

Abstract
Ligation of CD40 on chronic lymphocytic leukemia (CLL) cells induces phenotypic and biochemical changes that facilitate CLL cell–T cell interactions and enhances the sensitivity of CLL cells to clearance by adaptive and innate immune-effector mechanisms. CLL cells can be transduced to express CD40 ligand (CD154) using a replication-defective adenovirus vector, thereby cross-linking CD40 on transduced and non-transduced, bystander CLL cells. In a previous study, patients received infusions of autologous CLL cells, transduced to express murine CD154 (mCD154), which induced anti-leukemic immune responses, but also anti-mCD154 antibodies. In this study, we report a phase I study, in which patients were infused with 1×108, 3×108 or 1×109 autologous CLL cells transduced ex vivo to express ISF35, a humanized, membrane-stable CD154. Infusions were well tolerated and consistently followed by reductions in blood lymphocyte counts and lymphadenopathy. After infusion, circulating CLL cells had enhanced or de novo expression of CD95, DR5, p73 and Bid, which enhanced their susceptibility to death-receptor-mediated or drug-induced apoptosis, including CLL cells with deletions at 17p13.1 (del(17p)). Two patients who had CLL with del(17p) had subsequent chemoimmunotherapy and responded well to treatment. In summary, infusions of autologous, ISF35-transduced CLL cells were well tolerated, had biological and clinical activity, and might enhance the susceptibility of CLL cells with del(17p) to chemoimmunotherapy.
Keywords: CLL, CD154, gene therapy, adenovirus
Introduction
Chronic lymphocytic leukemia (CLL) is a malignancy of monoclonal, mature B cells that coexpress CD5, CD19 and CD23. CLL cells accumulate in the blood, marrow and secondary lymphoid tissues; over time, they cause suppression of adaptive immunity and hematopoietic function (reviewed in Wierda et al.1).
Treatment with chemoimmunotherapy regimens results in complete remission rates of >50%2,3 and in longer progression-free4–6 and overall survival5 compared with chemotherapy in randomized phase III trials. Unfortunately, these highly effective treatments deplete normal lymphocytes and hematopoietic cells, thereby impairing immune function and hematopoiesis; recovery can take months to years. Immune and hematopoietic dysfunctions constitute the major basis for morbidity and mortality in CLL. Moreover, most treated patients eventually relapse and require additional therapy. Therefore, new treatments that work by different mechanisms are needed.
Owing to their lacking expression of important immune co-stimulatory surface molecules required for cognate intercellular inactions, CLL cells generally are not recognized, even by normal allogeneic T cells. Ligation of CD40 on CLL cells can markedly modify the phenotype, resulting in de novo or enhanced expression of immune costimulatory and adhesion molecules.7,8 This can be achieved through transduction of CLL cells to express CD40 ligand (CD154) using an adenovirus vector. Indeed, transduced and CD40-activated CLL cells can induce autologous T-cell activation, leading to generation of anti-leukemia cellular and antibody immune responses.8
We previously conducted a clinical trial in which patients with CLL received an infusion of autologous leukemia cells transduced ex vivo with a replication-defective adenovirus encoding murine CD154 (mCD154),9 which was expressed more effectively and with greater stability on CLL cells than human CD154 (hCD154). This treatment was well tolerated; patients experienced acute reductions in the leukemia count and decreases in the size of enlarged lymph nodes and spleen. In vivo generation of autologous T cells against autologous CLL cells and generation of antibody against CLL antigens were demonstrated.9,10 However, some of the patients developed antibodies against the mCD154, but not hCD154. To mitigate this problem, we generated ISF35, a novel, recombinant human–murine chimeric CD40-binding protein created to maximize expression on B-cell plasma membrane and to resist cleavage. ISF35 has 91% homology to hCD154, does not have metalloproteinase cleavage sites, and does not contain the mCD154 antibody-binding domains targeted by neutralizing anti-mCD154 antibodies.
This is a phase I, dose-escalation trial of autologous CLL cells transduced ex vivo with a replication-defective adenovirus carrying ISF35 (Ad-ISF35). The primary objective was to determine tolerability and identify dose-limiting toxicities and maximum-tolerated dose. In addition, we focused on the correlative studies of the innate immune response, potentially accounting for the rapid reduction in leukemia count and lymph node size after treatment with ISF35.
Materials and methods
Patient eligibility and pretreatment work-up
Patients provided informed consent according to the MD Anderson Cancer Center Institutional Review Board guidelines; this study was conducted in accordance with the Declaration of Helsinki. Screening tests were carried out to confirm eligibility, including complete history and physical examination, and routine laboratory evaluation, including complete blood count with differential and chemical survey. CLL cell immunoglobulin heavy-chain variable IGHV gene mutation status, ZAP-70 expression and cytogenetic abnormalities by fluorescence in situ hybridization, including deletions at 13q14.3, 11q22.3 and 17p13.1, and for trisomy 12 were evaluated.
Patients must have had a diagnosis of CLL, documented by immunophenotype, and an indication for treatment by the 1996 National Cancer Institute Working Group guidelines;11 an Eastern Cooperative Oncology Group performance status of ≤2; and adequate hematological, renal, hepatic and coagulation function. Patients must have had platelets ≥50K/μl, hemoglobin (HGB) ≥10 g per 100 ml, serum creatinine ≤1.5×upper limit of normal, measured creatinine clearance ≥40, total bilirubin ≤2.5×upper limit of normal, alanine transaminase ≤2.5×upper limit of normal, prothrombin time-international normalized ratio ≤2 and partial thromboplastin time ≤1.66×upper limit of normal. The following were excluded: patients with >55% prolymphocytes; concurrent or chemotherapy within 4 weeks; previous gene therapy; previous allogeneic stem cell transplant; untreated autoimmune hemolytic anemia or immune thrombocytopenia; active infection requiring intravenous antibiotics; known infection with human immunodeficiency virus, hepatitis B or hepatitis C; and uncompensated hypothyroidism.
Treatment and follow-up
After eligibility was confirmed and baseline evaluations obtained, patients underwent leukapheresis to obtain a minimum of 50 ml volume of pheresis product for transduction. The pheresis product was taken to the MD Anderson Cancer Center Good Manufacturing Practice facility and cells were cultured with high-titer Ad-ISF35. Cells were monitored daily for expression of ISF35 by flow cytometry; once expression levels of ISF35 exceeded 40% of cells, the cells were harvested, washed, bacterial cultures taken for release testing, and then the transduced cells were cryopreserved until use. The transduced cells were released for infusion when 14-day bacterial culture was negative. Patients received their specified dose of non-irradiated cells as a single intravenous infusion of autologous ISF35-transduced cells, thawed at the bedside, in the outpatient infusion unit. Before receiving the full, intended dose, patients received a test dose of 3–6×105 autologous ISF35-transduced cells intravenously over 1 min, then were monitored for 10–15 min; if no reaction occurred, they received the remainder of their intended dose. The dose of transduced cells represented the absolute number of ISF35-positive cells and was calculated using the cell count and proportion of cells expressing ISF35, determined by flow cytometry; doses included 1×108, 3×108 and 1×109 ISF35-transduced cells. Patients were monitored for 4 h after infusion with frequent evaluation of vital signs, then released to home for follow-up. Follow-up visits were held on days 2, 7, 14, 21, 28, 56 and every 3 months thereafter and included history and physical examination, routine blood counts and chemistry panel, and samples for correlative laboratory studies. Dose-limiting toxicities were defined as any treatment-related grade ≥3 adverse events, according to the Common Terminology Criteria, version 3.
A follow-up clinical trial was opened to allow additional infusions of autologous ISF35-transduced CLL cells, provided a patient had cryopreserved-transduced cells available. Four of the original nine patients treated on the phase I study gave informed consent, were enrolled, and received additional infusions of autologous ISF35-transduced cells.
Correlative laboratory investigations
Correlative laboratory analyses included an immunophenotype of ISF35-transduced cells before infusion and immunophenotype of circulating leukemia cells before and after infusion at time points based on follow-up. Comparisons were made with pretreatment levels. Monoclonal antibodies used included antibodies against CD154, CD95, DR5, CD3, CD4 or CD8.
Immunoblot analysis for the BH3-interacting domain death agonist (Bid) was performed on prepared protein lysates using RIPA buffer (50 mm Tris/HCl (pH 7.4), 1% Nonidet P-40, 0.25% Na-deoxycholate, 0.1% SDS, 150 mm NaCl, 5 mm EDTA) supplemented with protease inhibitors (10 mg/ml aprotinin, 10 mg/ml leupeptin, 10 mg/ml pepstatin and 1 mm phenylmethylsulfonyl fluoride) from peripheral blood mononuclear cells harvested at the indicated time points for each experiment. After SDS-polyacrylamide gel electrophoresis, immunoblot membranes were probed with anti-Bid antibody (Cell Signaling Technology, Beverly, MA, USA) followed by goat anti-rabbit IgG–HRP (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Stripped membranes were probed with anti-β-actin (Sigma, St Louis, MO, USA) followed by goat anti-mouse IgG–HRP to monitor for protein loading. Immune detection was carried out using horseradish peroxidase (HRP)-conjugated secondary antibodies and the reaction was developed with Chemiluminescent Substrate kit-ECL Plus from Amersham-Biosciences (Piscataway, NJ, USA). As a negative control, we used a lysate of non-transduced CLL cells. Psfk-1 cell lysate (BD Biosciences, La Jolla, CA, USA) was used as positive control.
Immunoblotting for TAp73 and Mcl-1 at the indicated time points was performed with antibodies that specifically recognize the pro-apoptotic TAp73 (p73 in this article) isoforms (IMG-246, Imgenex, San Diego, CA, USA), Mcl-1 (Santa Cruz Biotechnology) and GAPDH (Upstate Biochemicals, Lake Placid, NY, USA). Proteins were quantitated by densitometry with the Odyssey-LiCor (Odyssey, Lincoln, NE, USA), normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) within the same sample and expressed as percent of control.
For Annexin V assay, blood mononuclear cell were obtained at the indicated times post infusion of ISF35; 1×106 primary CLL cells were incubated with Annexin V-FITC (BD Biosciences) for 15 min. Fluorescence of at least 10 000 cells was determined on a Becton Dickinson FACSCalibur flow cytometer (Becton Dickinson, Franklin Lakes, NY, USA) to determine the percentage of apoptotic cells within the population.
Results
Previously treated or chemotherapy-naïve patients were enrolled into this phase I clinical trial. Most patients had had previous treatment for CLL (Table 1). Some patients had previously received chemoimmunotherapy, others had received rituximab with granulocyte monocyte colony-stimulating factor. All patients had an indication for treatment by the 1996 National Cancer Institute Working Group guidelines.11 Four patients had CLL cells with an unmutated (≥98% homology to germ line) IGHV gene, six had leukemia cells that expressed ZAP-70 (≥20% positive) and four had CLL cells that harbored deletion in the short arm of chromosome 17 (del(17p)), demonstrated by fluorescence in situ hybridization. All patients had measurable disease with elevated absolute lymphocyte count (ALC) and palpable lymphadenopathy.
Table 1.
Patient characteristics
| Subject number (initials) | 1 (MP) | 2 (RL) | 3 (RG) | 4 (RT) | 5 (RM) | 6 (JC) | 7 (BF) | 8 (JW) | 9 (LH) |
| ISF35 dose level | 1×108 | 1×108 | 1×108 | 3×108 | 3×108 | 3×108 | 1×109 | 1×109 | 1×109 |
| Age (years) | 75 | 70 | 30 | 78 | 70 | 70 | 71 | 71 | 59 |
| Sex | F | M | M | M | M | M | F | F | F |
| Year of diagnosis | 1998 | 1999 | 2002 | 1998 | 1994 | 2001 | 2003 | 1995 | 2003 |
| No. of previous treatments | 0 | 0 | 1 | 1 | 2 | 2 | 1 | 3 | 2 |
| Previous treatments | — | — | FCR | R+GM-CSF | FCR; R+GM-CSF |
R; R+GM-CSF |
R+GM-CSF | CVP; T cells; R+GM-CSF |
R+GM-CSF X2 |
| Rai stage | II | I | IV | I | IV | IV | I | IV | I |
| ALC (K/μl) | 43 | 58 | 14 | 105 | 66 | 72 | 37 | 75 | 54 |
| LDT (mo) | 6.8 | 18 | 2.7 | 5.2 | 5.2 | 6 | 5.3 | 8.8 | 6.5 |
| β-2M (mg/l) | 2.8 | 4.8 | 2.4 | 4.4 | 2.9 | 6.7 | 4.8 | NA | 2.1 |
| IGHV % Mutated | 7.8 | 12 | 0 | 0 | 7.6 | 0 | 1.8 | 7.1 | 2.4 |
| ZAP70-IHC | Negative | Positive | Positive | Positive | Negative | Positive | Positive | Negative | Positive |
| FISH | None | +12 | del 17p | del 17p | del 17p | None | del 11q | del 13q | del 17p |
| Cytogenetics | Diploid | Complex | Diploid | Diploid | Diploid | Diploid | del 11q | del 13q | Diploid |
| %CD38 Positive | 0.7 | 56 | 91 | 35 | 1 | 5 | 3 | 26 | 1.9 |
Abbreviations: ALC, absolute lymphocyte count; CVP, cyclophosphamide, vincristine, prednisone; F, female; FCR, fludarabine, cyclophosphamide, rituximab; FISH, fluorescence in situ hybridization (hierarchical categorization); GM-CSF, granulocyte macrophage colony stimulating factor; IGHV, immunoglobulin heavy chain variable gene; IHC, immunohistochemistry; LDT, lymphocyte doubling time; M, male; β-2M; β-2 microglobulin; mo, months; NA, not available; R, rituximab.
The autologous ISF35-transduced products were evaluated prior to infusion for expression of ISF35 and CD95 (Fas), which is consistently induced on CLL cells after CD40 ligation (Table 2 and Supplementary Figure 1). For all patients, ex vivo transduction with Ad-ISF35 resulted in >40% of each patient’s leukemia cells expressing the ISF35 transgene. Each patient’s dose was the indicated absolute number of ISF35-positive cells. Free viral particle measurements indicated that patients received up to 1.3×108 free viral particles with their infusion (Table 2).
Table 2.
Ad-ISF35-transduced cell product
| Subject number (initials) | 1 (MP) | 2 (RL) | 3 (RG) | 4 (RT) | 5 (RM) | 6 (JC) | 7 (BF) | 8 (JW) | 9 (LH) |
| Intended dose (ISF35-cells) | 1×108 | 1×108 | 1×108 | 3×108 | 3×108 | 3×108 | 1×109 | 1×109 | 1×109 |
| Volume infused (ml) | 10 | 10 | 10 | 10 | 10 | 10 | 30 | 30 | 30 |
| % ISF35 transduction | 49 | 77 | 52 | 57 | 58 | 40 | 42 | 54 | 41 |
| ISF35 MFIR | 2.0 | 6.9 | 2.1 | 2.9 | 2.9 | 1.8 | 3.7 | 2.1 | 2.2 |
| %CD95+ in transduced | 83 | 96 | 94 | 97 | 96 | 93 | 90 | 78 | 93 |
| CD95 MFIR in transduced | 7.6 | 43.6 | 15.0 | 34.4 | 33.4 | 17.4 | 20.3 | 11.7 | 27.1 |
| Free Ad-ISF35 (p/ml) | 8.7×106 | 4.3×106 | 3.0×106 | 5.4×106 | 7.8×106 | 5.3×106 | 4.2×106 | 1.8×107 | 2.8×106 |
| Total free Ad-ISF35 infused | 8.7×107 | 4.3×107 | 3.0×107 | 5.4×107 | 7.8×107 | 5.3×107 | 1.3×108 | 5.4×108 | 8.4×107 |
Abbreviations: MFIR, mean fluorescence intensity ratio; p/ml, particles/ml.
The infusions were well tolerated. Toxicities consisted primarily of grade≤2 flu-like symptoms (for example, fevers, arthralgias, myalgias, nausea, vomiting and anorexia), with the exception of one patient (no. 7) with grade 3 headache on day 3 that resolved within 2 days (Table 3), which was the only dose-limiting toxicity observed. Symptoms generally lasted 2–4 days and subsequently resolved; there were no adverse events that lasted beyond day 8 of treatment. The patients who received the highest dose of autologous Ad-ISF35-transduced cells (for example, 1×109) generally experienced a higher incidence of grade 2 versus grade 1 events than patients treated at lower dose levels, although differences were not statistically significant because of the small sample size (Table 3).
Table 3.
Single dose, ISF35 treatment related toxicities (N = 9)
| Number of events | |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Group 1 (1×108 ISF35 CLL) | Group 2 (3×108 ISF3 CLL) | Group 3 (1×109 ISF35 CLL) | |||||
|
|
|
|
|||||
| Grade 1 | Grade 2 | Grade 1 | Grade 2 | Grade 1 | Grade 2 | Grade 3 | |
| Fever | 3 | 3 | 2 | 1 | |||
| Arthralgia | 2 | 2 | 1 | 2 | |||
| Fatigue | 1 | 1 | 2 | ||||
| Nausea/vomiting | 2 | 1 | 3 | 1 | 3 | ||
| Headache | 1 | 1 | 1 | 3 | 1 | ||
| Anorexia | 2 | 1 | 2 | ||||
| Dyspnea | 1 | 1 | |||||
| Herpes simplex/zoster | 1 | 1 | |||||
| Dehydration | 1 | ||||||
| Hypotension | 1 | ||||||
| Rash | 1 | ||||||
| Upper respiratory tract symptoms | 1 | 2 | |||||
| Stomatitis | 1 | ||||||
All patients experienced reduction in ALC after infusion of autologous Ad-ISF35-transduced cells; the nadir typically was at day 2 after infusion (Figure 1a). The mechanism for this acute decrease in ALC and lymph node size was most consistent with an innate versus adaptive immune response. A more protracted reduction would be expected for an antigen-specific, adaptive immune response. Over the subsequent 4 weeks, leukemia counts returned toward pretreatment level. There did not seem to be a dose–response relationship regarding decrease in ALC; however, patients who received 1×109 Ad-ISF35-modified cells seemed to have more durable reductions in ALC (Figure 1a).
Figure 1.

Complete blood counts with manual differential were performed before and after administration of autologous ISF35-transduced cells (a). ALC was normalized to the count obtained pre-treatment on the day of infusion (day 0) and plotted for each subject by dose level. The sum product of bidimensional lymph node measurements by palpation in three nodal regions was used as the measure of lymph node bulk (b). Measures were taken pre- (day 0) and post-infusion of autologous ISF35-transduced CLL cells. These data points were normalized to day 0 and presented for each patient by dose level. Consistent reductions in normalized ALC and lymph node bulk were noted acutely following treatment.
To study the innate immune response and confirm that the acute reduction in ALC was because of the in vivo apoptosis in circulating CLL cells triggered by infusion of ISF35-transduced cells, annexin V staining was performed on freshly isolated blood mononuclear cells after treatment in a subset of patients. This allowed us to monitor the increased levels of apoptosis in the non-transduced, circulating CLL cells following infusion of ISF35-transduced CLL cells (Figure 2). High levels of apoptosis in circulating CLL cells were observed in patients for up to 3 weeks after infusion of ISF35-transduced CLL cells.
Figure 2.
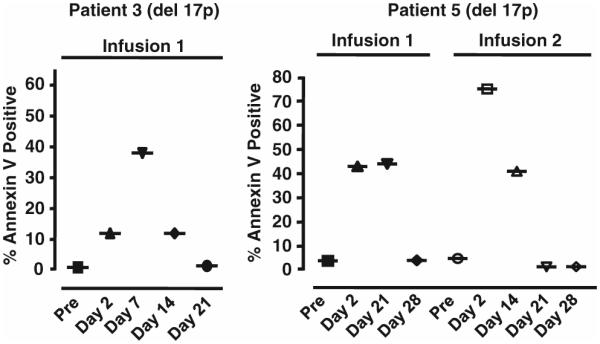
Blood was taken from patients 3 and 5 pre-treatment on the day of infusion (pre) and at the indicated days after infusion of autologous ISF35-transduced CLL cells. The freshly isolated blood mononuclear cells were immediately stained for annexin V to determine the proportion of circulating cells undergoing apoptosis. Patient 3 received a single infusion and patient 5 received two infusions. Both patients had del(17p) in their leukemia cells. Both patients had an increase in the percent of circulating leukemia cells undergoing apoptosis post-infusion demonstrated by staining for annexin V that lasted for up to 3 weeks after infusion.
We evaluated circulating CLL cells for expression of CD95, DR5, Bid and Mcl-1 after treatment to provide insight into in vivo leukemia cell death. Consistent with an innate immune response, circulating CLL cells were induced to express de novo or increased levels of the death receptors, CD95 and DR5, 1–2 days after infusion of autologous ISF35-transduced CLL cells (Table 4). Expression of these death receptors continued for 1–3 weeks after infusion. In addition, we found that circulating CLL cells were induced in vivo to express the pro-apoptotic protein Bid (Figure 3), which can enhance the susceptibility of CLL cells to death receptor-mediated apoptosis. Furthermore, decreased levels of the anti-apoptotic protein Mcl-1 were noted, further promoting apoptosis in circulating CLL cells after infusion of autologous ISF35-transduced cells (Figure 4). Finally, we evaluated CLL cells for expression of p73 before and after treatment (Figure 4); p73 can function like p53 to mediate chemotherapy-induced apoptosis. In each case examined, we noted in vivo increased expression, relative to pretreatment, of p73 in circulating CLL cells after infusion of ISF35-transduced cells (Figure 4). Collectively, these changes enhanced the susceptibility of CLL to apoptosis and are consistent with previously reported changes that occur in CLL cells after CD40 ligation in vitro.12,13
Table 4.
Increased expression of CD95 and DR5 in circulating CLL after ISF35
| Subject | CD95 MFIR | DR5 MFIR | ||
|---|---|---|---|---|
|
|
|
|||
| Fold increase | Peak day | Fold increase | Peak day | |
| 1 | 1.38 | 14 | 1.10 | 21 |
| 2 | 1.65 | 2 | 1.19 | 2 |
| 3 | 1.82 | 14 | 1.59 | 7 |
| 4 | 1.60 | 2 | 1.21 | 21 |
| 5 | 1.33 | 2 | 1.30 | 2 |
| 6 | 1.21 | 2 | 1.21 | 2 |
| 7 | 2.33 | 7 | 1.21 | 14 |
| 8 | 1.13 | 7 | 1.05 | 2 |
| 9 | 2.36 | 7 | 1.20 | 14 |
Abbreviations: CLL, chronic lymphocytic leukemia; MFIR, mean fluorescence intensity ratio.
Figure 3.
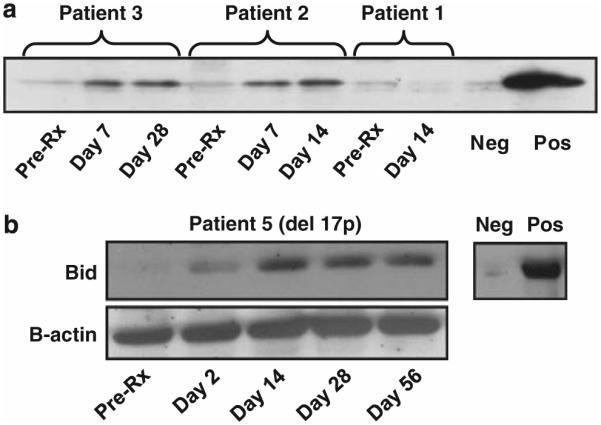
Freshly isolated blood mononuclear cell from patients 1, 2, 3 (a) and 5 (b) were obtained pre-treatment (Pre-Rx) and at the indicated days post infusion of autologous ISF35-transduced CLL cells. Immunoblot was performed to evaluate expression of the intracellular pro-apoptotic protein Bid. Circulating leukemia cell expression of Bid increased and persisted for several weeks after infusion, including for patients 3 and 5, who have leukemia cells with del(17p).
Figure 4.
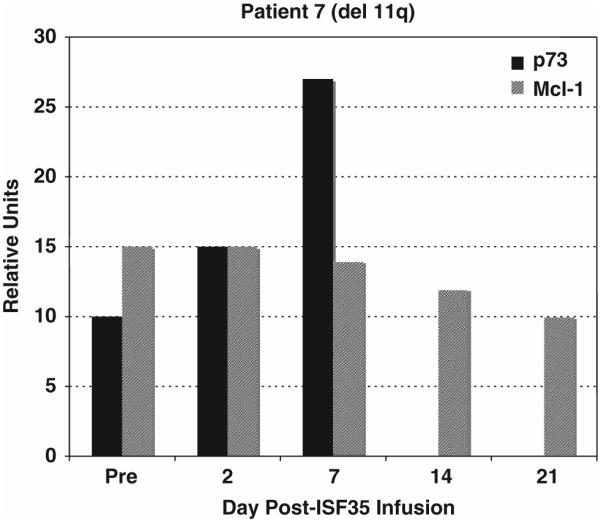
Circulating cells were evaluated for expression of p73 and Mcl-1 by immunoblot. Blood was obtained pre-treatment (Pre) and after infusion of autologous ISF35-transduced leukemia cells. Immediately, blood mononuclear cells were isolated and cell lysates were evaluated by immunoblot for protein expression for p73 and Mcl-1. Blots were scanned and data are expressed in relative units. Increased expression of p73 was observed in circulating cells following infusion of ISF35-transduced cells in this patient with del(17p). There was also reduction in the anti-apoptotic protein Mcl-1 after infusion.
In addition to experiencing reductions in ALC, the treated patients also experienced concomitant reductions in lymphadenopathy at all dose levels, some achieving complete resolution of all palpable lymphadenopathy (Figure 1b). There did not seem to be a dose–response relationship between the extent of lymph node reduction and the number of infused ISF35-transduced CLL cells. Nonetheless, the noted reductions in lymph node size after treatment seemed more durable than reductions in ALC noted after therapy (Figure 1). Consistent with our previous CD154 gene therapy trial, increases in absolute T-cell counts were noted for patients treated in this trial, despite an overall reduction in ALC (Supplementary Table 1). Nearly all patients had increases in absolute T-cell counts compared with pretreatment values. These increases in T-cell counts were most notable in the period 1–4 weeks after treatment. The maximum increase was 374%.
We examined the kinetics of increase in leukemia cell count before and after infusion of autologous ISF35-transduced CLL cells. Lymphocyte doubling times were calculated on the basis of all available pre-enrollment blood counts obtained during the previous treatment-free interval (Table 1). Using the pretreatment lymphocyte doubling time, we predicted the expected increase in lymphocyte count from the time of ISF35 treatment. These predictions were juxtaposed to the actual lymphocyte counts observed after treatment (Figure 5). For most patients, there were apparent reductions in the rates at which the ALCs were increasing after infusion of ISF35-transduced cells, resulting in deviations from the expected values. In several cases, disease progression seemed to be arrested after the infusion of a single dose of ISF35-transduced CLL cells (Figure 5). No patients developed anti-ISF35 antibodies.
Figure 5.

Actual (measured) ALC (solid line) for patients 1, 3, 4 and 6 are shown before and after infusion of autologous ISF35-transduced CLL cells. Day 0 was the count before the treatment on the day of infusion. The predicted ALC shown by the dashed line is the projected count for each patient based on actual blood counts obtained in the absence of treatment for the time period of at least 6 months immediately before receiving ISF35-transduced cells. These data demonstrate a reduction in actual lymphocyte counts following ISF35 infusion versus the projected counts.
Three patients received a second infusion of ISF35-transduced cells and the fourth patient received a total of three infusions of autologous ISF35-transduced cells. Repeat infusions were well tolerated; there were no grade 3 or 4 toxicities; therefore no dose-limiting toxicity was identified. Adverse events with repeated infusions were similar to those noted after the initial infusion; these included fever, chills, arthralgia, myalgia, nausea, vomiting or anorexia. Similar to the adverse events noted during the initial infusion, these symptoms resolved within 2–3 days after each infusion (Supplementary Table 2). No patient experienced adverse events lasting more than 3 days after repeated infusions. Similar to the initial infusion, the second and third infusions were associated with reduction in ALC and lymph node size. The reductions in circulating leukemia cells were greater with subsequent infusions and such responses seemed to be of longer duration (Supplementary Figure 2 and data not shown). The impact on lymph node size was also augmented in subsequent ISF35 therapy and the lymph nodes of two of four patients were no longer palpable. This reduction in lymph node bulk was durable and responses lasted several months after ISF35-transduced cell infusion (data not shown). No patients developed anti-ISF35 antibodies.
All patients enrolled have extended follow-up information with a range of 28–37 months (Supplementary Table 3). Two patients died during the follow-up period, both at 28 months. Eight of the nine patients received subsequent salvage treatment. The time between ISF35 and first salvage therapy was 4 to 20 months. One patient received one salvage regimen, four patients received two salvage regimens, one patient received three salvage regimens and two patients received four salvage treatments. Two patients proceeded to allogeneic stem cell transplant, including a cord blood stem cell transplant. All but one patient responded to salvage therapy, with the best responses seen with chemoimmunotherapy (fludarabine, cyclophosphamide and rituximab). These are favorable responses considering that four of the patients had CLL cells harboring del(17p), which is typically associated with disease that is refractory to chemotherapy (Table 1).
Discussion
This was the first phase I clinical trial in humans demonstrating the safety and clinical activity of autologous ISF35-transduced leukemia cells for patients with CLL; no maximum tolerated dose was identified. Treatment-related adverse events were flu-like symptoms of fever, chills, arthralgias, myalgias, nausea and anorexia. One patient experienced grade 3 headache. Symptoms resolved within 2–4 days after the infusion, and there were no lasting toxicities. No autoimmune hemolytic anemia or immune thrombocytopenia was observed, and absolute T lymphocyte counts were increased with treatment. After administration of ISF35-transduced cells, there was no evidence of liver function test abnormalities, unlike those seen in the earlier trial using leukemia cells transduced to express mCD154.9
In this phase I trial, a replication-defective adenovirus encoding ISF35 (Ad-ISF35), a recombinant CD40-ligand transgene, was used ex vivo to transduce CLL cells. Patients received an intravenous infusion of autologous ISF35-transduced cells, which resulted in acute and prolonged decrease in ALC and lymph node size with apoptosis and phenotypic and physiological changes in circulating, non-transduced CLL cells. The changes in circulating CLL cells were consistent with in vivo ligation of CD40, and were similar to those observed in a previous clinical trial involving infusions of autologous CLL cells transduced to express mCD154 in which correlative studies focused on demonstrating induction of T-cell-mediated adaptive immune responses against autologous CLL cells.9 In this study, we evaluated the primary end point of safety and also clinical activity and focused on evaluating whether the acute decline in leukemia cell counts was related to enhanced rates of CLL-cell apoptosis in vivo. We examined for in vivo induced expression of death receptors and pro-apoptotic proteins in bystander CLL cells that did not express the ISF35 transgene.12,14
Infusion of ISF35-transduced CLL cells was consistently followed by reductions in both blood lymphocyte count and lymph node size. The concurrent reductions in both leukemia cell count and lymph node size argues that treatment induced clearance, rather than margination, of CLL cells. Consistent with this, staining the leukemia cells with annexin V revealed that after treatment, there was a significantly higher proportion of circulating CLL cells undergoing apoptosis in vivo after treatment. Notably, the increased proportions of cells undergoing apoptosis were observed for up to 3 weeks after infusion of ISF35-transduced CLL cells.
The rapid reduction in leukemia count and lymph node size after treatment suggested clearance by innate immune effector mechanisms. Consistent with this, we observed that circulating CLL cells were induced to express death receptors (CD95 and DR5) and the pro-apoptotic molecule Bid, and had reduced levels of the anti-apoptotic protein Mcl-1 after infusion of autologous ISF35-transduced CLL cells. Expression of Bid by CLL cells after CD40 activation is associated with enhanced susceptibility to spontaneous and drug-induced apoptosis, even in CLL cells lacking functional p53.15 The capacity to induce these changes in CLL cells lacking functional p53 seems to be because of the induction by CD40 activation of another member of the p53 family, namely p73, which shares with p53 the capacity to induce expression of genes encoding proteins involved in apoptosis. That we observed these changes in vivo in circulating cells of patients for weeks after treatment with ISF35 indicates that activation via ligation of CD40 in vivo also can result in changes that enhance the susceptibility of CLL cells to spontaneous or drug-induced apoptosis. In addition, Bid allows for cross talk between the extrinsic death receptor-mediated signaling and the intrinsic mitochondrial-mediated apoptotic pathway, potentially allowing for more effective clearance by innate immune effector mechanisms.
The acute clearance of leukemia cells observed in ISF35-treated patients seemed to be independent of cytogenetic abnormalities, including patients who had CLL with del(17p). We previously demonstrated in vitro that CD40 activation upregulated expression of p73, a p53 surrogate, which allows p53-deficient cells to undergo apoptosis.15 p73 is rarely mutated in human cancer.16,17 Isoforms of p73 exist that can serve as a transactivator of gene expression (TAp73) or as a possible inhibitor to TAp73 (DN73), which are generated via differential splicing of the p73 transcript.18 The TAp73 (referred to as p73 in our article) proteins efficiently bind to p53 response elements to transcriptionally induce pro-apoptotic target genes and to induce apoptosis in a variety of tumor cells, whereas DNp73 isoforms largely exert their anti-apoptotic activity by interfering with the ability of p73 to transactivate downstream apoptotic genes. In this trial, we demonstrated that the bystander leukemia cells of treated patients were induced to express p73 and became sensitized to anti-leukemia drugs (data not shown). Importantly, such sensitization to chemotherapy was observed even in CLL cells with del(17p), which was associated with loss of P53 function. Two of four patients with CLL cells that had del(17p) were subsequently treated with the fludarabine, cyclophosphamide and rituximab regimen for salvage and achieved excellent responses to this therapy.
In many cases, the reduction in lymphocyte count and lymph node size were durable. Patients 1 and 6 had absolute lymphocyte doubling time previous to treatment of 6.8 and 6 months, respectively. Subsequent to treatment, these patients had stable blood lymphocyte counts and did not require additional treatment for more than 22 and 17 months, respectively. Patient 8 had a lymphocyte doubling time before treatment of 8.8 months and required salvage therapy 20 months after ISF35 treatment. The acute reductions in circulating lymphocytes and lymph node size, and the long-term disease stabilization, are consistent with biphasic activity (innate and adaptive immunity) of ISF35 treatment. In this manner, administration of the ISF35-transduced cells initially activates the total pool of circulating CLL cells, even though the proportions of ISF35-transduced cells infused were less than 1% of the total leukemia burden. Conceivably, the adaptive antibody and cellular immune responses8–10,19 against leukemia-associated antigens could account for the relatively long duration of response observed in some patients. However, identification of the factor(s) associated with durable responses to therapy will require treatment of larger numbers of patients than that used in a phase I study.
These results support two strategies for future clinical development of ISF35. First, as ISF35-transduced cells were well tolerated, particularly in this population with a median age of 70, we plan to initiate trials with repeated doses of ISF35-transduced CLL cells as monotherapy. The rationale with this strategy will be to achieve long-term disease control in this fragile population that is more susceptible to myelosuppression and immune suppression associated with standard chemotherapy. The proposed strategy is to administer repeated doses of ISF35-transduced CLL cells over a protracted period in order to convert progressive CLL into a more indolent course. With this strategy, delay in progression or stabilization of disease might obviate chemotherapy and provide for a lasting clinical benefit.
Along with the long-term responses, the ISF35-transduced cells also may have sensitized the CLL cells of treated patients to chemotherapy. This is a particularly important finding, especially for patients with leukemia cells that have del(17p) and loss of functional p53. Patients with CLL cells that have del(17p) typically are refractory to standard purine analogue-based treatments. As this population potentially has an unmet medical need, we plan to initiate trials with ISF35-transduced CLL cells followed by chemoimmunotherapy for patients with del(17p). Ultimately, we believe that pretreatment with ISF35-transduced cells may serve as an important adjuvant to cytotoxic agents, helping to sensitize the leukemia cell population to cytotoxic therapy through induction of p73.
Supplementary Material
Acknowledgements
This clinical trial and correlative laboratory studies were supported by the research funding provided by Memgen, LLC. This project was a collaboration formed under the CLL Research Consortium.
WG Wierda, JE Castro, D Sampath and J McMannis received research grant funding to complete the work included herein.
Footnotes
Conflict of interest
R Aguillon, A Jalayer and M Keating declare no conflict of interest. CE Prussak has a financial interest in MemGen, the sponsor of this trial. TJ Kipps has been a scientific advisor to Memgen and inventor of technology patented by the University of California and licensed to Memgen.
Author contributions
WG Wierda designed and performed the research, collected, analyzed and interpreted the data, performed statistical analysis and wrote the article. JE Castro, R Aguillon and D Sampath performed the research and contributed analytical tools. A Jalayer collected the data. J McMannis contributed analytical tools. CE Prussak designed the research. M Keating performed the research. TJ Kipps designed the research and wrote the article.
Supplementary Information accompanies the paper on the Leukemia website (http://www.nature.com/leu)
References
- 1.Wierda WG, Keating MJ, O’Brien S. Chronic lymphocytic leukemia. In: DeVita VT, Lawrence TS, Rosenberg SA, editors. Cancer: Principles & Practice of Oncology. 8th Lippincott Williams & Wilkins; Philadelphia, PA: 2008. pp. 2278–2291. [Google Scholar]
- 2.Keating MJ, O’Brien S, Albitar M, Lerner S, Plunkett W, Giles F, et al. Early results of a chemoimmunotherapy regimen of fludarabine, cyclophosphamide, and rituximab as initial therapy for chronic lymphocytic leukemia. J Clin Oncol. 2005;23:4079–4088. doi: 10.1200/JCO.2005.12.051. [DOI] [PubMed] [Google Scholar]
- 3.Tam CS, O’Brien S, Wierda W, Kantarjian H, Wen S, Do KA, et al. Long-term results of the fludarabine, cyclophosphamide, and rituximab regimen as initial therapy of chronic lymphocytic leukemia. Blood. 2008;112:975–980. doi: 10.1182/blood-2008-02-140582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hallek M, Fingerle-Rowson G, Fink A-M, Busch R, Mayer J, Hensel M, et al. Immunochemotherapy with fludarabine (F), cyclophosphamide (C), and rituximab (R) (FCR) versus fludarabine and cyclophosphamide (FC) improves response rates and progression-free survival (PFS) of previously untreated patients (pts) with advanced chronic lymphocytic leukemia (CLL) Blood. 2008;112 abstract no. 325. [Google Scholar]
- 5.Hallek M, Fingerle-Rowson G, Fink A-M, Busch R, Mayer J, Hensel M, et al. First-line treatment with fludarabine (F), cyclophosphamide (C), and rituximab (R) (FCR) improves overall survival (OS) in previously untreated patients (pts) with advanced chronic lymphocytic leukemia (CLL): results of a randomized phase III trial on behalf of an international group of investigators and the German CLL Study Group. Blood. 2009;114 abstract no. 535. [Google Scholar]
- 6.Robak T, Dmoszynska A, Solal-Celigny P, Warzocha K, Loscertales J, Catalano J, et al. Rituximab plus fludarabine and cyclophosphamide prolongs progression-free survival compared with fludarabine and cyclophosphamide alone in previously treated chronic lymphocytic leukemia. J Clin Oncol. 2010;28:1756–1765. doi: 10.1200/JCO.2009.26.4556. [DOI] [PubMed] [Google Scholar]
- 7.Ranheim EA, Kipps TJ. Activated T cells induce expression of B7/BB1 on normal or leukemic B cells through a CD40-dependent signal. J Exp Med. 1993;177:925–935. doi: 10.1084/jem.177.4.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kato K, Cantwell MJ, Sharma S, Kipps TJ. Gene transfer of CD40-ligand induces autologous immune recognition of chronic lymphocytic leukemia B cells. J Clin Invest. 1998;101:1133–1141. doi: 10.1172/JCI1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wierda WG, Cantwell MJ, Woods SJ, Rassenti LZ, Prussak CE, Kipps TJ. CD40-ligand (CD154) gene therapy for chronic lymphocytic leukemia. Blood. 2000;96:2917–2924. [PubMed] [Google Scholar]
- 10.Fukuda T, Chen L, Endo T, Tang L, Lu D, Castro JE, et al. Antisera induced by infusions of autologous Ad-CD154-leukemia B cells identify ROR1 as an oncofetal antigen and receptor for Wnt5a. Proc Natl Acad Sci USA. 2008;105:3047–3052. doi: 10.1073/pnas.0712148105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheson BD, Bennett JM, Grever M, Kay N, Keating MJ, O’Brien S, et al. National Cancer Institute-sponsored Working Group guidelines for chronic lymphocytic leukemia: revised guidelines for diagnosis and treatment. Blood. 1996;87:4990–4997. [PubMed] [Google Scholar]
- 12.Chu P, Deforce D, Pedersen IM, Kim Y, Kitada S, Reed JC, et al. Latent sensitivity to Fas-mediated apoptosis after CD40 ligation may explain activity of CD154 gene therapy in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2002;99:3854–3859. doi: 10.1073/pnas.022604399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chu P, Wierda WG, Kipps TJ. CD40 activation does not protect chronic lymphocytic leukemia B cells from apoptosis induced by cytotoxic T lymphocytes. Blood. 2000;95:3853–3858. [PubMed] [Google Scholar]
- 14.Dicker F, Kater AP, Fukuda T, Kipps TJ. Fas-ligand (CD178) and TRAIL synergistically induce apoptosis of CD40-activated chronic lymphocytic leukemia B cells. Blood. 2005;105:3193–3198. doi: 10.1182/blood-2003-10-3684. [DOI] [PubMed] [Google Scholar]
- 15.Dicker F, Kater AP, Prada CE, Fukuda T, Castro JE, Sun G, et al. CD154 induces p73 to overcome the resistance to apoptosis of chronic lymphocytic leukemia cells lacking functional p53. Blood. 2006;108:3450–3457. doi: 10.1182/blood-2006-04-017749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ozaki T, Nakagawara A. p73, a sophisticated p53 family member in the cancer world. Cancer Sci. 2005;96:729–737. doi: 10.1111/j.1349-7006.2005.00116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pluta A, Nyman U, Joseph B, Robak T, Zhivotovsky B, Smolewski P. The role of p73 in hematological malignancies. Leukemia. 2006;20:757–766. doi: 10.1038/sj.leu.2404166. [DOI] [PubMed] [Google Scholar]
- 18.Muller M, Schilling T, Sayan AE, Kairat A, Lorenz K, Schulze-Bergkamen H, et al. TAp73/Delta Np73 influences apoptotic response, chemosensitivity and prognosis in hepatocellular carcinoma. Cell Death Differ. 2005;12:1564–1577. doi: 10.1038/sj.cdd.4401774. [DOI] [PubMed] [Google Scholar]
- 19.Foster AE, Okur FV, Biagi E, Lu A, Dotti G, Yvon E, et al. Selective elimination of a chemoresistant side population of B-CLL cells by cytotoxic T lymphocytes in subjects receiving an autologous hCD40L/IL-2 tumor vaccine. Leukemia. 2010;24:563–572. doi: 10.1038/leu.2009.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.