | Up | carboxylesterase 2-like |
| CL754.Contig1_All | 628 | 5.0353 | 11.5911 | 1.2029 | Up | UDP-glycosyltransferase |
| Unigene24441_All | 654 | 0.7938 | 6.2386 | 2.9744 | Up | UDP-glycosyltransferase |
Fig. 10.
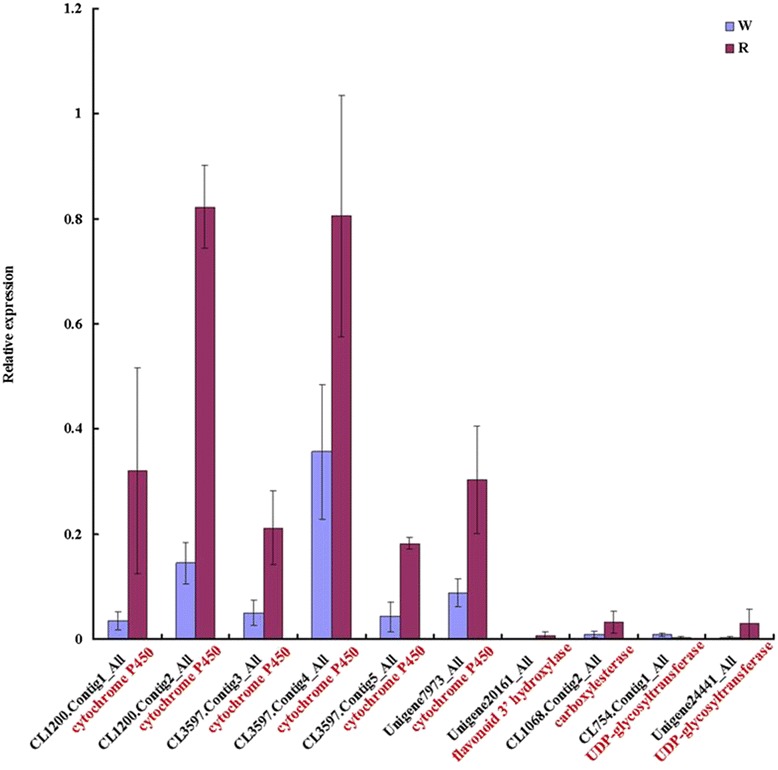
The expression profiles of 10 unigenes between W and R libraries in cranberry
Conclusions
A high-quality transcriptomic dataset of the cranberry fruit was obtained using the Illumina HiSeq 2000 sequencing platform. A significant number of important metabolic pathways and functions associated with the unigenes were identified. Moreover, a large number of SSRs were identified that can be used for subsequent marker development, genetic linkage and QTL analysis. Many candidate genes that are potentially involved in flavonoid biosynthesis, transport and regulation were identified and are worthy of further functional research. Our study provides the largest number of unigenes to date, and lays the groundwork for in-depth transcriptomic profiling of cranberry fruits.
Materials and methods
Plant material and tissue collection
Six-year-old cranberry plants (V. macrocarpon ‘Bergman’) were grown in Changchun, Jilin Province in northeast China. This cultivar is a cross between ‘Early Black’ and ‘Searles’ with a basic chromosome number x = 12 (2x = 2n = 24) [78]. It propagates by asexual propagation of cutting and micro-propagation, which permits homogeny among individual plants. Ten plants were selected as a sample. We removed the weak buds and opened the flowers, keeping the flowers at the full pink bud stage with forceps. The flowers can be thinned to one per stem to ensure a better fruit set. Fruit samples (almost 20 fruits of the same maturation degree) were collected based on the skin color of the fruit and the development period in days. Fruit sampling included two ripening stages, the white fruit (30 days after full bloom, stage W) and red fruit (60 days after full bloom, stage R). For both of the stages, all of the fruit, including pulp, skin and seeds, were mixed, and immediately frozen in liquid nitrogen and kept at −80 °C until they were used for sample preparation.
Library preparation and sequencing
Total RNA was isolated by TRIzol® Reagent (Invitrogen, Carlsbad, CA, USA). The RNA quantity and quality were assessed by a Nanodrop 2000 instrument (Thermo Scientific, USA) and an Agilent 2100 Bioanalyzer (Agilent Technologies, California, USA). The cDNA libraries were constructed by mixing equal quantities of RNA from each fruit stage, including pericarp, pulp and seeds, according to the manufacturer’s instructions (Illumina, San Diego, CA, USA). First, magnetic beads with oligo(dT) molecules were used to enrich for poly(A) mRNA from 20 μg of total RNA. Then, samples were fragmented into short pieces and reverse transcribed into cDNA with a PrimeScript™ 1st Strand cDNA Synthesis Kit (Takara). The second-strand cDNA was then synthesized using buffer, dNTPs, RNase H, and DNA polymerase I. Short fragments were purified using the QIAquick PCR Purification Kit (QIAGEN) and resolved with EB buffer for end repair and single nucleotide A (adenine) tailing. After this, short fragments were connected with sequencing adapters, and enriched by 15 cycles of PCR amplification to select suitable fragments for the final cDNA library. The quantification and qualification of the cDNA library were characterized by an Agilent 2100 Bioanalyzer and ABI StepOnePlus Real-Time PCR System, and sequenced on the Illumina HiSeq™ 2000 pair-end system. Illumina sequencing was performed at the Beijing Genomics (BGI) Center in Shenzhen, China.
Sequence assembling and analysis
The raw reads generated from the Illumina sequencing were pre-processed with the filter-fq software (unpublished software). The whole sequences of reads only with adaptor, reads containing more than 5 % unknown nucleotides, and low quality reads (reads containing more than 20 % bases with Q-value ≤ 10) were eliminated. The clean reads were assembled into contigs using the Trinity tool with the following parameters: seqType = fq, min_contig_length = 100, min_glue = 3, group_pairs_distance = 250, path_reinforcement_distance = 85, bfly_opts = ′-V 5 --edge-thr = 0.05 --stderr', min_kmer_cov = 3, CPU = 7 [79]. First, the RNA-Seq data were assembled into longer, gapless unique sequences with the Inchworm software. The resulting sequence from Inchworm was defined as contigs. Subsequently, the contigs were clustered and complete de Bruijn graphs were constructed for each cluster using the Chrysalis software. Then, the Butterfly software was used to track the paths that reads and pairs of reads took within the graphs. As multiple samples from the same species were sequenced, unigenes from each sample’s assembly were taken for further processing with the sequence clustering software TGICL, for sequence splicing and overlap removal to acquire non-redundant unigenes that were as long as possible [33]. At last, the unigenes from each sample were assembled for unique All-unigene sequences by TGICL software. For gene family clustering, the unigenes were divided into two classes: clusters and singletons. The former were prefixed ‘CL’ and the cluster id followed this prefix. In one cluster, the similarity between unigenes was more than 70 %. For the singletons, the prefix was ‘unigene’. To assess the quality of the de novo assembly, a similarity search against the cranberry genome was further conducted using the Mega BLAST algorithm with a score greater than 200 [36].
All unigene sequences were aligned by BLASTx (E-value < 0.00001) to protein databases such as NR, Swiss-Prot, GO, COG and KEGG, and to NT using the BlastN (E-value < 0.00001), retrieving proteins with the highest sequence similarity with the given unigenes, along with their protein functional annotations [80, 81]. When a unigene was not aligned to any of the above databases, ESTScan software was used to decide upon the sequence direction [82]. For NR annotation, the Blast2GO program was used to get the GO annotation of unigenes [37]. Then, WEGO software was used to conduct GO functional classification for all unigenes and to understand the distribution of the gene functions [83].
SSR detection and validation
MicroSAtellite (MISA) (http://pgrc.ipk-gatersleben.de/misa/misa.html) was used to identify SSRs in the assembled transcriptome of the cranberry fruit. SSR motifs were identified between one and six nucleotides in size and with 1, 2, 3, 4, 5 and 6 repeats for mono-, di-, tri-, quad-, penta-, and hexamers, respectively. The sequences with a minimum of 150 bp flanking region from SSR loci were used to design primers using PRIMER 3.0 software [84]. The major primer design parameters were as follows: a PCR product length of 80–300 bp; a primer size of 18–28 bp (optimum 23 bp); a melting temperature of 51–58 °C (optimum 60 °C); and a temperature difference between the forward primer and reverse primer less than 2 °C. Then, the final primers were obtained by filtering rules as follows: no SSRs in the primer; align the primers to the unigene sequence with the 5′ site allowed three mismatches and the 3′ site allowed one mismatch; remove the primers which aligned to more than one unigene; find SSRs on the product sequences by ssr_finder (http://www.fresnostate.edu/ssrfinder/) and keep the product for which the result of ssr_finder is the same as the result of MISA.
Thirteen cranberry cultivars were used to validate the SSR markers by PCR. Fresh leaves were collected for genomic DNA extraction by the cetyltrimethylammonium bromide (CTAB) method [85] with a minor modification. The PCR reaction was performed in a 20 μl reaction volume containing 2 μl of 10× PCR Buffer, 0.4 μl of dNTPs (10 mmol/L), 0.4 μl of genomic DNA (100 ng/μl), 0.8 μl of both the forward primer and reverse primer (10 μM), and 0.1 μl of Taq polymerase (5 U/μl). The PCR conditions used were as follows: initial denaturation for 2 min at 94 °C, followed by 35 cycles of denaturation for 30 s at 95 °C, annealing for 30 s at 55 °C (primer specific), and extension for 30 s at 72 °C, followed by a final extension for 5 min at 72 °C, ending at 4 °C. The amplification products were separated by 6 % native polyacrylamide gel electrophoresis and were visualized by silver staining.
Differential gene expression analysis
An analysis of statistical comparison was performed to predict genes with different expression levels using the method previously described by Audic and Claverie [86]. The calculation of unigene expression used the FPKM method and SOAP (http://soap.genomics.org.cn/soapaligner.html), which were able to eliminate the influence of different gene lengths and sequencing levels on the calculation of gene expression [76]. Therefore, the calculated gene expression could be directly used to compare the difference in gene expression between the samples. The FPKM formula was:
where C is the number of cleaned reads that were uniquely aligned to one unigene; N is the total number of cleaned reads that were uniquely aligned to all unigenes; and L is the base number in the CDS (Coding sequence) of one unigene. The expression between two samples can also be assessed with the FDR (False Discovery Rate) method, which is a statistical method used in multiple hypothesis testing to correct for P-value [87]. In our analysis, an estimated absolute value of log2-fold change of ≥2 and FDR adjusted P-value ≤ 0.001 were used as the threshold to judge the significance of DEGs. DEGs were subsequently carried into GO functional analysis and KEGG pathway analysis.
Confirmation of gene expression by qRT-PCR
Total RNA was extracted as described for the cDNA library preparation and sequencing. Each RNA sample was treated with PrimeScript™ RT Reagent Kit with gDNA Eraser (Takara), to remove residual genomic DNA and reverse transcribe into cDNA, according to the manufacturer’s protocol. The primers for qRT-PCR were designed using the Primer Express® Software v3.0.1 (Applied Biosystems), and are listed in Additional file 6. The actin gene of cranberry (CL7164.Contig2_All) was used as the internal housekeeping gene control. qRT-PCR was performed using the SYBR Premix Ex Taq Kit (Takara) according to the manufacturer’s instructions. The PCR was carried out on a Stratagene Mx3000P instrument (Agilent, USA) with the following conditions: denaturation at 95 °C for 15 s; 40 cycles of 95 °C for 15 s, and 60 °C for 30 s. All qRT-PCR reactions were repeated three times, with three technical replications per experiments. The results were normalized to the expression level of the constitutive actin gene. A relative quantitative method (2-ΔΔCt) was used to evaluate the quantitative variation.
Availability of supporting data
All clean reads generated by Illumina sequencing have been deposited in the Sequence Read Archive (SRA) data base (http://www.ncbi.nlm.nih.gov/sra) under the accession ID SRX1140359 for R, and SRX1140360 for W.
Acknowledgments
We thank Prof. Hanyan Li and Dr. Fawei Wang (Ministry of Education, Engineering Research Center of Bioreactor and Pharmaceutical Development, Jilin Agricultural University) for providing help in the experiments. This work was supported by the National Natural Science Foundation of China (31301755), the Special Fund for Agro-scientific Research in the Public Interest of China (201103037), the doctoral Program Foundation of Institutions of Higher Education of China (20132223120004), the Preferred Foundation of Scientific Research for Returned Overseas Chinese Scholar of the State Human Resource Ministry of China, the Project of International Science and Technology Cooperation of Jilin Science and Technology Department (20150414047GH).
Abbreviations
- 4CL
4-coumarate CoA ligase
- ABC
ATP-binding cassette
- ACT
Anthocyanin acyltransferase
- ANR
Anthocyanidin reductase
- ANS
Anthocyanidin synthase
- bHLH
basic helix–loop–helix
- BTL
Bilitranslocase
- C4H
Cinnamate 4-hydroxylase
- CDS
Coding sequence
- CHI
Chalcone isomerase
- CHS
Chalcone synthase
- COG
Clusters of orthologous group
- CTAB
Cetyltrimethylammonium bromide
- DEGs
Differentially expressed genes
- DFR
Dihydroflavonol 4-reductase
- F3′5′H
Flavonoid 3′,5′-hydroxylase
- F3′H
Flavonoid 3′-hydroxylase
- F3H
Flavanone 3-hydroxylase
- FDR
False discovery rate
- FLS
Flavonol synthase
- FPKM
Fragments per kb per million
- GO
Gene ontology
- GSH
c-Glu-cys-gly
- GST
Glutathione S-transferase
- HPLC
High-performance liquid chromatography
- KEGG
Kyoto encyclopedia of genes and genomes
- LAR
Leucoanthocyanidin reductase
- LDOX
Leucoanthocyanidin dioxygenase
- MATE
Multidrug and toxic compound extrusion protein
- MISA
MicroSAtellite
- MS
Mass spectrometer
- MTT
Membrane transporter-mediated transport
- MVT
Membrane vesicle-mediated transport
- NGS
Next-generation sequencing
- NR
Non-redundant protein
- NT
Nucleotide database
- OMT
O-methyltransferase
- PAL
Phenylalanine ammonia lyase
- PAs
Proanthocyanidins
- qRT-PCR
Quantitative real time PCR
- QTL
Quantitative trait loci
- RNA-Seq
RNA sequencing
- SNARE
Soluble N-ethylmaleimide-sensitive factor attachment protein receptors
- SSRs
Simple sequence repeats
- TFs
Transcription factors
- UFGT
UDP-Glucose flavonoid 3-O-glucosyl transferase
- UV/Vis
Ultraviolet–visible
- VSR
Vacuolar sorting receptor
- WD
Tryptophan-aspartate
Additional files
57,331 All-unigene sequences in cranberry fruit transcriptome, available at https://mynotebook.labarchives.com/, DOI: 10.6070/H4FB50ZC (FA 44805 kb)
21,898 cranberry unigenes assigning to 128 KEGG pathways. (XLS 50 kb)
Frequency of identified SSR motifs. (XLS 57 kb)
Primer sequences of cranberry selected for SSR marker validation. (XLS 19 kb)
1,134 DEGs assigning to 113 KEGG pathways. (XLS 53 kb)
Primer sequences of cranberry selected for qRT-PCR analysis. (XLS 16 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Conceived and designed the experiments: Haiyue Sun, Yadong Li. Performed the experiments: Yushan Liu, Yuzhuo Gai, Jinman Geng, Li Chen, Hongdi Liu, Limin Kang. Analyzed the data: Haiyue Sun, Yushan Liu. Contributed reagents/materials/tools: Yuzhuo Gai, Youwen Tian. Wrote the paper: Haiyue Sun. All authors read and approved the final manuscript.
Contributor Information
Haiyue Sun, Email: haiyue-sun@hotmail.com.
Yushan Liu, Email: crazy4321@sina.cn.
Yuzhuo Gai, Email: gallia@qq.com.
Jinman Geng, Email: gengjinman@2008.sina.com.
Li Chen, Email: chenlixx@yeah.net.
Hongdi Liu, Email: tang_tang1234@126.com.
Limin Kang, Email: kang15543270828@126.com.
Youwen Tian, Email: tyw5219@163.com.
Yadong Li, Email: blueberryli@163.com.
References
- 1.Trehane J. Blueberries, cranberries and other Vacciniums. Portland, Cambridge: Timber Press; 2004. [Google Scholar]
- 2.FAOSTAT. http://faostat3.fao.org/faostat-gateway/go/to/browse/Q/QC/E
- 3.Manganaris GA, Goulas V, Vicente AR, Terry LA. Berry antioxidants: small fruits providing large benefits. J Sci Food Agric. 2014;94(5):825–833. doi: 10.1002/jsfa.6432. [DOI] [PubMed] [Google Scholar]
- 4.Côté J, Caillet S, Doyon G, Sylvain JF, Lacroix M. Analyzing cranberry bioactive compounds. Crit Rev Food Sci Nutr. 2010;50(9):872–888. doi: 10.1080/10408390903042069. [DOI] [PubMed] [Google Scholar]
- 5.Brown PN, Shipley PR: Determination of anthocyanins in cranberry fruit and cranberry fruit products by high-performance liquid chromatography with ultraviolet detection: single-laboratory validation. J AOAC Int 2011, 94(2):459–466. Erratum in: J AOAC Int 2012, 95(1):286. [PMC free article] [PubMed]
- 6.Brown PN, Turi CE, Shipley PR, Murch SJ. Comparisons of large (Vaccinium macrocarpon Ait.) and small (Vaccinium oxycoccos L., Vaccinium vitis-idaea L.) cranberry in British Columbia by phytochemical determination, antioxidant potential, and metabolomic profiling with chemometric analysis. Planta Med. 2012;78(6):630–640. doi: 10.1055/s-0031-1298239. [DOI] [PubMed] [Google Scholar]
- 7.Neto CC, Vinson JA. Cranberry. In: Benzie IFF, Wachtel-Galor S, editors. Herbal Medicine: Biomolecular and Clinical Aspects. 2. Boca Raton (FL): CRC Press; 2011. [PubMed] [Google Scholar]
- 8.Wilson T, Porcari JP, Harbin D. Cranberry extract inhibits low density lipoprotein oxidation. Life Sci. 1998;62(24):L381–L386. doi: 10.1016/S0024-3205(98)00204-5. [DOI] [PubMed] [Google Scholar]
- 9.Terris MK, Issa MM, Tacker JR. Dietary supplementation with cranberry concentrate tablets may increase the risk of nephrolithiasis. Urology. 2001;57(1):26–29. doi: 10.1016/S0090-4295(00)00884-0. [DOI] [PubMed] [Google Scholar]
- 10.Howell AB. Cranberry proanthocyanidins and the maintenance of urinary tract health. Crit Rev Food Sci Nutr. 2002;42(3 Suppl):273–278. doi: 10.1080/10408390209351915. [DOI] [PubMed] [Google Scholar]
- 11.Yamanaka A, Kimizuka R, Kato T, Okuda K. Inhibitory effects of cranberry juice on attachment of oral streptococci and biofilm formation. Oral Microbiol Immunol. 2004;19(3):150–154. doi: 10.1111/j.0902-0055.2004.00130.x. [DOI] [PubMed] [Google Scholar]
- 12.Labrecque J, Bodet C, Chandad F, Grenier D. Effects of a high-molecular-weight cranberry fraction on growth, biofilm formation and adherence of Porphyromonas gingivalis. J Antimicrob Chemother. 2006;58(2):439–443. doi: 10.1093/jac/dkl220. [DOI] [PubMed] [Google Scholar]
- 13.Liu Y, Black MA, Caron L, Camesano TA. Role of cranberry juice on molecular-scale surface characteristics and adhesion behavior of Escherichia coli. Biotechnol Bioeng. 2006;93(2):297–305. doi: 10.1002/bit.20675. [DOI] [PubMed] [Google Scholar]
- 14.Goldman RD. Cranberry juice for urinary tract infection in children. Can Fam Physician. 2012;58(4):398–401. [PMC free article] [PubMed] [Google Scholar]
- 15.Déziel B, MacPhee J, Patel K, Catalli A, Kulka M, Neto C, et al. American cranberry (Vaccinium macrocarpon) extract affects human prostate cancer cell growth via cell cycle arrest by modulating expression of cell cycle regulators. Food Funct. 2012;3(5):556–564. doi: 10.1039/c2fo10145a. [DOI] [PubMed] [Google Scholar]
- 16.Simão TN, Lozovoy MA, Simão AN, Oliveira SR, Venturini D, Morimoto HK, et al. Reduced-energy cranberry juice increases folic acid and adiponectin and reduces homocysteine and oxidative stress in patients with the metabolic syndrome. Br J Nutr. 2013;110(10):1885–1894. doi: 10.1017/S0007114513001207. [DOI] [PubMed] [Google Scholar]
- 17.Kowalska K, Olejnik A, Rychlik J, Grajek W. Cranberries (Oxycoccus quadripetalus) inhibit adipogenesis and lipogenesis in 3T3-L1 cells. Food Chem. 2014;148:246–252. doi: 10.1016/j.foodchem.2013.10.032. [DOI] [PubMed] [Google Scholar]
- 18.Galletta GJ, Ballington JR. Blueberries, Cranberries, and Lingonberries. In: Janick J, Moore JN, editors. Fruit Breeding, Vine and Small Fruits (Volume 2) New York: John Wiley & Sons Incorporation; 1996. pp. 65–83. [Google Scholar]
- 19.Rhodes MJC. The maturation and ripening of fruits. In: Thimann KV, editor. Senescence in plants. Boca Raton, Florida: CRC Press; 1980. pp. 157–205. [Google Scholar]
- 20.Seymour GB, Taylor JE, Tucker GA: Biochemistry of fruit ripening, pp. 212–217. Chapman and Hall, London (1993)
- 21.Zalapa JE, Cuevas H, Zhu H, Steffan S, Senalik D, Zeldin E, et al. Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Am J Bot. 2012;99(2):193–208. doi: 10.3732/ajb.1100394. [DOI] [PubMed] [Google Scholar]
- 22.Zhu H, Senalik D, McCown BH, Zeldin EL, Speers J, Hyman J, et al. Mining and validation of pyrosequenced simple sequence repeats (SSRs) from American cranberry (Vaccinium macrocarpon Ait.) Theor Appl Genet. 2012;124(1):87–96. doi: 10.1007/s00122-011-1689-2. [DOI] [PubMed] [Google Scholar]
- 23.Georgi L, Johnson-Cicalese J, Honig J, Das SP, Rajah VD, Bhattacharya D, et al. The first genetic map of the American cranberry: exploration of synteny conservation and quantitative trait loci. Theor Appl Genet. 2013;126(3):673–692. doi: 10.1007/s00122-012-2010-8. [DOI] [PubMed] [Google Scholar]
- 24.Polashock J, Zelzion E, Fajardo D, Zalapa J, Georgi L, Bhattacharya D, et al. The American cranberry: first insights into the whole genome of a species adapted to bog habitat. BMC Plant Biol. 2014;14:165. doi: 10.1186/1471-2229-14-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schuster SC. Next-generation sequencing transforms today's biology. Nat Methods. 2008;5(1):16–18. doi: 10.1038/nmeth1156. [DOI] [PubMed] [Google Scholar]
- 26.Ansorge WJ. Next-generation DNA sequencing techniques. N Biotechnol. 2009;25(4):195–203. doi: 10.1016/j.nbt.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 27.Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11(1):31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 28.Jain M. Next-generation sequencing technologies for gene expression profiling in plants. Brief Funct Genomics. 2012;11(1):63–70. doi: 10.1093/bfgp/elr038. [DOI] [PubMed] [Google Scholar]
- 29.Buermans HP, den Dunnen JT. Next generation sequencing technology: Advances and applications. Biochim Biophys Acta. 2014;1842(10):1932-1941. [DOI] [PubMed]
- 30.Hyun TK, Lee S, Rim Y, Kumar R, Han X, Lee SY, et al. De-novo RNA sequencing and metabolite profiling to identify genes involved in anthocyanin biosynthesis in Korean black raspberry (Rubus coreanus Miquel) PLoS One. 2014;9(2) doi: 10.1371/journal.pone.0088292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao S, Tuan PA, Li X, Kim YB, Kim H, Park CG, et al. Identification of phenylpropanoid biosynthetic genes and phenylpropanoid accumulation by transcriptome analysis of Lycium chinense. BMC Genomics. 2013;14:802. doi: 10.1186/1471-2164-14-802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lai Z, Lin Y. Analysis of the global transcriptome of longan (Dimocarpus longan Lour) embryogenic callus using Illumina paired-end sequencing. BMC Genomics. 2013;14:561. doi: 10.1186/1471-2164-14-561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pertea G, Huang X, Liang F, Antonescu V, Sultana R, Karamycheva S, et al. TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics. 2003;19(5):651–652. doi: 10.1093/bioinformatics/btg034. [DOI] [PubMed] [Google Scholar]
- 34.Salzberg SL, Yorke JA. Beware of mis-assembled genomes. Bioinformatics. 2005;21(24):4320–4321. doi: 10.1093/bioinformatics/bti769. [DOI] [PubMed] [Google Scholar]
- 35.Li X, Sun H, Pei J, Dong Y, Wang F, Chen H, et al. De novo sequencing and comparative analysis of the blueberry transcriptome to discover putative genes related to antioxidants. Gene. 2012;511(1):54–61. doi: 10.1016/j.gene.2012.09.021. [DOI] [PubMed] [Google Scholar]
- 36.Morgulis A, Coulouris G, Raytselis Y, Madden TL, Agarwala R, Schäffer AA. Database indexing for production MegaBLAST searches. Bioinformatics. 2008;24(16):1757–1764. doi: 10.1093/bioinformatics/btn322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 38.Buer CS, Imin N, Djordjevic MA. Flavonoids: new roles for old molecules. J Integr Plant Biol. 2010;52(1):98–111. doi: 10.1111/j.1744-7909.2010.00905.x. [DOI] [PubMed] [Google Scholar]
- 39.Boss PK, Davies C, Robinson SP. Analysis of the expression of anthocyanin pathway genes in developing Vitis vinifera L. cv shiraz grape berries and the implications for pathway regulation. Plant Physiol. 1996;111:1059–1066. doi: 10.1104/pp.111.4.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bogs J, Ebadi A, McDavid D, Robinson SP. Identification of the flavonoid hydroxylases from grapevine and their regulation during fruit development. Plant Physiol. 2006;140:279–291. doi: 10.1104/pp.105.073262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Castellarin SD, Di Gaspero G. Transcriptional control of anthocyanin biosynthetic genes in extreme phenotypes for berry pigmentation of naturally occurring grapevines. BMC Plant Biol. 2007;7:46. doi: 10.1186/1471-2229-7-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jaakola L, Määttä K, Pirttilä AM, Törrönen R, Kärenlampi S, Hohtola A. Expression of genes involved in anthocyanin biosynthesis in relation to anthocyanin, proanthocyanidin, and flavonol levels during bilberry fruit development. Plant Physiol. 2002;130(2):729–739. doi: 10.1104/pp.006957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.He F, Mu L, Yan GL, Liang NN, Pan QH, Wang J, et al. Biosynthesis of anthocyanins and their regulation in colored grapes. Molecules. 2010;15(12):9057–9091. doi: 10.3390/molecules15129057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.He F, Pan QH, Shi Y, Duan CQ. Biosynthesis and genetic regulation of proanthocyanidins in plants. Molecules. 2008;13(10):2674–2703. doi: 10.3390/molecules13102674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stafford HA. Possible multienzyme complexes regulating the formation of C6-C3 phenolic compounds and lignins in higher plants. Recent Adv Phytochem. 1974;8:53–79. doi: 10.1016/B978-0-12-612408-8.50009-3. [DOI] [Google Scholar]
- 46.Winkel BSJ. Metabolic channeling in plants. Annu Rev Plant Biol. 2004;55:85–107. doi: 10.1146/annurev.arplant.55.031903.141714. [DOI] [PubMed] [Google Scholar]
- 47.Zhao J, Dixon RA. The ‘ins’ and ‘outs’ of flavonoid transport. Trends Plant Sci. 2010;15:72–80. doi: 10.1016/j.tplants.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 48.Gomez C, Terrier N, Torregrosa L, Vialet S, Fournier-Level A, Verries C, et al. Grapevine MATE-type proteins act as vacuolar H + −dependent acylated anthocyanin transporters. Plant Physiol. 2009;150:402–415. doi: 10.1104/pp.109.135624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Braidot E, Zancani M, Petrussa E, Peresson C, Bertolini A, Patui S, et al. Transport and accumulation of flavonoids in grapevine (Vitis vinifera L.) Plant Signal Behav. 2008;3(9):626–632. doi: 10.4161/psb.3.9.6686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Verrier PJ, Bird D, Burla B, Dassa E, Forestier C, Geisler M, et al. Plant ABC proteins--a unified nomenclature and updated inventory. Trends Plant Sci. 2008;13(4):151–159. doi: 10.1016/j.tplants.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 51.Klein M, Martinoia E, Hoffmann-Thoma G, Weissenböck G. A membrane-potential dependent ABC-like transporter mediates the vacuolar uptake of rye flavone glucuronides: regulation of glucuronide uptake by glutathione and its conjugates. Plant J. 2000;21(3):289–304. doi: 10.1046/j.1365-313x.2000.00684.x. [DOI] [PubMed] [Google Scholar]
- 52.Debeaujon I, Peeters AJM, Leon-Kloosterziel KM, Koornneef M. The TRANSPARENT TESTA12 gene of Arabidopsis encodes a multidrug secondary transporter-like protein required for flavonoid sequestration in vacuoles of the seed coat endothelium. Plant Cell. 2001;13:853–871. doi: 10.1105/tpc.13.4.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao J, Dixon RA. Mate transporters facilitate vacuolar uptake of epicatechin 3'-O-glucoside for proanthocyanidin biosynthesis in Medicago truncatula and Arabidopsis. Plant Cell. 2009;21:2323–2340. doi: 10.1105/tpc.109.067819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grotewold E, Davies K. Trafficking and sequestration of anthocyanins. Nat Prod Commun. 2008;3:1251–1258. [Google Scholar]
- 55.Conn S, Curtin C, Bézier A, Franco C, Zhang W. Purification, molecular cloning, and characterization of glutathione S-transferases (GSTs) from pigmented Vitis vinifera L. cell suspension cultures as putative anthocyanin transport proteins. J Exp Bot. 2008;59(13):3621–3634. doi: 10.1093/jxb/ern217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gomez C, Conejero G, Torregrosa L, Cheynier V, Terrier N, Ageorges A. In vivo grapevine anthocyanin transport involves vesicle-mediated trafficking and the contribution of ANTHOMATE transporters and GST. Plant J. 2011;67:960–970. doi: 10.1111/j.1365-313X.2011.04648.x. [DOI] [PubMed] [Google Scholar]
- 57.Passamonti S, Cocolo A, Braidot E, Petrussa E, Peresson C, Medic N, et al. Characterization of electrogenic bromosulfophthalein transport in carnation petal microsomes and its inhibition by antibodies against bilitranslocase. FEBS J. 2005;272:3282–3296. doi: 10.1111/j.1742-4658.2005.04751.x. [DOI] [PubMed] [Google Scholar]
- 58.Braidot E, Petrussa E, Bertolini A, Peresson C, Ermacora P, Loi N, et al. Evidence for a putative flavonoid translocator similar to mammalian bilitranslocase in grape berries (Vitis vinifera L.) during ripening. Planta. 2008;228:203–213. doi: 10.1007/s00425-008-0730-4. [DOI] [PubMed] [Google Scholar]
- 59.Petrussa E, Braidot E, Zancani M, Peresson C, Bertolini A, Patui S, et al. Plant flavonoids––biosynthesis, transport and involvement in stress responses. Int J Mol Sci. 2013;14(7):14950–14973. doi: 10.3390/ijms140714950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Quattrocchio F, Bauldry A, Lepiniec L, Grotewold E. The Science of Flavonoids. In: Grotewold E, editor. The Regulation of Flavonoid Biosynthesis. New York: Springer; 2006. pp. 97–122. [Google Scholar]
- 61.Hichri I, Barrieu F, Bogs J, Kappel C, Delrot S, Lauvergeat V. Recent advances in the transcriptional regulation of the flavonoid biosynthetic pathway. J Exp Bot. 2011;62:2465–2483. doi: 10.1093/jxb/erq442. [DOI] [PubMed] [Google Scholar]
- 62.Jones S. An overview of the basic helix-loop-helix proteins. Genome Biol. 2004;5(6):226. doi: 10.1186/gb-2004-5-6-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Heim MA, Jakoby M, Werber M, Martin C, Weisshaar B, Bailey PC. The basic helix-loop-helix transcription factor family in plants: A genome-wide study of protein structure and functional diversity. Mol Biol Evol. 2003;20:735–747. doi: 10.1093/molbev/msg088. [DOI] [PubMed] [Google Scholar]
- 64.Stracke R, Werber M, Weisshaar B. The R2R3-MYB gene family in Arabidopsis thaliana. Curr Opin Plant Biol. 2001;4:447–456. doi: 10.1016/S1369-5266(00)00199-0. [DOI] [PubMed] [Google Scholar]
- 65.Czemmel S, Heppel SC, Bogs J. R2R3 MYB transcription factors: Key regulators of the flavonoid biosynthetic pathway in grapevine. Protoplasma. 2012;249:109–118. doi: 10.1007/s00709-012-0380-z. [DOI] [PubMed] [Google Scholar]
- 66.Laitinen RA, Ainasoja M, Broholm SK, Teeri TH, Elomaa P. Identification of target genes for a MYB-type anthocyanin regulator in Gerbera hybrida. J Exp Bot. 2008;59(13):3691–3703. doi: 10.1093/jxb/ern216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jia L, Clegg MT, Jiang T. Evolutionary dynamics of the DNA-binding domains in putative R2R3-MYB genes identified from rice subspecies indica and japonica genomes. Plant Physiol. 2004;134(2):575–585. doi: 10.1104/pp.103.027201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Deluc L, Barrieu F, Marchive C, Lauvergeat V, Decendit A, Richard T, et al. Characterization of a grapevine R2R3-MYB transcription factor that regulates the phenylpropanoid pathway. Plant Physiol. 2006;140(2):499–511. doi: 10.1104/pp.105.067231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Albert NW, Lewis DH, Zhang H, Schwinn KE, Jameson PE, Davies KM. Members of an R2R3-MYB transcription factor family in Petunia are developmentally and environmentally regulated to control complex floral and vegetative pigmentation patterning. Plant J. 2011;65(5):771–784. doi: 10.1111/j.1365-313X.2010.04465.x. [DOI] [PubMed] [Google Scholar]
- 70.DeVetten N, Quattrocchio F, Mol J, Koes R. The AN11 locus controlling flower pigmentation in petunia encodes a novel WD-repeat protein conserved in yeast, plants, and animals. Gen Dev. 1997;11:1422–1434. doi: 10.1101/gad.11.11.1422. [DOI] [PubMed] [Google Scholar]
- 71.Ramsay NA, Glover BJ. MYB-bHLH-WD40 protein complex and the evolution of cellular diversity. Trends Plant Sci. 2005;10:63–70. doi: 10.1016/j.tplants.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 72.Qi T, Song S, Ren Q, Wu D, Huang H, Chen Y, et al. The Jasmonate-ZIM-domain proteins interact with the WD-Repeat/bHLH/MYB complexes to regulate Jasmonate-mediated anthocyanin accumulation and trichome initiation in Arabidopsis thaliana. Plant Cell. 2011;23(5):1795–1814. doi: 10.1105/tpc.111.083261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nesi N, Jond C, Debeaujon I, Caboche M, Lepiniec L. The Arabidopsis TT2 gene encodes an R2R3 MYB domain protein that acts as a key determinant for proanthocyanidin accumulation in developing seed. Plant Cell. 2001;13:2099–2114. doi: 10.1105/TPC.010098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Goodman CD, Casati P, Walbot V. A multidrug resistance-associated protein involved in anthocyanin transport in Zea mays. Plant Cell. 1812;2004:16. doi: 10.1105/tpc.022574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sledge MK, Wang L, May GD, Chekhovskiy K, Zwonitzer JC, Mian MA. Medicago truncatula EST-SSRs reveal cross-species genetic markers for Medicago spp. Eujayl I Theor Appl Genet. 2004;108(3):414–422. doi: 10.1007/s00122-003-1450-6. [DOI] [PubMed] [Google Scholar]
- 76.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 77.Hamberger B, Bak S. Plant P450s as versatile drivers for evolution of species-specific chemical diversity. Philos Trans R Soc Lond B Biol Sci. 2013;368(1612):20120426. doi: 10.1098/rstb.2012.0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Camp WH. A preliminary consideration of the biosystematy of Oxycoccus. Bul Torrey Bot Club. 1944;71:426–437. doi: 10.2307/2481315. [DOI] [Google Scholar]
- 79.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 81.Gish W, States DJ. Identification of protein coding regions by database similarity search. Nat Genet. 1993;3(3):266–272. doi: 10.1038/ng0393-266. [DOI] [PubMed] [Google Scholar]
- 82.Iseli C, Jongeneel CV, Bucher P. ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc Int Conf Intell Syst Mol Biol. 1999;138–148. [PubMed]
- 83.Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006;34:W293–W297. doi: 10.1093/nar/gkl031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132(3):365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- 85.Doyle JJ, Doyle JL. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical Bulletin. 1987;19:11–15. [Google Scholar]
- 86.Audic S, Claverie JM. The significance of digital gene expression profiles. Genome Res. 1997;7(10):986–995. doi: 10.1101/gr.7.10.986. [DOI] [PubMed] [Google Scholar]
- 87.Kim KI, van de Wiel MA. Effects of dependence in high-dimensional multiple testing problems. BMC Bioinformatics. 2008;9:114. doi: 10.1186/1471-2105-9-114. [DOI] [PMC free article] [PubMed] [Google Scholar]