

Abstract
Chemotherapeutic resistance in breast cancer, whether acquired or intrinsic, remains a major clinical obstacle. Thus, increasing tumor cell sensitivity to chemotherapeutic agents will be helpful in improving the clinical management of breast cancer. In the present study, we found an induction of HO-1 expression in doxorubicin (DOX)-treated MDA-MB-231 human breast adenocarcinoma cells, which showed insensitivity to DOX treatment. Knockdown HO-1 expression dramatically upregulated the incidence of MDA-MB-231 cell death under DOX treatment, indicating that HO-1 functions as a critical contributor to drug resistance in MDA-MB-231 cells. We further observed that DOX exposure induced a cytoprotective autophagic flux in MDA-MB-231 cells, which was dependent on HO-1 induction. Moreover, upregulation of HO-1 expression required the activation of both signal transducer and activator of transcription (STAT)3 and its upstream regulator, protein kinase Src. Abrogating Src/STAT3 pathway activation attenuated HO-1 and autophagy induction, thus increasing the chemosensitivity of MDA-MB-231 cells. Therefore, we conclude that Src/STAT3-dependent HO-1 induction protects MDA-MB-231 breast cancer cells from DOX-induced death through promoting autophagy. In the following study, we further demonstrated the contribution of Src/STAT3/HO-1/autophagy pathway activation to DOX resistance in another breast cancer cell line, MDA-MB-468, which bears a similar phenotype to MDA-MB-231 cells. Therefore, activation of Src/STAT3/HO-1/autophagy signaling pathway might play a general role in protecting certain subtypes of breast cancer cells from DOX-induced cytotoxicity. Targeting this signaling event may provide a potential approach for overcoming DOX resistance in breast cancer therapeutics.
Keywords: Autophagy, chemoresistance, heme oxygenase-1, Src, STAT3
Breast cancer is the most common malignant disease for Western women.1 Early detection of breast cancer has improved the prognosis for patients with primary disease confined to the breast but the prognosis for patients with advanced metastatic breast cancer remains poor, which is often due to poor response to standard chemotherapy that is mainly based on anthracyclines and taxanes. Significant advances in the understanding of the mechanisms of anti-cancer drug resistance have not been paralleled by the introduction of novel strategies to circumvent or avoid resistance in clinical practice. Thus, increasing tumor cell sensitivity to these chemotherapeutic agents is an attractive goal towards improving the therapeutic efficacy of breast cancer, and modulation of tumor-specific signaling pathways may provide a different and complementary approach.
Heme oxygenase (HO), the rate-limiting enzyme in heme degradation, catalyzes the oxidation of heme to generate several biologically active molecules: carbon monoxide (CO), biliverdin and ferrous ion.2 Among the three isoforms of the HO family, HO-1 is widely expressed at low levels in most tissues under steady state and is highly inducible under a variety of chemical and physical cellular stresses.3 The transcriptional upregulation of ho-1 gene and the subsequent de novo synthesis of the corresponding protein play a critical role in antioxidative defense, anti-inflammatory and anti-apoptotic effects.4 Due to its cytoprotective properties, the roles of HO-1 in maintaining tumor cell survival and mediating chemotherapeutic resistance have attracted great attention. Increased expression of HO-1 has been observed in several cancers, including brain tumors, melanomas, chronic myeloid leukemia and lymphosarcoma, suggesting possible contribution of HO-1 to tumor progression through promotion of angiogenesis, metastases and proliferation.5–8 HO-1 expression is also believed to contribute to resistance to chemotherapeutic agents in AML cells, and pancreatic and lung cancer cells.9,10 On the contrary, few reports have demonstrated the anti-proliferative role of HO-1 in prostate and breast cancer.11,12 These contrasting observations have undoubtedly increased the significance of HO-1 in the field of cancer biology.
Autophagy is a highly conserved process during which parts of the cytoplasm, including damaged, superfluous organelles or long-lived proteins, are sequestered into double-membrane vesicles known as autophagosomes.13 Under steady state, this provides a quality-control mechanism, removing damaged organelles and proteins. Under stress conditions, the autophagic digestion recovers energy in an attempt to maintain/restore metabolic homeostasis. It is believed that autophagy plays a critical role in the pathogenesis of diverse diseases, such as inflammatory bowel disease, neuronal degeneration, aging and cancer.14,15 Among them, the role of autophagy in cancer has been extensively studied and discussed. While most studies suggest a protective role for autophagy, some reports show that autophagy may act as a cell death mechanism in response to stress.16,17 Recent studies have struggled to reveal the complex paradoxical role of autophagy in cancer development as well as in cancer therapy.
In the current study, we found that DOX-insensitive MDA-MB-231 and MDA-MB-468 breast cancer cells exhibited increased autophagy accompanied by HO-1 induction following DOX treatment. Furthermore, Src-STAT3 signaling pathway’s activation mediated the induction of HO-1 expression and the subsequent upregulation of autophagy. Blocking STAT3 or Src kinase activation or inhibition of autophagy or HO-1 induction increased the sensitivity of these cells to DOX treatment, suggesting that Src/STAT3/HO-1/autophagy pathway activation is a novel mechanism for mediating chemoresistance in breast cancer cells.
Materials and Methods
Plasmids, antibodies and reagents
STAT3-dependent luciferase reporter plasmid was provided by Dr Ming Shi from our institute. Human HO-1, STAT3, ATG5 and Src siRNA and their control siRNA were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA), Invitrogen-Life Technology (Beijing, China) and Ruibo Biotechnology (Guangzhou, China), respectively. The antibodies against Beclin-1, LC3B, phospho-Tyr416-Src, Src, phospho-Tyr705-STAT3 and STAT3 were purchased from Cell Signaling Technology (Beverly, MA, USA). The antibodies against HO-1 and β-actin were obtained from Santa Cruz Biotechnology. The anti-ATG5 antibody, DOX, 3-Methyladenine (3-MA) and chloroquine were purchased from Sigma (St. Louis, MO, USA).
Cell culture and transfection
The human breast adenocarcinoma cell lines MDA-MB-231 and MDA-MB-453 were obtained from ATCC (Rockville, MD, USA). MDA-MB-468 cells were kindly provided by Dr Lihua Ding (Beijing Institute of Biotechnology). All the cells were maintained in DMEM supplemented with 10% FBS at 37°C, in an atmosphere of 5% CO2. The transfections were performed with the LipofectAMINE 2000 or LipofectAMINE RNAi MAX reagents (Life Technologies, Rockville, MD, USA) according to the manufacturer’s instructions.
Western blot assay
Cellular protein extracts were prepared with cell lysis buffer (10 mM Tris-HCl, pH 7.4, 1% SDS, 1 mM Na3VO4) and resolved by SDS-PAGE. After blocking, blots were probed with the appropriate primary antibodies overnight at 4°C and then washed and incubated with HRP-conjugated secondary antibodies. Bands were detected as described in our previous studies.18
Luciferase reporter assay
Cells were transiently transfected with the STAT3-dependent luciferase reporter construct and then treated with DOX 36 h after transfection. The luciferase activities were tested at the indicated time periods after DOX treatment. The luciferase activities were presented as relative activities normalized to the luciferase activity in the control cells without treatment, as described previously.19
Apoptosis assay
Cellular apoptosis was measured by propidium iodide (PI) staining of the nuclei followed by flow cytometric analysis on a FACSCalibur as described previously.18
Autophagy assay
Cellular autophagy was monitored using a Cyto-ID Autophagy Detection Kit (Enzo Life Sciences; New York, USA) following the manufacturer’s protocol. The 488-nm excitable Cyto-ID Green Autophagy Detection Reagent (dye) supplied in the kit becomes brightly fluorescent in vesicles produced during autophagy and, thus, serves as a convenient tool to detect autophagy at the cellular level under confocal microscopy. Following treatment, the cells were trypsinized, washed in assay buffer that was supplied in the kit, and re-suspended in the assay buffer. We then analyzed the samples in the green (FL1) channel of a flow cytometer to obtain the quantitative fluorescent data (mean of fluorescence intensity).
Results
Induction of heme oxygenase-1 expression mediated doxorubicin resistance in MDA-MB-231 cells
To determine the mechanisms involved in chemoresistance in breast cancer cells, two breast cancer cell lines, MDA-MB-453 and MDA-MB-231, which showed quite different sensitivity to doxorubicin (DOX) treatment, were first selected for analysis in this study. As shown in Figure1(a), a significant increase in cellular apoptosis (from 1.7 ± 0.01% to 36.8 ± 0.03%) was observed in MDA-MB-453 cells after 48 h of DOX (0.2 μM) treatment. However, the percentage of apoptotic 231 cells only increased from 2.1 ± 0.01% to 7.8 ± 0.015% under the same DOX exposure conditions. These data indicate that MDA-MB-231 cells are more resistant to DOX treatment than MDA-MB-453 cells.
Figure 1.
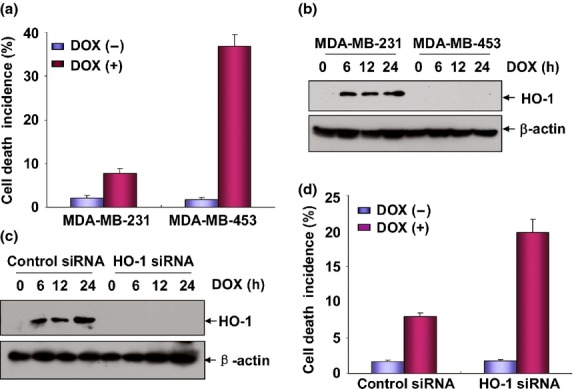
Induction of heme oxygenase-1 (HO-1) expression mediated doxorubicin (DOX) resistance in MDA-MB-231 cells. (a) MDA-MB-231 and MDA-MB-453 cells were left untreated or treated with DOX (0.2 μM) for 48 h and then cell death incidence was determined by flow cytrometric assay after propidium iodide (PI) staining of the nuclei. (b) MDA-MB-231 and MDA-MB-453 cells were treated with DOX (0.2 μM) for the indicated time periods and then the induction of HO-1 expression was detected by western-blot assay. (c) MDA-MB-231 cells were transfected with HO-1 siRNA or control siRNA and then treated with DOX (0.2 μM) for the indicated time periods 36 h after transfection. The efficiency of HO-1 siRNA was determined by western-blot assay. To avoid the off-target effect, two different siRNA against HO-1 were purchased and tested. Data shown here were obtained from the most effective one. (d) MDA-MB-231 cells were transfected and treated with DOX as described in (c) and then cell death incidence was detected 48 h after DOX treatment.
Heme oxygenase-1 (HO-1) is a key enzyme that can exert potential cytoprotective properties under multiple stress conditions.20 Therefore, it functions as a critical mediator for drug resistance in a variety of tumor cell lines. However, its role in the chemotherapeutic agents-induced responses in breast cancer cells has not been clearly defined.21,22 To examine whether HO-1 is involved in regulating DOX sensitivity in MDA-MB-453 and MDA-MB-231 cells, the cells were incubated with 0.2 μM of DOX and then HO-1 expression was examined at the indicated time periods after DOX exposure. We found a significant increase of HO-1 expression in DOX-treated MDA-MB-231 cells, but these responses were totally absent in the MDA-MB-453 cells under the same conditions (Fig.1b). Furthermore, knockdown of HO-1 induction obviously increased the DOX sensitivity in MDA-MB-231 cells (Fig.1c,d), indicating that HO-1 induction contributes to mediate DOX resistance in MDA-MB-231 cells.
Doxorubicin-induced autophagy protected MDA-MB-231 cells from apoptosis
In the following analysis, we attempted to determine the downstream signaling events following HO-1 induction in mediating DOX resistance in MDA-MB-231 cells. Autophagy is a metabolic process which can exert either an anti-apoptotic or a pro-apoptotic role under different stress conditions.23 One of the novel findings regarding autophagy regulation is that HO-1 has been illustrated to function as an upstream regulator by promoting or antagonizing autophagy.24,25 Therefore, we next investigated whether autophagy was induced upon DOX exposure and was involved in regulating DOX-induced apoptosis in MDA-MB-231 cells. As shown in Figure2(a), when DOX-treated MDA-MB-231 and MDA-MB-453 cells were stained with the Cyto-ID Green Autophagy Detection Reagent, we observed an obvious induction of autophagic flux in MDA-MB-231 cells, evidenced by a green fluorescence signal accumulated in spherical vacuoles in the perinuclear region of the cells; in contrast, no signal was detected in the MDA-MB-453 cells under the same DOX exposure conditions. To further confirm the above results, we next performed flow cytometry-based quantitative analysis of the cell populations loaded with Cyto-ID Green Autophagy Detection Reagent. As shown in Figure2(b), control MDA-MB-231 and MDA-MB-453 cells were stained only faintly, revealing low fluorescence signal intensity. After treatment with DOX for 24 h, the Cyto-ID Green Reagent signal increased dramatically in MDA-MB-231 cells. No similar phenomena were observed in the MDA-MB-453 cells under the same conditions, indicating that DOX caused an increase in autophagic vesicles only in MDA-MB-231 cells. In the following test, we observed a time-dependent increase of LC3B-I/II and Beclin-1 expression levels in the DOX-treated MDA-MB-231 cells, but not in MDA-MB-453 cells, further confirming that drug treatment enhanced autophagy activation in MDA-MB-231 cells.
Figure 2.
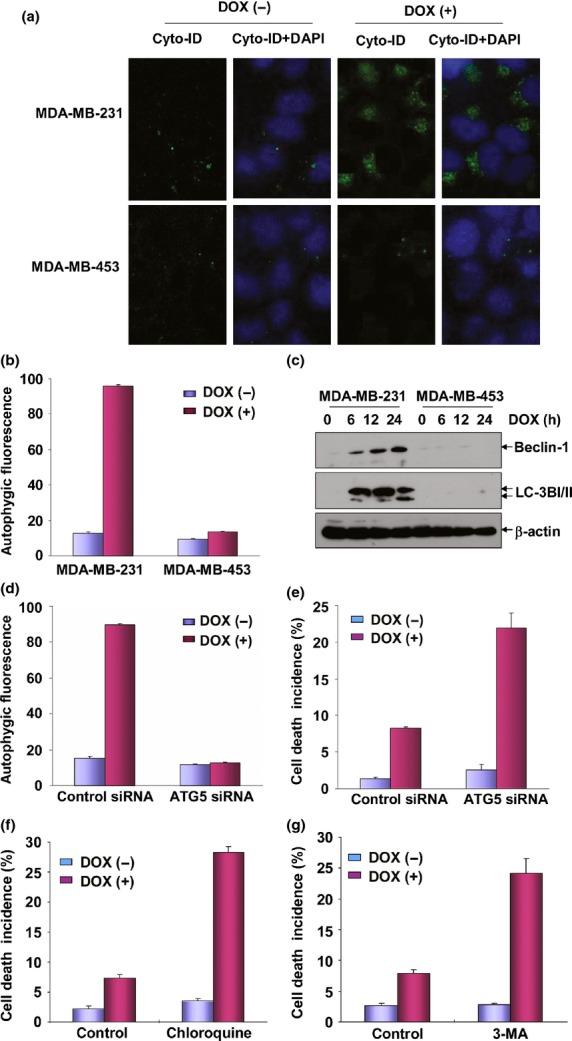
Doxorubicin (DOX)-induced autophagy protected MDA-MB-231 cells from apoptosis. (a) MDA-MB-231 and MDA-MB-453 cells were left untreated or treated with DOX (0.2 μM) for 24 h and then autophagy was detected under confocal microscopy after staining the cells with Cyto-ID Green Autophagy Detection Reagent (ENZO Biotechnology). (b) The cells in (a) were trypsinized, collected and subjected to flow cytometric analysis to obtain the quantitative data indicating the autophagic flux inside the cells. (c) MDA-MB-231 and MDA-MB-453 cells were treated with DOX as described in Figure1(b) and then the expressions of Beclin-1 and LC3BI/II were detected. (d) MDA-MB-231 cells were transfected with ATG5 siRNA or control siRNA and then treated with DOX (0.2 μM) 36 h after transfection. Then the cells were collected and subjected to flow cytometric analysis to test the autophagic flux inside the cells. To avoid the off-target effect, two different siRNA against ATG5 were purchased and tested. Data shown here were obtained from the most effective one. (e) MDA-MB-231 cells were transfected and treated with DOX as described in (d) and then cell death incidence was detected 48 h after DOX treatment. (f) MDA-MB-231 cells were pretreated with chloroquine and then exposed to DOX (0.2 μM). The cell death incidence was detected 48 h after DOX treatment. (g) MDA-MB-231 cells were pretreated with 3-MA and then exposed to DOX (0.2 μM). The cell death incidence was detected 48 h after DOX treatment.
To determine whether autophagy is able to regulate the sensitivity of breast cancer cells to DOX, MDA-MB-231 cells were transfected with the specific siRNA targeted ATG5, the critical component of autophagosome, and then cell apoptosis was detected in the presence or absence of ATG5 expression. As shown in Figure2(d,e), knockdown ATG5 levels blocked the induction of autophagy, while increasing the sensitivity of MDA-MB-231 cells to DOX treatment, indicating that autophagy functions as a protective mechanism in antagonizing apoptosis induced by DOX. To further confirm these results, MDA-MB-231 cells were pretreated with autophagy inhibitor, 3-MA, or lysosome inhibitor, chloroquine, and cell susceptibility to DOX-induced apoptosis was determined. Inhibition of DOX-induced autophagy in MDA-MB-231 cells by 3-MA or chloroquine was verified by reduction of LC3BI/II and Beclin-1 expression in the 3-MA or chloroquine-pretreated cells compared with the control cells (data not shown). Under the same conditions, an increase of MDA-MB-231 cell sensitivity in response to DOX exposure was observed upon 3-MA or chloroquine pretreatment (Fig.2f,g). These findings clearly suggest that upregulation of autophagy promotes anti-tumor drug resistance in MDA-MB-231 cells.
Doxorubicin-induced autophagy was dependent on heme oxygenase-1 expression in MDA-MB-231 cells
To investigate whether HO-1 regulates autophagy induction in DOX-treated MDA-MB-231 cells, the cells were transfected with HO-1 siRNA and then DOX-induced autophagy flux was examined in the absence or presence of HO-1 expression. We observed that the induction of autophagy-related proteins, Beclin-1 and LC3BI/II, were almost totally blocked by siRNA knockdown of HO-1 expression (Fig.3a). Furthermore, the fluorescence signals from the Cyto-ID Autophagy Detection Reagent-stained MDA-MB-231 cells greatly decreased after HO-1 siRNA transfection (Fig.3b,c). These data together indicate that DOX-induced autophagy is dependent on HO-1 expression in MDA-MB-231 cells.
Figure 3.
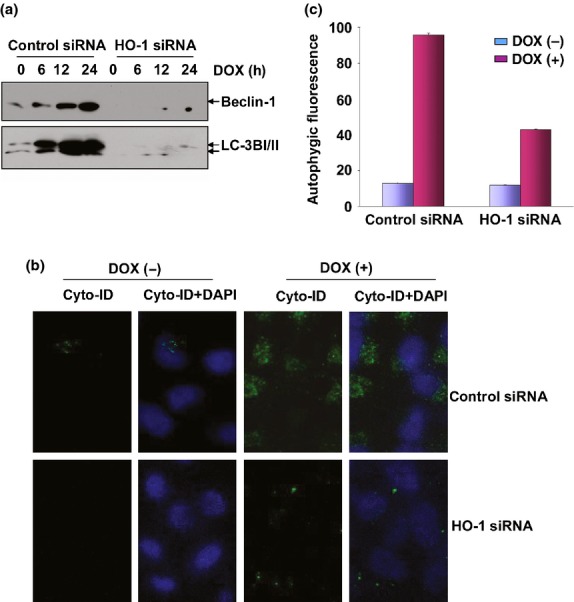
Doxorubicin (DOX)-induced autophagy was dependent on heme oxygenase-1 (HO-1). (a) MDA-MB-231 cells were transfected and treated as described in Figure1(c) and then the expression levels of Beclin-1 and LC3BI/II were detected. The image shown in this figure and Figure1(c) were developed from the same membrane. (b,c) MDA-MB-231 cells were transfected and treated as described in Figure1(c) and then autophagy was detected either under confocal microscopy (b) or by flow cytometric analysis (c) after staining the cells with Cyto-ID Green Autophagy Detection Reagent.
STAT3 was responsible for heme oxygenase-1 induction in doxorubicin-treated MDA-MB-231 cells
After revealing HO-1-dependent autophagy in mediating the cytoprotective effect, we next focused on elucidating the upstream signaling events leading to HO-1 induction in DOX-induced responses. According to previous reports, induction of HO-1 expression occurs exclusively at the transcriptional levels, which potentially involves the coordinated interaction of multiple transcription factors, such as Nrf2, NF-κB, Egr-1, AP-1 and STAT3.26–29 However, the functional significance of each factor is different under various stress conditions. Therefore, in the next study, we compared the activation status of the potential transcriptional factors involved in regulating HO-1 induction in DOX-treated MDA-MB-231 and MDA-MB-453 cells. We found that STAT3 was strongly activated in MDA-MB-231 cells after treatment with DOX, evidenced by dramatic upregulation of levels on tyrosine705-phosphorylated STAT3. No signaling indicating STAT3 activation was observed in MDA-MB-453 cells under the same DOX exposure conditions (Fig.4a). Furthermore, an enhancement of STAT3-dependent luciferase activities was readily detected in MDA-MB-231 cells after DOX stimulation (Fig.4b). These data indicate that although MDA-MB-231 cells are not sensitive to DOX treatment, DOX induces upregulation of STAT3 transcriptional activities in these cells.
Figure 4.
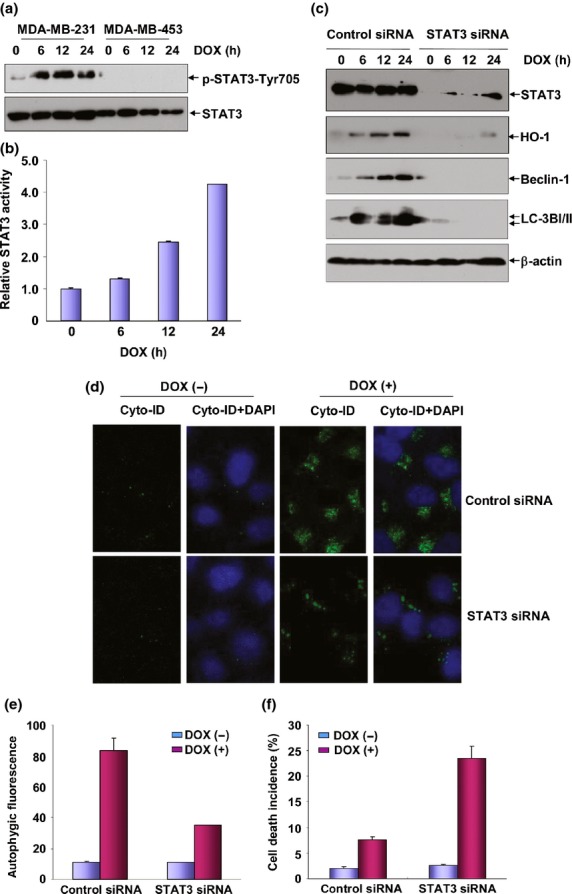
STAT-3 was responsible for heme oxygenase-1 (HO-1) induction under doxorubicin (DOX) exposure. (a) MDA-MB-231 and MDA-MB-453 cells were treated with DOX (0.2 μM) for the indicated time periods and then the induction of STAT3 activation was detected by western-blot assay. (b) MDA-MB-231 cells were transfected with STAT3-dependent luciferase reporter plasmid and then exposed to DOX (0.2 μM) 36 h after transfection. Then the induction of STAT3 luciferase activities was determined at the indicated time periods after DOX treatment. (c) MDA-MB-231 cells were transfected with STAT3 siRNA or control siRNA and then treated with DOX (0.2 μM) 36 h after transfection. Then the expressions of HO-1, Beclin-1 and LC3BI/II were determined 12 h after DOX exposure. To avoid the off-target effect, two different siRNA against STAT3 were purchased and tested. Data shown here were obtained from the most effective one. (d,e) MDA-MB-231 cells were transfected and treated as described in figure (c) and then autophagy was detected as described in Figure3(b,c). (f) MDA-MB-231 cells were transfected and treated as described in figure (c) and then cell death incidence was detected 48 h after DOX treatment.
To determine whether STAT3 activation contributes to HO-1-dependent autophagy induction in the DOX-treated MDA-MB-231 cells, STAT3 siRNA was transfected into the MDA-MB-231 cells and then the induction of HO-1 expression and the subsequent signal transduction pathways activation were examined in the absence or presence of STAT3 expression. As shown in Figure4(c), HO-1 induction and the upregulation of the autophagy-related proteins (Beclin-1 and LC3BI/II) were totally blocked in STAT3 siRNA-transfected cells compared to the control siRNA-transfected group. In addition, DOX-induced autophagic fluorescence intensity and the signals indicating the autophagic flux inside the cells captured under confocal microscopy greatly reduced upon knockdown of STAT3 expression (Fig.4d,e). These data clearly demonstrate the dependence of HO-1 expression and the subsequent induction of autopahgy on STAT3 activation in the DOX-treated MDA-MB-231 cells. We further observed the significant increase of DOX-induced cellular apoptosis upon knockdown of STAT3 expression in MDA-MB-231 cells (Fig.4f), confirming that STAT3 activation is responsible for mediating the induction HO-1-dependent autophagy to deliver the cytoprotective effect in the DOX responses.
Src functioned as the upstream protein kinase responsible for STAT3 activation in doxorubicin-treated MDA-MB-231 cells
Both JAK2 and Src kinases are well-known cytoplasmic tyrosine kinases that are responsible for phosphorylating and activating STAT3.30,31 Therefore, to determine the upstream signal molecules leading to STAT3 activation in MDA-MB-231 cells under DOX exposure, we next detected the activation status of Src and JAK2 in the DOX-treated MDA-MB-231 and MDA-MB-453 cells. As shown in Figure5(a), after treatment with DOX, phosphorylation of Src was only significantly induced in MDA-MB-231 cells, but not in MDA-MB-453 cells. No signal of JAK2 activation was detected in either MDA-MB-231 or MDA-MB-453 cells under the same DOX exposure conditions (data not show).
Figure 5.
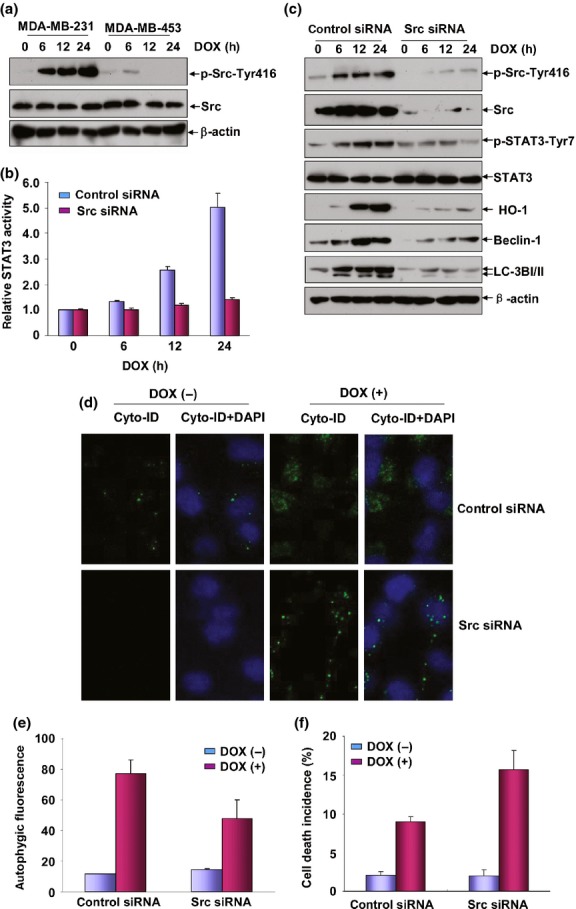
Src was the upstream protein kinase responsible for STAT3 activation under doxorubicin (DOX) treatment. (a) MDA-MB-231 and MDA-MB-453 cells were treated with DOX (0.2 μM) for the indicated time periods and then the induction of Src activation was detected by western-blot assay. (b) MDA-MB-231 cells were co-transfected with STAT3-dependent luciferase reporter plasmid and Src siRNA and then exposed to DOX (0.2 μM) 36 h after transfection. Then the induction of STAT3 luciferase activities was determined at the indicated time periods after DOX treatment. (c) MDA-MB-231 cells were transfected with Src siRNA or control siRNA and then treated with DOX (0.2 μM) 36 h after transfection. Then the activation of STAT3 and the expressions of heme oxygenase-1 (HO-1), Beclin-1 and LC3BI/II were determined 12 h after DOX exposure. To avoid the off-target effect, two different siRNA against Src were purchased and tested. Data shown here were obtained from the most effective one. (d,e) MDA-MB-231 cells were transfected and treated as described in figure (c) and then autophagy was detected as described in Figure3(b,c). (f) MDA-MB-231 cells were transfected and treated as described in figure (c) and then cell death incidence was detected 48 h after DOX treatment.
To determine whether Src is responsible for STAT3 phosphorylation in response to DOX treatment, MDA-MB-231 cells were transfected with Src siRNA or control siRNA. We found that knockdown Src expression significantly inhibited the upregulation of STAT3-dependent luciferase activities and the levels of STAT3 phosphorylation induced by DOX (Fig.5b,c). Furthermore, Dox-induced HO-1, Beclin-1 and LC3BI/II expressions were greatly reduced by inhibition of Src expression (Fig.5c). Consistent with these data, we also observed a decrease of autophagy in Src siRNA-transfected MDA-MB-231 cells according to the results from the quantitative autophagic fluorescence assay (Fig.5d) and the Cyto-ID Autophagy Detection Reagent-stained signals captured by confocal microscopy (Fig.5e). These data together indicate that Src is the critical protein kinase that is responsible for mediating the induced activation of STAT3 and the subsequent HO-1-dependent autophagy in the DOX-treated MDA-MB-231 cells.
Finally, we analyzed whether Src was also involved in regulating DOX sensitivity in the MDA-MB-231 cells. As expected, cell death incidence was significantly increased in the Src siRNA-transfected MDA-MB-231 cells compared with that in the control siRNA-transfected cells (Fig.5f). The results together suggest that activation of Src/STAT3/HO-1/autophagy pathway functions as a protective mechanism to antagonize the cytotoxicity of DOX in the MDA-MB-231 cells.
Src/STAT3/HO-1-dependent autophagy protected MDA-MB-468 cells from doxorubicin-induced apoptosis
To determine whether the Src/STAT3/HO-1/autophagy pathway activation plays a general cytoprotective role in breast cancer cells under DOX treatment, we next detected the activation status of this pathway in the MDA-MB-468 cells, which also bear ER, PR and Her2-negative phenotype as MDA-MB-231 cells (data not shown) and are also resistant to DOX-induced apoptosis compared with MDA-MB-453 cells (Fig.6a). We observed that DOX treatment not only induced the activation of Src and STAT3, but also upregulated the expression levels of HO-1, Beclin-1 and LC-3I/II in the MDA-MB-468 cells; in contrast, no signal indicating the activation of these signaling molecules was detected in the MDA-MB-453 cells under the same DOX exposure conditions (Fig.6b). Moreover, an induction of autophagic flux in MDA-MB-468 cells was observed under DOX exposure in the flow cytometry-based analysis (Fig.6c), further confirming the induction of autophagy in the DOX-treated MDA-MB-468 cells. In the following analyses, MDA-MB-468 cells were transfected with Src, STAT3 or HO-1 siRNA, respectively, to test whether signal transduction of Src/STAT3/HO-1 cascade contributed to autophagy. As shown in Figure6(d–f), knockdown Src, STAT3 or HO-1 expression almost totally blocked the activation of their downstream targets as well as the signaling event indicating autophagy induction (Beclin-1 and LC3I/II upregulations). Finally, abrogating autophagy induction by 3-MA obviously increased the sensitivity of MDA-MB-468 cells to DOX-induced cytotoxicity (Fig.6g). Taken together, these data indicate that Src/STAT3/HO-1/autophagy pathway activation also plays a cytoprotective role in MDA-MB-468 cells under DOX treatment.
Figure 6.
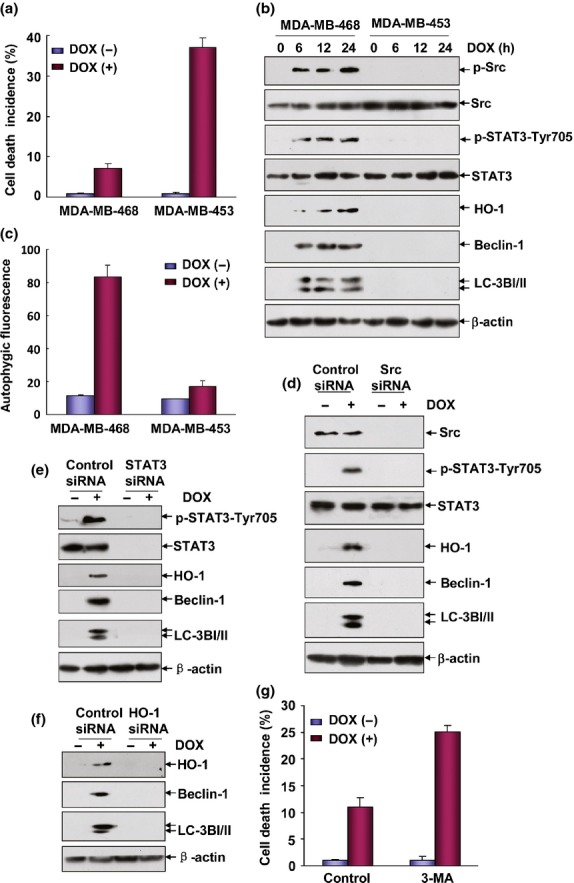
Src/STAT3/heme oxygenase-1 (HO-1)-dependent autophagy protected MDA-MB-468 cells from doxorubicin (DOX)-induced apoptosis. (a) MDA-MB-468 and MDA-MB-453 cells were left untreated or treated with DOX (0.2 μM) for 48 h and then cell death incidence was determined as described in Figure1(a). (b) MDA-MB-468 and MDA-MB-453 cells were treated with DOX (0.2 μM) for the indicated time periods and then the induction of Src and STAT3 activation and HO-1, Beclin-1 and IC3I/II expression were detected by western-blot assay. (c) MDA-MB-468 and MDA-MB-453 cells were left untreated or treated with DOX (0.2 μM) for 24 h and then autophagy was detected as described in Figure2(a). (d–f) MDA-MB-468 cells were transfected with Src, STAT3, HO-1 siRNA or their control siRNA, respectively. Then the cells were treated with DOX (0.2 μM) 36 h after transfection. The activation of Src, STAT3 and the expressions of HO-1, Beclin-1 and LC3BI/II were determined 12 h after DOX exposure. (g) MDA-MB-468 cells were pretreated with 3-MA followed by exposure to DOX (0.2 μM) and then cell death incidence was detected 48 h after DOX treatment.
Discussion
Acquired resistance to chemotherapeutic agents is a serious problem in breast cancer patients. Upregulation of drug transporters, which efflux the drugs, has been linked to both intrinsic and acquired chemoresistance.32 Adaptive activation of proliferative and survival signal pathways also contributes to resistance to the chemotherapeutic agents.33 In the current study, we have identified a novel mechanism for the chemoresistance of breast cancer cells to DOX, which is mediated by Scr/STAT3/HO-1/autophagy pro-survival pathway activation. Blocking the activity of either molecule in this pathway significantly overcomes DOX resistance in the breast cancer cells, indicating the feasibility of targeting this pathway to increase the efficacy of breast cancer chemotherapeutics.
There is considerable evidence that HO-1 provides robust protection against various cellular stresses.34 However, the role of HO-1 in the tumor therapeutic agent-induced responses in the breast cancer cells seems more complicated. Some reports confirm the contribution of HO-1 induction to angiogenesis and metastasis of breast cancer cells under certain types of drugs treatment,35 while others demonstrate the function of HO-1 expression in suppressing breast tumor cell proliferation and migration in response to other anticancer agents.12,36 The data in the present study contribute evidence on the tumor protective function of HO-1 induction, which constitutes an adaptive defense mechanism against breast cancer cell death induced by DOX. Due to these facts, we believe that the opposite actions of HO-1 implicated in either breast cancer promotion or prevention may be specific to differences in stimuli. Based on the current understanding, HO-1 confers protective actions by regulating a variety of downstream signaling molecules or signal transduction events. Modulation of autophagy appears to be one of the most important mechanisms underlying the cytoprotective role of HO-1.24,37
Autophagy is a complicated process that exerts opposing effects on tumorigenesis and tumor progression processes.38 It is believed that autophagy suppresses the growth of early tumors by increasing genomic stability, eliminating cells with defective proteins or inhibiting inflammation.14 At later or advanced stages, autophagy supports the survival of established tumors and promotes cancer progression by antagonizing various forms of cellular stress, such as oxidative stress, starvation and DNA damage.39,40 The dual role of autophagy in cancer development is further augmented by conflicting reports about the effect of autophagy on chemotherapeutic treatment. Some groups suggest that autophagy augments the toxicity of drugs to facilitate apoptosis of cancer cells,41 while others propose that autophagy is induced in cancer cells as a survival strategy against these drugs.42,43 We speculate that this discrepancy might be related to the nature and duration of the treatment-induced metabolic stress, the anticancer drugs used as well as tumor type. As one of the novel upstream regulators for autophagy, HO-1 has been shown to function as either a positive mediator for the cytoprotective autophagy or a negative regulator to inhibit the cytotoxic autophagy in the different tumor therapeutic agent-induced responses.37,44 Under both circumstances, the final outcome of HO-1 induction displays a defense mechanism against cell death induced by these drugs. The data in the current study provide evidence for the HO-1-dependent cytoprotective autophagy in the DOX-treated breast cancer cells and its role in overcoming DOX-induced cytotoxicity. Therefore, our data have provided new evidence for the mechanism underlying HO-1 induction and autophagy regulation in the chemotherapeutic agent-induced response in the breast cancer cells. However, the signaling transduction cascade by which HO-1 induces autophagy is currently unknown. A previous report demonstrated that HO-1 modulates p38K phosphorylation to induce autophagy signaling, which is critical for protecting hepatocyte cell death.24 In fact, we found the activation of p38K in MDA-MB-231 and 468 cells under DOX exposure (data not shown). Therefore, whether HO-1’s action on regulating autophagy is associated with p38K activation requires further investigation.
In the investigations to determine the upstream signaling events leading to HO-1 induction in the DOX-treated breast cancer cells, we found the activation of both STAT3 and AP-1 in the MDA-MB-231 cells in response to DOX exposure. However, AP-1 seemed not to be involved in HO-1 induction due to the comparable levels of HO-1 expression before and after AP-1 transactivation was blocked (data not shown). In contrast, STAT3 obviously played a major role in this process. As a critical member of the STAT transcriptional factor family, STAT3 has been established to possess a pivotal function in the initiation, progression, maintenance and metastasis of multiple cancer types by transactivation of a host of target genes involved in cell proliferation, survival, angiogenesis and invasiveness.45 For breast cancer, the involvement of STAT3 in early mammary tumorigenesis of ER-negative and triple-negative (ER, PR and Her2-negative) subtypes has been well-defined. In these cells, persistent activation of STAT3 is readily observed and inhibition of STAT3 activation has been proved to suppress cell proliferation and to promote apoptosis.46 Most importantly, accumulative evidence indicates that STAT3 activation is required for the maintenance and self-renewal of stem cell-like breast cancer cells (CD44+ CD24−),47 which have been shown to be related to tumor recurrence, metastasis and chemoresistance. Therefore, targeting STAT3 represents a promising strategy for prevention of breast cancer. Data in the current study have provided novel evidence on the contribution of STAT3 activation to breast cancer cell resistance to DOX toxicity, further emphasizing the importance of and demand to develop STAT3 inhibitors for efficient breast cancer treatment.
Because phosphorylation is essential for STAT3 biological activity, blocking the upstream protein kianses responsible for STAT3 activation also represents a rational approach to prevent carcinogenesis. For this reason, the effective inhibitors targeting JAK2 and Src have been extensively investigated. By understanding of that 70% of breast cancer cells overexpress c-Src cellular tyrosine kinase, pharmacological inhibitors of Src are being actively developed in breast cancer.48 Preclinical studies have identified that breast cancer cell lines representing the basal/triple-negative group are uniquely sensitive to growth inhibition by Dasatinib, the only approved Src inhibitor, therefore shedding light on the therapy of triple-negative breast cancer (TNBC), which is a clinically important subtype with limited approved treatment options other than chemotherapy.49 In the current study, we have identified Src activation-dependent signaling events in mediating DOX resistance in MDA-MB-231 and MDA-MS-468 cells, both of which bear triple-negative phenotypes, suggesting that targeting Src is not only essential to block proliferation, invasion and metastasis of TNBC, but also critical for overcoming the resistance and recurrence of this subgroup after therapy. In fact, Src overactivation is frequently observed in Her2-positive breast cancer cells. How Src is activated in the Her2-negative cells is an interesting question. Because the numerous receptor tyrosine kinases (such as EGFR, PDGFR and EGFR) and the focal adhesion kinase (FAK) have been reported to be involved in Src activation under different conditions,50 whether targeting these upstream protein kinases for Src is also contributive to overcoming anti-cancer drugs resistance is worth exploring.
In conclusion, data in this study demonstrate that HO-1-dependent autophagy is able to attenuate DOX toxicity in breast cancer cells, which is largely mediated by Src/STAT3 pathway activation. Therefore, therapeutic strategies aimed at reducing Src/STAT3/HO-1 pathway activation and autophagy may provide a potential approach to overcome DOX resistance, leading to improved clinical use of DOX in breast cancer cells.
Acknowledgments
This project was supported by the National Natural Science Foundation of China (Nos 31171342 and 31270797), the National Key Research and Development Programs on Fundamental Sciences (973 Project, 2011CB503803) to Dr Lun Song and the National Natural Science Foundation of China (No. 81360396) to Dr Changyuan Wei.
Disclosure Statement
The authors have no conflict of interest to declare.
References
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- Shekhawat G, Verma K. Haem oxygenase (HO): an overlooked enzyme of plant metabolism and defence. J Exp Bot. 2010;61:2255–70. doi: 10.1093/jxb/erq074. [DOI] [PubMed] [Google Scholar]
- Ferrandiz M, Devesa I. Inducers of heme oxygenase-1. Curr Pharm Des. 2008;14:473–86. doi: 10.2174/138161208783597399. [DOI] [PubMed] [Google Scholar]
- Gozzelino R, Jeney V, Soares MP. Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol Toxicol. 2010;50:323–54. doi: 10.1146/annurev.pharmtox.010909.105600. [DOI] [PubMed] [Google Scholar]
- Jozkowicz A, Was H, Dulak J. Heme oxygenase-1 in tumors: is it a false friend? Antioxid Redox Signal. 2007;9:2099–118. doi: 10.1089/ars.2007.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu F, Yeh C, Sun Y, et al. Signal peptide peptidase-mediated nuclear localization of heme oxygenase-1 promotes cancer cell proliferation and invasion independent of its enzymatic activity. Oncogene. 2015;34:2360–70. doi: 10.1038/onc.2014.166. [DOI] [PubMed] [Google Scholar]
- Gandini NA, Fermento ME, Salomón DG, et al. Heme oxygenase-1 expression in human gliomas and its correlation with poor prognosis in patients with astrocytoma. Tumour Biol. 2014;35:2803–15. doi: 10.1007/s13277-013-1373-z. [DOI] [PubMed] [Google Scholar]
- Tibullo D, Barbagallo I, Giallongo C, et al. Nuclear translocation of heme oxygenase-1 confers resistance to Imatinib in chronic myeloid leukemia cells. Curr Pharm Des. 2013;19:2765–70. doi: 10.2174/1381612811319150012. [DOI] [PubMed] [Google Scholar]
- Lee JG, McKinney KQ, Mougeot JL, Bonkovsky HL, Hwang SI. Proteomic strategy for probing complementary lethality of kinase inhibitors against pancreatic cancer. Proteomics. 2013;13:3554–62. doi: 10.1002/pmic.201300248. [DOI] [PubMed] [Google Scholar]
- Jeon W-K, Hong H-Y, Seo WC, et al. Smad7 sensitizes A549 lung cancer cells to cisplatin-induced apoptosis through heme oxygenase-1 inhibition. Biochem Biophys Res Commun. 2012;420:288–92. doi: 10.1016/j.bbrc.2012.02.151. [DOI] [PubMed] [Google Scholar]
- Gueron G, Giudice J, Valacco P, et al. Heme-oxygenase-1 implications in cell morphology and the adhesive behavior of prostate cancer cells. Oncotarget. 2014;5:4087–102. doi: 10.18632/oncotarget.1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill M, Pereira V, Chauveau C, et al. Heme oxygenase-1 inhibits rat and human breast cancer cell proliferation: mutual cross inhibition with indoleamine 2, 3-dioxygenase. FASEB J. 2005;19:1957–68. doi: 10.1096/fj.05-3875com. [DOI] [PubMed] [Google Scholar]
- Autophagy Mizushima N. Process and function. Genes Dev. 2007;21:2861–73. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7:961–7. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:651–62. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- Ogata M, Hino S, Saito A, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26:9220–31. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C, Jung JU. Autophagy genes as tumor suppressors. Curr Opin Cell Biol. 2010;22:226–33. doi: 10.1016/j.ceb.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Dong W, Hu M, et al. GADD45α mediates arsenite induced cell apoptotic effect in human hepatoma cells via JNKs/AP-1-dependent pathway. J Cell Biochem. 2010;109:1264–73. doi: 10.1002/jcb.22509. [DOI] [PubMed] [Google Scholar]
- Song L, Dong W, Gao M, et al. A novel role of IKKα in the mediation of UVB-induced G0/G1 cell cycle arrest response by suppressing Cyclin D1 expression. Biochim Biophys Acta. 2010;1803:323–32. doi: 10.1016/j.bbamcr.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Was H, Dulak J, Jozkowicz A. Heme oxygenase-1 in tumor biology and therapy. Curr Drug Targets. 2010;11:1551–70. doi: 10.2174/1389450111009011551. [DOI] [PubMed] [Google Scholar]
- Zhong Y, Zhang F, Sun Z, et al. Drug resistance associates with activation of Nrf2 in MCF-7/DOX cells, and wogonin reverses it by down-regulating Nrf2-mediated cellular defense response. Mol Carcinog. 2013;52:824–34. doi: 10.1002/mc.21921. [DOI] [PubMed] [Google Scholar]
- Park SY, Jin ML, Kim YH, Lee S-J, Park G. Sanguinarine inhibits invasiveness and the MMP-9 and COX-2 expression in TPA-induced breast cancer cells by inducing HO-1 expression. Oncol Rep. 2014;31:497–504. doi: 10.3892/or.2013.2843. [DOI] [PubMed] [Google Scholar]
- Brech A, Ahlquist T, Lothe RA, Stenmark H. Autophagy in tumour suppression and promotion. Mol Oncol. 2009;3:366–75. doi: 10.1016/j.molonc.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carchman EH, Rao J, Loughran PA, Rosengart MR, Zuckerbraun BS. Heme oxygenase-1–mediated autophagy protects against hepatocyte cell death and hepatic injury from infection/sepsis in mice. Hepatology. 2011;53:2053–62. doi: 10.1002/hep.24324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CW, Lin YF, Liu TT, Wang JY. Heme oxygenase-1 aggravates heat stress-induced neuronal injury and decreases autophagy in cerebellar Purkinje cells of rats. Exp Biol Med. 2013;238:744–54. doi: 10.1177/1535370213493705. [DOI] [PubMed] [Google Scholar]
- Chen J-S, Huang P-H, Wang C-H, et al. Nrf-2 mediated heme oxygenase-1 expression, an antioxidant-independent mechanism, contributes to anti-atherogenesis and vascular protective effects of Ginkgo biloba extract. Atherosclerosis. 2011;214:301–9. doi: 10.1016/j.atherosclerosis.2010.11.010. [DOI] [PubMed] [Google Scholar]
- Gabunia K, Ellison SP, Singh H, et al. Interleukin-19 (IL-19) induces heme oxygenase-1 (HO-1) expression and decreases reactive oxygen species in human vascular smooth muscle cells. J Biol Chem. 2012;287:2477–84. doi: 10.1074/jbc.M111.312470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paine A, Eiz-Vesper B, Blasczyk R, Immenschuh S. Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem Pharmacol. 2010;80:1895–903. doi: 10.1016/j.bcp.2010.07.014. [DOI] [PubMed] [Google Scholar]
- Reichard JF, Motz GT, Puga A. Heme oxygenase-1 induction by NRF2 requires inactivation of the transcriptional repressor BACH1. Nucleic Acids Res. 2007;35:7074–86. doi: 10.1093/nar/gkm638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedvat M, Huszar D, Herrmann A, et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell. 2009;16:487–97. doi: 10.1016/j.ccr.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot JJ, Song X, Wang X, et al. The cleaved cytoplasmic tail of polycystin-1 regulates Src-dependent STAT3 activation. J Am Soc Nephrol. 2014;25:1737–48. doi: 10.1681/ASN.2013091026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–26. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- Alexander S, Friedl P. Cancer invasion and resistance: interconnected processes of disease progression and therapy failure. Trends Mol Med. 2012;18:13–26. doi: 10.1016/j.molmed.2011.11.003. [DOI] [PubMed] [Google Scholar]
- Bakhautdin B, Das D, Mandal P, et al. Protective role of HO-1 and carbon monoxide in ethanol-induced cell death in hepatocytes and liver injury in mice. J Hepatol. 2014;61:1029–37. doi: 10.1016/j.jhep.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Song NY, Kim EH, et al. 15-Deoxy-Δ12, 14-prostaglandin J2 induces p53 expression through upregulation of heme oxygenase-1 in human breast cancer (MCF-7) cells. Free Radic Res. 2014;48:1018–27. doi: 10.3109/10715762.2014.897343. [DOI] [PubMed] [Google Scholar]
- Ruiz-Ramos R, Lopez-Carrillo L, Rios-Perez AD, De Vizcaya-Ruíz A, Cebrian ME. Sodium arsenite induces ROS generation, DNA oxidative damage, HO-1 and c-Myc proteins, NF-κB activation and cell proliferation in human breast cancer MCF-7 cells. Mutat Res. 2009;674:109–15. doi: 10.1016/j.mrgentox.2008.09.021. [DOI] [PubMed] [Google Scholar]
- Banerjee P, Basu A, Wegiel B, et al. Heme oxygenase-1 promotes survival of renal cancer cells through modulation of apoptosis-and autophagy-regulating molecules. J Biol Chem. 2012;287:32113–23. doi: 10.1074/jbc.M112.393140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Debnath J. Autophagy and tumorigenesis. Semin Immunopathol. 2010;32:383–96. doi: 10.1007/s00281-010-0213-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryter SW, Choi KM. Regulation of autophagy in oxygen-dependent cellular stress. Curr Pharm Des. 2013;19:2747–56. doi: 10.2174/1381612811319150010. [DOI] [PubMed] [Google Scholar]
- Pursiheimo J, Rantanen K, Heikkinen P, Johansen T, Jaakkola P. Hypoxia-activated autophagy accelerates degradation of SQSTM1/p62. Oncogene. 2008;28:334–44. doi: 10.1038/onc.2008.392. [DOI] [PubMed] [Google Scholar]
- Notte A, Leclere L, Michiels C. Autophagy as a mediator of chemotherapy-induced cell death in cancer. Biochem Pharmacol. 2011;82:427–34. doi: 10.1016/j.bcp.2011.06.015. [DOI] [PubMed] [Google Scholar]
- Qadir M, Kwok B, Dragowska W, et al. Macroautophagy inhibition sensitizes tamoxifen-resistant breast cancer cells and enhances mitochondrial depolarization. Breast Cancer Res Treat. 2008;112:389–403. doi: 10.1007/s10549-007-9873-4. [DOI] [PubMed] [Google Scholar]
- Han J, Hou W, Goldstein LA, et al. Involvement of protective autophagy in TRAIL resistance of apoptosis-defective tumor cells. J Biol Chem. 2008;283:19665–77. doi: 10.1074/jbc.M710169200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolisetty S, Traylor AM, Kim J, et al. Heme oxygenase-1 inhibits renal tubular macroautophagy in acute kidney injury. J Am Soc Nephrol. 2010;21:1702–12. doi: 10.1681/ASN.2010030238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamran MZ, Patil P, Gude RP. Role of STAT3 in cancer metastasis and translational advances. Biomed Res Int. 2013;2013:421821. doi: 10.1155/2013/421821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng XS, Wang S, Deng A, et al. Metformin targets Stat3 to inhibit cell growth and induce apoptosis in triple-negative breast cancers. Cell Cycle. 2012;11:367–76. doi: 10.4161/cc.11.2.18813. [DOI] [PubMed] [Google Scholar]
- Marotta LL, Almendro V, Marusyk A, et al. The JAK2/STAT3 signaling pathway is required for growth of CD44+ CD24–stem cell–like breast cancer cells in human tumors. J Clin Invest. 2011;121:2723–35. doi: 10.1172/JCI44745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiró G, Ortiz-Martínez F, Gallardo A, et al. Src, a potential target for overcoming trastuzumab resistance in HER2-positive breast carcinoma. Br J Cancer. 2014;111:689–95. doi: 10.1038/bjc.2014.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tryfonopoulos D, Walsh S, Collins D, et al. Src: a potential target for the treatment of triple-negative breast cancer. Ann Oncol. 2011;22:2234–40. doi: 10.1093/annonc/mdq757. [DOI] [PubMed] [Google Scholar]
- Aleshin A, Finn RS. SRC: a century of science brought to the clinic. Neoplasia. 2010;12:599–607. doi: 10.1593/neo.10328. [DOI] [PMC free article] [PubMed] [Google Scholar]