

Abstract
Background and Purpose
cAMP plays an important role in the transduction of signalling pathways involved in neuroprotection and immune regulation. Control of the levels of this nucleotide by inhibition of cAMP-specific PDEs such as PDE7 may affect the pathological processes of neuroinflammatory diseases like multiple sclerosis (MS). In the present study, we evaluated the therapeutic potential of the selective PDE7 inhibitor, TC3.6, in a model of primary progressive multiple sclerosis (PPMS), a rare and severe variant of MS.
Experimental Approach
Theiler’s murine encephalomyelitis virus-induced demyelinated disease (TMEV-IDD) is one of the models used to validate the therapeutic efficacy of new drugs in MS. As recent studies have analysed the effect of PDE7 inhibitors in the EAE model of MS, here the TMEV-IDD model was used to test their efficacy in a progressive variant of MS. Mice were subjected to two protocols of TC3.6 administration: on the pre-symptomatic phase and once the disease was established.
Key Results
Treatment with TC3.6 ameliorated the disease course and improved motor deficits of infected mice. This was associated with down-regulation of microglial activation and reduced cellular infiltrates. Decreased expression of pro-inflammatory mediators such as COX-2 and the cytokines, IL-1β, TNF-α, IFN-γ and IL-6 in the spinal cord of TMEV-infected mice was also observed after TC3.6 administration.
Conclusion
These findings support the importance of PDE7 inhibitors, and specifically TC3.6, as a novel class of agents with therapeutic potential for PPMS. Preclinical studies are needed to determine whether their effects translate into durable clinical benefits.
Tables of Links
| TARGETS | |
|---|---|
| COX-2 | PDE7A |
| PDE3 | PDE7B |
| PDE4 |
| LIGANDS | |||
|---|---|---|---|
| BRL50481 | Glutamate | IL-2 | Nitric oxide (NO) |
| cAMP | IFN-β | IL-6 | Rp-cAMPs |
| cGMP | IFN-γ | IL-12 | TNF-α |
| H-89 | IL-1β | Rolipram |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Multiple sclerosis (MS) is a chronic inflammatory disease of the CNS associated with demyelination and axonal damage that impair the normal neurotransmission leading to sensory deficits and deteriorated motor coordination. While the aetiology of MS remains elusive, myelin destruction may be a direct consequence of the autoimmune response against myelin epitopes, or may be a neurodegenerative disease where oligodendrocytes get progressively impaired and ultimately die (Ludwin, 2006; Kipp et al., 2012).
Therapeutic strategies for the treatment of MS mainly include immune-based therapies that may have severe drawbacks such as lack of efficacy in long term treatment (Curtin and Hartung, 2014). Moreover, clinical trials of such agents in primary progressive multiple sclerosis (PPMS) have failed and there is limited evidence of their efficacy in secondary progressive disease (Comi, 2013). Today, there is an urgent need to discover new neuroprotective treatments able to modify the course of MS, both in the primary and secondary progressive forms, as this is a major cause of neurological disability in young adults (Karussis, 2014).
The best animal models used to validate the therapeutic effect of new drugs in MS are experimental allergic encephalomyelitis (EAE) and Theiler’s murine encephalomyelitis virus-induced demyelinating disease (TMEV-IDD; McCarthy et al., 2012). Specifically, IFN-β was one of the first molecules investigated in EAE models, which gave satisfactory results in terms of its therapeutic activity in patients. IFN-β short treatment was also effective in TMEV-IDD modulating the expression of the major histocompatibility complex class II (MHC-II) and hence the action of CD8+ T lymphocytes, but problems emerged when it was administered as a long-term treatment (Njenga et al., 2000). TMEV-IDD-infected mice exhibit several clinical deficits such as progressive impaired motor coordination, incontinence, and paralysis associated with axonal loss and electrophysiological abnormalities (Tsunoda and Fujinami, 2010; Sato et al., 2011). This mice model closely resembles the primary and progressive clinical form of MS (PPMS; Mecha et al., 2013), being an excellent pharmacological tool to discover new treatments with innovative mechanisms of action for this rare and severe variant of MS.
PDEs are a family of enzymes that hydrolyse the nucleotides cAMP and cGMP and are classified into 11 groups on the basis of their sequence homology, cellular distribution, substrate specificity and sensitivity to different PDE inhibitors (Bender and Beavo, 2006). cAMP is a second messenger involved in a variety of cellular responses including inflammation and inmmunomodulation, and the intracellular levels of this nucleotide should be closely controlled. Specifically, an increase in intracellular levels of cAMP is usually accompanied by an inhibition of certain functions of different cell types involved in the immune response and inflammation (Tasken and Stokka, 2006; Brudvik and Tasken, 2012). Thus, cAMP-specific PDEs inhibitors, such PDE4 and PDE7 inhibitors, have recently emerged as promising therapeutic agents (Martinez and Gil, 2014; Maurice et al., 2014). Moreover, with regard to MS, the cAMP cascade regulates myelin phagocytosis in microglia and macrophages (Makranz et al., 2006) and is also involved in myelin formation (Malone et al., 2013).
The enzymes most studied in relation to their regulatory effects on the immune system and the CNS are the cAMP-specific PDE4 and PDE7. In immune cells, inhibition of PDE4 limits the production and release of pro-inflammatory cytokines from activated peripheral mononuclear cells, including but not limited to, TNF-α, IL-2, IL-12 and IFN-γ (Kaminuma et al., 1998; Muise et al., 2002; Claveau et al., 2004). In addition, the prototypical PDE4 inhibitor, rolipram, has been demonstrated to be a potent anti-inflammatory agent in the EAE model of MS (Sommer et al., 1995; Sanchez et al., 2005; Paintlia et al., 2009; Gonzalez-Garcia et al., 2013) but its secondary emetic effects in humans have hindered its clinical development (Robichaud et al., 2001). An alternative approach is to inhibit other cAMP-specific PDE isoenzymes expressed in immune and neural cells such as PDE7 (Lee et al., 2002). PDE7 participates in the modulation of critical immune processes such as T-cell proliferation (Goto et al., 2009) and, PDE7 isoenzymes, PDE7A and PDE7B, have been found in the CNS of rodents (Miro et al., 2001; Morales-Garcia et al., 2011; Johansson et al., 2012). Moreover, recent studies showed that PDE7 inhibitors could play an anti-inflammatory and neuroprotective role in cellular and animal models of Parkinson’s disease (Morales-Garcia et al., 2011), spinal cord injury (Paterniti et al., 2011), Alzheimer’s disease (Perez-Gonzalez et al., 2013) and stroke (Redondo et al., 2012c). In addition, different chemically diverse PDE7 inhibitors have shown efficacy in the EAE model of MS (Redondo et al., 2012a,b,) without inducing emesis (Garcia et al., 2014) pointing to a new era of innovative drugs. In this regard, the therapeutical potential for MS treatment of the thioxoquinazoline derivative TC3.6 synthesized in our laboratory has been recently reported. TC3.6 is able to prevent EAE, reduce IL-17 levels, prevent infiltration in the CNS and increase the expression of the T regulator cell marker Foxp3 (Gonzalez-Garcia et al., 2013). Moreover, TC3.6 has recently been shown to be an effective drug for promoting oligodendrocyte differentiation (Medina-Rodriguez et al., 2013).
With the aim of completing the therapeutic profile of TC3.6 for MS, here, we present its biological behaviour regarding its modulation of cAMP levels and the inflammatory response in microglial cells; its neuroprotective effects in PC12 cells and its efficacy in the primary progressive TMEV-IDD model. In particular, we assessed the following endpoints: neurological deficits, histological damage, leukocyte infiltration and pro-inflammatory mediators with the aim of showing the therapeutic potential of PDE7 inhibitors, more specifically TC3.6, for this unmet and severe disease.
Methods
PDE inhibitors
The PDE inhibitors used here were purchased from Calbiochem (Darmstadt, Germany; BRL50481) or synthesized according reported procedures (TC3.6; Redondo et al., 2012c). The purity of the compound (>95%) was assessed by HPLC.
Primary cultures of glial cells
Rat primary microglia were prepared from neonatal (P2) rat cerebral cortex as previously described (Mecha et al., 2011). Briefly, after removal of the meninges, the cerebral cortex was dissected and mechanically dissociated by gently pipetting through tips of small diameter. After centrifugation, the pellet was washed with cold HBSS (Gibco, Carlsbad, CA, USA) and the cells were plated on 75 cm2 flasks and allowed to grow in DMEM medium containing 10% bovine serum (FBS), 10% horse serum and 1% penicillin/streptomycin at 37°C in a humidified incubator (5% CO2). After 7 days, cultures were confluent and the flasks were agitated on an orbital shaker for 4 h at 230 r.p.m. at 37°C and microglial cells collected from the supernatant. After centrifugation at 168× g for 10 min, microglial cells were seeded in 96-well dishes. The purity of the cultures was >95%, as determined by immunofluorescence analysis using anti-CD11b antibody.
cAMP assay
To determinate the levels of cAMP, microglial cultures were incubated with TC3.6 or BRL50481 (all at 10 or 30 μM) for 1 h and cAMP quantified as described previously (Morales-Garcia et al., 2011).
Nitrite determinations
TC3.6 (30 μM) was added to the culture medium of microglial cells 1 h before exposure to LPS (10 μg·mL−1), and cells were harvested 16 h later for evaluation of nitrite levels. To analyse the role of cAMP, some microglial-containing plates were also pre-incubated with the cAMP antagonist Rp-cAMP (100 μM; BIOMOL Research Laboratories, New York, NY, USA) or the PKA inhibitor H-89 (20 μM; BIOMOL Research Laboratories) 2 h before the addition of the compounds. The production of nitric oxide was monitored by measuring the content of nitrite in media by the standard Griess reaction as described previously (Morales-Garcia et al., 2011).
PC12 cell cultures
The PC12 cell line was a kind gift from Dr. Lorenzo Romero (Experimental Neurology Unit, Hospital Nacional de Parapléjicos, Toledo, Spain). The cell line was maintained in DMEM (Invitrogen, Carlsbad, CA, USA) supplemented with 10% inactivated FBS plus 1% v v-1 penicillin/streptomycin (Life Technologies, Carlsbad, CA, USA) at 37°C in a 5% humidified incubator. Cells were maintained for 6 days until the experiment. Cells were seeded in 96 plate wells (2 × 104 cells per well). First, cells were treated with TC3.6 (10, 30 or 60 μM) to assess the effect of the compound on basal cell survival. In another set of experiments, cells were exposed to glutamate (10 mM) for 24 h incubation and 30 min before treated or not with TC3.6 at the doses of 10 or 30 μM.
Cytotoxicity assay
PC12 cell death was quantified by measurement of LDH release from damaged cells into the bathing medium by using the LDH kit (Roche, Life Science, Manheim, Germany). Cells were exposed to glutamate (10 mM) in the absence or presence of TC3.6 (10 and 30 μM) and 24 h later supernatants were collected and analysed. TC3.6 was added 30 min before glutamate. Background LDH levels were determined in paralleled cultures subjected to sham washes and subtracted from experimental values. Percentage of cell death in experimental conditions was referred to the value of glutamate alone (set as 100%).
Animal and Theiler’s virus inoculation
All experimental procedures followed the European Communities Council Directive 2010/63/EU and the Spanish regulations (BOE67/8509-12; BOE1201/2005) on the use and care of laboratory animals, and were approved by the local Animal Care and Ethics Committee of the CSIC. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). We used female SJL/J mice, susceptible to TMEV-IDD development, from Harlan Interfauna Ibérica (Barcelona, Spain). The mice were housed in the Animal Resource Facility at the Cajal Institute, CSIC (Madrid) and maintained on food and water ad libitum in a 12 h dark–light cycle. Four- to six-week-old mice were inoculated, intracerebrally, in the right cerebral hemisphere with 106 plaque forming units of Daniel’s (DA) TMEV strain as described previously (Arevalo-Martin et al., 2003). Another set of mice (4- to 6-week-old) were injected intracerebrally with 30 μL of DMEM supplemented with 10% FCS as sham animals.
Experimental in vivo procedure
Two protocols of TC3.6 administration were used: (i) during the pre-symptomatic phase (30 days post-infection), TMEV-infected mice received TC3.6 (10 mg·kg−1 i.p.; n = 6) or appropriate vehicle (0.2% DMSO, Tocrisolve 5% in PBS; n = 6) for 12 consecutive days and then, animals were maintained and then killed 70 days after TMEV infection; (ii) at 60 days after TMEV infection and once the disease had become established, the mice were treated daily for 14 days with TC3.6 (5 mg·kg−1, i.p.; n = 10) or appropriate vehicle (0.2% DMSO, Tocrisolve 5% in PBS; n = 10) and killed at day 75 post-infection.
Evaluation of symptomatology and motor function
General health conditions (weight and clinical score) and motor function of animals were periodically evaluated every 5 days from day 30 until day 50 and every 10 days from day 50 to 70 (pre-symptomatic treatment) or 75 (established disease) after TMEV infection. Clinical scores were assigned based on the classical scale of 0–5: score 1, mice showed waddling gait; score 2, mice showed a more severe waddling gait; score 3, mice had a loss of righting ability associated with spastic hind limbs; score 4, mice showed paralysis of hind limbs; and score 5, mice were moribund.
The screening for locomotor activity was performed using an activity cage (Activity Monitor System Omnitech Electronics, Inc., Colombus, OH, USA) coupled to a Digiscan Analyser. The number of times that the animals broke the horizontal or vertical sensor beams was measured in two 5 min sessions for horizontal and vertical activity.
Tissue processing and immunohistochemistry
Spinal cords were removed from sham and infected mice at the indicated days after the infection (n = 4 for each group). Animal tissue was processed as previously described (Mestre et al., 2009). Free-floating transversal thoracic spinal cord sections (30 μm thick) were processed as detailed to visualize the pro-inflammatory cytokines with specific anti-COX-2 antibody (Santa Cruz Biotechnology, Inc., CA, USA; goat polyclonal IgG, 1:500 in PBS, v v-1), anti-TNF-α ligand antibody (Santa Cruz Biotechnology, Inc.; goat polyclonal IgG 1:500 in PBS, v v-1), anti-IL-1β ligand antibody (Santa Cruz Biotechnology, Inc.; rabbit polyclonal IgG 1:500 in PBS, v v-1). Microglial cells were stained with a rabbit polyclonal anti-Iba-1 antibody (1:1000; Wako Chemical Pure Industry, GmbH, Neuss, Germany); CD4- and CD8-positive T-cells were stained with rat anti-mouse polyclonal antibodies (1:1000, BD Pharmingen, BD Biosciences, Madrid, Spain). Axonal density was visualized with pan-neurofilament primary antibody (rabbit polyclonal antiserum cocktail to neurofilaments, NA1297, 1:2000; Biomol International, Madrid, Spain) and axonal neurofilaments in longitudinal sections of spinal cord were stained with a rabbit polyclonal antibody anti-Neurofilament H (1:1000; Merck Millipore, Darmstadt, Germany). Longitudinal sections were also stained with RIP, a monoclonal antibody isotype IgG1 (1:1000; DSHB University of Iowa, Ames, IA, USA).
In all cases, specificity of staining was confirmed by omitting the primary antibody. Six spinal cord sections per animal (n = 4 for each group) were analysed and two microphotographs per section were taken. Quantification of staining was performed using the Image J software designed by the National Institutes of Health.
Eriochrome cyanine staining
Myelin staining was performed as described previously (Arevalo-Martin et al., 2010). Six spinal cord sections per animal (n = 4 of each group) were analysed and two microphotographs per section were taken. Briefly, the slides were dried and warmed at 37°C, immersed in acetone for 5 min, and stained in eriochrome cyanine solution for 30 min at room temperature (RT). Slides were rinsed in water, differentiated in 5% iron alum for 10 min at RT, rinsed again in water and fully differentiated in borax-ferricyanide solution for 10 min at RT. Slides were then dehydrated through graded ethanol solutions, immersed in xylene and coverslipped using DePeX mounting medium.
Mouse neurofilament elisa assay
Levels of mouse neurofilament were determined in tissue homogenates of spinal cord (n = 6 per group) by a commercial Kit (My Biosource, San Diego, CA, USA) following the manufacturer’s instructions. The detection range was between 20 and 0.312 ng·mL−1. The sensitivity of the assay was 0.06 ng·mL−1 and the intra- and inter-assay coefficients of variations were 8 ± 1.2% and 12 ± 2.3%, respectively. Results are expressed as ng·mg−1 protein.
RNA extraction
After saline perfusion, tissue samples were frozen in dry ice and stored at −70°C until required (n = 6 for each group). Total RNA was extracted using the RNeasy Lipid RNA extraction kit (Qiagen, Manchester, UK), given the high content of lipids found in the spinal cord. Contaminating genomic DNA was degraded by a treatment with DNaseI (Qiagen). The yield of RNA was determined using a Nanodrop® spectrophotometer (Nanodrop Technologies, Wilmington, DE, USA).
Reverse transcription and real-time PCR
Total RNA (1 μg in 20 μL) was reverse transcribed into cDNA using the Promega reverse transcription kit (Promega, Madrid, Spain) with poly-dT primers and amplified with the primers summarized in Table 2013. Gene expression was quantified using SYBR Green. Cycling conditions were as follows: 50°C for 2 min, 95°C for 10 min, followed by 40 cycles of amplification (95°C for 15 s, 60°C for 1 min). Samples were assayed on the Applied Biosystems PRISM 7500 Sequence detection system. Each sample was assayed in duplicate and a 6-point standard curve was run in parallel. To ensure the absence of genomic DNA contamination, a control sample of non-reverse-transcribed RNA was run for each set of RNA extractions. Relative quantification was obtained by calculating the ratio between the values obtained for each gene of interest and the housekeeping gene GAPDH.
Table 1.
Primers used in SYBR PCR
| Gene | Sense (5′-3′) | Antisense (5′-3′) |
|---|---|---|
| IL-1β | TGGTGTGTGACGTTCCCATT | TCCATTGAGGTGGAGAGCTTTC |
| TNF-α | AGAGGCACTCCCCCAAAAGA | CGATCACCCCGAAGTTCAGT |
| IL-6 | TCCAGAAACCGCTATGAAGTTC | CACCAGCATCAGTCCCAAGA |
| IFN-γ | GGCCATCAGCAACAACATAAGCGT | TGGGTTGTTGACCTCAAACTTGGC |
| GAPDH | TGATGCTGGTGCTGAGTATGTCGT | TCTCGTGGTTCACACCCATCACAA |
Statistical analysis
For the in vitro microglia experiments, values represent the means ± SD of six replications in three different experiments and were analysed by one-way anova and Tukey’s test for multiple comparisons. For experiments with PC12 cells, values represent the mean ± SEM of three independent experiments in triplicate. For in vivo experiments, values represent the mean ± SD and anova for repeated measures followed by an appropriate post hoc test were used to determine statistical significance (95%; P < 0.05). The statistical analysis in the first in vivo experiment (treatment in pre-symptomatic phase) assessed differences in the day of disease onset between TMEV + vehicle and TMEV + TC3.6 groups, as well as the clinical score and motor activity deficit progression of each group. The statistical analysis of the second in vivo experiment (treatment in established disease) showed the differences in the clinical score and in the motor performance in the activity cage at day 75 after TMEV infection.
Results
TC3.6 increases the levels of cAMP in microglia cultures to induce an anti-inflammatory effect
TC3.6 is a synthetic heterocyclic small molecule with an IC50 of 0.55 μM on PDE7 inhibition (Figure 1A). It is rather selective regarding other isoenzymes such as PDE3, PDE4B, PDE4D and PDE10, with IC50 values of 70.7, 57.9, 23.9 and 50.1 μM respectively. Here, we tested if TC3.6 has the expected cellular behaviour being able to modulate cAMP levels. Briefly, cells were seeded at 3 × 104 per well in 96-well dishes and incubated overnight before the assay. After 15 min incubation with 10 or 30 μM of TC3.6, intracellular levels of cAMP were significantly increased (Figure 1B). In line with this result, the treatment with a commercial PDE7 inhibitor (BRL50481) used as standard reference produced the same increase in intracellular cAMP at doses of 10 and 30 μM. The anti-inflammatory potential of TC3.6 was also determined on microglial cells exposed to LPS by measuring nitrites accumulation. To analyse the role of cAMP in nitrite changes, some microglial-containing plates were also pre-incubated with the cAMP antagonist Rp-cAMP (100 μM) or the PKA inhibitor, H-89 (20 μM) for 24 h before the addition of the different compounds. In these conditions, the anti-inflammatory properties of TC3.6 (30 μM) were completely abolished (Figure 1C).
Figure 1.
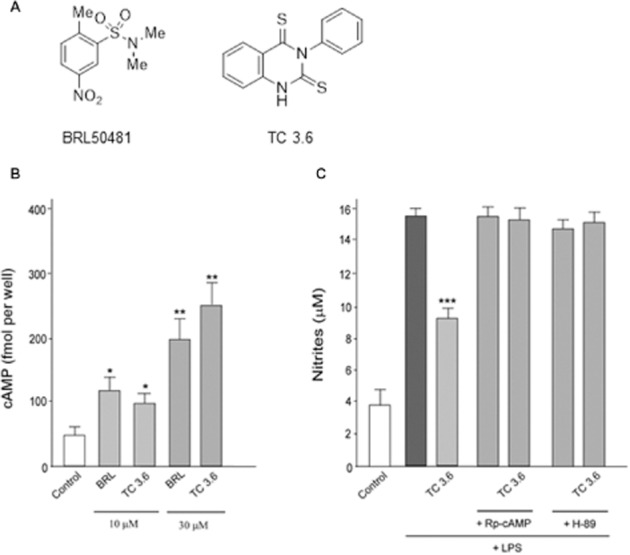
TC3.6 increases the levels of cAMP in microglia culture and modulates the anti-inflammatory response. (A) Chemical compounds used. (B) Intracellular levels of cAMP measured in rat primary microglial cultures in the presence of TC3.6 and BRL50481 (10 or 30 μM). Values represent the means ± SD from three different experiments. *P < 0.05, **P < 0.01 versus control. (C) Nitrite production in rat primary microglial cultures treated with LPS (10 μg·mL−1) in the absence or presence of TC3.6 and BRL50481 (30 μM). Some of the cultures were pretreated with the PKA inhibitor H-89 or the cAMP antagonist Rp-cAMP. Values represent the means ± SD of six replications in three different experiments. ***P < 0.001 versus LPS-treated cells.
TC3.6 mediates neuroprotective effects
Since TC3.6 was found to have anti-inflammatory effects, we decided to explore its potential neuroprotective properties by using PC12 cell cultures subjected to glutamate-induced cell death. PC12 cells were treated with glutamate (10 mM) and LDH release was quantified as a cell death read-out. First, we assessed whether TC3.6 per se could affect the viability of PC12 cells. As shown in Figure 2A, the higher dose of TC3.6 (60 μM) reduced PC12 cell survival; thus, we discarded the use of 60 μM for glutamate experiments. Exposure to glutamate induced the LDH release to a level that we set as the highest cell death reached (100%). When the cells were pretreated with 10 or 30 μM TC3.6, glutamate-induced cell death was diminished by 40 and 60% respectively, thus, TC3.6 exerted dose–dependent neuroprotective activity in PC12 cells (Figure 2B).
Figure 2.
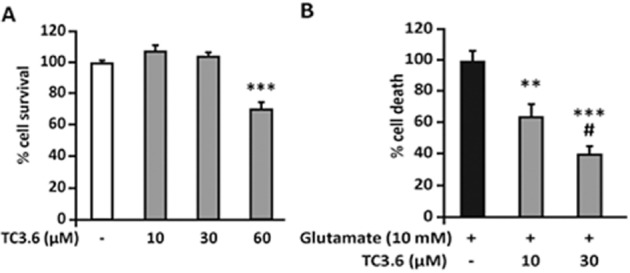
Neuroprotective activity of TC3.6. (A) PC12 cells were incubated for 24 h with TC3.6 (10, 30 and 60 μM) and cell viability was quantified; results are shown as mean ± SEM from three independent experiments in triplicate. ***P < 0.001 versus control. (B) PC12 cells were cultured and incubated with 10 mM glutamate to induce cell death in the absence or presence of TC3.6 (10 and 30 μM) added 30 min before glutamate; results are shown as mean ± SEM from three independent experiments in triplicate. **P < 0.01 versus control and #P < 0.05 versus glutamate +TC3.6 (10 μM).
Inhibition of PDE7 improves motor function during established neurological deficits
TMEV-IDD is a model of chronic progressive MS in which following virus inoculation, there is a latency period until the symptoms and motor deficits appear. In our initial studies, TC3.6 administration during the pre-symptomatic phase (30 days post-infection) for 12 consecutive days (10 mg·kg−1, i.p.) significantly delayed the TMEV-IDD onset to 60 days post-infection instead of 45 days in the case of TMEV-vehicle mice (Figure 3A). Moreover, at 50, 60 and 70 days post-infection, the treatment with TC3.6 reduced the clinical scores in comparison with those observed in TMEV-vehicle mice at the same day indicating a delayed progression of the disease.
Figure 3.
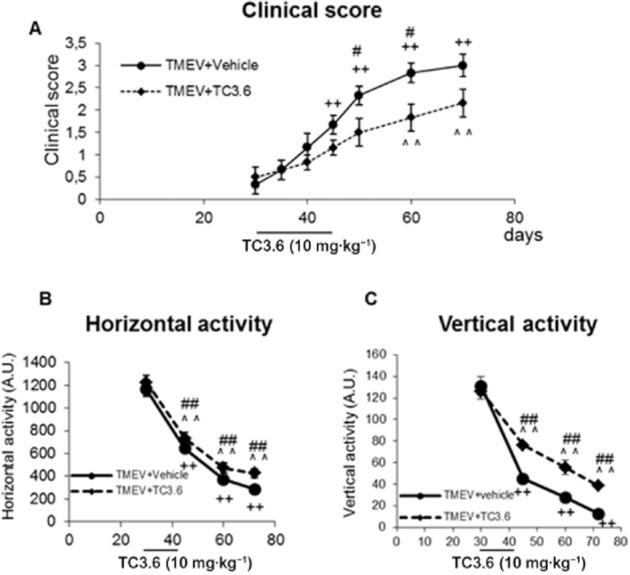
The treatment with TC3.6 delays TMEV-IDD progression. TMEV-infected mice were treated with TC3.6 (10 mg·kg−1; n = 6) or appropriate vehicle (0.2% DMSO, Tocrisolve 5% in PBS; n = 6) during pre-symptomatic phase (30 days post-infection) for 12 consecutive days, then were maintained and killed at 70 days after TMEV infection. (A) Clinical score of animals were recorded every 5 days from day 30 until day 50 and every 10 days from day 50 to 70 after TMEV infection. The motor function was analysed (days 30, 45, 60 and 70 post-infection) in an activity cage, including the horizontal activity (B) and the vertical activity (C) for 10 min. ++P < 0.001 versus TMEV + vehicle (30 days); ∧∧P < 0.01 versus TMEV + TC3.6 (30 days), anova for repeated measures #P < 0.05 and ##P < 0.01 versus TMEV + vehicle (on the same day).
Data from the motor performance in the activity cage revealed an improvement in motor function in the TC3.6 treated animals (Figure 3B and C). Deambulatory activity displayed by TMEV-infected mice receiving TC3.6 was significantly higher at 45, 60 and 70 days post-infection when compared with the horizontal activity data obtained from TMEV-vehicle mice at the corresponding days (Figure 3B). This improved behaviour was also noticed when we assessed the vertical activity parameter (Figure 3C).
Therefore, in an attempt to study the efficacy of TC3.6 during established disease for the therapeutic interest of its potential utility, we designed a set of experiments in which the inhibitor of PDE7 was administered to diseased mice showing significant neurological deficits. Clinical symptoms and motor function of TMEV-infected mice were recorded every 5 or 10 days starting from day 30 post-infection. The treatment of TMEV-infected mice with TC3.6 (5 mg·kg−1), at day 60 post-infection for 14 consecutive days slowed the clinical score progression. Thus, the clinical score reached by TMEV-vehicle mice was significantly higher (score = 3) than that of TMEV-TC3.6-treated mice (score < 2; Figure 4A). In support of this, the evaluation of spontaneous motor activity, at day 75 post-infection, showed that the administration of TC3.6 significantly improved the performance of the activity cage test, as horizontal and vertical activity parameters were better in mice that received the inhibitor of PDE7 (Figure 4B and C respectively).
Figure 4.
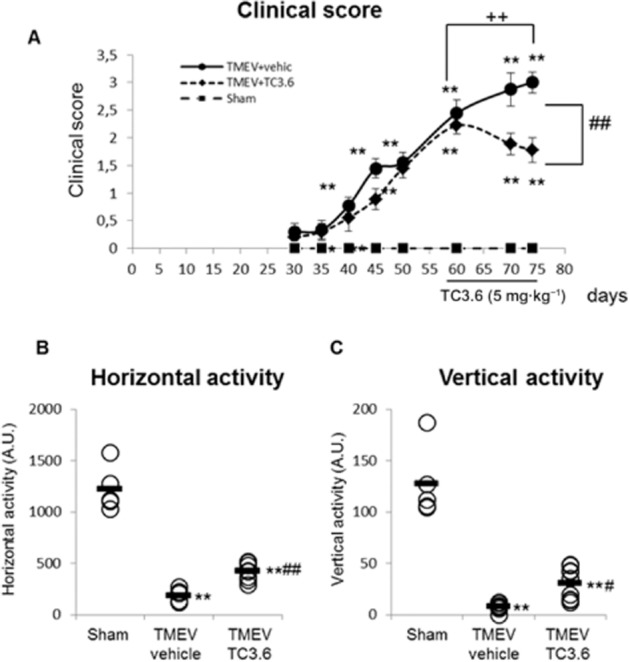
TC3.6 improves motor function during established neurological symptomatology. Once the disease had become established (60 days post-infection), TMEV-infected mice were treated with TC3.6 (5 mg·kg−1; n = 10) daily for 14 days or with appropriate vehicle (0.2% DMSO, Tocrisolve 5% in PBS; n = 10). (A) Clinical scores of animals were recorded every 5 days from day 30 until day 50 and every 10 days from day 50 to 75 after TMEV infection. The motor function at day 75 post-infection was assessed in an activity cage and included the horizontal activity (B) and the vertical activity (C) for 10 min. **P < 0.01 versus sham; ++P < 0.001 versus TMEV + vehicle (day 60), anova for repeated measures. #P < 0.05 and ##P < 0.01 versus TMEV + vehicle (day 75), anova. Each mouse is represented by a circle (O) and the mean of each group is represented by a line (—).
TC3.6 reduces inflammation in the spinal cord of TMEV-infected (disease established) mice
Immunohistochemical analysis of the spinal cord of TMEV-infected mice revealed that the treatment with TC3.6 induced a significant attenuation in the level of neuroinflammation. H&E staining revealed that infection with TMEV provoked the infiltration of immune cells into the spinal cord (Figure 5A). Treatment with TC3.6 partially diminished this infiltration (Figure 5A) and specifically decreased the accumulation of CD4 and CD8 lymphocytes into the parenchyma (Figure 5B). In Figure 5C, a reduction in microglial reactivity (P < 0.05) in the spinal cord of TC3.6-treated mice is shown in comparison with microglial images obtained from spinal cord sections of TMEV-infected mice that received vehicle. Quantification analysis revealed a significant reduction in cell infiltrates (Figure 5D; P < 0.01), CD4+ (Figure 5E; P < 0.05) and CD8+ T lymphocytes (Figure 5F; P < 0.05) in TMEV-infected mice treated with TC3.6 versus TMEV-infected mice treated with the vehicle. In addition, a reduction in the percentage of Iba-1 staining (Figure 5G; P < 0.05) in TMEV mice treated with TC3.6 versus TMEV + vehicle mice was observed. These findings support the data from our in vitro studies with primary microglial cultures.
Figure 5.

TC3.6 administered once the disease has become established reduces inflammation in the spinal cord of TMEV-infected mice. Representative sections of ventral columns of thoracic spinal cord (at day 75 post-infection) of TMEV-infected mice subjected to vehicle or TC3.6 treatment (dose schedule: 5 mg·kg−1 daily for 14 consecutive days). Sections were stained (A) to visualize leukocyte infiltrates ( haematoxylin/eosin); (B) CD4+ T-cells and (C) for microglia by Iba-1 staining. Quantification analysis of cell infiltrates (D), CD4+ T-cells (E), CD8+ T-cells (F) and percentage of area stained by Iba-1 (G; confocal images with constant laser beam). **P < 0.01; *P < 0.05 versus TMEV + vehicle. Scale bars: 50 or 100 μM.
PDE7 inhibition diminishes the expression of pro-inflammatory mediators in the spinal cord of TMEV-infected mice
Given the reduced microglia reactivity induced by TC3.6 treatment, we then assessed the expression of pro-inflammatory markers like COX-2 and pro-inflammatory cytokines in the TMEV-IDD model. As shown in Figure 6A, immunohistochemical staining of IL-1β, TNF-α and COX-2 was drastically reduced in the spinal cord of TMEV-infected mice treated with TC3.6. Analysis of mRNA expression, by real-time PCR, of the pro-inflammatory cytokines IL-1β, TNF-α, IL-6 and IFN-γ, showed a significant increase in the spinal cord of TMEV-infected mice. Accordingly with immunohistochemical analysis, transcripts for IL-1β and TNF-α were significantly reduced in the spinal cord of TMEV-infected mice subjected to TC3.6 administration (Figure 6B and C). The expression level of IL-6 mRNA was also significantly decreased (Figure 6D), but levels of IFN-γ, although lower than those observed in TMEV-vehicle mice, did not reach statistical significance (Figure 6E).
Figure 6.
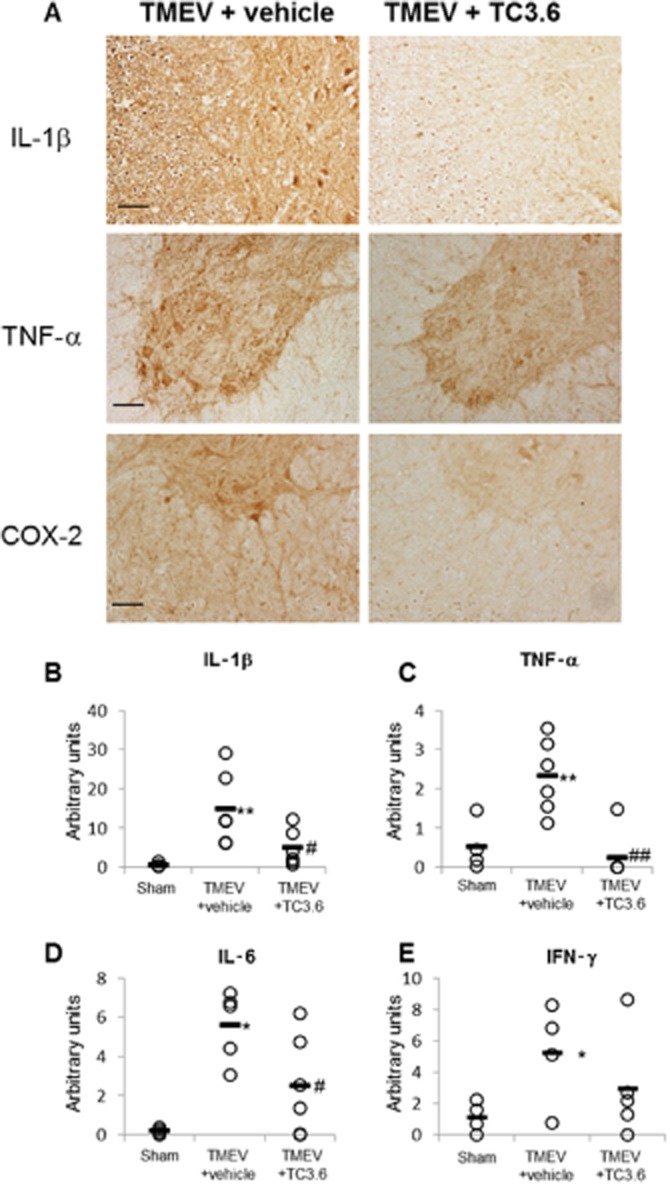
The treatment with TC3.6 diminishes the expression of pro-inflammatory mediators in spinal cord of TMEV-infected mice. (A) Representative sections of ventral columns of thoracic spinal cord (at day 75 post-infection) of TMEV-infected mice treated with TC3.6 or appropriate vehicle for 14 consecutive days were stained for IL-1β, TNF-α and COX-2. Scale bar 100 μm. The effect of TC3.6 on IL-1β (B), TNF-α (C), IL-6 (D) and IFN-γ (E) mRNA levels in thoracic sections of spinal cord were analysed by semi-quantitative PCR. Each mouse is represented by a circle (O) and the mean of each group is represented by a line (—) (n = 6). *P < 0.05 and **P < 0.01 versus sham; #P < 0.05 and ##P < 0.01 versus TMEV + vehicle.
PDE7 inhibition reduces demyelinated lesions and improves spinal cord white matter disorganization increasing neurofilament in the spinal cord of TMEV-infected mice
The reduced infiltrates and microglia reactivity induced by the treatment with TC3.6 were supported by histological examinations of spinal cord sections from experimental animals. Staining was performed using eriochrome of cyanine to visualize demyelination. Sections from the spinal cord of TMEV-infected mice given vehicle showed demyelination and vacuolization (Figure 7A) that was much less in spinal cord sections from TC3.6-treated TMEV mice. Previous studies from our group (Loria et al., 2010; Mecha et al., 2013) have shown that at the chronic symptomatic phases, there is a reduction in the axonal density; shown by using pan-neurofilament staining of transversal spinal cord sections. The same was observed in the present study (Figure 7B). However, the treatment with TC3.6 noticeably improved the white matter disorganization. The quantification of intensity of staining by pan-neurofilament revealed a significant effect (P < 0.05) of TC3.6 treatment on the preservation of the axonal package (Figure 7D). Similarly, the axonal swelling evident when longitudinal spinal cord sections of TMEV-infected animals were stained with Neurofilament-H and CNPase for myelin labelling (Figure 7C) was less frequent and milder following TC3.6 administration. Although the quantification of axonal loss by using electronic microscopy is considered to be the best technique, biochemical methods for measuring neurofilament levels in the spinal cord tissue homogenates were also employed to estimate neurodegeneration (Petzold et al., 2003). Here, using elisa assays, we showed that the treatment with TC3.6 increased the levels of neurofilament (P < 0.05) in spinal cord in comparison to the levels observed in TMEV-infected mice given vehicle (Figure 7E).
Figure 7.
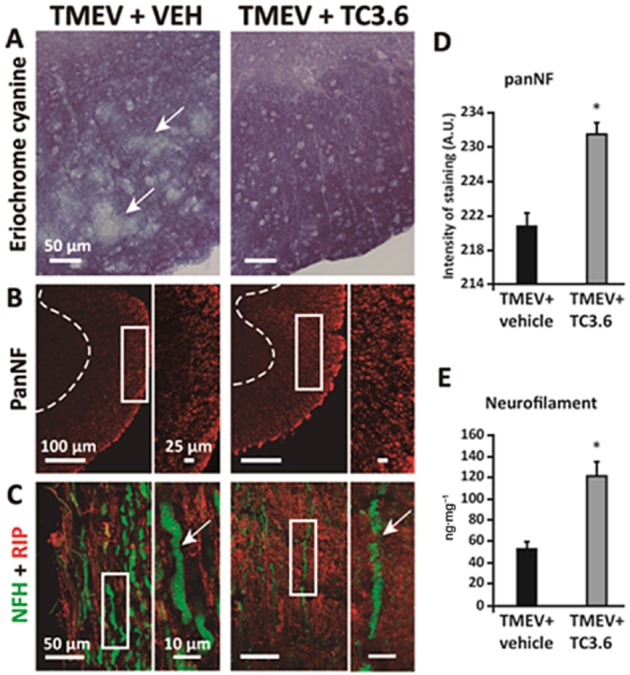
Effects of inhibition of PDE7 by TC3.6 on demyelination and neurodegeneration in the TMEV-IDD model. (A) Representative transverse sections of ventral columns of thoracic spinal cord of TMEV-infected mice treated with TC3.6 or appropriated vehicle (at day 75 post-infection) stained by eriochrome cyanine to visualize myelin; scale bar 50 μm. (B) Representative transverse sections of ventral columns of thoracic spinal cord of TMEV-infected mice treated with TC3.6 or vehicle (at day 75 post-infection) stained with pan-neurofilament; scale bars: 100 and 25 μm; confocal images with constant laser beam were analysed for quantification. (C) Representative longitudinal sections of thoracic spinal cord of TMEV-infected mice treated with TC3.6 or vehicle (at day 75 post-infection) stained with NFH and RIP (CNPase); scale bar 50 and 10 μm. (D) Intensity of pan-neurofilament staining in white matter was quantified. *P < 0.05 versus TMEV + vehicle. (E) Levels of neurofilament (at day 75 post-infection) in spinal cord tissue homogenates were increased by treatment with TC3.6 in TMEV-infected mice. *P < 0.05 versus TMEV + vehicle.
Discussion
Recent studies indicate PDE7 inhibition as a new therapeutic approach for neuroimmune diseases (Giembycz and Smith, 2006; Paterniti et al., 2011; Redondo et al., 2012a,b,; Gonzalez-Garcia et al., 2013). Our data showed that inhibition of PDE7 by the compound TC3.6 administered systemically (i.p.) ameliorates clinical signs and improves motor function in the TMEV-IDD progressive model of MS. A sub-chronic schedule of TC3.6 administration was efficacious in two windows of treatment, during the pre-symptomatic phase and during the established disease. Our results also show that inhibition of PDE7 results in a reduction of cellular infiltrates, microglial immunoreactivity as well as in the expression of COX-2 and pro-inflammatory cytokines in the spinal cord of TMEV-infected mice. This was accompanied by reduced demyelination, preservation of axons package and increased neurofilament levels. These observations suggest that inhibition of PDE7 may have potential for alleviating MS-like pathology, and especially in the progressive state of the disease, which is severe and without any treatment at present.
The importance of controlling cAMP levels in the regulation of critical aspects of immune responses is well known (Giembycz and Dent, 1992; Van Wauwe et al., 1995). Here, we showed that TC3.6 elevates cAMP levels and significantly attenuates nitrite release in activated microglial cells. The prevention of the anti-inflammatory effect of TC3.6 by inhibitors of PKA suggests that the activity of PDE7 inhibitors is regulated by microglial cAMP concentration, and that its anti-inflammatory activity involves cAMP-dependent PKA activation. The neuroprotective profile of TC3.6 in glutamate-induced cytotoxicity in PC12 cells is also supported by previous findings about the family of PDEs as PDE4 and more recently PDE7 have been shown to play a role in neuroprotection and neuroinflammation (Paterniti et al., 2011). Previous studies have shown that inhibition of PDE4 with the selective inhibitor rolipram in EAE models ameliorated the clinical course of the disease (Robichaud et al., 2001; Paintlia et al., 2009). Unfortunately, clinical trials with PDE4 inhibitors revealed major adverse effects of these drugs, namely nausea and vomiting (Robichaud et al., 2001). PDE7 inhibitors are considered a good strategy to overcome these adverse effects with their more subtle modulation of cAMP levels (Giembycz, 2005) without emetogenic activity (Garcia et al., 2014). We showed in the present study that TC3.6, a selective PDE7 inhibitor able to penetrate into the CNS (unpublished data), is efficacious at diminishing the symptoms and neuroinflammation in the Theiler’s model of MS. Several studies have described changes in the expression of several PDE7 isoforms in the brain of rodents and humans in neurodegenerative diseases (Miro et al., 2001; Perez-Torres et al., 2003; Reyes-Irisarri et al., 2005). Therefore, PDE7 might be a pharmacological target in the context of neurological diseases such as MS.
PDE7 inhibitors have been shown to alleviate EAE symptomatology but the underlying histopathological changes were not described (Redondo et al., 2012a; Gonzalez-Garcia et al., 2013). A mechanism by which the compound TC3.6 ameliorated TMEV symptomatology may be related to its anti-inflammatory and neuroprotective profile. Recent data seem to support this notion. Thus, in a comparative study of the effects of rolipram and TC3.6 in EAE, it was found that both compounds prevented the clinical signs of EAE after MBP inoculation by reducing IL-17 and increasing the T regulator cell marker Foxp-3 (Gonzalez-Garcia et al., 2013). Here, in a viral model of MS, we found that transcript levels of TNF-α, IL-1β and IL-6 were significantly decreased in the TC3.6-treated groups and immunohistological analysis also supported this anti-inflammatory activity. In agreement with the above study (Gonzalez-Garcia et al., 2013), we found reduced cellular infiltrates in TMEV-infected mice treated with TC3.6. In addition, COX-2 expression was reduced in the spinal cord of TMEV-infected mice subjected to PDE7 inhibition. COX-2 immunoreactivity has been found in MS and in animal models of MS (Mestre et al., 2006; Yiangou et al., 2006). In the Theiler’s virus model, COX-2 immunoreactivity has been mainly associated with the demyelinating process (Carlson et al., 2006). Selective PDE7 inhibitors promote oligodendrocyte precursor survival and differentiation towards myelin-forming phenotypes in both murine and human OPCs (Medina-Rodriguez et al., 2013). In the present study, by using immunohistochemistry techniques, confocal microscopy and biochemical assays, we demonstrated an improvement in the organization of axon package and a partial recovery in neurofilament levels together with a diminished demyelination in TMEV-IDD after the treatment with a selective inhibitor of PDE7. Further studies using electronic microscopy would add support to these initial findings.
In summary, recent data in EAE underline PDE7 inhibition as a new therapeutic target for inflammatory diseases. Here, we observed that the quinazoline derivative PDE7 inhibitor TC3.6 exhibits anti-inflammatory and neuroprotective activity, reduces neuroinflammation, decreases axonal damage and improves symptomatology and motor function in an autoimmune viral model of progressive MS. Collectively, our data concur and support the consideration of TC3.6 as a promising new candidate for MS management, including primary progressive MS, as it has both immunomodulatory and neuroprotective properties. Development of this small molecule to clinical trials will be the only way to finally assess its therapeutic potential for MS patients.
Acknowledgments
The authors are grateful to Dr. Moses Rodríguez (Department of Immunology and Neurology, Mayo Clinic/Foundation, Rochester, MN, USA) for gentile gift of Theiler’s virus DA strain. We gratefully appreciated Dr. Miriam Mecha (Cajal Institute) for her advices and critical reading of the manuscript. We are also grateful to Joaquín Sancho and Laura Ramos for their excellent technical assistance.
The authors gratefully acknowledge the financial support of the Ministry of Science and Innovation (MICINN, projects no. SAF2010-16365 and SAF2012-33600), Instituto de Salud Carlos III (project no. RD07/0060/0015, and RD07/0060/0010 RETICS Program) and Fundación Española para la Ciencia y la Tecnología (FECYT), project no. FCT-09-INC-0367. M. R. acknowledges pre-doctoral fellowship from CSIC (JAE program).
Glossary
- PPMS
primary progressive multiple sclerosis
- TMEV-IDD
Theiler’s murine encephalomyelitis virus-induced demyelinating disease
Author contributions
L M, M R, F J C-S, J.A M-G and S A-G performed the research and evaluated the results. A P-C and C G supervised experimental tasks, analysed the data and manuscript correction. C G and A M designed and supervised the study, discussed the experimental setting and results and wrote the manuscript.
Conflict of interest
The authors declare no conflict of interest.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: enzymes. Br J Pharmacol. 2013;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arevalo-Martin A, Vela JM, Molina-Holgado E, Borrell J, Guaza C. Therapeutic action of cannabinoids in a murine model of multiple sclerosis. J Neurosci. 2003;23:2511–2516. doi: 10.1523/JNEUROSCI.23-07-02511.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arevalo-Martin A, Garcia-Ovejero D, Molina-Holgado E. The endocannabinoid 2-arachidonoylglycerol reduces lesion expansion and white matter damage after spinal cord injury. Neurobiol Dis. 2010;38:304–312. doi: 10.1016/j.nbd.2010.02.002. [DOI] [PubMed] [Google Scholar]
- Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev. 2006;58:488–520. doi: 10.1124/pr.58.3.5. [DOI] [PubMed] [Google Scholar]
- Brudvik KW, Tasken K. Modulation of T cell immune functions by the prostaglandin E(2) – cAMP pathway in chronic inflammatory states. Br J Pharmacol. 2012;166:411–419. doi: 10.1111/j.1476-5381.2011.01800.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson NG, Hill KE, Tsunoda I, Fujinami RS, Rose JW. The pathologic role for COX-2 in apoptotic oligodendrocytes in virus induced demyelinating disease: implications for multiple sclerosis. J Neuroimmunol. 2006;174:21–31. doi: 10.1016/j.jneuroim.2006.01.008. [DOI] [PubMed] [Google Scholar]
- Claveau D, Chen SL, O’Keefe S, Zaller DM, Styhler A, Liu S, et al. Preferential inhibition of T helper 1, but not T helper 2, cytokines in vitro by L-826,141 [4-[2-(3,4-Bisdifluromethoxyphenyl)-2-[4-(1,1,1,3,3,3-hexafluoro-2-hydroxypropan- 2-yl)-phenyl]-ethyl]3-methylpyridine-1-oxide], a potent and selective phosphodiesterase 4 inhibitor. J Pharmacol Exp Ther. 2004;310:752–760. doi: 10.1124/jpet.103.064691. [DOI] [PubMed] [Google Scholar]
- Comi G. Disease-modifying treatments for progressive multiple sclerosis. Mult Scler. 2013;19:1428–1436. doi: 10.1177/1352458513502572. [DOI] [PubMed] [Google Scholar]
- Curtin F, Hartung HP. Novel therapeutic options for multiple sclerosis. Expert Rev Clin Pharmacol. 2014;7:91–104. doi: 10.1586/17512433.2014.865517. [DOI] [PubMed] [Google Scholar]
- Garcia AM, Brea J, Morales-Garcia JA, Perez DI, Gonzalez A, Alonso-Gil S, et al. Modulation of cAMP-specific PDE without emetogenic activity: new sulfide-like PDE7 inhibitors. J Med Chem. 2014;57:8590–8607. doi: 10.1021/jm501090m. [DOI] [PubMed] [Google Scholar]
- Giembycz MA. Life after PDE4: overcoming adverse events with dual-specificity phosphodiesterase inhibitors. Curr Opin Pharmacol. 2005;5:238–244. doi: 10.1016/j.coph.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Giembycz MA, Dent G. Prospects for selective cyclic nucleotide phosphodiesterase inhibitors in the treatment of bronchial asthma. Clin Exp Allergy. 1992;22:337–344. doi: 10.1111/j.1365-2222.1992.tb03095.x. [DOI] [PubMed] [Google Scholar]
- Giembycz MA, Smith SJ. Phosphodiesterase 7A: a new therapeutic target for alleviating chronic inflammation? Curr Pharm Des. 2006;12:3207–3220. doi: 10.2174/138161206778194123. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Garcia C, Bravo B, Ballester A, Gomez-Perez R, Eguiluz C, Redondo M, et al. Comparative assessment of PDE 4 and 7 inhibitors as therapeutic agents in experimental autoimmune encephalomyelitis. Br J Pharmacol. 2013;170:602–613. doi: 10.1111/bph.12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto M, Murakawa M, Kadoshima-Yamaoka K, Tanaka Y, Inoue H, Murafuji H, et al. Phosphodiesterase 7A inhibitor ASB16165 suppresses proliferation and cytokine production of NKT cells. Cell Immunol. 2009;258:147–151. doi: 10.1016/j.cellimm.2009.04.005. [DOI] [PubMed] [Google Scholar]
- Johansson EM, Reyes-Irisarri E, Mengod G. Comparison of cAMP-specific phosphodiesterase mRNAs distribution in mouse and rat brain. Neurosci Lett. 2012;525:1–6. doi: 10.1016/j.neulet.2012.07.050. [DOI] [PubMed] [Google Scholar]
- Kaminuma O, Mori A, Wada K, Kikkawa H, Ikezawa K, Suko M, et al. A selective type 4 phosphodiesterase inhibitor, T-440, modulates intracellular cyclic AMP level and interleukin-2 production of Jurkat cells. Immunopharmacology. 1998;38:247–252. doi: 10.1016/s0162-3109(97)00071-4. [DOI] [PubMed] [Google Scholar]
- Karussis D. The diagnosis of multiple sclerosis and the various related demyelinating syndromes: a critical review. J Autoimmun. 2014;48–49:134–142. doi: 10.1016/j.jaut.2014.01.022. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipp M, van der Valk P, Amor S. Pathology of multiple sclerosis. CNS Neurol Disord Drug Targets. 2012;11:506–517. doi: 10.2174/187152712801661248. [DOI] [PubMed] [Google Scholar]
- Lee R, Wolda S, Moon E, Esselstyn J, Hertel C, Lerner A. PDE7A is expressed in human B-lymphocytes and is up-regulated by elevation of intracellular cAMP. Cell Signal. 2002;14:277–284. doi: 10.1016/s0898-6568(01)00250-9. [DOI] [PubMed] [Google Scholar]
- Loria F, Petrosino S, Hernangomez M, Mestre L, Spagnolo A, Correa F, et al. An endocannabinoid tone limits excitotoxicity in vitro and in a model of multiple sclerosis. Neurobiol Dis. 2010;37:166–176. doi: 10.1016/j.nbd.2009.09.020. [DOI] [PubMed] [Google Scholar]
- Ludwin SK. The pathogenesis of multiple sclerosis: relating human pathology to experimental studies. J Neuropathol Exp Neurol. 2006;65:305–318. doi: 10.1097/01.jnen.0000225024.12074.80. [DOI] [PubMed] [Google Scholar]
- Makranz C, Cohen G, Reichert F, Kodama T, Rotshenker S. cAMP cascade (PKA, Epac, adenylyl cyclase, Gi, and phosphodiesterases) regulates myelin phagocytosis mediated by complement receptor-3 and scavenger receptor-AI/II in microglia and macrophages. Glia. 2006;53:441–448. doi: 10.1002/glia.20303. [DOI] [PubMed] [Google Scholar]
- Malone M, Gary D, Yang IH, Miglioretti A, Houdayer T, Thakor N, et al. Neuronal activity promotes myelination via a cAMP pathway. Glia. 2013;61:843–854. doi: 10.1002/glia.22476. [DOI] [PubMed] [Google Scholar]
- Martinez A, Gil C. cAMP-specific phosphodiesterase inhibitors: promising drugs for inflammatory and neurological diseases. Expert Opin Ther Pat. 2014;24:1311–1321. doi: 10.1517/13543776.2014.968127. [DOI] [PubMed] [Google Scholar]
- Maurice DH, Ke H, Ahmad F, Wang Y, Chung J, Manganiello VC. Advances in targeting cyclic nucleotide phosphodiesterases. Nat Rev Drug Discov. 2014;13:290–314. doi: 10.1038/nrd4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DP, Richards MH, Miller SD. Mouse models of multiple sclerosis: experimental autoimmune encephalomyelitis and Theiler’s virus-induced demyelinating disease. Methods Mol Biol. 2012;900:381–401. doi: 10.1007/978-1-60761-720-4_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecha M, Iñigo PM, Mestre L, Hernangómez M, Borrell J, Guaza C. An easy and fast way to obtain a high number of glial cells from rat cerebral tissue: a beginners approach. Protocol Exchange. 2011 doi: 10.1038/protex.2011.1218. [Google Scholar]
- Mecha M, Carrillo-Salinas FJ, Mestre L, Feliu A, Guaza C. Viral models of multiple sclerosis: neurodegeneration and demyelination in mice infected with Theiler’s virus. Prog Neurobiol. 2013;101–102:46–64. doi: 10.1016/j.pneurobio.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina-Rodriguez EM, Arenzana FJ, Pastor J, Redondo M, Palomo V, Garcia de Sola R, et al. Inhibition of endogenous phosphodiesterase 7 promotes oligodendrocyte precursor differentiation and survival. Cell Mol Life Sci. 2013;70:3449–3462. doi: 10.1007/s00018-013-1340-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mestre L, Correa F, Docagne F, Clemente D, Guaza C. The synthetic cannabinoid WIN 55,212-2 increases COX-2 expression and PGE2 release in murine brain-derived endothelial cells following Theiler’s virus infection. Biochem Pharmacol. 2006;72:869–880. doi: 10.1016/j.bcp.2006.06.037. [DOI] [PubMed] [Google Scholar]
- Mestre L, Docagne F, Correa F, Loria F, Hernangomez M, Borrell J, et al. A cannabinoid agonist interferes with the progression of a chronic model of multiple sclerosis by downregulating adhesion molecules. Mol Cell Neurosci. 2009;40:258–266. doi: 10.1016/j.mcn.2008.10.015. [DOI] [PubMed] [Google Scholar]
- Miro X, Perez-Torres S, Palacios JM, Puigdomenech P, Mengod G. Differential distribution of cAMP-specific phosphodiesterase 7A mRNA in rat brain and peripheral organs. Synapse. 2001;40:201–214. doi: 10.1002/syn.1043. [DOI] [PubMed] [Google Scholar]
- Morales-Garcia JA, Redondo M, Alonso-Gil S, Gil C, Perez C, Martinez A, et al. Phosphodiesterase 7 inhibition preserves dopaminergic neurons in cellular and rodent models of Parkinson disease. PLoS ONE. 2011;6:e17240. doi: 10.1371/journal.pone.0017240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muise ES, Chute IC, Claveau D, Masson P, Boulet L, Tkalec L, et al. Comparison of inhibition of ovalbumin-induced bronchoconstriction in guinea pigs and in vitro inhibition of tumor necrosis factor-alpha formation with phosphodiesterase 4 (PDE4) selective inhibitors. Biochem Pharmacol. 2002;63:1527–1535. doi: 10.1016/s0006-2952(02)00903-6. [DOI] [PubMed] [Google Scholar]
- Njenga MK, Coenen MJ, DeCuir N, Yeh HY, Rodriguez M. Short-term treatment with interferon-alpha/beta promotes remyelination, whereas long-term treatment aggravates demyelination in a murine model of multiple sclerosis. J Neurosci Res. 2000;59:661–670. doi: 10.1002/(SICI)1097-4547(20000301)59:5<661::AID-JNR9>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Paintlia AS, Paintlia MK, Singh I, Skoff RB, Singh AK. Combination therapy of lovastatin and rolipram provides neuroprotection and promotes neurorepair in inflammatory demyelination model of multiple sclerosis. Glia. 2009;57:182–193. doi: 10.1002/glia.20745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterniti I, Mazzon E, Gil C, Impellizzeri D, Palomo V, Redondo M, et al. PDE 7 inhibitors: new potential drugs for the therapy of spinal cord injury. PLoS ONE. 2011;6:e15937. doi: 10.1371/journal.pone.0015937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Gonzalez R, Pascual C, Antequera D, Bolos M, Redondo M, Perez DI, et al. Phosphodiesterase 7 inhibitor reduced cognitive impairment and pathological hallmarks in a mouse model of Alzheimer’s disease. Neurobiol Aging. 2013;34:2133–2145. doi: 10.1016/j.neurobiolaging.2013.03.011. [DOI] [PubMed] [Google Scholar]
- Perez-Torres S, Cortes R, Tolnay M, Probst A, Palacios JM, Mengod G. Alterations on phosphodiesterase type 7 and 8 isozyme mRNA expression in Alzheimer’s disease brains examined by in situ hybridization. Exp Neurol. 2003;182:322–334. doi: 10.1016/s0014-4886(03)00042-6. [DOI] [PubMed] [Google Scholar]
- Petzold A, Baker D, Pryce G, Keir G, Thompson EJ, Giovannoni G. Quantification of neurodegeneration by measurement of brain-specific proteins. J Neuroimmunol. 2003;138:45–48. doi: 10.1016/s0165-5728(03)00092-4. [DOI] [PubMed] [Google Scholar]
- Redondo M, Brea J, Perez DI, Soteras I, Val C, Perez C, et al. Effect of phosphodiesterase 7 (PDE7) inhibitors in experimental autoimmune encephalomyelitis mice. Discovery of a new chemically diverse family of compounds. J Med Chem. 2012a;55:3274–3284. doi: 10.1021/jm201720d. [DOI] [PubMed] [Google Scholar]
- Redondo M, Palomo V, Brea J, Perez DI, Martin-Alvarez R, Perez C, et al. Identification in silico and experimental validation of novel phosphodiesterase 7 inhibitors with efficacy in experimental autoimmune encephalomyelitis mice. ACS Chem Neurosci. 2012b;3:793–803. doi: 10.1021/cn300105c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redondo M, Zarruk JG, Ceballos P, Perez DI, Perez C, Perez-Castillo A, et al. Neuroprotective efficacy of quinazoline type phosphodiesterase 7 inhibitors in cellular cultures and experimental stroke model. Eur J Med Chem. 2012c;47:175–185. doi: 10.1016/j.ejmech.2011.10.040. [DOI] [PubMed] [Google Scholar]
- Reyes-Irisarri E, Perez-Torres S, Mengod G. Neuronal expression of cAMP-specific phosphodiesterase 7B mRNA in the rat brain. Neuroscience. 2005;132:1173–1185. doi: 10.1016/j.neuroscience.2005.01.050. [DOI] [PubMed] [Google Scholar]
- Robichaud A, Savoie C, Stamatiou PB, Tattersall FD, Chan CC. PDE4 inhibitors induce emesis in ferrets via a noradrenergic pathway. Neuropharmacology. 2001;40:262–269. doi: 10.1016/s0028-3908(00)00142-8. [DOI] [PubMed] [Google Scholar]
- Sanchez AJ, Puerta C, Ballester S, Gonzalez P, Arriaga A, Garcia-Merino A. Rolipram impairs NF-kappaB activity and MMP-9 expression in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2005;168:13–20. doi: 10.1016/j.jneuroim.2005.03.024. [DOI] [PubMed] [Google Scholar]
- Sato F, Tanaka H, Hasanovic F, Tsunoda I. Theiler’s virus infection: pathophysiology of demyelination and neurodegeneration. Pathophysiology. 2011;18:31–41. doi: 10.1016/j.pathophys.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer N, Loschmann PA, Northoff GH, Weller M, Steinbrecher A, Steinbach JP, et al. The antidepressant rolipram suppresses cytokine production and prevents autoimmune encephalomyelitis. Nat Med. 1995;1:244–248. doi: 10.1038/nm0395-244. [DOI] [PubMed] [Google Scholar]
- Tasken K, Stokka AJ. The molecular machinery for cAMP-dependent immunomodulation in T-cells. Biochem Soc Trans. 2006;34:476–479. doi: 10.1042/BST0340476. [DOI] [PubMed] [Google Scholar]
- Tsunoda I, Fujinami RS. Neuropathogenesis of Theiler’s murine encephalomyelitis virus infection, an animal model for multiple sclerosis. J Neuroimmune Pharmacol. 2010;5:355–369. doi: 10.1007/s11481-009-9179-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Wauwe J, Aerts F, Walter H, de Boer M. Cytokine production by phytohemagglutinin-stimulated human blood cells: effects of corticosteroids, T cell immunosuppressants and phosphodiesterase IV inhibitors. Inflamm Res. 1995;44:400–405. doi: 10.1007/BF01797868. [DOI] [PubMed] [Google Scholar]
- Yiangou Y, Facer P, Durrenberger P, Chessell IP, Naylor A, Bountra C, et al. COX-2, CB2 and P2X7-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol. 2006;6:12. doi: 10.1186/1471-2377-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]