

Abstract
Breast cancer (BC) is the second most common cause of cancer deaths. Triple-negative breast cancer (TNBC) does not show immunohistochemical expression of oestrogen receptors, progesterone receptors or HER2. At present, no suitable treatment option is available for patients with TNBC. This dearth of effective conventional therapies for the treatment of advanced stage breast cancer has provoked the development of novel strategies for the management of patients with TNBC. This review presents recent information associated with different therapeutic options for the treatment of TNBC focusing on promising targets such as the Notch signalling, Wnt/β-catenin and Hedgehog pathways, in addition to EGFR, PARP1, mTOR, TGF-β and angiogenesis inhibitors.
Tables of Links
| TARGETS | |
|---|---|
| GPCRsa | Enzymesc |
| FZD7 receptor | ADAM |
| SMO receptor | ADAM17 |
| Catalytic receptorsb | Akt (PKB) |
| EGFR | Aspartyl protease |
| Fas | GSK3β |
| HER2 | mTOR |
| TGFBR1 | PARP1 |
| VEGFR2 | p70S6kinase |
| PKCα | |
| SGK1 | |
| ULK1 |
| LIGANDS | |
|---|---|
| β-catenin | Lapatinib |
| Angiopoietin-1 | LY2157299 |
| Angiopoietin-2 | Neratinib |
| Cisplatin | Olaparib |
| Erlotinib | Rapamycin |
| Everolimus | Rucaparib |
| Gefitinib | Temsirolimus |
| IFN-γ | TGFβ |
| IGF-1 | TNF-α |
| IL-1α | Veliparib |
| IL-1β | Wnt |
| IL-2 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14
Alexander et al., 2013a,b,c,,).
Introduction
Triple-negative breast cancer (TNBC) is a discrete subset of breast cancer (BC) characterized by a lack of immunohistochemical expression of oestrogen receptors (ER), progesterone receptors (PR) and HER2 and accounts for approximately 15–20% of BC patients (Curigliano and Goldhirsch, 2011; Penault-Llorca and Viale, 2012). TNBC has been shown to be more prevalent in African, American and Hispanic women, and it is mostly younger women that are susceptible to it (Ismail-Khan and Bui, 2008; Chacon and Costanzo, 2010; de Ruijter et al., 2011). Undoubtedly, scientific advancements made in the field of breast cancer research have resulted in a tremendous increase in survival rate of BC patients. However, this is applicable only if it is diagnosed at an early stage and without metastasis. TNBC remains the hardest breast cancer subtype to treat because the disease is an assemblage of different breast cancer subtypes. Researchers are keenly attempting to classify these subtypes and identify new treatments (Kumar et al., 2013).Therapeutic treatment options for patients with stage IV TNBC are very limited and often unsuccessful. TNBC is commonly an aggressive tumour and when compared with other breast cancers, it grows faster and is less likely to be seen on an annual mammogram and it is more likely to spread to other sites in the body at an early stage.
Metastatic TNBC is a destructive condition that is associated with a high proliferation index, ensuing visceral and CNS metastases (Otvos and Surmacz, 2011) and poor outcome in spite of treatment. Average survival of advanced TNBC is 12 months, much shorter than the duration of survival observed in other subtypes of advanced BC. Therefore, the identification of specific targets and more effective, dynamic and promising therapies for TNBC patients remains an important clinical challenge. In this review, we have focused on recently discovered therapeutic targets for TNBC, such as the Notch signalling pathway, Wnt/β-catenin pathway, Hedgehog (Hh) pathway, EGFR inhibitors, PARP1 inhibitors, mammalian target of rapamycin (mTOR) inhibitors, chondroitin sulfate proteoglycan 4 (CSPG4) protein targeted monoclonal antibody, angiogenesis inhibitors and TGF-β inhibitors, as well as a few therapeutic options that have recently been developed for the treatment of TNBC based on these targets along with their progress in clinical trials (Table 2011).
Table 1.
Different pathways and how they can be used as targets, with some examples
| Pathways | Mechanism of action | Examples | Trial phase | References |
|---|---|---|---|---|
| Notch signalling pathway | γ-secretase inhibitors or aspartyl protease inhibitors | RO-4929097 | Phase I/II | Shih and Wang, 2007; Olsauskas-Kuprys et al. 2013 |
| Hedgehog signalling pathway | SMO antagonist and Pathed1 antagonists | Cyclopamine | Phase II | Merchant et al. 2010 |
| Wnt/b-catenin pathway | FZD7 receptor and Wnt co-receptor the low-density lipoprotein receptor-related protein-6 inhibitors | Salinomycin | Phase I/II | King et al. 2012 |
| PARP inhibitors | Inhibits repair of single stranded DNA breaks | Iniparib | Phase II/III | Tutt et al. 2010, Lancet 2010; 376:235–244 |
| Olaparib | Phase II | |||
| Rucaparib | Phase II | |||
| Veliparib | Phase I | |||
| EGFR inhibitors | Inhibits overexpressed epidermal growth factor | GefItinib | Phase II | Ueno et al. 2011 |
| mTOR inhibitors | Inhibits FK506 binding protein 12-rapamycin associated protein 1 | Everolumus | Phase II | http://clinicaltrials.gov/ct2/show/NCT00998036 |
| Temsirolimus | Phase II | |||
| TGF-β signalling pathway | TGFBR1 inhibitor | LY2157299 | Phase I | Bhola et al. 2013 |
| CSPG4 proteins | Chondroitin sulfate proteoglycan 4 targeted monoclonal antibody | CSPG4-specific monoclonal antibody | Tested in a few patients with breast cancer | Wang et al. 2010a |
Notch signalling pathway
The Notch signalling pathway is a highly conserved signalling pathway for cell-to-cell communication, regulating key cellular processes (Al-Hussaini et al., 2011). This pathway is involved in the development as well as progression of breast cancer. Speiser and his co-workers found that TNBC has shown overexpression of Notch 1 and Notch 4 receptors in vascular endothelial cells and tumours with a subcellular location different from that of hormone-positive breast cancer (Reedijk et al., 2005; Speiser et al., 2012).
Activation of the Notch signalling pathway requires the binding of a Notch ligand to a Notch receptor on an adjacent cell. So far, five Notch ligands [Delta-like (Dll) 1, 3, 4, and Jagged (JAG)1, 2] and four Notch receptors have been identified. Notch ligands are single transmembrane proteins occurring naturally, and have a typical extracellular DSL domain that mediates receptor binding and multiple EGF-like repeats. JAG ligands possess an extra cysteine-rich domain, which is not present in the Dll ligands. This binding of a Notch ligand to a Notch receptor results in the formation of a Notch ligand-receptor complex, which undergoes several key cellular processes including cleavage by proteolytic enzymes, initiated by ADAM/TACE proteases at an extracellular site and results in the formation of Notch extracellular truncation (NEXT). Finally, the Notch intracellular domain (ICD) is formed by a very specific enzyme called γ-secretase, which helps to get the Notch ICD translocated from the cytoplasm into the nucleus where it binds to the DNA-binding protein ubiquitous transcription factor CSL [CBF1 (C-promoter binding factor 1), suppressor of hairless and Lag-1] and this orchestrates the activation of a CSL complex, which eventually remodels it from a transcriptional repressor into an activator, which initiates the activation of several downstream effectors (Shih and Wang, 2007).
Recently, one of the new approaches for curbing the Notch signalling pathway is to stop the entry of Notch ICD into the nucleus, which can be achieved by using aspartyl protease inhibitors or γ-secretase inhibitors like RO-4929097, which is in a phase II clinical trial for recurrent TNBC. Furthermore, the trio combination of RO-4929097, paclitaxel and carboplatin is in a phase I clinical trial for stage I and II TNBC (Olsauskas-Kuprys et al., 2013). Activation of the Notch signalling pathway necessitates the binding of a Notch ligand with a Notch receptor, which generates a Notch ligand-receptor complex and gets converted into NEXT and then into Notch ICD (intracellular domain) by γ-secretase. This enzyme cleaves Notch ICD into two proteins presenilin and nicastrin. Presenilin is catalytic in nature whereas nicastrin stimulates the maturation of genes. Finally, the Notch ICD gets translocated into the nucleus and binds with transcriptional activator CSL and induces the transcription of downstream targets including several genes such as VEGFR3, ER, Hes and Hey. These transcriptional targets include transcription factors (NF-κB2 and c-Myc), cell-cycle regulators (cyclin D1 and p21), and growth factor receptors (HER2) and regulators of angiogenesis and apoptosis. So the interruption of the Notch pathway can thus have noteworthy downstream effects on differentiation, angiogenesis and apoptosis, cell growth (Figure 1). Hence, targeting the Notch signalling pathway with γ-secretase inhibitors should be meticulously studied for further improving the treatment options for patients with TNBC.
Figure 1.

Activation of Notch signalling pathway: γ-secretase and aspartyl protease serve as primary targets for Notch-specific investigational drug design.
Hh signalling pathway
This is a highly conserved developmental pathway and serves as a key signalling cascade involved in the correct development of an embryo. This signalling pathway has been implicated in tumour initiation, progression, angiogenesis and metastasis across different malignancies. Hh signalling is known to regulate the self-renewal of stem cells in the nervous system and human embryonic skin (Palma and Ruiz i Altaba, 2004).This pathway has three different gene homologues like Sonic Hh (Shh), Desert Hh and Indian Hh but the most targeted gene homologue is the Shh pathway (Wismar et al., 2000).
The Hh pathway is an extremely organized and orchestrated cascade involving the inhibition of the twelve transmembrane protein called Pathed1 by binding a Hh ligand, subsequently it activates the seven transmembrane protein known as Smoothened (SMO; Taipale et al., 2002; Kasper et al., 2009). The activated protein SMO releases a five-zinc finger transcription factor Gli from a large protein complex and also actively participates in the nuclear translocation of Gli as well as in the transcription of target genes (Jiang and Hui, 2008). From the results of various researchers, it is concluded that Glia is one of the hallmarks in the activation of the Hh pathway (Cayuso et al., 2006). The activation of Glia is a crucial step in the Hh pathway (Figure 2), which is mediated by the zinc finger transcription factors like Gli1, Gli2 and Gli3, where Gli1 and Gli2 represent pathway activator genes and Gli3 acts as an inhibitor of this cascade (Kogerman et al., 1999; Bai et al., 2004; Kasper et al., 2006) but the precise mechanism of signal transduction of this cascade from SMO to the Gli proteins is not yet clear. However, data are rapidly accumulating suggesting that the primary cilium provides a good platform for dispatching the signals from the cell membrane to the nucleus (Oro, 2007). The primary cilia have been considered to serve as the processing sites for Gli transcription factors. The activated Glia targeted genes get transformed in the nucleus and participate in transcription that follows metastasis, apoptosis and angiogenesis by shifting the balance between transcriptional factors and proteins, which result in the development of TNBC. Likewise, this transcription elevates SNAIL protein and angiopoietin-1, 2, which are responsible for metastasis and angiogenesis respectively (Katoh and Katoh, 2008; Merchant and Matsui, 2010). However, SMO directly takes part in the activation of MYCN, which leads to proliferation by elevating the expressions of cyclin D and FOXM 1 that is itself a transcription factor associated with the development and progression of TNBC (Polkinghorn and Tarbell, 2007). Also, FOXM1 regulates the expression of cell cycle-related genes essential for coordinated DNA synthesis and mitosis (Teh et al., 2002; Schuller et al., 2007).
Figure 2.
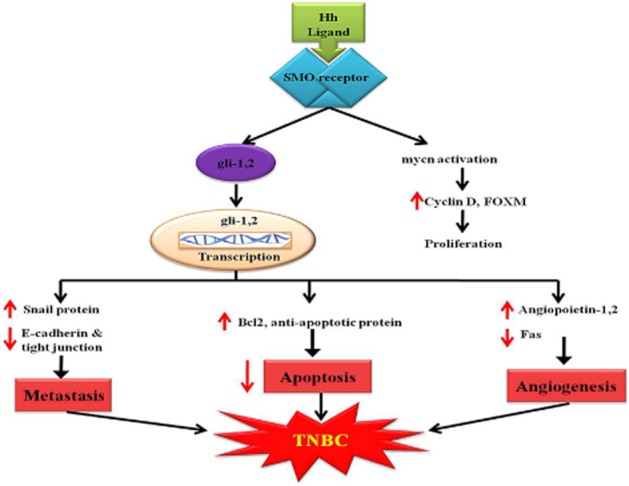
Hedgehog signalling pathway showing development of TNBC.
The possibility that the Hh signalling pathway could be pharmacologically inhibited was suggested from the identification of cyclopamine, a steroidal alkyloid isolated from Veratrum californicum and an SMO antagonist, with oral bioavailability of 33% and t1/2 of 4 h found in rodents, dogs and cynomolgus. Cyclopamine formulations and its derivatives with improved specificity, bioavailability and pharmacokinetics are in phase II trials for TNBC (Merchant and Matsui, 2010). Hence, SMO protein and Pathed1 protein can be used as further targets for effective treatment of TNBC.
Wnt/β-catenin pathway
The Wnt/β-catenin signalling pathway plays a major role in embryonic growth and can lead to tumour formation when aberrantly activated. There is much evidence indicating that this pathway is abnormally up-regulated in tumourigenesis of different types of cancers including TNBC (Barker and Clevers, 2006; Bayet-Robert et al., 2010). Recently, the Wnt receptor frizzled-7 (FZD7) and the Wnt co-receptor the low-density lipoprotein receptor-related protein-6 (LRP6) were found to be up-regulated in TNBC. Moreover, it has been established that transcriptional knockdown of FZD7 or LRP6 in TNBC cells restrains tumour growth in vivo (King et al., 2012). The characteristic of Wnt/b-catenin signalling (Figure 3) activation is the stabilization of cytosolic β-catenin, which goes into the nucleus to activate Wnt-targeted genes by binding transcription factors of the T cell factor/lymphoid enhancing factor (TCF/LEF) family (Lu et al., 2011b; King et al., 2012).
Figure 3.

Delineation of different pathways leading to the development of TNBC, which can be addressed as targets of TNBC.
In the absence of Wnt ligands, β-catenin levels are effectively synchronized by a supramolecular complex containing adenomatous polyposis coli (APC), axin and glycogen synthase kinase 3β (GSK3β). CK1 and GSK3β sequentially phosphorylate the amino terminal region of β-catenin. Phosphorylated β-catenin turns into multi-ubiquitinated (Ub) and broken by the 26S proteasome. Furthermore, the binding of Wnt to its receptors on the cell surface leads to inhibition of the action of this complex (Lu et al., 2011b; King et al., 2012). Several Wnt/β-catenin target genes have been identified, including those that control cell proliferation and apoptosis, thus mediating cancer initiation and progression (Barker and Clevers, 2006; Bayet-Robert et al., 2010). In addition to this, salinomycin and nigericin, act as inhibitors of the Wnt/β-catenin signalling pathway by inducing degradation of LRP6, and are selective breast cancer stem cell killers (Lu et al., 2011a). Silinomycin, is a well-known anti-coccidial drug whose pharmacokinetic properties as an anticancer agent are under investigation in a phase I/II trial for TNBC (Naujokat and Steinhart, 2012). Therefore, the Wnt/β-catenin signalling pathway especially the Wnt receptors, which are present on the cell surface, is a promising therapeutic target for the treatment of TNBC.
PARP inhibitors
PARP1, a poly ADP-ribose polymerase, is a member of a family of enzymes that remove damaged and (or) incorrect DNA sequences by different excision repair pathways, involving base excision repair, nucleotide excision repair and mismatch repair by filling the ensuing gap using the complementary DNS strand as a template (Veuger et al., 2004). This repair of single stranded DNA breaks is achieved by synthesizing poly ADP-ribose (Figure 3). However, if DNA breaks occur in the absence of PARP1, during DNA replication, the replication fork stalls resulting in the accumulation of double-stranded DNA (dsDNA) breaks that are repaired (Figure 3) via homologous recombination (HR), which is considered to be an error-free repair mechanism that involves BRAC1 and BRAC2 proteins (Waldman and Waldman, 1991; Schultz et al., 2003; Godon et al., 2008). Therefore, a cancer that involves a mutation in these BRAC1 and BRAC2 genes (BRCAness) can be effectively treated with PARP1 inhibitors, which may represent very efficient therapies for TNBC because of the high sensitivity of these tumours to the inhibitor and more importantly, it is devoid of harmful effects on the remaining healthy cells. This contrasts with conventional chemotherapies, which are highly toxic to all cells and can provoke DNA damage in healthy cells that may further lead to secondary cancer generation (Bryant et al., 2005; Farmer et al., 2005). At present various PARP1 inhibitors are under development in different phases of clinical trial but, as yet, no major breakthrough has been achieved.
EGFR-targeted therapies
EGFR is a cell surface transmembrane tyrosine kinase receptor, encoded by the cell erythroblastosis virus oncogene B1(C-erbB1) and is a member of the HER/erythroblastosis virus oncogene B (ErbB) family (Herbst, 2004; Zhang et al., 2007). It was discovered by Stanley Cohen of Vanderbilt University who won the Nobel Prize for medicine with Rita Levi-Montalcini for discovering these growth factors in 1986. The EGFR pathway generates Akt (PKB) and MAPK causing drug resistance. In addition, it regulates cell proliferation, differentiation, invasion, angiogenesis and apoptosis through various signalling pathways (Figure 3) and serves as a poor prognostic factor in cancer (Gauthier et al., 2003; Tabach et al., 2005). EGFRs are overexpressed in various types of malignancy including TNBC (Sarrio et al., 2008; Gluz et al., 2009). Gene expression profiling and immunohistochemical studies have demonstrated that 40 to 60% of TNBCs are associated with an increased expression of EGFRs and the EGFR gene was up-regulated in 18% of this subgroup. However, EGFR mutation was infrequent in TNBC. In October 2010, for the first time, researchers revealed that the EGFR could be a therapeutic target for women with TNBC (Ueno and Zhang, 2011). Gefitinib, an approved EGFR inhibitor with oral bioavailability of 59% and a half-life of 6–49 h, is currently undergoing a phase II clinical trial for TNBC and EGFR-positive metastatic breast cancer (http://clinicaltrials.gov/ct2/show/NCT01732276). Many combinations, such as lapatinib with everolimus (mTOR inhibitor: agent that targets a protein in the cancerours cells), are in phase II clinical trial for advanced TNBC patients (http://clinicaltrials.gov/show/NCT01272141). Furthermore, cetuxumab in combination with ixabepilone (http://clinicaltrials.gov/ct2/show/NCT01097642), temsirolimus plus neratinib (http://clinicaltrials.gov/ct2/show/NCT01111825) and a trio combination of panitumumab, gemcitabine and carboplatin are undergoing phase II clinical trials for TNBC (http://clinicaltrials.gov/ct2/show/NCT00894504) and stage 4 breast cancers (Ferraro et al., 2013).
mTOR inhibitors
The mTOR, also known as FK506 binding protein 12-rapamycin associated protein 1 (FRAP1) is a serine/threonine protein kinase type of protein encoded by FRAP1 gene in humans (Moore et al., 1996). mTOR belongs to the PI3K-related protein family, which controls cell proliferation, cell growth, cell motility, cell survival, protein synthesis and transcription (Hay and Sonenberg, 2004; Beevers et al., 2006). mTOR combines the input from upstream pathways, involving insulin growth factors (IGF-1 and IGF-2) and amino acids (Hay and Sonenberg, 2004). The mTOR pathway is deregulated in various human diseases including breast cancer.
mTOR is a catalytic subunit consisting of two distinct complexes – mTORC1 and mTORC2 (Wullschleger et al., 2006) that induce S-phase kinase association protein causing protein synthesis, proliferation, growth, metastasis and angiogenesis (Figure 3). mTOR complex 1 (mTORC1) is made up of mTOR, Raptor, GβL (mLST8), and Deptor and is to some extent inhibited by rapamycin whereas, mTOR complex 2 (mTORC2) is composed of mTOR, Rictor, GβL, Sin1, PRR5/Protor-1 and Deptor (Figure 4) and supports cellular survival by activating Akt. mTORC1 integrates several signals indicating the presence of growth factors, nutrients or energy to uphold either cellular growth when conditions are favourable or catabolic processes in stress or when conditions are unfavourable. The signal from growth factors and hormones (e.g. insulin) is transferred to mTORC1 via Akt, which causes the inactivation of TSC2 and prevents the inhibition of mTORC1. In contrast, low ATP levels cause AMPK-dependent activation of TSC2 and phosphorylation of raptor to reduce mTORC1 signalling. Amino acid availability is signalled to mTORC1 via a pathway that includes the Rag and Ragulator (LAMTOR1-3) proteins. In its active state, mTORC1 has numerous downstream biological effects involving the translation of mRNA through the phosphorylation of downstream targets (p70 S6 kinase and 4E-BP1), ribosome biogenesis, suppression of autophagy (Atg13, ULK1) and activation of transcription causing mitochondrial metabolism or adipogenesis. mTORC2 also controls cytoskeletal dynamics by activating PKCα and maintains ion transport and growth via SGK1.
Figure 4.
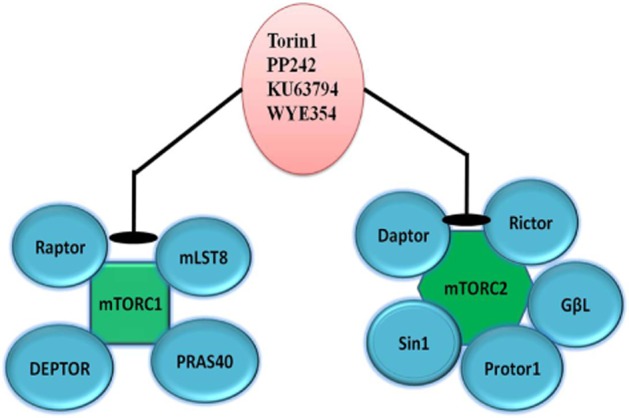
Components of mTOR complex, which can be targeted for treatment of TNBC.
mTOR inhibitors are being tried in combinations for effective treatment of TNBC. As mentioned earlier, everolumus with lapatinib is in phase II of a clinical trial. Also, the triple combination of temsirolimus, cisplatin, erlotinib is undergoing clinical trials (http://clinicaltrials.gov/ct2/show/NCT00998036).
TGF-β signalling pathway inhibitors
The TGF-β signalling pathway is involved in various cellular processes in embryonic cell growth, cell differentiation, cellular homeostasis, apoptosis and also some cellular functions in adult organs. TGF-β1 is member of the TGF-β superfamily of cytokines, which is encoded by the TGF-β1 gene (Ghadami et al., 2000; Vaughn et al., 2000) and it was first discovered in human platelets as a protein having a 25 KDa molecular weight with a key role in wound healing (Assoian et al., 1983). TGF-β1 is also involved in regulating the immune system (Letterio and Roberts, 1998). TGF-β1 has been shown to inhibit the secretion as well as activities of various cytokines like IFN-γ, TNF-α and IL-2 (Wahl et al., 1988; Tiemessen et al., 2003). However, TGF-β1 has exactly opposite actions on cells of myeloid origin where it increases secretion and the expression of monocytic cytokines such as IL-1α, IL-1β and TNF-α (Wahl et al., 2006). Recent research has proposed that TGF-β1 plays a vital role in breast cancer stem cells as these cells have been shown to overexpress TGF-β1 and the TGF-β receptor 1 (TGFBR1; Bhola et al., 2013).
Very recently, Bhola and his co-worker for the first time found TGF-β inhibitors have an ability to block the expansion of chemotherapy-resistant tumour initiating cells (TIC) in vivo (Bhola et al., 2013). This may be the basis for upcoming clinical studies and their function in combinational chemotherapy for patients suffering from TNBC should be evaluated. Furthermore, an epithelial-to-mesenchymal transition (EMT) is potentially induced by TGF-β within mammary cells and this transformation results in the acquisition of tumour-like properties (Mani et al., 2008). In fact, EMT can be reversed by using TGFBR1/2 inhibitors and inducing a mesenchymal-to-epithelial differentiation inside mammary epithelial cells (Shipitsin et al., 2007; Bhola et al., 2013). TGF-β ligands are frequently augmented in the TNBC tumour microenvironment, which may be formed by tumour cells or by tumour-associated immune and stromal cells (Gilbert et al., 1997; Wahl et al., 2006). Also the TGF-β pathway generates SMAD2/3 and SMAD4 and finally Inh causing similar effects, such as protein synthesis, proliferation, growth, metastasis and angiogenesis, as that of earlier pathways (Figure 3). Thus it is possible that the TGF-β pathway is involved in the development of breast carcinomas. TGF-β inhibitors are now been developed as anti-metastatic therapies in patients with cancer.
CSPG4 protein
The CSPG4, also known as non-glial antigen or high molecular weight melanoma-associated antigen or melanoma chondroitin sulfate proteoglycan, is a cell surface proteoglycan, which has been recently found to be expressed not only by melanoma cells, but also by different types of human carcinoma and sarcoma including basal breast carcinoma (Wang et al., 2010b). This is a major breakthrough, as inhibiting it has been found to be therapeutically effective for immunotherapy of breast cancer (Campoli et al., 2010). CSPG4 causes the spreading of protein on the endothelial basement membrane and stabilizes the cell substratum interaction causing similar effects, such as protein synthesis, proliferation, growth, metastasis and angiogenesis, as those that occur in TNBC (Figure 3). The anti-tumour effects of a CSPG4-specific mAb probably reflect the blocking of essential migratory, mitogenic and survival signalling pathways in tumour cells (Wang et al., 2011). Recently, Ferrone et al. discovered CSPG4 as new target for the antibody-based immunotherapy of TNBC (Wang et al., 2010a). In this study, they found, for the first time, that the expression of membrane-bound CSPG4 varies considerably in BC subtypes, being significantly higher in TNBC and basal-like breast cancer than in other luminal subtypes. Moreover, they observed inhibition of tumour growth with no migration when TNBC cells were targeted with a CSPG4-specific monoclonal antibody. Furthermore, the findings confirmed the potential use of a pleiotropic CSPG4-specific monoclonal antibody as not only a diagnostic biomarker but also as a promising therapeutic target, which has resulted in inhibition of the activation of numerous signalling pathways crucial for the malignant development of TNBC (Wang et al., 2010a; Cattaruzza et al., 2013).
Conclusion
TNBC is growing rapidly in countries such as America and Africa, so to avert this condition, the availability of a proper treatment option is needed. Before accessing proper treatment, we must have proper targets that may be any receptor, protein or an enzyme. The deregulation of various signalling pathways (Notch, Hh, Wnt/β-catenin and TGF-β), EGFR, CSPG4 protein have been confirmed in patients suffering from TNBC, and have recently come under development as new treatment options. The use of conventional therapeutics in order to maximize the therapeutic effects and simultaneously to minimize the adverse effects in subjects suffering from TNBC is essential in order to show the tumour growth inhibition induced by curbing these signalling pathways. In spite of the increasing challenges, it is conjectured that in the near future, the enormous efforts to identify new precise inhibitors of these pathways will be rewarded and new clinical trials instigated for testing these novel, potential anti-cancer drugs.
A lot of research to discover new targets that are expressed in TNBC patients and can be restricted or minimized to reduce the risk of further developments is ongoing. We have attempted to assemble the current targets of TNBC that have been proved to be promising foci for the treatment of TNBC. This review will definitely aid further research in identifying a suitable treatment for TNBC.
Acknowledgments
The first author expresses his sincere thanks to the National Institute of Pharmaceutical Education and Research (NIPER), Guwahati for providing financial assistance to carry out this work. The funding source had no involvement in the study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the article for publication. We sincerely acknowledge Nityanand Bolshette and Krishan kumar Thakur for their helpful discussions and technical assistance.
Glossary
- CSL
CBF1[C-promoter binding factor 1], suppressor of hairless and Lag-1
- CSPG4
chondroitin sulfate proteoglycan 4
- Dll
Delta-like
- FZD7
Frizzled-7
- Hh
Hedgehog
- JAG
jagged
- LRP6
low-density lipoprotein receptor-related protein-6
- mTOR
mammalian target of rapamycin
- NEXT
Notch extracellular truncation
- Notch ICD
Notch intracellular domain
- Shh
Sonic Hh
- SMO
Smoothened
Conflict of interest
The authors declare that they have no competing interests or conflict of interests.
References
- Al-Hussaini H, Subramanyam D, Reedijk M, Sridhar SS. Notch signaling pathway as a therapeutic target in breast cancer. Mol Cancer Ther. 2011;10:9–15. doi: 10.1158/1535-7163.MCT-10-0677. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: catalytic receptors. Br J Pharmacol. 2013b;170:1676–1705. doi: 10.1111/bph.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: enzymes. Br J Pharmacol. 2013c;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assoian RK, Komoriya A, Meyers CA, Miller DM, Sporn MB. Transforming growth factor-beta in human platelets. Identification of a major storage site, purification, and characterization. J Biol Chem. 1983;258:7155–7160. [PubMed] [Google Scholar]
- Bai CB, Stephen D, Joyner AL. All mouse ventral spinal cord patterning by hedgehog is Gli dependent and involves an activator function of Gli3. Dev Cell. 2004;6:103–115. doi: 10.1016/s1534-5807(03)00394-0. [DOI] [PubMed] [Google Scholar]
- Barker N, Clevers H. Mining the Wnt pathway for cancer therapeutics. Nat Rev Drug Discov. 2006;5:997–1014. doi: 10.1038/nrd2154. [DOI] [PubMed] [Google Scholar]
- Bayet-Robert M, Kwiatkowski F, Leheurteur M, Gachon F, Planchat E, Abrial C, et al. Phase I dose escalation trial of docetaxel plus curcumin in patients with advanced and metastatic breast cancer. Cancer Biol Ther. 2010;9:8–14. doi: 10.4161/cbt.9.1.10392. [DOI] [PubMed] [Google Scholar]
- Beevers CS, Li F, Liu L, Huang S. Curcumin inhibits the mammalian target of rapamycin-mediated signaling pathways in cancer cells. Int J Cancer. 2006;119:757–764. doi: 10.1002/ijc.21932. [DOI] [PubMed] [Google Scholar]
- Bhola NE, Balko JM, Dugger TC, Kuba MG, Sanchez V, Sanders M, et al. TGF-beta inhibition enhances chemotherapy action against triple-negative breast cancer. J Clin Invest. 2013;123:1348–1358. doi: 10.1172/JCI65416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- Campoli M, Ferrone S, Wang X. Functional and clinical relevance of chondroitin sulfate proteoglycan 4. Adv Cancer Res. 2010;109:73–121. doi: 10.1016/B978-0-12-380890-5.00003-X. [DOI] [PubMed] [Google Scholar]
- Cattaruzza S, Nicolosi PA, Braghetta P, Pazzaglia L, Benassi MS, Picci P, et al. NG2/CSPG4-collagen type VI interplays putatively involved in the microenvironmental control of tumour engraftment and local expansion. J Mol Cell Biol. 2013;5:176–193. doi: 10.1093/jmcb/mjt010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayuso J, Ulloa F, Cox B, Briscoe J, Marti E. The Sonic hedgehog pathway independently controls the patterning, proliferation and survival of neuroepithelial cells by regulating Gli activity. Development. 2006;133:517–528. doi: 10.1242/dev.02228. [DOI] [PubMed] [Google Scholar]
- Chacon RD, Costanzo MV. Triple-negative breast cancer. Breast Cancer Res. 2010;12(Suppl 2):S3. doi: 10.1186/bcr2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curigliano G, Goldhirsch A. The triple-negative subtype: new ideas for the poorest prognosis breast cancer. J Natl Cancer Inst Monogr. 2011;2011:108–110. doi: 10.1093/jncimonographs/lgr038. [DOI] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- Ferraro DA, Gaborit N, Maron R, Cohen-Dvashi H, Porat Z, Pareja F, et al. Inhibition of triple-negative breast cancer models by combinations of antibodies to EGFR. Proc Natl Acad Sci U S A. 2013;110:1815–1820. doi: 10.1073/pnas.1220763110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier ML, Torretto C, Ly J, Francescutti V, O’Day DH. Protein kinase C alpha negatively regulates cell spreading and motility in MDA-MB-231 human breast cancer cells downstream of epidermal growth factor receptor. Biochem Biophys Res Commun. 2003;307:839–846. doi: 10.1016/s0006-291x(03)01273-7. [DOI] [PubMed] [Google Scholar]
- Ghadami M, Makita Y, Yoshida K, Nishimura G, Fukushima Y, Wakui K, et al. Genetic mapping of the Camurati-Engelmann disease locus to chromosome 19q13.1-q13.3. Am J Hum Genet. 2000;66:143–147. doi: 10.1086/302728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert KM, Thoman M, Bauche K, Pham T, Weigle WO. Transforming growth factor-beta 1 induces antigen-specific unresponsiveness in naive T cells. Immunol Invest. 1997;26:459–472. doi: 10.3109/08820139709022702. [DOI] [PubMed] [Google Scholar]
- Gluz O, Liedtke C, Gottschalk N, Pusztai L, Nitz U, Harbeck N. Triple-negative breast cancer – current status and future directions. Ann Oncol. 2009;20:1913–1927. doi: 10.1093/annonc/mdp492. [DOI] [PubMed] [Google Scholar]
- Godon C, Cordelieres FP, Biard D, Giocanti N, Megnin-Chanet F, Hall J, et al. PARP inhibition versus PARP-1 silencing: different outcomes in terms of single-strand break repair and radiation susceptibility. Nucleic Acids Res. 2008;36:4454–4464. doi: 10.1093/nar/gkn403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- Herbst RS. Review of epidermal growth factor receptor biology. Int J Radiat Oncol Biol Phys. 2004;59(2 Suppl):21–26. doi: 10.1016/j.ijrobp.2003.11.041. [DOI] [PubMed] [Google Scholar]
- Ismail-Khan R, Bui MM. A review of triple-negative breast cancer. Cancer Control. 2008;17:173–176. doi: 10.1177/107327481001700305. [DOI] [PubMed] [Google Scholar]
- Jiang J, Hui CC. Hedgehog signaling in development and cancer. Dev Cell. 2008;15:801–812. doi: 10.1016/j.devcel.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasper M, Regl G, Frischauf AM, Aberger F. GLI transcription factors: mediators of oncogenic Hedgehog signalling. Eur J Cancer. 2006;42:437–445. doi: 10.1016/j.ejca.2005.08.039. [DOI] [PubMed] [Google Scholar]
- Kasper M, Jaks V, Fiaschi M, Toftgard R. Hedgehog signalling in breast cancer. Carcinogenesis. 2009;30:903–911. doi: 10.1093/carcin/bgp048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh Y, Katoh M. Hedgehog signaling, epithelial-to-mesenchymal transition and miRNA (review) Int J Mol Med. 2008;22:271–275. [PubMed] [Google Scholar]
- King TD, Suto MJ, Li Y. The Wnt/beta-catenin signaling pathway: a potential therapeutic target in the treatment of triple negative breast cancer. J Cell Biochem. 2012;113:13–18. doi: 10.1002/jcb.23350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogerman P, Grimm T, Kogerman L, Krause D, Unden AB, Sandstedt B, et al. Mammalian suppressor-of-fused modulates nuclear-cytoplasmic shuttling of Gli-1. Nat Cell Biol. 1999;1:312–319. doi: 10.1038/13031. [DOI] [PubMed] [Google Scholar]
- Kumar P, Bolshette NB, Jamdade VS, Mundhe NA, Thakur KK, Saikia KK, et al. Breast cancer status in India: an overview. Biomed Prev Nutr. 2013;3:177–183. [Google Scholar]
- Letterio JJ, Roberts AB. Regulation of immune responses by TGF-beta. Annu Rev Immunol. 1998;16:137–161. doi: 10.1146/annurev.immunol.16.1.137. [DOI] [PubMed] [Google Scholar]
- Lu D, Choi MY, Yu J, Castro JE, Kipps TJ, Carson DA. Salinomycin inhibits Wnt signaling and selectively induces apoptosis in chronic lymphocytic leukemia cells. Proc Natl Acad Sci U S A. 2011a;108:13253–13257. doi: 10.1073/pnas.1110431108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Lin C, Roberts MJ, Waud WR, Piazza GA, Li Y. Niclosamide suppresses cancer cell growth by inducing Wnt co-receptor LRP6 degradation and inhibiting the Wnt/beta-catenin pathway. PLoS ONE. 2011b;6:e29290. doi: 10.1371/journal.pone.0029290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merchant AA, Matsui W. Targeting Hedgehog – a cancer stem cell pathway. Clin Cancer Res. 2010;16:3130–3140. doi: 10.1158/1078-0432.CCR-09-2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore PA, Rosen CA, Carter KC. Assignment of the human FKBP12-rapamycin-associated protein (FRAP) gene to chromosome 1p36 by fluorescence in situ hybridization. Genomics. 1996;33:331–332. doi: 10.1006/geno.1996.0206. [DOI] [PubMed] [Google Scholar]
- Naujokat C, Steinhart R. Salinomycin as a drug for targeting human cancer stem cells. J Biomed Biotechnol. 2012;2012:950658. doi: 10.1155/2012/950658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsauskas-Kuprys R, Zlobin A, Osipo C. Gamma secretase inhibitors of Notch signaling. Onco Targets Ther. 2013;6:943–955. doi: 10.2147/OTT.S33766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oro AE. The primary cilia, a ‘Rab-id’ transit system for hedgehog signaling. Curr Opin Cell Biol. 2007;19:691–696. doi: 10.1016/j.ceb.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otvos L, Jr, Surmacz E. Targeting the leptin receptor: a potential new mode of treatment for breast cancer. Expert Rev Anticancer Ther. 2011;11:1147–1150. doi: 10.1586/era.11.109. [DOI] [PubMed] [Google Scholar]
- Palma V, Ruiz i Altaba A. Hedgehog-GLI signaling regulates the behavior of cells with stem cell properties in the developing neocortex. Development. 2004;131:337–345. doi: 10.1242/dev.00930. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penault-Llorca F, Viale G. Pathological and molecular diagnosis of triple-negative breast cancer: a clinical perspective. Ann Oncol. 2012;23(Suppl 6):vi19–vi22. doi: 10.1093/annonc/mds190. [DOI] [PubMed] [Google Scholar]
- Polkinghorn WR, Tarbell NJ. Medulloblastoma: tumorigenesis, current clinical paradigm, and efforts to improve risk stratification. Nat Clin Pract Oncol. 2007;4:295–304. doi: 10.1038/ncponc0794. [DOI] [PubMed] [Google Scholar]
- Reedijk M, Odorcic S, Chang L, Zhang H, Miller N, McCready DR, et al. High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res. 2005;65:8530–8537. doi: 10.1158/0008-5472.CAN-05-1069. [DOI] [PubMed] [Google Scholar]
- de Ruijter TC, Veeck J, de Hoon JP, van Engeland M, Tjan-Heijnen VC. Characteristics of triple-negative breast cancer. J Cancer Res Clin Oncol. 2011;137:183–192. doi: 10.1007/s00432-010-0957-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrio D, Rodriguez-Pinilla SM, Hardisson D, Cano A, Moreno-Bueno G, Palacios J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 2008;68:989–997. doi: 10.1158/0008-5472.CAN-07-2017. [DOI] [PubMed] [Google Scholar]
- Schuller U, Zhao Q, Godinho SA, Heine VM, Medema RH, Pellman D, et al. Forkhead transcription factor FoxM1 regulates mitotic entry and prevents spindle defects in cerebellar granule neuron precursors. Mol Cell Biol. 2007;27:8259–8270. doi: 10.1128/MCB.00707-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz N, Lopez E, Saleh-Gohari N, Helleday T. Poly(ADP-ribose) polymerase (PARP-1) has a controlling role in homologous recombination. Nucleic Acids Res. 2003;31:4959–4964. doi: 10.1093/nar/gkg703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih M, Wang TL. Notch signaling, gamma-secretase inhibitors, and cancer therapy. Cancer Res. 2007;67:1879–1882. doi: 10.1158/0008-5472.CAN-06-3958. [DOI] [PubMed] [Google Scholar]
- Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J, et al. Molecular definition of breast tumor heterogeneity. Cancer Cell. 2007;11:259–273. doi: 10.1016/j.ccr.2007.01.013. [DOI] [PubMed] [Google Scholar]
- Speiser J, Foreman K, Drinka E, Godellas C, Perez C, Salhadar A, et al. Notch-1 and Notch-4 biomarker expression in triple-negative breast cancer. Int J Surg Pathol. 2012;20:139–145. doi: 10.1177/1066896911427035. [DOI] [PubMed] [Google Scholar]
- Tabach Y, Milyavsky M, Shats I, Brosh R, Zuk O, Yitzhaky A, et al. The promoters of human cell cycle genes integrate signals from two tumor suppressive pathways during cellular transformation. Mol Syst Biol. 2005;1:2005.0022. doi: 10.1038/msb4100030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale J, Cooper MK, Maiti T, Beachy PA. Patched acts catalytically to suppress the activity of Smoothened. Nature. 2002;418:892–897. doi: 10.1038/nature00989. [DOI] [PubMed] [Google Scholar]
- Teh MT, Wong ST, Neill GW, Ghali LR, Philpott MP, Quinn AG. FOXM1 is a downstream target of Gli1 in basal cell carcinomas. Cancer Res. 2002;62:4773–4780. [PubMed] [Google Scholar]
- Tiemessen MM, Kunzmann S, Schmidt-Weber CB, Garssen J, Bruijnzeel-Koomen CA, Knol EF, et al. Transforming growth factor-beta inhibits human antigen-specific CD4+ T cell proliferation without modulating the cytokine response. Int Immunol. 2003;15:1495–1504. doi: 10.1093/intimm/dxg147. [DOI] [PubMed] [Google Scholar]
- Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, et al. Oral poly (ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof of concept trial. Lancet. 2010;376:235–244. doi: 10.1016/S0140-6736(10)60892-6. [DOI] [PubMed] [Google Scholar]
- Ueno NT, Zhang D. Targeting EGFR in triple negative breast cancer. J Cancer. 2011;2:324–328. doi: 10.7150/jca.2.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughn SP, Broussard S, Hall CR, Scott A, Blanton SH, Milunsky JM, et al. Confirmation of the mapping of the Camurati-Englemann locus to 19q13. 2 and refinement to a 3.2-cM region. Genomics. 2000;66:119–121. doi: 10.1006/geno.2000.6192. [DOI] [PubMed] [Google Scholar]
- Veuger SJ, Curtin NJ, Smith GC, Durkacz BW. Effects of novel inhibitors of poly(ADP-ribose) polymerase-1 and the DNA-dependent protein kinase on enzyme activities and DNA repair. Oncogene. 2004;23:7322–7329. doi: 10.1038/sj.onc.1207984. [DOI] [PubMed] [Google Scholar]
- Wahl SM, Hunt DA, Wong HL, Dougherty S, McCartney-Francis N, Wahl LM, et al. Transforming growth factor-beta is a potent immunosuppressive agent that inhibits IL-1-dependent lymphocyte proliferation. J Immunol. 1988;140:3026–3032. [PubMed] [Google Scholar]
- Wahl SM, Wen J, Moutsopoulos N. TGF-beta: a mobile purveyor of immune privilege. Immunol Rev. 2006;213:213–227. doi: 10.1111/j.1600-065X.2006.00437.x. [DOI] [PubMed] [Google Scholar]
- Waldman AS, Waldman BC. Stimulation of intrachromosomal homologous recombination in mammalian cells by an inhibitor of poly(ADP-ribosylation) Nucleic Acids Res. 1991;19:5943–5947. doi: 10.1093/nar/19.21.5943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Osada T, Wang Y, Yu L, Sakakura K, Katayama A, et al. CSPG4 protein as a new target for the antibody-based immunotherapy of triple-negative breast cancer. J Natl Cancer Inst. 2010a;102:1496–1512. doi: 10.1093/jnci/djq343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang Y, Yu L, Sakakura K, Visus C, Schwab JH, et al. CSPG4 in cancer: multiple roles. Curr Mol Med. 2010b;10:419–429. doi: 10.2174/156652410791316977. [DOI] [PubMed] [Google Scholar]
- Wang X, Katayama A, Wang Y, Yu L, Favoino E, Sakakura K, et al. Functional characterization of an scFv-Fc antibody that immunotherapeutically targets the common cancer cell surface proteoglycan CSPG4. Cancer Res. 2011;71:7410–7422. doi: 10.1158/0008-5472.CAN-10-1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wismar J, Habtemichael N, Warren JT, Dai JD, Gilbert LI, Gateff E. The mutation without children(rgl) causes ecdysteroid deficiency in third-instar larvae of Drosophila melanogaster. Dev Biol. 2000;226:1–17. doi: 10.1006/dbio.2000.9811. [DOI] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Zhang H, Berezov A, Wang Q, Zhang G, Drebin J, Murali R, et al. ErbB receptors: from oncogenes to targeted cancer therapies. J Clin Invest. 2007;117:2051–2058. doi: 10.1172/JCI32278. [DOI] [PMC free article] [PubMed] [Google Scholar]