

Abstract
Multiple sclerosis (MS) has been thought to be a complex and indecipherable disease, and poorly understood with regards to aetiology. Here, we suggest an emphatically positive view of progress over several decades in the understanding and treatment of MS, particularly focusing on advances made within the past 20 years. As with virtually all complex disorders, MS is caused by the interaction of genetic and environmental factors. In recent years, formidable biochemical, bioinformatic, epidemiological and neuroimaging tools have been brought to bear on research into the causes of MS. While susceptibility to the disease is now relatively well accounted for, disease course is not and remains a salient challenge. In the therapeutic realm, numerous agents have become available, reflecting the fact that the disease can be attacked successfully at many levels and using varied strategies. Tailoring therapies to individuals, risk mitigation and selection of first-line as compared with second-line medications remain to be completed. In our view, the MS landscape has been comprehensively and irreversibly transformed by this progress. Here we focus on MS therapeutics—the most meaningful outcome of research efforts.
Introduction
Multiple sclerosis (MS) is an inflammatory demyelinating and neurodegenerative disorder of the human CNS. The essential clinical and neuropathological features of MS were captured in the mid-19th century: the disease clasically manifested in episodic, partially reversible symptomatic attacks and exhibited demyelination out of proportion to axonal loss in autopsy tissue sections. Inflammation, gliosis and axonal injury were additional prominent neuropathological characteristics, as was the clinical evolution from intermittent attacks to slow, steady progressive worsening. Patients with MS typically present between the ages of 20–40 years, with affected women outnumbering men 2:1, and the progressive phase of disease manifests anytime between 5–35 years after onset. Although 10–15% of cases occur in first-degree relatives of patients with MS, there is no interpretable pattern of inheritance. Given these unique features, early hypotheses about the aetiology of MS proposed it to be infectious, vascular or metabolic, according to the predisposition of the speculator.
The often cited landmarks in our comprehension of MS include Charcot’s definition and naming of MS,1 Dawson’s reports about MS neuropathology,2 the description of cerebrospinal fluid abnormalities in patients with MS,3 and Rivers’ discovery of experimental autoimmune encephalomyelitis (EAE).4 These recognitions, while thoroughly merit-based, are somewhat arbitrary, as other investigators working at about the same time also contributed equally valuable insights. Current concepts of MS pathogenesis derive from advances in neurobiology (such as electrophysiology, and the cellular and molecular bases of myelination), neuroimaging (particularly MRI), neuropathology, immunology and genetics of complex diseases.
On 23 July 1993, subcutaneous IFN-β1b was approved for the treatment of relapsing forms of MS. In retrospect, the contemporary literature clearly illustrates how little was known about MS at the time. A definitive contemporary review noted vaguely: “epidemiological data suggest an aetiological role for both genetic and environmental factors in MS, but these have yet to be fully characterized.”5 A summary of treatment options stated simply that “therapies directed at altering the natural history of the underlying disease process … are controversial or experimental.”6 We now survey a changed landscape: names have been assigned to major environmental risk factors and genetic susceptibility determinants. Gene–environment interactions have become apparent. MS pathogenesis can be specified with increased confidence. Most importantly, patients and practitioners now face the welcome but formidable challenge of rationally ordering a wide spectrum of treatment options.
Here, we review the progress made over these two decades of MS therapeutics and indicate work yet to be done. This Review primarily focuses on disease-modifying drugs. Although advances in MS treatment have proceeded impressively, in parallel with increased knowledge about mechanisms of tissue injury coming mainly from neuropathology and MRI, approved drugs uniformly incorporate anti-inflammatory properties and aim to prevent rather than repair tissue injury.
Current concept of MS
Clinical MS follows a course unusual among neurological disorders but frequently observed in other autoimmune diseases.7 New onset MS is usually (~85% of the time) typified by relapses: abrupt symptomatic episodes followed by recovery of varying extent. This phase of disease is termed relapsing–remitting MS (RRMS). Relapses occur, on average, about once per 2 years. Between relapses, most patients remain at a stable level of function determined by recovery from the last relapse. After 5–25 years, the pattern often changes: the number of acute worsenings is greatly reduced and replaced with a slow, steady increase in symptom severity. This disease phase is designated secondary progressive MS (SPMS), and responds to immunotherapy only in so far as there are residual inflammatory events: either clinical relapses or MRI lesions. Loss of mobility (restriction to wheelchair or bed) is not the most common outcome but remains the most feared major complication. Total relapse number does not predict loss of mobility, but numerous and severe relapses within the first few years of disease carry adverse prognostic implications.
A small minority (<10%) of patients do not experience symptoms of RRMS and present only with progression, a pattern called primary progressive MS (PPMS). Although the symptom pattern is different, no genetic, imaging or pathological features distinguish PPMS from SPMS. The apparent lack of a relapsing–remitting phase in patients with PPMS could plausibly be attributed to individual lesions localizing in clinically silent regions, which summate to eventually produce disability.8,9 This concept is supported by observations in patients who serendipitously present with MRI lesions that strongly suggest the presence of MS but without any clinical symptoms or signs. More than a third of these persons with radiologically isolated syndrome (RIS) will develop clinical evidence for MS within 5 years. Of those, 10% will meet criteria for primary progressive MS.10
In another subgroup of patients (~10–15%), relapses are mild and SPMS—as defined by motor impairment—does not occur. Historically, these cases have been termed ‘benign MS’. Further follow-up has disclosed that only a subpopulation of this group remain functionally unaffected by MS, with the remainder undergoing delayed motor progression or cognitive impairment.11–16 A tiny, unfortunate, minority of all patients with MS develop very severe symptoms quite quickly (within 5 years), and a small proportion of these individuals, who may have clinical or neuropathologically defined disease patterns referred to as Marburg variant or Balo concentric sclerosis (BCS), die from this aggressive form of MS.
MS disease manifestations occur only in the CNS, so blood tests are unhelpful for diagnosis or monitoring of patients, but essential for excluding MS mimics such as vitamin B12 deficiency, human T lymphotropic virus 1 infection, systemic lupus erythematosus and neuromyelitis optica. Recent studies suggest that evidence of disease could be found early in the cervical lymph nodes,17 raising the possibility that some form of peripheral indicator of disease activity might yet be uncovered. At present, disease activity and severity in patients are assessed clinically by counting relapses and quantifying neurological impairments. Major insights have come from examining the CNS using MRI, which encompasses numerous techniques, each of which captures distinct disease features. Frustratingly, MRI findings fail to correlate strongly to concurrent clinical state. MRI changes do, however, carry quite impressive prognostic implications. Presently, MRI and clinical metrics are used to qualify drugs as effective for restraining MS-associated inflammation and tissue injury through a rather uniform approach to clinical trial design.
Disease-modifying drugs for MS
Disease-modifying drugs are aimed at the pathogenic processes that underlie MS, and can change patients’ outcomes, as well as provide data that test hypotheses about disease pathogenesis. In retrospect, it is possible to discern three phases of the development of disease-modifying drugs to treat MS (see Figure 1: Timeline).
Figure 1.
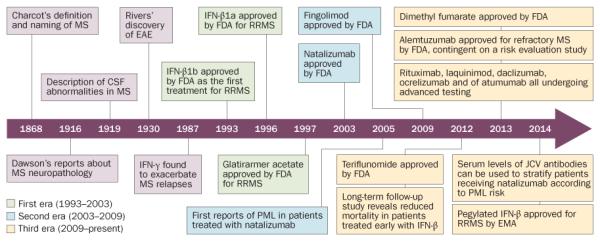
Timeline for the development of disease-modifying drugs for MS. Abbreviations: CSF, cerebrospinal fluid; EAE, experimental autoimmune encephalomyelitis; JCV, polyomavirus JC; MS, multiple sclerosis; PML, progressive multifocal leukoencephalopathy; RRMS, relapsing–remitting MS.
The first era (1993–2003)
The first era, beginning in 1993, was preceeded by important investigations indicating that intensive immunotherapy could alter the course of MS, and that MS exacerbations could be treated with intravenous methylprednisolone.18 However, the modern era of MS treatment began in 1993 with FDA approval of IFN-β1b to treat relapsing forms of MS.19,20 This watershed moment introduced the era in which MS became a treatable disorder. Further, approval of IFN-β1b relied in part on MRI, opening the era of radiographic disease monitoring. Medications developed during this period comprised three formulations of IFN-β and glatiramer acetate. In placebo-controlled and double-blind clinical trials monitoring 1000–2000 participants for 2 years, all these agents produced slightly more than 30% reductions in annual relapse rate, with complementary lessening of disease severity scores at trial completion (as reviewed else-where21). MRI-visible disease activity was reduced by a degree comparable to or greater than the clinical activity.
These therapeutics, given by self-injection, are expensive, inconvenient and exhibit variable adverse effects. Subsequent research confirmed excellent long-term safety profiles for glatiramer acetate and the different formulations of IFN-β. None of these first-line agents has emerged as superior. Although longitudinal prospective comparisons of treated and untreated patients have not been possible, studies using the best available techniques to control relevant confounds have shown that these agents reduce disease severity quite well for some patients, and might delay progression or lessen its impact.22,23 A 21-year follow-up study of nearly every participant in the first clinical trial of IFN-β showed a survival benefit for those receiving active drug during the trial, as compared with those given placebo.24,25
There are no predictive biomarkers that can aid selection among these drugs. Risk–benefit calculations for these agents initially seemed simple: if the patient’s MS required treatment, one of these drugs could be recommended with confidence that it might help and would do no harm. Although this view was accurate as a broad-stroke statement, some countervailing evidence has emerged over time. Long-term follow-up of patients in a pivotal trial of intramuscular IFN-β1a revealed a poor prognosis for patients who exhibited active radiographic disease during the 2 years on drug.26 These patients, a small minority, seemed to fare worse than those receiving placebo during the trial, which was consistent with the interindividual heterogeneity of MS. This finding raised the hypothesis that patients with MS might exhibit a similarly mixed response to therapies as do patients with a range of other autoimmune disorders. Observational studies using different IFN-β preparations confirmed that this effect was shared among all members of the IFN-β drug class: the minority of patients with MS who showed new or active MRI lesions shortly after beginning IFN-β bore an unfavourable prognosis, whereas the majority, whose disease was radiographically quiescent during this period, showed substantially better outcomes.27
Lessons were learned during this initial phase of MS therapeutic development. First and most importantly, the natural history of MS could be modified by treatment; second, treatment effects could be detected via MRI; third, long-term studies incorporating MRI were essential for optimal application of these novel medications; and fourth, beginning treatment at the earliest confirmation of the MS diagnosis seemed to produce better medium-term outcomes. For glatiramer acetate, distinctions between good responders and poor responders were not as clear-cut as with IFN-β.21 Interferons are endogenous regulatory cytokines that increase or decrease transcriptional initiation for hundreds of genes in a cell-type-dependent fashion.28 Therefore, it seemed plausible that, in patients with MS who had good or poor responses to treatment, bioinformatic analysis of patterns in interferon-induced gene expression might predict clinical responses to IFN-β. These data could, in turn, yield insights into pathogenetic mechanisms.29 It was recently reported that untreated patients with MS can be divided into two groups by RNA profiling: one that does not respond to IFN-β therapy and has more-aggressive disease, and another that responds to IFN-β with reduction in relapses following treatment.30 However, at present we have neither established predictive biomarkers (beyond MRI or the presence of neutralizing antibodies to IFN-β) nor confirmed mechanisms for the first-line MS treatments.
The second era (2003–2009)
The second phase of MS therapeutic development is characterized by the approvals of natalizumab in late 2003 and fingolimod in 2009.31–33 Natalizumab, a monoclonal antibody to integrin-α4 in leukocytes, has undergone a complex, and continuing, process of integration into clinical practice.34,35 This agent arose from preclinical studies showing that neutralization of α4-integrin suppressed disease activity in the rodent model of neuroinflammatory demyelination, EAE.36 Administered by monthly intravenous infusion, natalizumab exerts impressive inhibitory effects for the inflammatory aspects of MS, with >65% reduction in relapses during 2 years of treatment, and >90% suppression of new inflammatory MRI lesions.37,38 Although early safety profiles were favourable, two participants in the MS clinical trials (that is, 1:1,000) developed progressive multifocal leukoencephalopathy (PML), a rare, often fatal, opportunistic brain infection.39 Subsequent developments illustrate the complexity of developing novel disease treatments.
Historically, PML was invariably observed in patients with severely impaired cell-mediated immunity, and always kept company with other opportunistic infections.40 Given the overall rarity of PML, this entirely unexpected complication was clearly caused by natalizumab, provoking immediate voluntary suspension of the drug’s distribution, though eventually no other PML cases were found to be incubating in the study population.41
The mechanism by which natalizumab causes PML remains unknown.42 PML is caused by polyomavirus JC (JCV), a widespread commensal that is present in 55% of the population, prompting development of a biomarker—measurement of JC virus antibodies—that proved highly predictive for PML susceptibility. Incorporation of this biomarker into therapeutic decision making allows the routine, though carefully monitored, use of natalizumab. Natalizumab-PML is not associated with generalized immunosuppression, and may be mechanism-driven.43 The search for host factors that predispose to PML besides JCV infection continues.
The other second-era drug, fingolimod, is a prodrug that is converted in vivo to a sphingosine-1-phosphate (S1P) analogue. Fingolimod downregulates S1P receptor 1 on leukocytes and the endothelium, trapping naive and central memory T lymphocytes in lymph nodes. Treatment with fingolimod thereby suppresses MS disease activity, with 55–60% lower relapse rates and an impressive reduction of MRI-visible activity compared with placebo.31,32,44 Fingolimod lacks target specificity, downregulating four of the five S1P receptors, including those on vascular endothelium, arterial smooth muscle cells, atrial myocytes, bronchial smooth muscle, and CNS astrocytes and oligodendrocytes.44 For this reason, treatment is associated with a spectrum of potential safety concerns, each of which is highly uncommon, and requires a relatively onerous safety-monitoring protocol. Hypothetical therapeutic effects of fingolimod treatment involving glial or endothelial cells might also be attributed to its action towards other S1P receptors.45,46 Fingolimod produces lymphopenia, unassociated with opportunistic infections beyond disseminated herpes zoster. For this reason, patients must have documented varicella zoster virus (VCV) immunity to be considered for fingolimod.47
The third era (2009–present)
The third stage of MS therapeutic development is now upon us. This period is characterized by the introduction (either recent or imminent) of medications including both small-molecules and biologics. For example, two oral immunomodulatory medications (teriflunomide and dimethyl fumarate) have been approved.48–51 After phase III trials,52–54 alemtuzumab—a leukocyte-depleting monoclonal antibody—was approved in the EU, Australia, Canada and subsequently in the USA. Other agents, including the small molecule laquinimod55,56 and the biologics daclizumab (a CD25 antibody), ocrelizumab and ofatumumab (both CD20 antibodies), are undergoing advanced clinical testing.57–59
Each agent tested so far in the third era has had a distinct mechanism of action, and yet has shown efficacy in double-blinded controlled trials. The variety of agents shown to reduce relapse frequency and MRI-monitored disease activity is consistent with recent chromatid mapping studies of allelic variants associated with MS. These studies have indicated that multiple cell types—including type 17 T helper cells (TH17), FOXP3 regulatory T cells, B cells and macrophages—are involved in MS disease pathogenesis.60 Each clinically effective agent blunts inflammation, which lays a strong foundation for the hypothesis that inflammation drives MS disease expression.
Failed clinical trials: surprises and lessons
Perhaps equally informative to the discovery of effective novel agents in these three eras, some therapeutic trials have produced negative results, revealing drugs that increased disease activity.61 The first trial, reported in 1987, used intravenous infusion of IFN-γ that led promptly to disease worsening in seven of 18 study patients.62,63 The trial was terminated and the agent deemed inappropriate for MS treatment. Contemporary scientific rationale for the trial seemed sound, as IFN-γ was well documented as beneficial for EAE.64,65 With time and further research, this discordance between MS and EAE provided insight into the pathophysiology of disorders, mediated by TH1 and TH2 lymphocytes, and helped to discriminate the induction and effector phases of MS.
Following success in treating rheumatoid arthritis and inflammatory bowel disease via inhibition of tumour necrosis factor (TNF), and promising preclinical results in EAE models, in 1996 there was an open-label safety trial of TNF-neutralizing antibodies in two patients with MS. Both patients showed heightened disease activity after treatment.66 Another group of investigators conducted a 1-year placebo-controlled study of a soluble TNF-receptor–IgG fusion protein in 168 patients with MS.67 After 6 months, recipients of the TNF-blocking drug experienced significantly more and worse relapses, without increased MRI activity, compared with controls. Later, individuals receiving TNF blockade for other disorders rarely, but unequivocally, manifested clinical and MRI-defined inflammatory demyelination despite not having MS.68 Notably, all TNF-blockers were implicated. Detailed analysis of a type 1 TNF receptor variant, associated with increased risk of MS, proved that it encodes a protein capable of giving rise to a soluble truncated receptor, which would presumably lower the availability of TNF.69 Interestingly, the associated haplotype was linked with the risk of developing MS but not rheumatoid arthritis, inflammatory bowel disease or psoriasis, suggesting that genetic differences between these diseases underlie these seemingly paradoxical responses to TNF blockade.70 A recent proposal attributed the unexpected adverse consequences of TNF blockade to effects of TNF on regulatory T cells,71 highlighting the complex immunopathology underlying these conditions.
Other drugs that were predicted to be active against MS on the basis of preclinical studies have proven to be inert. In some EAE models, the dimeric cytokine IL-23 has a determinative role for disease pathogenesis, and this research helped identify and characterize the TH17 lymphocyte phenotype.72,73 It was, therefore, unexpected that ustekinumab—an antibody against IL-12 and IL-23—proved ineffective for patients with MS, while carrying potent therapeutic benefit for patients with other inflammatory conditions.74,75
The CD20 antibodies rituximab and ocrelizumab deplete mature B lymphocytes, and positive results from clinical trials of these agents strongly suggested that B cells play an unexpected critical part in disease pathogenesis.76 Further studies showed that the pathogenetic role(s) of B cells in MS (and in EAE) were not limited to antibody production. In particular, a definitive genetic modelling study pointed to the crucial role of B cells as antigen-presenting cells.77 Therefore, it was surprising that an elegant intervention using atacicept, a fusion protein that suppresses activity of two B cell growth factors, increased disease activity (both relapses and MRI) in a phase II trial.78,79
Present day treatment of MS
The treatment of patients with MS is undergoing a rapid transition, moving from the routine use of earlier first era therapies to a more rational approach based on effectiveness versus safety and tolerability. MS exacerbations are treated with 3–5 days of intravenous methylprednisolone, as oral prednisone alone is an ineffective treatment that increases the risk of new attacks.80 Older treatments such as intramuscular adrenocorticotropic hormone have not been investigated in the modern era. The rationale behind treating MS relapses with the remarketed and expensive preparation Acthar® (Mallinckrodt Pharmaceuticals, Ireland), instead of with the less expensive methylprednisolone, is currently lacking.
Although there has not been a head-to-head comparison of the newer therapies, broadly speaking the blockade of T cell traffic into the CNS with anti-integrin-α4 monoclonal antibodies (natalizumab) seems to be the most efficacious therapy, followed by the S1P agonist fingolimod, and then dimethyl fumarate. The β-interferons, glatiramer acetate and the oral pyrimidine-synthesis-blocker teriflunomide seem to have efficacies similar to each other but lesser than those of the agents noted above (Table 1).
Table 1.
Efficacy ranking of approved therapies for multiple sclerosis*
| Drug | Era of development |
Mechanism of action | Key considerations |
|---|---|---|---|
| Most effective | |||
|
| |||
| Natalizumab | Second | Monoclonal antibody against integrin-α4 in leukocytes |
Risk of PML must be assessed via presence of JCV antibodies Substantial risk of relapse after discontinuation |
|
| |||
| Highly effective | |||
|
| |||
| Fingolimod | Second | Sphingosine S1P receptor modulator |
Cardiac complications preclude use in individuals aged ≥50 years, and in those with history of cardiac disease VZV antibody testing must be conducted to mitigate risk of disseminated herpes zoster |
|
| |||
| Dimethyl fumarate |
Third | Immunomodulator | Necessary to monitor lymphocyte count as risk mitigation against PML Gastrointestinal complications may limit use |
|
| |||
| Moderately effective | |||
|
| |||
| IFN-β | First | Immunomodulator | Well characterized long-term safety and efficacy profiles Patients should not be required to ‘fail’ treatment before receiving alternatives |
|
| |||
| Glatiramer acetate |
First | Immunomodulator | Best safety profile for pregnant women with mild disease Patients should not be required to ‘fail’ treatment before receiving alternatives |
|
| |||
| Teriflunomide | Third | Pyrimidine synthesis inhibitor |
Risk of teratogenicity precludes use in women who are, or intend to become, pregnant |
Rankings are estimated on the basis of clinical trials, postapproval studies and few head-to-head comparisons. The factors that determine drug efficacy in any individual patient are largely undefined, and good clinical judgement is essential for treatment selection, especially in women of childbearing age. Abbreviations: JCV, polyomavirus JC; PML, progressive multifocal leukoencephalopathy; VZV, varicella zoster virus.
Some critical points must be made here given the present state of knowledge. First, our efficacy ranking is based on results from clinical trials and postapproval clinical observations, and it does not account for adverse effects, which must be considered during the selection of a first-line agent. Some head-to-head clinical trials have been conducted, following the judgement that the availability of active agents made placebo-controlled efficacy trials unethical. Where available, these data have been considered in our rankings, as head-to-head trials constitute the gold standard for efficacy comparisons. Second, the statement that ‘X agent is more efficacious than Y agent, and their safety profiles are equivalent’ does not imply that all patients should receive agent X. If agent Y completely controls the disease in an individual patient, agent Y is an entirely appropriate therapeutic selection. The underlying point is that patients are individuals and that drug efficacy in an individual patient is determined by factors that remain largely undefined.
Risk mitigation strategies dictate measurement of JCV antibodies and VZV antibodies.81 Patients negative for JCV are often recommended to begin natalizumab, whereas JCV-positive individuals are tested for VZV to evaluate the suitability of fingolimod treatment.44 If the result is positive, indicating VZV immunity, fingolimod can be begun. If not, fingolimod treatment should follow VZV immunization. Regarding safety, natalizumab seems to be safe in JCV-antibody-negative patients. In patients with cardiac issues—particularly with bundle branch blocks—fingolimod should be avoided. So far, dimethyl fumarate seems to be safe, though gastrointestinal adverse effects can occasionally cause difficulties in use of this drug.
As there is no biomarker that can predict response to therapy, clinical assessment is important in deciding how to initiate therapy. Thus, for female patients, particularly those wishing to have children, who have mild disease and no brainstem or spinal cord lesions, it is reasonable to treat with glatiramer acetate because of the safety profile in pregnant women. By contrast, male patients, African Americans of either sex, and patients with substantial brainstem or spinal cord disease who have had multiple exacerbations and have accumulated disability should be treated with the more-efficacious therapies from the onset.
There is no rationale for the notion—often promulgated by insurance companies—that all patients, regardless of their clinical state, should fail β-interferons or glatiramer acetate before receiving a second-line treatment. Indeed, rigid adherence to the approach that dictates failure of one or more ‘first-line’ medications for all individuals with MS is poor practice: ‘time is brain’ (and spinal cord) in MS just as it is in stroke. As discussed above, it is critical to carry out careful and frequent monitoring of patients with MRI to assess disease activity and guide subsequent modification or escalation of therapy.
The development of this early-stage treatment algorithm for MS represents persistent effort involving thousands of patients and practitioners over a period of 20 years. The increasing complexity of MS treatments makes a compelling argument for patients to be seen for consultation or ongoing care at comprehensive MS centres with expertise in the use of immunotherapies.
MS is primarily an autoimmune disorder
The clinical, radiographic and pathological severities of MS often seem dissociated. At early disease stages, inflammation dominates the picture, but all too commonly neurodegenerative progression relentlessly ensues. During this phase of disease, currently available treatments that target inflammation are ineffective. These attributes of MS have led some to conclude that MS might be a primarily neurodegenerative disorder that is complicated by inflammatory epiphenomena. However, the disease’s genetic architecture presents compelling evidence for a primary autoimmune model of MS. Genome-wide association studies (GWAS) and subsequent targeted genomic studies have identified 108 variants associated with MS susceptibility.82,83 Though each of these variants contributes only a small increase in the complex phenotype of disease risk, the biological functions associated with individual allelic variants have been striking. Many of these variants fall within specific signalling cascades, which suggests that alterations in pathways—rather than individual genes—might be the key to understanding how individual variants result in disease susceptibility. Over half of genetic variants associated with MS risk are also found in other putative autoimmune diseases, and risk alleles are primarily associated with genes that regulate immune function.84
Integration of genetic and epigenetic fine-mapping has identified causal variants in MS-associated loci, and the functions of these variants has been explored via generation of cis-regulatory element maps for a spectrum of immune cell types. Approximately 60% of probable causal variants mapped to enhancer-like elements, with preferential correspondence to stimulus-dependent CD4+ T cell enhancers. By overlapping causal single nucleotide variants with 31 transcription factor binding maps generated by ENCODE (the Encyclopaedia of DNA Elements), one study revealed that single nucleotide variants were strongly enriched within binding sites for immune-related transcription factors.60 Furthermore, variants associated with individual immune–inflammatory diseases correlated to different combinations of transcription factors that control immune cell identity and response to stimulation. In patients with MS, single nucleotide variants preferentially coincided with recognition sites for NF-κB, EBF1 and MEF2A.60
Key pathological observations also argue against the hypothesis that MS could primarily be a neurodegenerative disorder. First, inflammation and neurodegeneration are tightly linked.83,84 Beyond the obvious inflammatory clinical–pathological character of early MS, dense meningeal infiltrates are found at autopsy in the subarachnoid spaces of patients with long-standing MS, and these infiltrates are intimately associated with subpial demyelination, neuronal and neuritic damage, oligodendrocyte loss, cortical atrophy, and parenchymal microglial activation in the outer cortical layers.85–87
Recent neuropathological studies using brain biopsies obtained ‘en passant’ early in the disease have confirmed that inflammatory cortical demyelination occurs early in MS, preceding the appearance of classic white matter plaques in some patients with MS.88,89 Notably, neurodegenerative changes—including oligodendrocyte loss, reactive astrocytosis, and axonal and neuronal injury within these cortical plaques—occurred on a background of inflammation. These findings suggested that, at its onset, MS was a primarily inflammatory demyelinating disorder rather than a neurodegenerative disorder, because neurodegeneration was not found to be dissociated from inflammation. Absence of infiltrating leukocytes in chronic cortical lesions might be explained by the rapid resolution of cortical inflammation and by the extremely fast and efficient remyelination of cortical demyelinated plaques in early disease stages.90,91
The clinical trials of the past two and a half decades carry twofold scientific importance: first, they revealed that manipulation of inflammation can either enhance or suppress MS disease activity, thereby strengthening the link between inflammatory pathways and disease expression. Second, they made it clear that there are numerous intricacies of MS pathogenesis that remain to be clarified. As far as our patients are concerned, the most prominent challenge of the next decade(s) will be to integrate existing and forthcoming therapeutic options and scientific knowledge into a coherent strategy to guide research, treatment and monitoring.
Future directions
From our perspective, the MS community—patients, families, clinicians and researchers—now finds itself at a watershed moment. Nearly two decades of ‘therapeutics enthusiasm’ has yielded consensus-effective MS agents that unquestionably help in the short term. However, given limited post-marketing research, the long-term efficacy of these agents remains to be conclusively proven, which represents the current major challenge to clinicians and researchers. For example, numerous agents are widely available that show adequate disease control at the level of relapses. It is plausible, but unproven, that application of these treatments with appropriate monitoring and regimen modification will prevent, delay or attenuate disease progression in the later stages. Given the cost and inconvenience of the available agents, determining the extent to which MS disease progression is affected by contemporary therapies represents an urgent task.
Efforts to test current treatments in patients with progressive MS should have one of several outcomes: MS anti-inflammatory therapeutics may strongly reduce the likelihood or severity of progression in most patients; or these agents may be useful for impairing progression for some but not all patients; or the overall effect on progression may be disappointingly unimpressive. Our next task will be defined by the outcome of this research: if the first, highly-encouraging, result is obtained, then continuation of the present therapeutic strategy is appropriate. If the second observation is made, then individualized therapy becomes paramount and the major imperative will be to identify biomarkers for this purpose. Finally, if our therapeutic endeavours are off-base and MS progression is largely unaffected, we may find ourselves back near square one: with a large accumulation of genetic and environmental associations for MS risk, but without the crucial insights to determine disease outcome and to drive therapy.
In any scenario, progressive MS is the next frontier for MS research (Box 1). Several attractive schemata can explain MS progression at the tissue level, but none of them has strong support in vivo because of difficulty addressing progression via neuroimaging or histopathology. Therefore, the principal task before us is to understand progressive MS, and then to devise effective treatment. This universal strategy for attacking disease is no less valuable for being a cliché.
Box 1. Research agenda for progressive MS.
Long-term follow-up of clinical trial participants to determine whether early, definitive suppression of inflammation associated with MS delays or ameliorates progression
Studies to define genes, environmental factors and mechanistic pathways that underlie MS disease course
Development of monitoring tools for patients with progressive MS that will enable clinical trials and pathogenesis research
Abbreviation: MS, multiple sclerosis.
Conclusions
Fifteen decades of MS research have culminated in the past two, with potent insights into the genetic basis of the disease, its environmental associations, its tissue pathology, its characterization by MRI, and its susceptibility to treatment by immunomodulation. This progress now poises the MS community for advancement to the prized objective of alleviating the burden exerted by progressive MS.
MS disease progression remains difficult to treat. It is worth hoping that the present therapeutic armamentarium will make a decisive difference in the occurrence or severity of progressive MS. At the same time, it is incumbent on the clinical and research communities to press forward to model and decipher MS progression, which will help both to develop therapeutics and generate knowledge about mechanisms of neurodegeneration.
Key points.
■ MS has been a treatable disease for approximately 20 years, and incremental improvements in treatment options have culminated in remarkable progress for the amelioration of inflammatory aspects of MS
■ Traditionally, three patterns of disease evolution (relapsing–remitting, secondary progressive and primary progressive) were recognized, but present evidence suggests that these differing clinical phenotypes share common pathophysiology
■ A major unmet medical need in MS therapeutics is to define biomarkers to aid selection from the several treatment options so that individual patients can receive optimal personalized therapy
■ The requirement that patients fail IFN-β and glatiramer acetate therapy before being offered alternatives risks irreversible neural tissue injury during the process of initiating appropriate medication
■ Early, general statements that implicated a genetic component in multiple sclerosis (MS) susceptibility have been replaced by the identification of more than 100 genetic variants associated with disease susceptibility, ~90% of which are noncoding
■ Over half of genetic variants associated with MS risk are also found in other autoimmune diseases, and are primarily associated with genes that regulate immune function
Acknowledgements
D.A.H.’s work was supported by a National MS Society Collaborative Research Centre Award CA1061-A-18, NIH grants P01 AI045757, U19 AI046130, U19 AI070352, and P01 AI039671, the Penates Foundation and the Nancy Taylor Foundation for Chronic Diseases, Inc.
Footnotes
Competing interests
R.M.R. is a full-time employee of Biogen. However, this article was written and submitted while he worked at the Cleveland Clinic. D.A.H. and C.F.L. declare no competing interests.
Author contributions
The authors contributed equally to the researching of data for and writing of the article, and each made substantial contributions to the discussion of content and the revision/editing of the manuscript before submission.
Contributor Information
Richard M. Ransohoff, Biogen Idec, 14 Cambridge Centre, Cambridge, MA 02142, USA
David A. Hafler, Departments of Neurology and Immunobiology, Yale School of Medicine, 15 York Street, New Haven, CT 06520, USA
Claudia F. Lucchinetti, Department of Neurology, Mayo Clinic College of Medicine, 200 First Street SW, Rochester, MN 55905, USA
References
- 1.Charcot JM. Histologie de la sclérose en plaques [French] Gazette des Hopitaux. 1868;41:554–555. [Google Scholar]
- 2.Dawson JD. The histology of disseminated sclerosis. Trans. Royal Soc. Edin. 1916;50:517–740. [Google Scholar]
- 3.Fishman RA. In: Cerebrospinal fluid in diseases of the nervous system. 2nd Saunders WB, editor. 1992. [Google Scholar]
- 4.Rivers TM, Sprunt DH, Berry GP. Observations on attempts to produce acute disseminated encephalomyelitis in monkeys. J. Exp. Med. 1933;58:39–56. doi: 10.1084/jem.58.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Compston A, Sadovnick AD. Epidemiology and genetics of multiple sclerosis. Curr. Opin. Neurol. Neurosurg. 1992;5:175–181. [PubMed] [Google Scholar]
- 6.Rudick RA, Goodkin DE, Ransohoff RM. Pharmacotherapy of multiple sclerosis: current status. Cleve. Clin. J. Med. 1992;59:267–277. doi: 10.3949/ccjm.59.3.267. [DOI] [PubMed] [Google Scholar]
- 7.Weinshenker BG. The natural history of multiple sclerosis. Neurol. Clin. 1995;13:119–146. [PubMed] [Google Scholar]
- 8.Okuda DT, et al. Asymptomatic spinal cord lesions predict disease progression in radiologically isolated syndrome. Neurology. 2011;76:686–692. doi: 10.1212/WNL.0b013e31820d8b1d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amato MP, et al. Association of MRI metrics and cognitive impairment in radiologically isolated syndromes. Neurology. 2012;78:309–314. doi: 10.1212/WNL.0b013e31824528c9. [DOI] [PubMed] [Google Scholar]
- 10.Okuda DT, et al. Radiologically isolated syndrome: 5-year risk for an initial clinical event. PloS ONE. 2014;9:e90509. doi: 10.1371/journal.pone.0090509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calabrese M, et al. Low degree of cortical pathology is associated with benign course of multiple sclerosis. Mult. Scler. 2013;19:904–911. doi: 10.1177/1352458512463767. [DOI] [PubMed] [Google Scholar]
- 12.Correale J, Ysrraelit MC, Fiol MP. Benign multiple sclerosis: does it exist? Curr. Neurol. Neurosci. Rep. 2012;12:601–609. doi: 10.1007/s11910-012-0292-5. [DOI] [PubMed] [Google Scholar]
- 13.Calabrese M, et al. Evidence for relative cortical sparing in benign multiple sclerosis: a longitudinal magnetic resonance imaging study. Mult. Scler. 2009;15:36–41. doi: 10.1177/1352458508096686. [DOI] [PubMed] [Google Scholar]
- 14.Portaccio E, et al. Neuropsychological and MRI measures predict short-term evolution in benign multiple sclerosis. Neurology. 2009;73:498–503. doi: 10.1212/WNL.0b013e3181b351fd. [DOI] [PubMed] [Google Scholar]
- 15.Benedict RH, Fazekas F. Benign or not benign MS: a role for routine neuropsychological assessment? Neurology. 2009;73:494–495. doi: 10.1212/WNL.0b013e3181b35225. [DOI] [PubMed] [Google Scholar]
- 16.Hawkins SA, McDonnell GV. Benign multiple sclerosis? Clinical course, long term follow up, and assessment of prognostic factors. J. Neurol. Neurosurg. Psychiatry. 1999;67:148–152. doi: 10.1136/jnnp.67.2.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stern JN, et al. B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Sci. Transl. Med. 2014;6:248ra107. doi: 10.1126/scitranslmed.3008879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. I. Clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. The IFNB Multiple Sclerosis Study Group. Neurology. 1993;43:655–661. doi: 10.1212/wnl.43.4.655. [DOI] [PubMed] [Google Scholar]
- 19.Hauser SL, et al. Intensive immunosuppression in progressive multiple sclerosis—a randomized, three-arm study of high-dose intravenous cyclophosphamide, plasma exchange, and ACTH. N. Engl. J. Med. 1983;308:173–180. doi: 10.1056/NEJM198301273080401. [DOI] [PubMed] [Google Scholar]
- 20.Paty DW, Li DK. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. II. MRI analysis results of a multicenter, randomized, double-blind, placebo-controlled trial. UBC MS/MRI Study Group and the IFNB Multiple Sclerosis Study Group. Neurology. 1993;43:662–667. doi: 10.1212/wnl.43.4.662. [DOI] [PubMed] [Google Scholar]
- 21.McGraw CA, Lublin FD. Interferon beta and glatiramer acetate therapy. Neurotherapeutics. 2013;10:2–18. doi: 10.1007/s13311-012-0163-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trojano M, et al. Real-life impact of early interferonβ therapy in relapsing multiple sclerosis. Ann. Neurol. 2009;66:513–520. doi: 10.1002/ana.21757. [DOI] [PubMed] [Google Scholar]
- 23.Trojano M, et al. New natural history of interferon-β-treated relapsing multiple sclerosis. Ann. Neurol. 2007;61:300–306. doi: 10.1002/ana.21102. [DOI] [PubMed] [Google Scholar]
- 24.Scalfari A, et al. Mortality in patients with multiple sclerosis. Neurology. 2013;81:184–192. doi: 10.1212/WNL.0b013e31829a3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goodin DS, et al. Survival in MS: a randomized cohort study 21 years after the start of the pivotal IFNβ-1b trial. Neurology. 2012;78:1315–1322. doi: 10.1212/WNL.0b013e3182535cf6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bermel RA, et al. Predictors of long-term outcome in multiple sclerosis patients treated with interferon β. Ann. Neurol. 2013;73:95–103. doi: 10.1002/ana.23758. [DOI] [PubMed] [Google Scholar]
- 27.Prosperini L, et al. Interferon beta failure predicted by EMA criteria or isolated MRI activity in multiple sclerosis. Mult. Scler. 2014;20:566–576. doi: 10.1177/1352458513502399. [DOI] [PubMed] [Google Scholar]
- 28.Borden EC, et al. Interferons at age 50: past, current and future impact on biomedicine. Nat. Rev. Drug Discov. 2007;6:975–990. doi: 10.1038/nrd2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Comabella M, et al. A type I interferon signature in monocytes is associated with poor response to interferon-β in multiple sclerosis. Brain. 2009;132:3353–3365. doi: 10.1093/brain/awp228. [DOI] [PubMed] [Google Scholar]
- 30.Ottoboni L, et al. An RNA profile identifies two subsets of multiple sclerosis patients differing in disease activity. Sci. Transl. Med. 2012;4:153ra131. doi: 10.1126/scitranslmed.3004186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cohen JA, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N. Engl. J. Med. 2010;362:402–415. doi: 10.1056/NEJMoa0907839. [DOI] [PubMed] [Google Scholar]
- 32.Kappos L, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N. Engl. J. Med. 2010;362:387–401. doi: 10.1056/NEJMoa0909494. [DOI] [PubMed] [Google Scholar]
- 33.Kappos L, et al. Oral fingolimod (FTY720) for relapsing multiple sclerosis. N. Engl. J. Med. 2006;355:1124–1140. doi: 10.1056/NEJMoa052643. [DOI] [PubMed] [Google Scholar]
- 34.Brown BA. Natalizumab in the treatment of multiple sclerosis. Ther. Clin. Risk Manag. 2009;5:585–594. doi: 10.2147/tcrm.s5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ransohoff RM. Natalizumab for multiple sclerosis. N. Engl. J Med. 2007;356:2622–2629. doi: 10.1056/NEJMct071462. [DOI] [PubMed] [Google Scholar]
- 36.Yednock TA, et al. Prevention of experimental autoimmune encephalomyelitis by antibodies against α4β1 integrin. Nature. 1992;356:63–66. doi: 10.1038/356063a0. [DOI] [PubMed] [Google Scholar]
- 37.Rudick RA, et al. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N. Engl. J. Med. 2006;354:911–923. doi: 10.1056/NEJMoa044396. [DOI] [PubMed] [Google Scholar]
- 38.Polman CH, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 2006;354:899–910. doi: 10.1056/NEJMoa044397. [DOI] [PubMed] [Google Scholar]
- 39.Langer-Gould A, Atlas SW, Green AJ, Bollen AW, Pelletier D. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N. Engl. J. Med. 2005;353:375–381. doi: 10.1056/NEJMoa051847. [DOI] [PubMed] [Google Scholar]
- 40.Koralnik IJ. Progressive multifocal leukoencephalopathy revisited: has the disease outgrown its name? Ann. Neurol. 2006;60:162–173. doi: 10.1002/ana.20933. [DOI] [PubMed] [Google Scholar]
- 41.Yousry TA, et al. Evaluation of patients treated with natalizumab for progressive multifocal leukoencephalopathy. N. Engl. J. Med. 2006;354:924–933. doi: 10.1056/NEJMoa054693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ransohoff RM. PML risk and natalizumab: more questions than answers. Lancet Neurol. 2010;9:231–233. doi: 10.1016/S1474-4422(10)70025-9. [DOI] [PubMed] [Google Scholar]
- 43.Bloomgren G, et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. N. Engl. J. Med. 2012;366:1870–1880. doi: 10.1056/NEJMoa1107829. [DOI] [PubMed] [Google Scholar]
- 44.Pelletier D, Hafler DA. Fingolimod for multiple sclerosis. N. Engl. J. Med. 2012;366:339–347. doi: 10.1056/NEJMct1101691. [DOI] [PubMed] [Google Scholar]
- 45.Hu Y, et al. Sphingosine 1-phosphate receptor modulator fingolimod (FTY720) does not promote remyelination in vivo. Mol. Cell. Neurosci. 2011;48:72–81. doi: 10.1016/j.mcn.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 46.Jung CG, et al. Functional consequences of S1P receptor modulation in rat oligodendroglial lineage cells. Glia. 2007;55:1656–1667. doi: 10.1002/glia.20576. [DOI] [PubMed] [Google Scholar]
- 47.Stecchi S, Scandellari C, Gabrielli L, Lazzarotto T. Recommendations for fingolimod treated patients vacinated for varicella zoster virus. Neurology. 2014;82(Suppl.):P7.217. [Google Scholar]
- 48.Gold R, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N. Engl. J. Med. 2012;367:1098–1107. doi: 10.1056/NEJMoa1114287. [DOI] [PubMed] [Google Scholar]
- 49.Fox RJ, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N. Engl. J. Med. 2012;367:1087–1097. doi: 10.1056/NEJMoa1206328. [DOI] [PubMed] [Google Scholar]
- 50.Kappos L, et al. Effect of BG-12 on contrast-enhanced lesions in patients with relapsing–remitting multiple sclerosis: subgroup analyses from the phase 2b study. Mult. Scler. 2012;18:314–321. doi: 10.1177/1352458511421054. [DOI] [PubMed] [Google Scholar]
- 51.Kappos L, et al. Efficacy and safety of oral fumarate in patients with relapsing–remitting multiple sclerosis: a multicentre, randomised, double-blind, placebo-controlled phase IIb study. Lancet. 2008;372:1463–1472. doi: 10.1016/S0140-6736(08)61619-0. [DOI] [PubMed] [Google Scholar]
- 52.Cohen JA, et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing–remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet. 2012;380:1819–1828. doi: 10.1016/S0140-6736(12)61769-3. [DOI] [PubMed] [Google Scholar]
- 53.Coles AJ, et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet. 2012;380:1829–1839. doi: 10.1016/S0140-6736(12)61768-1. [DOI] [PubMed] [Google Scholar]
- 54.CAMMS223 Trial Investigators et al. Alemtuzumab vs. interferon beta-1a in early multiple sclerosis. N. Engl. J. Med. 2008;359:1786–1801. doi: 10.1056/NEJMoa0802670. [DOI] [PubMed] [Google Scholar]
- 55.Comi G, et al. Placebo-controlled trial of oral laquinimod for multiple sclerosis. N. Engl. J. Med. 2012;366:1000–1009. doi: 10.1056/NEJMoa1104318. [DOI] [PubMed] [Google Scholar]
- 56.Comi G, et al. Effect of laquinimod on MRI-monitored disease activity in patients with relapsing–remitting multiple sclerosis: a multicentre, randomised, double-blind, placebo-controlled phase IIb study. Lancet. 2008;371:2085–2092. doi: 10.1016/S0140-6736(08)60918-6. [DOI] [PubMed] [Google Scholar]
- 57.Gold R, et al. Daclizumab high-yield process in relapsing–remitting multiple sclerosis (SELECT): a randomised, double-blind, placebo-controlled trial. Lancet. 2013;381:2167–2175. doi: 10.1016/S0140-6736(12)62190-4. [DOI] [PubMed] [Google Scholar]
- 58.Wynn D, et al. Daclizumab in active relapsing multiple sclerosis (CHOICE study): a phase 2, randomised, double-blind, placebo-controlled, add-on trial with interferon beta. Lancet Neurol. 2010;9:381–390. doi: 10.1016/S1474-4422(10)70033-8. [DOI] [PubMed] [Google Scholar]
- 59.Bielekova B, et al. Humanized anti-CD25 (daclizumab) inhibits disease activity in multiple sclerosis patients failing to respond to interferon β. Proc. Natl Acad. Sci. USA. 2004;101:8705–8708. doi: 10.1073/pnas.0402653101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Farh KK, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. doi: 10.1038/nature13835. http://dx.doi.org/10.1038/nature13835. [DOI] [PMC free article] [PubMed]
- 61.Wiendl H, Hohlfeld R. Therapeutic approaches in multiple sclerosis: lessons from failed and interrupted treatment trials. BioDrugs. 2002;16:183–200. doi: 10.2165/00063030-200216030-00003. [DOI] [PubMed] [Google Scholar]
- 62.Panitch HS, Hirsch RL, Haley AS, Johnson KP. Exacerbations of multiple sclerosis in patients treated with gamma interferon. Lancet. 1987;1:893–895. doi: 10.1016/s0140-6736(87)92863-7. [DOI] [PubMed] [Google Scholar]
- 63.Panitch HS, Hirsch RL, Schindler J, Johnson KP. Treatment of multiple sclerosis with gamma interferon: exacerbations associated with activation of the immune system. Neurology. 1987;37:1097–1102. doi: 10.1212/wnl.37.7.1097. [DOI] [PubMed] [Google Scholar]
- 64.Glabinski AR, Krakowski M, Han Y, Owens T, Ransohoff RM. Chemokine expression in GKO mice (lacking interferon-gamma) with experimental autoimmune encephalomyelitis. J. Neurovirol. 1999;5:95–101. doi: 10.3109/13550289909029750. [DOI] [PubMed] [Google Scholar]
- 65.Krakowski M, Owens T. Interferon-γ confers resistance to experimental allergic encephalomyelitis. Eur. J. Immunol. 1996;26:1641–1646. doi: 10.1002/eji.1830260735. [DOI] [PubMed] [Google Scholar]
- 66.van Oosten BW, et al. Increased MRI activity and immune activation in two multiple sclerosis patients treated with the monoclonal anti-tumor necrosis factor antibody cA2. Neurology. 1996;47:1531–1534. doi: 10.1212/wnl.47.6.1531. [DOI] [PubMed] [Google Scholar]
- 67.TNF neutralization in MS: results of a randomized, placebo-controlled multicenter study. The Lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. Neurology. 1999;53:457–465. [PubMed] [Google Scholar]
- 68.Solomon AJ, Spain RI, Kruer MC, Bourdette D. Inflammatory neurological disease in patients treated with tumor necrosis factor alpha inhibitors. Mult. Scler. 2011;17:1472–1487. doi: 10.1177/1352458511412996. [DOI] [PubMed] [Google Scholar]
- 69.Ottoboni L, et al. Clinical relevance and functional consequences of the TNFRSF1A multiple sclerosis locus. Neurology. 2013;81:1891–1899. doi: 10.1212/01.wnl.0000436612.66328.8a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dendrou CA, Bell JI, Fugger L. A clinical conundrum: the detrimental effect of TNF antagonists in multiple sclerosis. Pharmacogenomics. 2013;14:1397–1404. doi: 10.2217/pgs.13.140. [DOI] [PubMed] [Google Scholar]
- 71.Chen X, Oppenheim JJ. Contrasting effects of TNF and anti-TNF on the activation of effector T cells and regulatory T cells in autoimmunity. FEBS Lett. 2011;585:3611–3618. doi: 10.1016/j.febslet.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cua DJ, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 73.Ivanov II, et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 74.Segal BM, et al. Repeated subcutaneous injections of IL12/23 p40 neutralising antibody, ustekinumab, in patients with relapsing–remitting multiple sclerosis: a phase II, double-blind, placebo-controlled, randomised, dose-ranging study. Lancet Neurol. 2008;7:796–804. doi: 10.1016/S1474-4422(08)70173-X. [DOI] [PubMed] [Google Scholar]
- 75.Ryan C, Thrash B, Warren RB, Menter A. The use of ustekinumab in autoimmune disease. Expert Opin. Biol. Ther. 2010;10:587–604. doi: 10.1517/14712591003724670. [DOI] [PubMed] [Google Scholar]
- 76.Kappos L, et al. Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet. 2011;378:1779–1787. doi: 10.1016/S0140-6736(11)61649-8. [DOI] [PubMed] [Google Scholar]
- 77.Ransohoff RM. A mighty mouse: building a better model of multiple sclerosis. J. Clin. Invest. 2006;116:2313–2316. doi: 10.1172/JCI29834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lulu S, Waubant E. Humoral-targeted immunotherapies in multiple sclerosis. Neurotherapeutics. 2013;10:34–43. doi: 10.1007/s13311-012-0164-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Meinl E, Derfuss T, Krumbholz M, Probstel AK, Hohlfeld R. Humoral autoimmunity in multiple sclerosis. J. Neurol. Sci. 2011;306:180–182. doi: 10.1016/j.jns.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 80.Beck RW, et al. A randomized, controlled trial of corticosteroids in the treatment of acute optic neuritis. The Optic Neuritis Study Group. N. Engl. J. Med. 1992;326:581–588. doi: 10.1056/NEJM199202273260901. [DOI] [PubMed] [Google Scholar]
- 81.Major EO, Frohman E, Douek D. JC viremia in natalizumab-treated patients with multiple sclerosis. N. Engl. J. Med. 2013;368:2240–2241. doi: 10.1056/NEJMc1214233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.International Multiple Sclerosis Genetics Consortium (IMSGC) et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 2013;45:1353–1360. doi: 10.1038/ng.2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hafler DA, et al. Risk alleles for multiple sclerosis identified by a genomewide study. N. Engl. J. Med. 2007;357:851–862. doi: 10.1056/NEJMoa073493. [DOI] [PubMed] [Google Scholar]
- 84.Cotsapas C, Hafler DA. Immune-mediated disease genetics: the shared basis of pathogenesis. Trends Immunol. 2013;34:22–26. doi: 10.1016/j.it.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 85.Frischer JM, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain. 2009;132:1175–1189. doi: 10.1093/brain/awp070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hochmeister S, et al. Dysferlin is a new marker for leaky brain blood vessels in multiple sclerosis. J. Neuropathol. Exp. Neurol. 2006;65:855–865. doi: 10.1097/01.jnen.0000235119.52311.16. [DOI] [PubMed] [Google Scholar]
- 87.Howell OW, et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain. 2011;134:2755–2771. doi: 10.1093/brain/awr182. [DOI] [PubMed] [Google Scholar]
- 88.Magliozzi R, et al. A gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann. Neurol. 2010;68:477–493. doi: 10.1002/ana.22230. [DOI] [PubMed] [Google Scholar]
- 89.Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004;14:164–174. doi: 10.1111/j.1750-3639.2004.tb00049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Popescu BF, Bunyan RF, Parisi JE, Ransohoff RM, Lucchinetti CF. A case of multiple sclerosis presenting with inflammatory cortical demyelination. Neurology. 2011;76:1705–1710. doi: 10.1212/WNL.0b013e31821a44f1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lucchinetti CF, et al. Inflammatory cortical demyelination in early multiple sclerosis. N. Engl. J. Med. 2011;365:2188–2197. doi: 10.1056/NEJMoa1100648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chang A, et al. Cortical remyelination: a new target for repair therapies in multiple sclerosis. Ann. Neurol. 2012;72:918–926. doi: 10.1002/ana.23693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Albert M, Antel J, Bruck W, Stadelmann C. Extensive cortical remeylination in patients with chronic multiple sclerosis. Brain Pathol. 2007;17:129–138. doi: 10.1111/j.1750-3639.2006.00043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]