

Abstract
BRCA1 and 53BP1 antagonistically regulate homology-directed repair (HDR) and non-homologous end-joining (NHEJ) of DNA double-strand breaks (DSB). The histone deacetylase (HDAC) inhibitor trichostatin A directly inhibits the retention of 53BP1 at DSB sites by acetylating histone H4 (H4ac), which interferes with 53BP1 binding to dimethylated histone H4 Lys20 (H4K20me2). Conversely, we recently found that the retention of the BRCA1/BARD1 complex is also affected by another methylated histone residue, H3K9me2, which can be suppressed by the histone lysine methyltransferase (HKMT) inhibitor UNC0638. Here, we investigate the effects of the class I HDAC inhibitors MS-275 and FK228 compared to UNC0638 on histone modifications and the DNA damage response. In addition to H4ac, the HDAC inhibitors induce H3K9ac and inhibit H3K9me2 at doses that do not affect the expression levels of DNA repair genes. By contrast, UNC0638 selectively inhibits H3K9me2 without affecting the levels of H3K9ac, H3K56ac or H4ac. Reflecting their effects on histone modifications, the HDAC inhibitors inhibit ionizing radiation-induced foci (IRIF) formation of BRCA1 and BARD1 as well as 53BP1 and RIF1, whereas UNC0638 suppresses IRIF formation of BRCA1 and BARD1 but not 53BP1 and RIF1. Although HDAC inhibitors suppressed HDR, they did not cooperate with the poly(ADP-ribose) polymerase inhibitor olaparib to block cancer cell growth, possibly due to simultaneous suppression of NHEJ pathway components. Collectively, these results suggest the mechanism by that HDAC inhibitors inhibit both the HDR and NHEJ pathways, whereas HKMT inhibitor inhibits only the HDR pathway; this finding may affect the chemosensitizing effects of the inhibitors.
Keywords: BRCA1, DNA damage response, histone deacetylase inhibitor, histone modifications, 53BP1
Histone deacetylase (HDAC) inhibitors are promising compounds for the treatment of cancer and are known to sensitize cells to DNA damage-inducing agents.1,2 Inhibition of HDAC increases histone acetylation levels and, thus, modifies the chromatin structure and alters gene expression. The expression levels of DNA double-strand break (DSB) repair proteins including 53BP1, BRCA1, BRCA2, RAD51 and ATM are repressed by HDAC inhibitors, resulting in failure of homology-directed repair (HDR) and of DSB non-homologous end-joining (NHEJ).2,3 In addition to their transcriptional effects, HDAC inhibitors directly modify the local response at the DSB sites. TIP60-dependent acetylation of H4K16 diminishes 53BP1 binding to H4K20me2,4,5 a critical interaction for 53BP1 recruitment to DSB sites in the NHEJ pathway.6 HDAC1 and HDAC2 are recruited to the DSB sites to promote hypoacetylation of H3K56 and H4K16, and they play essential roles in NHEJ.7 Importantly trichostatin A (TSA), a class I/II HDAC inhibitor, induces hyperacetylation of H3K56ac and H4ac and reduces 53BP1 retention at DSB sites.4 Because 53BP1 and BRCA1 antagonize each other at DSB sites to promote NHEJ and HDR, respectively,8 the inhibition of 53BP1 by TSA can reverse BRCA1 recruitment defects.4 However, it was recently reported that TSA-induced chromatin condensation perturbs stable BRCA1 retention at later times after DSB,9 suggesting that HDAC inhibitors have polyphenic effects.
We recently found that in response to DNA damage, H3K9me2 interacts with BARD1,10 a RING- and BRCT-containing protein that forms a stable E3 complex with BRCA1.11,12 The interaction is primarily mediated by heterochromatin protein 1 (HP1)γ through an HP1-binding motif in the BARD1-BRCT domain and the chromoshadow domain of HP1, and it is critical for the stable retention of BRCA1/BARD1 at DSB sites. The BRCA1/BARD1 retained through H3K9me2/HP1γ induces HDR by inhibiting the accumulation of RIF1, a downstream effector of 53BP1 on NHEJ.10 Interestingly, an H3K9-specific histone lysine methyltransferase (HKMT) inhibitor, UNC0638, abolishes BRCA1/BARD1 retention.10 These results prompted us to investigate whether HDAC inhibitors also inhibit the retention of BRCA1/BARD1, accompanied by alteration of this modification at H3K9.
In this study, we employed MS-275 and FK228, class I HDAC inhibitors that specifically inhibit HDAC1 and HDAC2,13–15 to examine the correlation between their effects on DNA repair protein retention and histone modifications.
Materials and Methods
Cell culture
All cells were cultured in DMEM supplemented with 10% FBS and 1% antibiotic-antimycotic agent (Life Technologies, USA). The HeLa DR-GFP cells were provided by Dr Maria Jasin (Memorial Sloan Kettering Cancer Center). For chemical treatments, cells were incubated with 2.5 μM of MS-275 (Focus Biomolecules, USA), 2.5 nM of FK228 (TOCRIS Bioscience, UK), 3 μM of UNC0638 (Cayman Chemicals, USA) or the indicated doses of olaparib (JS Research Chemical Trading, Germany) for 24 h unless otherwise indicated. For IR, cells were exposed to X-ray irradiation (10 Gy) and further cultured for 1 h before indirect immunofluorescence unless otherwise indicated.
Antibodies
The antibodies used were rabbit polyclonal antibodies to BRCA1 (C20; Santa Cruz Biotechnology, USA), BARD1 (BL518; Bethyl Laboratories, USA), H3K9ac (07-352; Millipore, Germany), H3K56ac (2134-1; EPITMICS, USA), H4ac (06-866; Millipore), 53BP1 (Novus Biologicals, USA) RIF1, (A300-569A; Bethyl Laboratories) and RAD51 (Bio Academia, Japan), and mouse monoclonal antibodies to H3K9me2 (CMA307; Millipore), γH2AX (JBW301; Millipore) and α-tubulin and β-tubulin (DMIA+BMIB; Neomarkers, USA).
Western blotting
Cells were lysed with radioimmunoprecipitation assay (RIPA) buffer, clarified, adjusted for protein concentration and subjected to western blotting as described.16
RT-PCR
The total RNA from each sample was isolated with TRIzol reagent (Invitrogen, USA) according to the manufacturer’s protocol. RT-PCR was carried out with a PrimeScript High Fidelity RT-PCR kit (Takara, Japan) and the following primers:
| BRCA2 | Forward primer | 5′-CAGTGGTATGTGGGAGTTTGT-3′ |
| Reverse primer | 5′-ATCCATGACTTGCAGCTTCTC-3′, | |
| ΑΤΜ | Forward primer | 5′-CTTGTGCCTTGGCTACAGAT-3′ |
| Reverse primer | 5′-AACTATACTGGTGGTCAGTGCC-3′, | |
| RIF1 | Forward primer | 5′- ATTTCGTAGTGGAGCACCCATG-3′ |
| Reverse primer | 5′-AGCTACATGACACGAATTGCCC-3′, | |
| GAPDH | Forward primer | 5′-GACCCCTTCATTGACCTCAAC-3′ |
| Reverse primer | 5′-CTTCTCCATGGTGGTGAAGAC-3′ |
Immunofluorescence microscopy
Indirect immunofluorescence labeling of cells and fluorescence detection with a confocal laser scanning microscope (LSM 510; Carl Zeiss, Germany) was performed as previously described.10 For staining of BRCA1 and BARD1, cells were pre-extracted using incubation in cytoskeleton buffer with RNase A as described.10 Nuclear foci were mechanically counted using the Cellomics Image Analyzer (Thermo Fisher, Japan), except for RIF1, which was visually counted.
DR-GFP homology-directed repair assay
HeLa cells containing an integrated DR-GFP reporter construct were transfected with I-SceI nuclease. Twelve hours after the transfection, the cells were treated with HDAC inhibitors or UNC0638 for 24 h and further incubated without the inhibitors for 12 h. The cells were trypsinized, and the percentage of GFP positive cells was determined by flow cytometry.
Clonogenic survival assay
Cells were seeded at a concentration of 500 cells/well in six-well plates and after 6 h treated with the chemical agents. After 24 h of incubation, the cells were washed and further cultured in fresh medium without the chemicals for 9 days. The cells were then fixed and stained with crystal violet. The colonies were scanned and counted using an ImageQuant LAS-4000 instrument (GE Healthcare, USA).
Statistics
Statistical analyses were performed using the two-tailed Student’s t-test. P < 0.05 was considered significant.
Results
Effects of histone deacetylase inhibitors and UNC0638 on histone modifications and the expression levels of DNA repair genes
Inhibition of H3K9me2 with HKMT inhibitor UNC0638 dramatically suppressed ionizing irradiation-induced foci (IRIF) formation of BRCA1 and BARD1.10 To explore whether class I HDAC inhibitors exhibit similar effects, we first analyzed the effects of MS-275 and FK228 on the H3K9 modification. A time course analysis in U2OS cells suggested that H3K9me2 was significantly reduced in response to the HDAC inhibitors by 24 h, whereas H3K9ac was markedly enhanced (Fig.1a). To avoid effects on the expression levels of DNA repair genes, we searched for an appropriate dose of the HDAC inhibitors to reduce the H3K9me2 but not affect the expression levels of the DNA repair genes during the time period. H3K9me2 was significantly reduced after treatment with 2.5 μM MS-275 and 2.5 nM FK228, accompanied by substantial upregulation of H3K9ac (Fig.1b). Although the protein expression levels of BRCA1 and BARD1 were reduced at higher doses (data not shown), they were unchanged in response to 2.5 μM MS-275 or 2.5 nM FK228 (Fig.1b). The HDAC inhibitor effects on histone modifications using the doses at concentrations that did not alter BRCA1 and BARD1 expression were reproducible in MCF-7 cells (Fig.1c). Because TSA promotes H3K56ac and H4K16ac, which inhibit the interaction between 53BP1 and H4K20me2,4,7 we tested whether MS-275 and FK228, at our trial doses, also accelerated this modification. An anti-H4ac antibody that recognizes acetylation of N-terminal lysines including H4K16ac was used, as in a previous study.4 Similar to TSA, MS-275 and FK228 effectively elevated H3K56ac and H4ac in U2OS cells (Fig.2a), as well as in MCF-7 cells (Fig.2b). Conversely, the HKMT inhibitor UNC0638 reduced H3K9me2 without enhancing H3K56ac, H4ac or H3K9ac (Fig.2c). The expression levels of other DNA repair genes, including the genes that have been reported to be reduced by HDAC inhibitors,2 RAD51, 53BP1, BRCA2, ATM and RIF1, were not affected by treatment with HDAC inhibitors or UNC0638 (Figs1b,c and 2c,d).
Figure 1.
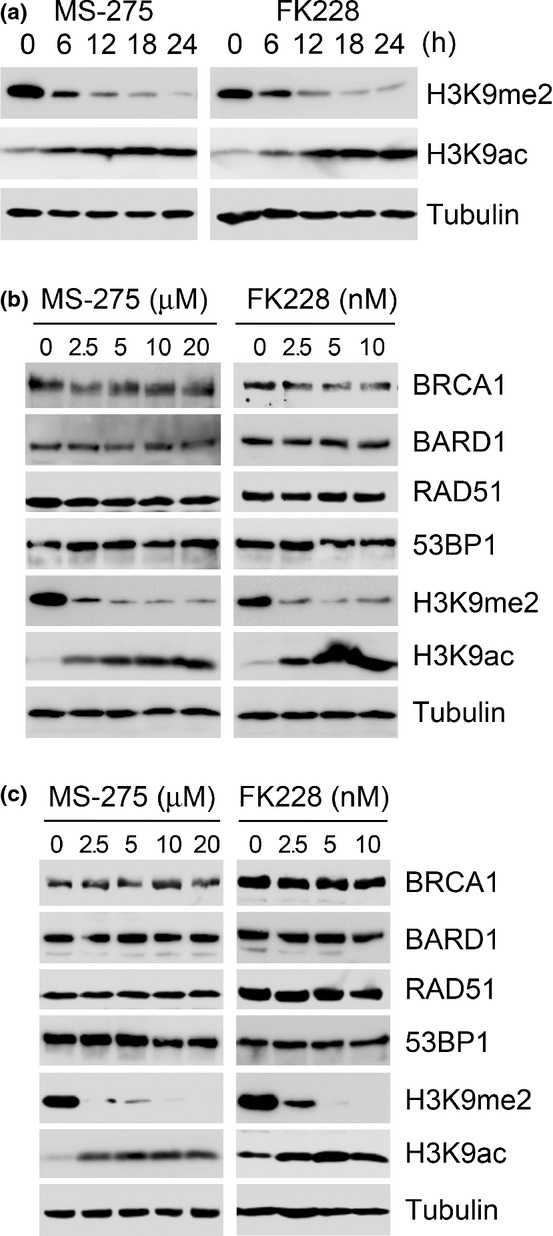
Alteration of H3K9 modifications induced by the histone deacetylase (HDAC) inhibitors MS-275 and FK228. (a) U2OS cells were incubated with MS-275 (2.5 μM) or FK228 (2.5 nM) for the indicated times. Whole cell lysates were immunoblotted with the indicated antibodies. (b, c) U2OS (b) or MCF-7 (c) cells were incubated for 24 h with the indicated doses of MS-275 or FK228 and immunoblotted with the indicated antibodies. MS-275 or FK228 did not inhibit the expression levels of DNA repair genes BRCA1, BARD1, RAD51 and 53BP1 at the doses tested.
Figure 2.
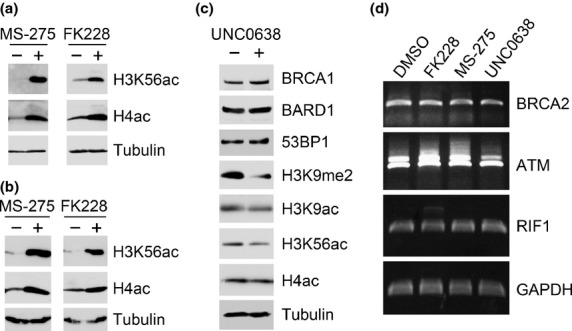
Induction of H3K56ac and H4ac by the histone deacetylase (HDAC) inhibitors, but not the HKMT inhibitor at doses that do not affect the expression levels of DNA repair genes. (a, b) U2OS (a) or MCF-7 (b) cells were incubated for 24 h with DMSO (−), MS-275 (2.5 μM) or FK228 (2.5 nM) and immunoblotted with the indicated antibodies. (c) U2OS cells were incubated for 24 h with DMSO (−) or UNC0638 (3 μM) and immunoblotted with the indicated antibodies. UNC0638 did not inhibit the expression levels of DNA repair genes BRCA1, BARD1 and 53BP1 at the doses tested. (d) U2OS cells were treated with MS-275 (2.5 μM), FK228 (2.5 nM) or UNC0638 (3 μM) for 24 h. Gene expression levels for of BRCA2, ATM, RIF1 and the loading control, GAPDH, were analyzed by RT-PCR.
Class I histone deacetylase inhibitors and UNC0638 inhibit irradiation-induced foci formation of BRCA1 and BARD1
Using the doses that we determined as above, we analyzed the HDAC inhibitor effect on IRIF formation of BRCA1 and BARD1. U2OS cells were treated with the inhibitors for 24 h, exposed to IR and, 1 h later, were immunostained for BRCA1 and BARD1 with the DSB marker γH2AX. Both MS-275 and FK228 increased the intensity of γH2AX-IRIF (Suppl. Fig. S1) and significantly reduced the formation of IRIF of BRCA1 (Fig.3a) and BARD1 (Fig.3b). UNC0638 also significantly reduced the retention of BRCA1 (Fig.3a) and BARD1 (Fig.3b). Low-magnification views are shown in Figure3(c). The BRCA1-IRIF-positive fractions in cells treated with vehicle DMSO, MS-275, FK228 and UNC0638 were 66.9%, 7.5%, 32.9% and 7.4%, respectively (Fig.3a, right panel). The BARD1-IRIF-positive fractions in cells treated with DMSO, MS-275, FK228 and UNC0638 were 80.8%, 22.0%, 18.2% and 8.6%, respectively (Fig.3b, right panel). The reduction in IRIF formation was not due to G1 arrest or decreased S phase population in the cells treated with the HDAC inhibitors (Suppl. Fig. S2). UNC0638 at the dose does not affect cell cycle either.10 The reduction in the total nuclear staining of BRCA1 and BARD1 in HDAC inhibitor-treated or UNC0638-treated cells was not due to reduced protein expression levels because cells without pre-extraction treatment exhibited equivalent staining (Suppl. Fig. S3). We also tested whether TSA inhibits the retention of BRCA1 and BARD1 in addition to 53BP1. Similar to MS-275 and FK228, TSA inhibits the retention of BRCA1 and BARD1 accompanied by the reduction of H3K9me2 (Suppl. Fig. S4).
Figure 3.
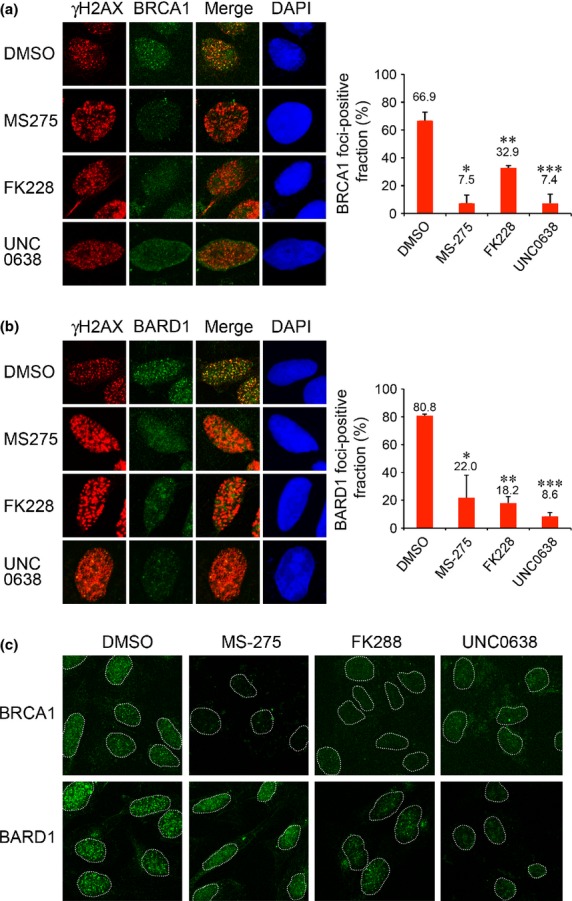
Histone deacetylase (HDAC) inhibitors suppress IRIF formation of BRCA1 and BARD1. (a) U2OS cells treated with DMSO, MS-275, FK228 or UNC0638 were exposed to IR and immunostained with γH2AX and BRCA1. Right panel: Quantification of the cells displaying more than 30 BRCA1 foci. Error bars, SD of two independent experiments, each based on more than 100 cells. Significant differences were calculated compared to DMSO-treated cells: *P = 0.009, **P = 0.015, ***P = 0.011. (b) U2OS cells treated as in (a) were immunostained with γH2AX and BARD1. Error bars, SD of two independent experiments, each based on more than 60 cells. *P = 0.035, **P = 0.003, ***P = 0.001. (c) Low-magnification views. Nuclei are outlined with dashed lines.
Class I histone deacetylase inhibitors but not UNC0638 inhibit ionizing irradiation-induced foci formation of 53BP1 and RIF1
We next tested whether MS-275 and FK228 would inhibit IRIF formation of the NHEJ proteins 53BP1 and RIF1. MS-275 significantly inhibited 53BP1-IRIF formation (Fig.4a,c). The 53BP1-IRIF-positive fractions in cells treated with DMSO and MS-275 were 92.5% and 44.9%, respectively (Fig.4a, right panel). However, FK228 did not dramatically inhibit the formation of 53BP1-IRIF (83.0%; Fig.4a, right panel). Conversely, RIF1-IRIF, an effector for NHEJ that acts downstream of 53BP1, was significantly reduced by both MS-275 and FK228 (Fig.4b,c). The RIF1-IRIF-positive fractions in cells treated with DMSO, MS-275 and FK228 were 79.2%, 14.4% and 27.2%, respectively (Fig.4b, right panel). Although FK228 only minimally affected the formation of 53BP1-IRIF when assessed by focus number, it substantially reduced the intensity of each focus (Fig.4,c). The 53BP1 foci with low intensity may not have been fully functional and resulted in a significant reduction in RIF1-IRIF. In contrast, UNC0638 did not affect IRIF formation of 53BP1 in number and intensity (Fig.4a). UNC0638 did not dramatically reduce the retention of RIF1 either (Fig.4b,c), suggesting that NHEJ is not disturbed by UNC0638.
Figure 4.
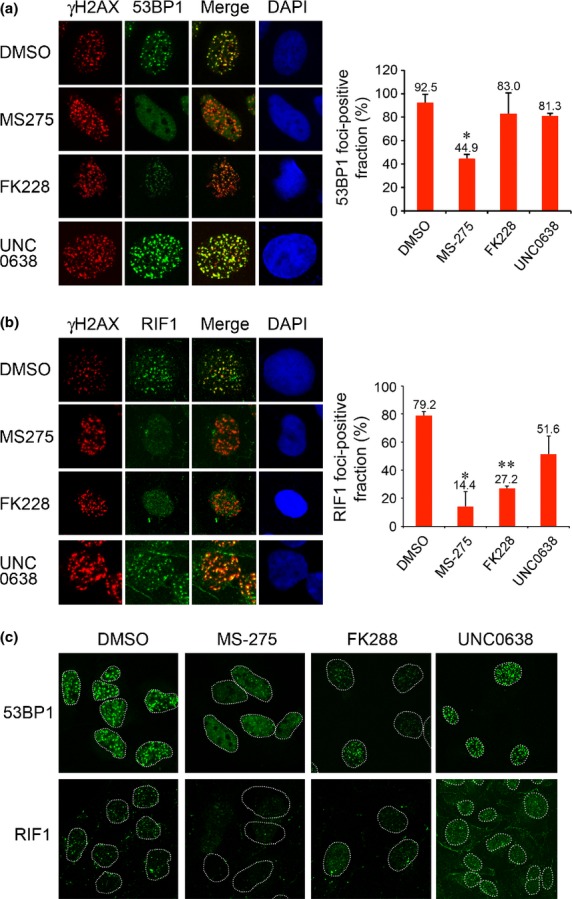
Histone deacetylase (HDAC) inhibitors suppress IRIF formation of 53BP1 and RIF1. (a) U2OS cells treated with DMSO, MS-275, FK228 or UNC0638 were exposed to IR and immunostained with γH2AX and 53BP1. Right panel: Quantification of the cells displaying more than 20 53BP1 foci. Error bars, SD of two independent experiments, each based on more than 40 cells. Significant differences were calculated compared to DMSO-treated cells: *P = 0.013. (b) U2OS cells treated as in (a) were immunostained with γH2AX and RIF1. Error bars, SD of two independent experiments, each based on more than 120 cells. *P = 0.024, **P = 0.002. (c) Low-magnification views. Nuclei are outlined with dashed lines.
Class I histone deacetylase inhibitors and UNC0638 inhibit homology-directed repair
To evaluate the effects of HDAC inhibitors and UNC0638 on HDR, we used the gene conversion assay with DR-GFP reporter cells. Treatment with HDAC inhibitors reduced HDR at I-SceI nuclease-induced DSB (Fig.5a,b). The percentages of homologous recombination in cells treated with MS-275 and FK228 relative to that in cells treated with DMSO were 53.7% and 68.8%, respectively (Fig.5b). The effect was modest when compared to UNC0638, which reduced HDR to 38.9% (Fig.5a,b). We also analyzed the IRIF composed of RAD51, the effector of HDR. MS-275, FK228 and UNC0638 all reduced the formation of IRIF of RAD51, although the effect of FK228 was modest at the doses tested. The RAD51-IRIF-positive fractions in cells treated with DMSO, MS-275, FK228 and UNC0638 were 71.4%, 20.7%, 47.0% and 30.6%, respectively (Fig.5c).
Figure 5.
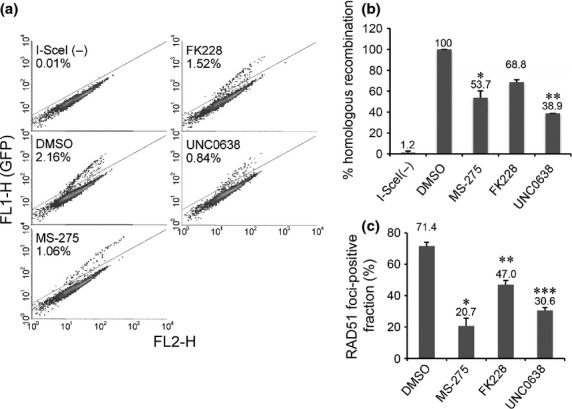
Histone deacetylase (HDAC) and HKMT inhibitors suppress homology-directed repair (HDR). (a) Fluorescence-activated cell sorting (FACS) data on HDR of I-SceI-induced DSB in HeLa DR-GFP reporter cells treated with DMSO, MS-275, FK228 or UNC0638. (b) Quantification of the GFP-positive fraction. Averages ± SD normalized to cells with DMSO were derived from two independent experiments. Significant differences were calculated compared to DMSO-treated cells: *P = 0.002, **P < 0.001. (c) U2OS cells treated with DMSO, MS-275, FK228 or UNC0638 were exposed to infrared (IR) and immunostained with γH2AX and RAD51 6 h after IR. Quantification of the cells displaying more than 20 RAD51 foci. Error bars represent SD from two independent experiments, each based on more than 100 cells. Significant differences were calculated compared to DMSO-treated cells: *P = 0.006, **P = 0.012, ***P = 0.003.
Class I histone deacetylase inhibitors do not show synthetic lethality with a poly(ADP-ribose) polymerase inhibitor
The ability of MS-275 and FK228 to inhibit HDR with disruption of stable retention of BRCA1/BARD1 at DSB sites prompted us to test whether the HDAC inhibitors induce a synergetic cytotoxic effect with a PARP inhibitor, which inhibits single-strand DNA break repair. We tested four common cell lines, U2OS, HCT116, MCF-7 and HeLa, for cytotoxicity with clonogenic survival assay. The cells were exposed to a range of doses of the PARP inhibitor olaparib for 24 h in the presence or absence of the HDAC inhibitors, and the clonogenic survival was measured 9 days after culture in the agent-free medium. The dose of MS-275 or FK228 for each cell line was based on the amount needed for 10% to 20% cell death when used as a single agent (Suppl. Fig. S5). Interestingly, despite the effect of the HDAC inhibitors on HDR, the exposure to MS-275 or FK228 with olaparib did not show synergistic cytotoxicity of U2OS, HCT116, MCF-7 or HeLa cells (Fig.6).
Figure 6.
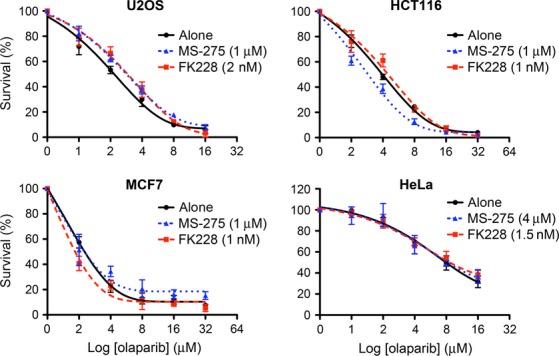
Histone deacetylase (HDAC) inhibitors do not show synthetic lethality with a PARP inhibitor. U2OS, HCT116, MCF7 or HeLa cells were exposed to the indicated doses of olaparib with DMSO (alone), MS-275 or FK228 and analyzed for clonogenic survival. The data are shown with nonlinear regression fit curves of one phase decay (GraphPad Prism). Averages ± SEM, normalized to cells without olaparib, were derived from triplicate experiments.
Discussion
Collectively, our findings revealed distinct effects of HDAC inhibitors and HKMT inhibitors on repair pathways in response to DSB. HDAC inhibitors suppressed both BRCA1/BARD1-mediated and 53BP1/RIF1-mediated pathways, which was accompanied by a reduction in H3K9me2 and an increase in H3K56 and H4 acetylation. However, UNC0638 selectively decreased H3K9me2, BRCA1/BARD1 retention and HDR pathways (Suppl. Table S1). It has been reported that MS-275 preferentially inhibits HDAC1 versus HDAC3 and has no effect toward HDAC8,14 and FK228 inhibits HDAC1 and HDAC2 much more effectively than HDAC4 and HDAC6 in vitro.15 However, direct comparison of the effects on histone modification of the two inhibitors in vivo has not been demonstrated. We showed that MS-275 and FK228 exert similar effects on the histone modification. For DNA damage response we observed some difference between the effects of MS-275 and FK228. When counted as number of foci, MS-275 inhibited 53BP1 IRIF, whereas FK228 did not. Although the reason for the different effects is currently unknown, the effect on NHEJ by FK228 could be similar to MS-275, because the intensity of 53BP1 foci and its downstream target RIF1 were significantly inhibited.
Selective disruption of HDR caused by BRCA1 deficiency is a target for synthetic lethality in combination with PARP inhibitors. Therefore, the mechanism of BRCA1/BARD1 retention at DSB sites mediated by BARD1-HP1-H3K9me2 interaction that is required for HDR could be an ideal target for the synthetic lethality. Indeed, cells in which endogenous BARD1 is replaced with BARD1 mutant that disrupt the BARD1-Hp1 interaction are more sensitive to olaparib than are the wild-type cells.10 Inhibition of H3K9me2 by UNC0638 also demonstrates synthetic lethality with olaparib.10 Because HDAC inhibitors also reduce H3K9me2 and perturb HDR, similar effects are possible. However, exposure to MS-275 or FK228 with olaparib for 24 h did not show synergistic cytotoxicity of U2OS, MCF7, HCT116 or HeLa cells in clonogenic survival assays. We interpret this result as evidence of additional NHEJ defects. As a contradictory finding, synergistic loss of cell viability by combinations of PARP inhibitors and the class I/II inhibitors suberoylanilide hydroxamic acid (SAHA)17,18 or PCI-24781 (abexinostat)19 has been shown. The synthetic lethality of PARP inhibitor and HDR defects has been proposed as the mechanism underlying the synergistic effects. The discrepancy could be due to the distinct class of the inhibitors. However, TSA, another class I/II inhibitor, inhibited the retention of BRCA1, BARD1, 53BP1 and RIF1, accompanied by the reduction of H3K9me2 and enhancement of H4ac, suggesting that the early effects of the class I/II inhibitors on HR and NHEJ are similar to class I inhibitors. We presume that the contradictory effects between our results for MS-275 or FK228 and the previous study on the synergistic loss of cell viability could be due to the different time length of the exposure to the inhibitors in the previous studies. Whereas we employed clonogenic survival assay with 24 h exposure to the inhibitors, the time length to exposure in previous studies ranged from 3 days to 6 weeks.17–19 The longer exposure to HDAC inhibitors may result in additional transcriptional effects that affect the synthetic lethal effect.
In conclusion, the direct effects of the class I HDAC inhibitors on histone H3K9, H3K56 and H4 suppress both HR and NHEJ pathways after DSB. While the transcriptional effect of the inhibitors may further affect the DNA damage response after longer exposure, the failure of the two pathways did not exhibit a synthetic lethal effect with PARP inhibitors. The inhibition of HR and NHEJ pathways early after treatment with HDAC inhibitors should be considered when their effects on the DNA damage response is applied in cancer therapy.
Acknowledgments
We are grateful to Yukiko Togashi and Shinko Hanaki for technical support. We thank Dr Maria Jasin for providing U2OS DR-GFP reporter cells. This study was supported by grants from the Japan Society for the Promotion of Science, the Ministry of Education, Science, Sports, Culture and Technology of Japan, the Ministry of Health, Labour and Welfare of Japan, and the Japan Private School Promotion Foundation.
Disclosure Statement
The authors have no conflict of interest to declare.
Supporting Information
Table S1. Summary of the effects of histone deacetylase (HDAC) inhibitors and UNC0638.
Fig. S1. MS-275, FK228 and UNC0638 increase the intensity of γH2AX IRIF.
Fig. S2. Cell cycle progression of cells treated with histone deacetylase (HDAC) inhibitors.
Fig. S3. Immunostain of BRCA1 and BARD1 without pre-extraction.
Fig. S4. Trichostatin A (TSA) inhibits H3K9me2 and suppresses IRIF formation of BRCA1 and BARD1 in addition to 53BP1 and RIF1.
Fig. S5. Clonogenic survival of cells exposed to class I histone deacetylase (HDAC) inhibitors.
References
- Spiegel S, Milstien S, Grant S. Endogenous modulators and pharmacological inhibitors of histone deacetylases in cancer therapy. Oncogene. 2011;31:537–51. doi: 10.1038/onc.2011.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groselj B, Sharma NL, Hamdy FC, Kerr M, Kiltie AE. Histone deacetylase inhibitors as radiosensitisers: effects on DNA damage signalling and repair. Br J Cancer. 2013;108:748–54. doi: 10.1038/bjc.2013.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuzon CT, Spektor T, Kong X, et al. Concerted activities of distinct H4K20 methyltransferases at DNA double-strand breaks regulate 53BP1 nucleation and NHEJ-directed repair. Cell Rep. 2014;8:430–8. doi: 10.1016/j.celrep.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Cho NW, Cui G, et al. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat Struct Mol Biol. 2013;20:317–25. doi: 10.1038/nsmb.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K-Y, Mizzen CA. Histone H4 deacetylation facilitates 53BP1 DNA damage signaling and double-strand break repair. J Mol Cell Biol. 2013;5:157–65. doi: 10.1093/jmcb/mjs066. [DOI] [PubMed] [Google Scholar]
- Sanders SL, Portoso M, Mata J, Bähler J, Allshire RC, Kouzarides T. Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell. 2004;119:603–14. doi: 10.1016/j.cell.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Miller KM, Tjeertes JV, Coates J, et al. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat Struct Mol Biol. 2010;17:1144–51. doi: 10.1038/nsmb.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley JM, Sung P. 53BP1, BRCA1, and the choice between recombination and end joining at DNA double-strand breaks. Mol Cell Biol. 2014;34:1380–8. doi: 10.1128/MCB.01639-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana S, Kruhlak MJ, Kim J, et al. A Macrohistone variant links dynamic chromatin compaction to BRCA1-dependent genome maintenance. Cell Rep. 2014;8:1049–62. doi: 10.1016/j.celrep.2014.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Nishikawa H, Fukuda T, et al. nteraction of BARD1 and HP1 is required for BRCA1 retention at sites of DNA damage. Cancer Res. 2015;75:1311–21. doi: 10.1158/0008-5472.CAN-14-2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashizume R, Fukuda M, Maeda I, et al. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J Biol Chem. 2001;276:14537–40. doi: 10.1074/jbc.C000881200. [DOI] [PubMed] [Google Scholar]
- Brzovic PS, Keeffe JR, Nishikawa H, et al. Binding and recognition in the assembly of an active BRCA1/BARD1 ubiquitin-ligase complex. Proc Natl Acad Sci U S A. 2003;100:5646–51. doi: 10.1073/pnas.0836054100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol. 2009;27:5459–68. doi: 10.1200/JCO.2009.22.1291. [DOI] [PubMed] [Google Scholar]
- Hu E, Dul E, Sung C-M, et al. Identification of novel isoform-selective inhibitors within class I histone deacetylases. J Pharmacol Exp Ther. 2003;307:720–8. doi: 10.1124/jpet.103.055541. [DOI] [PubMed] [Google Scholar]
- Furumai R, Matsuyama A, Kobashi N, et al. FK228 (Depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002;62:4916–21. [PubMed] [Google Scholar]
- Nishikawa H, Ooka S, Sato K, et al. Mass spectrometric and mutational analyses reveal Lys-6-linked polyubiquitin chains catalyzed by BRCA1-BARD1 ubiquitin ligase. J Biol Chem. 2004;279:3916–24. doi: 10.1074/jbc.M308540200. [DOI] [PubMed] [Google Scholar]
- Zhang J-X, Li D-Q, He AR, et al. Synergistic inhibition of hepatocellular carcinoma growth by cotargeting chromatin modifying enzymes and poly (ADP-ribose) polymerases. Hepatology. 2012;55:1840–51. doi: 10.1002/hep.25566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao OS, Goodman OB. Synergistic loss of prostate cancer cell viability by co-inhibition of HDAC and PARP. Mol Cancer Res. 2014;12:1755–66. doi: 10.1158/1541-7786.MCR-14-0173. [DOI] [PubMed] [Google Scholar]
- Adimoolam S, Sirisawad M, Chen J, Thiemann P, Ford JM, Buggy JJ. HDAC inhibitor PCI-24781 decreases RAD51 expression and inhibits homologous recombination. Proc Natl Acad Sci U S A. 2007;104:19482–7. doi: 10.1073/pnas.0707828104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Summary of the effects of histone deacetylase (HDAC) inhibitors and UNC0638.
Fig. S1. MS-275, FK228 and UNC0638 increase the intensity of γH2AX IRIF.
Fig. S2. Cell cycle progression of cells treated with histone deacetylase (HDAC) inhibitors.
Fig. S3. Immunostain of BRCA1 and BARD1 without pre-extraction.
Fig. S4. Trichostatin A (TSA) inhibits H3K9me2 and suppresses IRIF formation of BRCA1 and BARD1 in addition to 53BP1 and RIF1.
Fig. S5. Clonogenic survival of cells exposed to class I histone deacetylase (HDAC) inhibitors.