

Abstract
Background and Purpose
Cyclosporine (CSA) and non-steroidal anti-inflammatory drugs (NSAIDs) are co-prescribed for some arthritic conditions. We tested the hypothesis that this combined regimen elicits exaggerated nephrotoxicity in rats via the up-regulation of endothelin (ET) receptor signalling.
Experimental Approach
The effects of a 10 day treatment with CSA (20 mg·kg−1·day−1), indomethacin (5 mg·kg−1·day−1) or their combination on renal biochemical, inflammatory, oxidative and structural profiles were assessed. The roles of ETA receptor and COX-2 pathways in the interaction were evaluated.
Key Results
Oral treatment with CSA or indomethacin elevated serum urea and creatinine, caused renal tubular atrophy and interstitial fibrosis, increased renal TGF-β1, and reduced immunohistochemical expressions of ETA receptors and COX-2. CSA, but not indomethacin, increased renal ET-1, the lipid peroxidation product malondialdehyde (MDA) and GSH activity. Compared with individual treatments, simultaneous CSA/indomethacin exposure caused: (i) greater elevations in serum creatinine and renal MDA; (ii) loss of the compensatory increase in GSH; (iii) renal infiltration of inflammatory cells and worsening of fibrotic and necrotic profiles; and (iv) increased renal ET-1 and decreased ETA receptor and COX-2 expressions. Blockade of ETA receptors by atrasentan ameliorated the biochemical, structural, inflammatory and oxidative abnormalities caused by the CSA/indomethacin regimen. Furthermore, atrasentan partly reversed the CSA/indomethacin-evoked reductions in the expression of ETA receptor and COX-2 protein.
Conclusions and Implications
The exaggerated oxidative insult and associated dysregulation of the ETA receptor/COX-2/TGF-β1 signalling might account for the aggravated nephrotoxicity caused by the CSA/indomethacin regimen. The potential renoprotective effect of ETA receptor antagonism might be exploited therapeutically.
Tables of Links
| LIGANDS | |
|---|---|
| Cyclosporine | Indomethacin |
| ET-1 | TGF-β1 |
| GSH | Urea |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14
Introduction
The clinical immunosuppressant effect of cyclosporine (CSA) is often limited by the concomitant deterioration in kidney function. Manifestations of acute and chronic nephrotoxicity have been reported with CSA therapy. The acute reversible form of nephrotoxicity is due to renal vasoconstriction (Tedesco and Haragsim, 2012), whereas the chronic toxicity is triggered by vasoconstriction plus the development of irreversible arteriolopathy and tubulointerstitial fibrosis (Musson et al., 2012). Although the mechanisms of the vasoconstrictor effect of CSA are not precisely known, a role for renal vasoconstrictor/vasodilator imbalances has been proposed. These imbalances include increases in vasoconstrictor factors, such as endothelin-1 (ET-1) (Cauduro et al., 2005; Nasser et al., 2014) and thromboxane A2 (Hardy et al., 2000), and decreases in vasodilator factors, such as prostacyclin (Miller, 2002), nitric oxide (El-Mas et al., 2004a; 2007,) and hyperpolarizing factor (El-Mas et al., 2004b). Other factors involved include altered arachidonic acid metabolism and increased oxidative stress and free radical production (Amudha et al., 2007). Reported findings also implicate the ET-1/TGF-β1 signalling in renal dysfunction caused by CSA. CSA enhances the generation of ET-1 (Papachristou et al., 2009), which accentuates vascular derangements and promotes the synthesis and activation of profibrotic molecules such as TGF-β1 and adhesion molecules (Lüscher and Barton, 2000; Leask, 2009).
Clinically, CSA is commonly used with non-steroidal anti-inflammatory drugs (NSAIDs) for the management of several arthritic conditions such as rheumatoid arthritis, juvenile systemic lupus erythematosus and psoriatic arthritis (Gerloni et al., 2001). Like CSA, nephrotoxicity is one of the main side effects caused by NSAIDs and is manifested as salt retention and reduced glomerular filtration rate (Perazella and Tray, 2001). However, little information is available regarding whether additive or synergistic renal damage could emerge upon the concurrent CSA/NSAID exposure. Earlier studies showed that the use of CSA together with naproxen, sulindac or diclofenac causes additive deterioration in renal function (Deray et al., 1987; Altman et al., 1992). In addition, experimental studies demonstrated enhanced mortality upon concurrent use of CSA and indomethacin (Indo) (Williamson, 1988). The mechanisms of these interactions have not been explored.
In the current study, the hypothesis was tested that the deteriorated oxidative state that follows the dysregulation of ETA receptor-coupled COX-2/TGF-β1 signalling accounts for the exaggerated nephrotoxicity caused by the concomitant exposure to CSA and Indo in rats. We evaluated the biochemical, inflammatory, oxidative, and structural renal profiles in rats treated chronically with CSA, Indo or their combination. Immunohistochemical protein expression and receptor pharmacological studies were also employed to verify the role of downstream effectors of ETA receptors in the interaction.
Methods
Animals
Male Sprague-Dawley rats (Faculty of Pharmacy, Pharos University, Alexandria, Egypt) weighing 180–200 g were used in this study. All experiments were performed in strict accordance with institutional animal care and use guidelines. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Experimental protocols
A total of eight groups of rats (n = 6 each) were used in the current study. Rats were treated for 10 days (Yılmaz et al., 2011; El-Mas et al., 2015) with one of the following drug regimens via oral gavage: (i) vehicle (cremophor, 1 mL·kg−1·day−1); (ii) CSA (20 mg·kg−1·day−1; El-Mas et al., 2011); (iii)–(v) Indo (1, 3 or 5 mg·kg−1·day−1; Wong et al., 2008); (vi) CSA + Indo (1 mg·kg−1·day−1), (vii) CSA + Indo (5 mg·kg−1·day−1); and (viii) atrasentan (5 mg·kg−1·day−1) + CSA + Indo (5 mg·kg−1·day−1). Drugs were administered individually via separate oral gavages. Afterwards, overnight-fasted rats were anaesthetized with thiopental sodium (50 mg·kg−1, i.p.; El-Mas et al., 2003) and blood samples were collected from the orbital plexus and spun at 1200× g for 10 min. The serum was aspirated, divided into aliquots and stored at −70°C until used for biochemical analyses. Rats were then killed with an overdose of thiopental, the abdomen was opened, the internal viscera pulled aside, and the right kidney was quickly removed, weighed and homogenized in ice-cold PBS (pH = 7.4) to give 40% homogenate. The homogenate was divided into aliquots and stored at −70°C until used for the measurement of renal ET-1, TGF-β1, malondialdehyde (MDA) (Mihara and Uchiyama, 1978; Nasr et al., 2010), GSH (Richardson and Murphy, 1975) and superoxide dismutase (SOD) activity (Marklund and Marklund, 1974) in the renal tissues. Total protein was determined by using the Bradford assay (Compton and Jones, 1985). For histopathological or immunohistochemical studies, the left kidney was fixed in 10% formaldehyde for 18 h at 4°C and then embedded in paraffin blocks.
Biochemical analysis
Serum urea and creatinine levels were determined by the Randox® assay kit (Randox Laboratories Ltd., Crumlin, UK). The blood urea nitrogen (BUN) was computed from the urea levels. The elisa was used for measuring renal TGF-β1 (Invitrogen Incorporations, Carlsbad, CA, USA) and ET-1 (Antibodies online®, Aachen, Germany) as instructed by the manufacturer.
Histopathological studies
Renal histopathological changes were assessed in kidney sections using the haematoxylin and eosin (H&E) stain. Masson’s trichrome staining was used for detection of renal fibrosis (Drury and Wallington, 1980). Total histological damage in the kidney was assessed semi-quantitatively as described in previous studies including ours (Chatterjee et al., 2000; Helmy and El-Gowelli, 2012; Koca et al., 2013) by examining 10 random sections (×400) from each kidney and scoring tubular necrosis, inflammatory cell infiltration and interstitial fibrosis on a scale from 0 to 3 for each criterion (Figure 1). The mean value of all 10 scores was computed for each kidney. For tubular necrosis, the tubular profile was assessed according to the arbitrary scale, where (0) denoted normal histology, (1) denoted tubular cell swelling, brush border loss with up to one-third of the tubular profile showing nuclear loss, (2) denoted from one-third to two-thirds of the tubular profile showing nuclear loss, and (3) denoted more than two-thirds of the tubular profile showing nuclear loss (Figure 1). Similarly, inflammatory cell infiltration and interstitial fibrosis were assessed in H&E and Masson’s trichrome-stained slides, respectively, and each was scored as absent (0), minimal (1), moderate (2) and severe (3). The total histology score for these three criteria ranged from 0 (normal) to 9 (severely damaged) (Koca et al., 2013).
Figure 1.
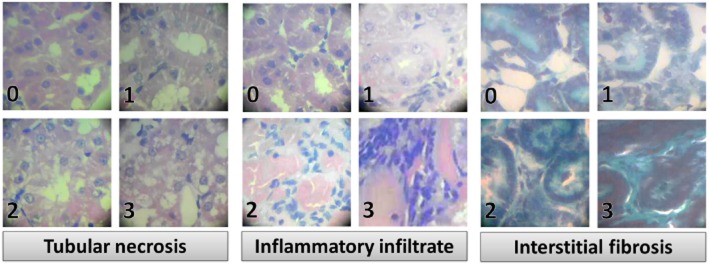
Photomicrographs (400×) showing progressive stages and scores for tubular necrosis, inflammatory cells infiltration (haematoxylin and eosin stain) and interstitial fibrosis (Masson’s trichrome stain).
Immunostaining
The technique described in our previous studies and others (Abassi et al., 2001; Chen and Sun, 2006; El-Mas et al., 2006) was employed for immunohistochemical determination of the protein expression of ETA receptors and COX-2 in the renal tissues. Kidney sections (5 μm) were placed on positively charged adhesion microscope slides (Thermo Scientific®, Berlin, Germany), deparaffinized in xylene and rehydrated in a series of decreasing ethanol concentration (100, 95 and 70%). Slides were rinsed gently with PBS and drained. The antigenic determinants in the cells were unblocked by incubating the sections at (95–98°C) for 20 min in citrate buffer (pH 6, Thermo Scientific) for heat-induced epitope retrieval. The sections were then rinsed with 1× TBST (50 mM Tris/HCl, pH 7.4, 150 mM NaCl, 0.1% Tween 20, Thermo Scientific). Endogenous peroxidases were blocked by adding 3% hydrogen peroxide and then washed with 1× TBST. A universal protein block was applied for 20 min. The appropriate primary monoclonal antibodies, rabbit anti-ETA and rabbit anti-COX-2 (Bioss Inc., Woburn, MA, USA), were diluted (1:200) as instructed by the manufacturer and applied to the slides for 45 min at 37°C. Negative controls were processed without applying the primary antibodies. The slides were then washed with 1× TBST, rinsed and incubated for 30 min with the secondary antibody (polyvalent HRP detection kit, Spring Bioscience®, Pleasanton, CA, USA). The chromogen 3,3′-diaminobenzidine (DAB) was prepared and applied as instructed by the manufacturer for protein visualization. Each slide was counterstained with haematoxylin and dipped in ascending concentrations of alcohol and then xylene. The immunohistochemical signals of ETA receptors and COX-2 were quantified by the Image J software (version 1.45s), and computer-assisted microscopy was employed for this purpose using the greyscale thresholding as described previously (Kaczmarek et al., 2004).
Data analysis and statistics
Data are expressed as means ± SEM. Multiple comparisons were analysed by one-way anova followed by Tukey’s post hoc test. The analysis was performed using GraphPad Prism, software release 3.02 (La Jolla, CA, USA). Probability levels less than 0.05 were considered significant.
Materials
CSA (Novartis Pharma, AG, Basel, Switzerland), Indo (European Egyptian Pharmaceutical Industries, Alexandria, Egypt), cremophor EL (Sigma-Aldrich, MO, USA) and thiopental sodium (Biochemie GmbH, Vienna, Austria) were purchased from commercial vendors. Atrasentan was generously supplied by Abbott Laboratories (Abbott Park, IL, USA). Cremophor (vehicle for CSA) was mixed with saline to a final dilution of 40%. CSA was freshly dissolved in 40% cremophor. Indo, atrasentan and thiopental sodium were dissolved/dispersed in saline. The drug/molecular target nomenclature employed in this study follows Alexander et al. (2013a,b,).
Results
Indo aggravates biochemical and structural indices of CSA nephrotoxicity
The effects of CSA, Indo or their combination on indices of renal function (serum creatinine and BUN) are shown in Figure 2. Compared with control (vehicle-treated) values, oral treatment of rats with CSA (20 mg·kg−1·day−1, 10 days) produced significant increases in serum creatinine and BUN. Similarly, Indo (1, 3 and 5 mg·kg−1·day−1) caused dose-related increases in serum creatinine and BUN levels. The co-administration of CSA and the lower dose of Indo (1 mg·kg−1·day−1) increased creatinine and BUN to levels that were not statistically different from the effect of each drug when used alone (Figure 2). On the other hand, the combined treatment with CSA and Indo (5 mg·kg−1·day−1) elevated serum creatinine, but not BUN, to levels that were slightly but significantly higher than those seen in rats receiving individual treatments (Figure 2). The rises in serum creatinine and BUN seen in CSA/Indo-treated rats were significantly reduced after selective blockade of ETA receptors by atrasentan (5 mg·kg−1·day−1).
Figure 2.
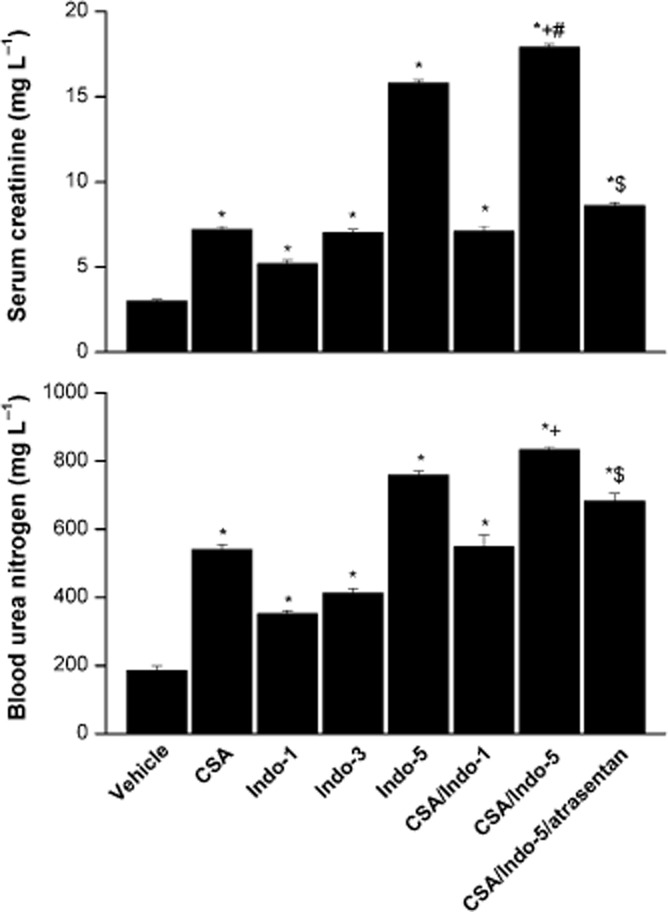
Effects of the 10 day treatment with CSA (20 mg·kg−1·day−1), Indo (1, 3 or 5 mg·kg−1·day−1) or their combination on serum creatinine and BUN in Sprague-Dawley rats. The effect of endothelin ETA receptor blockade by atrasentan (5 mg·kg−1·day−1) on the CSA–Indo combination is also shown. Values are means ± SEM of six observations. *P < 0.05 versus vehicle; +P < 0.05 versus CSA; #P < 0.05 versus Indo-5; $P < 0.05 versus CSA/Indo-5.
Histopathological changes caused by individual or combined treatments with CSA and Indo in the absence and presence of atrasentan are illustrated in Figures 3 and 4. Kidneys obtained from rats treated with CSA showed tubular atrophy and vacuolization (Figure 3B). The glomeruli exhibited slight to moderate mesangial matrix expansion with partial obliteration of Bowman’s space (Figure 3A). Staining with the Masson’s trichrome demonstrated interstitial fibrosis in kidneys of CSA-treated rats (Figure 3C). Renal tissues of Indo (5 mg·kg−1·day−1)-treated rats showed moderate obliteration of Bowman’s space and vacuolated tubules (Figure 3A and B) and slight interstitial fibrosis (Figure 3C). Combined administration of CSA plus Indo induced more intense renal damage manifested as patchy cortical necrosis, tubular atrophy, focal infiltration of inflammatory cells (visual determination) and interstitial fibrosis (Figure 3A–C). The treatment with CSA or Indo caused significant increases in tubular necrosis and interstitial fibrosis scores compared with cremophor-treated rats (Figure 4). Individual scores as well as the total histology severity score showed further increases in rats receiving the combined CSA/Indo regimen compared with either drug when used alone (Figure 4). The glomerular and tubular structural damage and the increases in all histology scores caused by the CSA/Indo regimen were dramatically reduced in rats treated concomitantly with the ETA receptor antagonist atrasentan (Figures 3 and 4).
Figure 3.

Photomicrographs (400×, haematoxylin and eosin) of renal cortical glomeruli (panel A) and tubules (panel B) obtained from Sprague-Dawley rats treated for 10 days with vehicle, CSA (20 mg·kg−1·day−1), Indo (5 mg·kg−1·day−1), CSA + Indo-5 and atrasentan + CSA + Indo-5. Panel (C) shows photomicrographs (400×) of renal cortex stained with Masson’s trichrome. Blue arrows point to tubular vacuolization, black arrows point to interstitial infiltration of inflammatory cells and red arrows point to necrotic areas. Black arrow heads point to deposition of collagen in the interstitial spaces. The scale bar in panel (A) (control image) corresponds to 10 μm.
Figure 4.
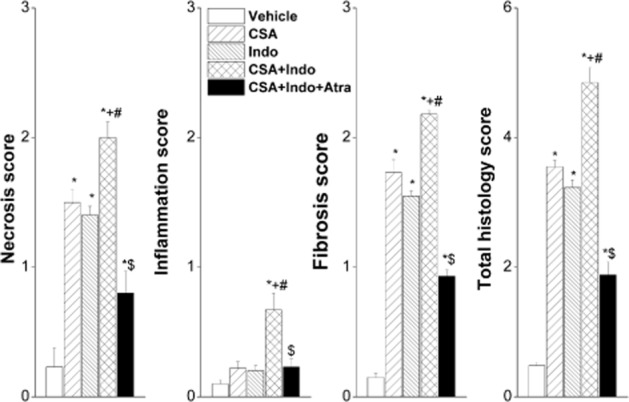
Effects of the 10 day treatment with CSA (20 mg·kg−1·day−1), Indo (5 mg·kg−1·day−1) or their combination on renal necrosis, inflammation, fibrosis and total histology scores. The effect of endothelin ETA receptor blockade by atrasentan (5 mg·kg−1·day−1) on the CSA–Indo combination is also shown. Values are means ± SEM of 6 observations. *P < 0.05 versus vehicle; +P < 0.05 versus CSA; #P < 0.05 versus Indo; $P < 0.05 versus CSA/Indo values.
Effect of CSA/Indo regimen on inflammatory and oxidative renal profiles
As shown in Figures 5 and 6, CSA significantly increased renal ET-1, TGF-β1 and lipid peroxidation (MDA). Renal GSH level was also increased by CSA, whereas SOD activity remained unchanged. Compared with CSA alone, the simultaneous treatment with CSA and Indo elicited similar increases in renal ET-1 and TGF-β1, and significantly higher renal MDA levels. The compensatory increase in renal GSH disappeared in CSA/ Indo-treated rats. The co-administration of atrasentan abolished almost completely the increases in TGF-β1 and MDA evoked by the CSA/ Indo regimen, whereas the concomitant increases in ET-1 were preserved (Figures 5 and 6). Except for significant increases in renal TGF-β1, none of the measured inflammatory or oxidative markers was affected by the single use of Indo.
Figure 5.
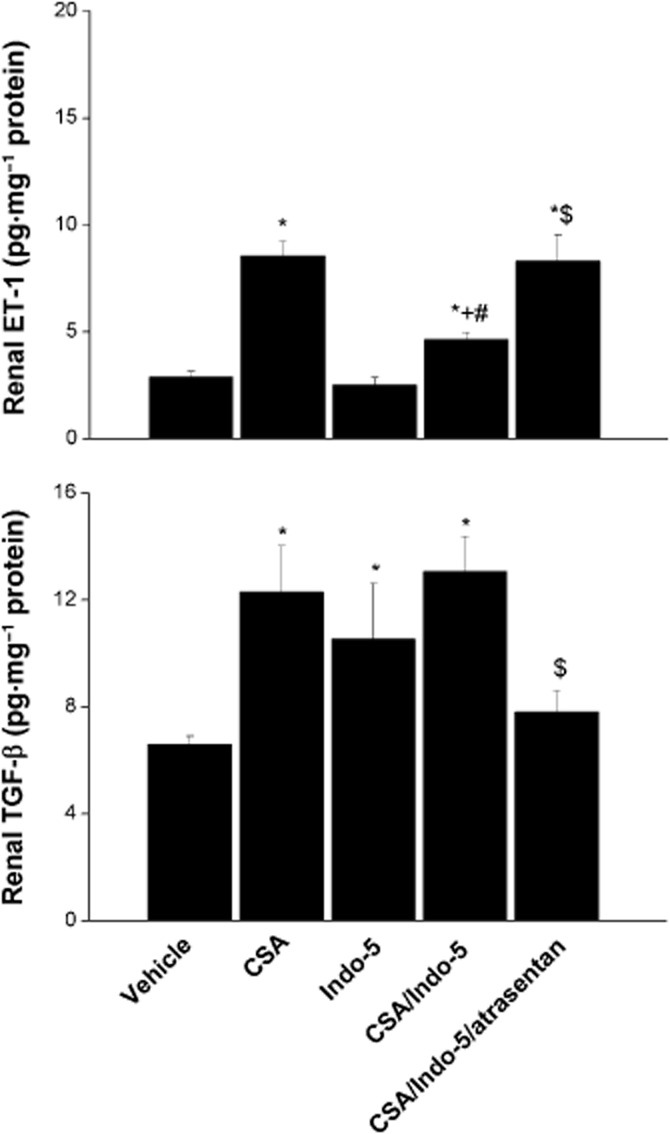
Effects of the 10 day treatment with CSA (20 mg·kg·1·day−1), Indo (5 mg·kg−1·day−1) or their combination on renal ET-1 and TGF-β1 in Sprague-Dawley rats. The effect of endothelin ETA receptor blockade by atrasentan (5 mg·kg−1·day−1) on the CSA–Indo combination is also shown. Values are means ± SEM of six observations. *P < 0.05 versus vehicle; +P < 0.05 versus CSA; #P < 0.05 versus Indo-5; $P < 0.05 versus CSA/Indo-5.
Figure 6.
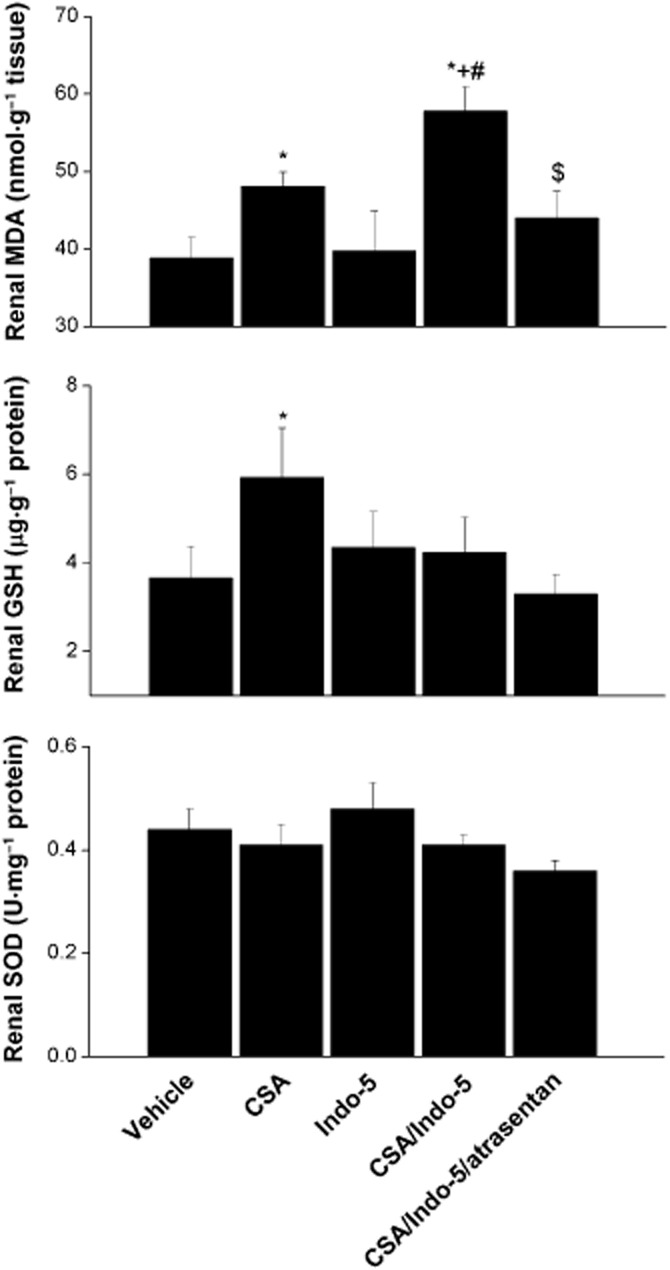
Effects of the 10 day treatment with CSA (20 mg·kg−1·day−1), Indo (5 mg·kg−1·day−1) or their combination on the renal MDA, GSH and SOD in Sprague-Dawley rats. The effect of endothelin ETA receptor blockade by atrasentan (5 mg·kg−1·day−1) on the CSA–Indo combination is also shown. Values are means ± SEM of six observations. *P < 0.05 versus vehicle; +P < 0.05 versus CSA; #P < 0.05 versus Indo-5; $P < 0.05 versus CSA/Indo-5.
Effect of CSA/Indo regimen on renal ETA receptors and COX-2 expressions
The immunohistochemical data are shown in Figures 7 and 8. Compared with the vehicle-treated group, CSA caused significant decreases in the protein expression of glomerular ETA receptors, whereas the medullary tubular ETA receptor expression remained unaffected (Figure 7). By contrast, Indo given alone or combined with CSA caused significant and similar reductions in the ETA receptor expression in both glomerular and tubular sites (Figure 7). The CSA/Indo-evoked decreases in glomerular ETA receptor were increased after ETA receptor blockade by atrasentan, but its levels remained significantly lower than the respective vehicle-treated values (Figure 7). On the other hand, COX-2 expression in glomerular and tubular tissues was comparably reduced by CSA, Indo or their combination and these effects were significantly alleviated in the presence of atrasentan (Figure 8).
Figure 7.
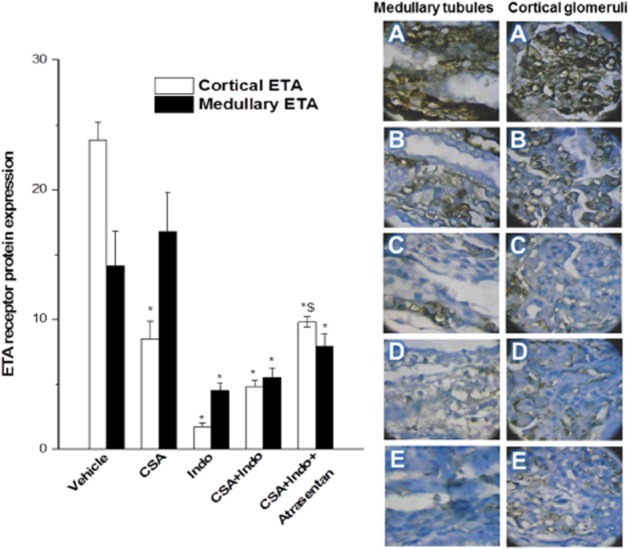
Immunohistochemical ETA receptor expression in renal cortical glomeruli and medullary tubules of Sprague-Dawley rats treated for 10 days with CSA (20 mg·kg−1·day−1), Indo (5 mg·kg−1·day−1) or their combination. The effect of endothelin ETA receptor blockade by atrasentan (5 mg·kg−1·day−1) with the CSA–Indo combination on ETA expression is also shown. Values are means ± SEM of six observations. *P < 0.05 versus vehicle; $P < 0.05 versus CSA/Indo-5. Representative images of immunostained tissues are also shown (400×, A: vehicle; B: CSA; C: Indo; D: CSA + Indo, E: atrasentan + CSA + Indo).
Figure 8.
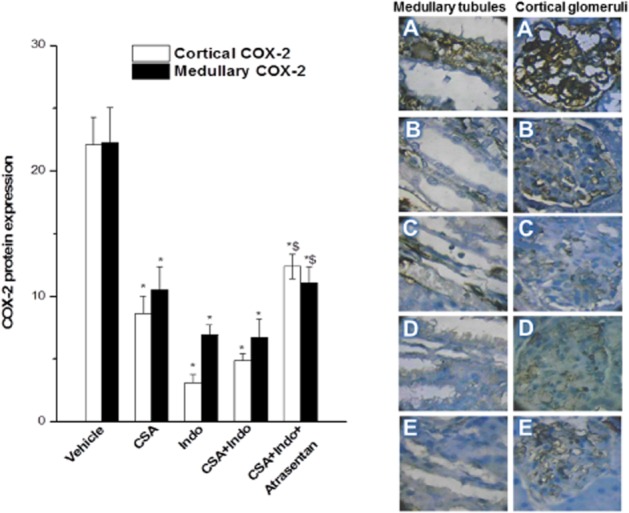
Immunohistochemical COX-2 expression in renal cortical glomeruli and medullary tubules of Sprague-Dawley rats treated for 10 days with CSA (20 mg·kg−1·day−1), Indo (5 mg·kg−1·day−1) or their combination. The effect of endothelin ETA receptor blockade by atrasentan (5 mg·kg−1·day−1) with the CSA – Indo combination on COX-2 expression is also shown. Values are means ± SEM of six observations. *P < 0.05 versus vehicle; $P < 0.05 versus CSA/Indo-5. Representative images of immunostained tissues are also shown (400×, A: vehicle; B: CSA; C: Indo; D: CSA + Indo, E: atrasentan + CSA + Indo).
Discussion
The current study is the first to report on the potential molecular mechanism of the exacerbated nephrotoxicity evoked by simultaneous exposure to CSA and Indo in rats. The most important observations can be summarized as follows. First, compared with individual treatments, the combined CSA/Indo regimen caused more deterioration in the renal profile as indicated by functional (increases in serum creatinine and BUN) and structural indices (renal cortical necrosis, tubular atrophy and interstitial fibrosis). Second, these effects were associated with exacerbated redox state as suggested by the excessive increases in the renal content of the lipid peroxidation product, MDA, and loss of the counter-regulatory elevation in GSH. Third, the oxidative damage caused by CSA/Indo seems to be facilitated by the concomitant elevations in TGF-β1 and decreases in COX-2 expression. Fourth, the blockade of ETA receptors by atrasentan significantly mitigated functional, structural and inflammatory derangements caused by the CSA/Indo regimen. Together, it is likely that the oxidative insult and associated dysregulation of the ETA receptor/COX-2/TGF-β1 signalling accounts, at least partly, for the exaggerated nephrotoxicity evoked by concurrent CSA/Indo administration.
Earlier studies demonstrated more prominent renal dysfunction in rats treated with the combined CSA/Indo regimen compared with each drug when used alone (Whiting et al., 1987; Williamson, 1988). Nonetheless, the latter studies were limited in scope and their conclusions were based solely on the ability of the combined regimen to elicit greater reductions in creatinine clearance (Williamson, 1988) and elevations in serum urea and creatinine (Whiting et al., 1987). In the current study, in addition to the elevated serum creatinine, biochemical markers of renal function, in rats treated with the same drug regimen, our histopathological data showed intensified renal structural damage in CSA/Indo-treated rats. The latter comprised more intense cortical necrosis, tubular atrophy, interstitial fibrosis and inflammatory cell infiltration. Nonetheless, the type of inflammatory cells (macrophages, CD4+ helper or CD8+ cytotoxic) that might be contributing to the inflammatory response elicited by the CSA/Indo treatment cannot be ascertained from the current data. Immunohistochemical studies will be undertaken in our laboratory to address this important issue.
Obviously, the morphological changes in the renal architecture caused by the combined CSA/Indo regimen may explain the associated rises in serum creatinine observed in the current study and the reduced creatinine clearance caused by the same treatment in earlier studies (Williamson, 1988). Notwithstanding, unlike our current histopathological data, Whiting et al. (1987) found no exaggeration of the renal structural damage on concurrent administration of CSA and Indo. Although the exact reasons for these discrepancies are not clear, one possible explanation might relate to the differences in the dose of Indo employed in the two studies (5 mg·kg−1·day−1 in the current study vs. 2 mg·kg−1·day−1 in the study by Whiting et al., 1987). In fact, our data showed that Indo at a dose of 1 mg·kg−1·day−1 failed to accentuate the CSA-evoked rises in blood creatinine or BUN (see Figure 2). Therefore, the strength (dose) of Indo seems critical for its capacity to exacerbate of CSA nephrotoxicity.
Oxidative stress is a key contributor to the pathogenesis of CSA-induced organ damage and dysfunction (El-Mas et al., 2011; 2012,; de Arriba et al., 2013). The current observation that CSA caused significant elevations in the renal content of MDA, the lipid peroxidation product of oxidative destruction of polyunsaturated fatty acids of biological membranes, is in line with the established role of oxidative stress caused by lipid peroxides in CSA-induced nephrotoxicity (Ishikawa and Homma, 2012). Alternatively, the concomitant increase in renal GSH might presumably constitute a feedback mechanism to offset the progression in oxidative damage caused by CSA (Helmy and El-Gowelli, 2012). This assumption gains support from the observation that N-acetyl cysteine, the precursor of GSH, protected against oxidative stress induced by CSA and reversed the CSA-evoked decreases in SOD activity (Duru et al., 2008). In another study (Gökçe et al., 2009), the increases in renal MDA caused by CSA were counterbalanced by elevations in the SOD activity. Notably, while the current study revealed no influence for CSA on renal SOD, the reported effects of CSA on renal SOD were variable and depended upon the CSA regimen. For example, while renal MDA and SOD were elevated by a CSA regimen of 15 mg·kg−1·day−1 for 10 days (Gökçe et al., 2009), the SOD activity was reduced by CSA at a dose of 20 mg·kg−1·day−1 given for 21 days (Anjaneyulu et al., 2003).
Evidently, the worsened renal profile in CSA/Indo-treated rats infers additive effects for the two drugs on cellular events predisposing to renal dysfunction. In the present study, we investigated the postulate that such exacerbated nephrotoxicity is mediated via the development of more deterioration in the redox state. Consistent with this view, two important findings are reported here. First, while Indo had no effect on renal MDA when used alone, the concurrent administration of CSA and Indo increased renal MDA to levels that were significantly higher than those caused by CSA alone (see Figure 6). The generation of excessive amounts of MDA denotes the ability of Indo to prime renal tissues to the lipid-peroxidating effect of CSA. Another factor that might have incited the renal oxidative damage of the CSA/Indo regimen was its ability to block the compensatory elevation in renal GSH induced by CSA, thus depriving renal tissues of one fundamental antioxidant (defence) mechanism against oxidative damage.
The renal oxidative damage directly correlates with increased renal arteriolar tone (Gao et al., 2015), which is a primary event that could subsequently lead, if not corrected, to irreversible arteriolopathy and tubulointerstitial fibrosis (Musson et al., 2012; Tedesco and Haragsim, 2012). The present data also suggest that several other cellular events might have intervened to intensify renal damage caused by oxidative stress. One potential mechanism relates perhaps to the divergent actions of the CSA/Indo regimen on the renal COX-2 expression (decreases) and TGF-β1 (increases). COX-2 inhibition is believed to reduce renal blood flow, up-regulate TGF-β1 gene and enhance fibrosis as a consequence of diminished generation of prostaglandins and related vasodilator and antifibrotic properties (Harris and Breyer, 2001; Rosenberger et al., 2003; Liu et al., 2010). Further, the improvement of the renal profile of CSA-treated rats after concomitant administration of dimethyl PGE2 suggests an important role for COX-derived PG deficiency in CSA nephrotoxicity (Whiting et al., 1987). The increased TGF-β1 promotes interstitial fibrosis by decreasing the degradation and increasing the production of extracellular matrix proteins (Wolf, 2006). Others also showed that the renal fibrotic response elicited by TGF-β1 is enhanced by lipid peroxidation in hyperlipidaemic rats (Dai et al., 2014). It is noteworthy, however, that contradictory data of positive and negative regulatory actions for TGF-β1 on COX-2 expression are available (Matsumura et al., 2009; Warner et al., 2011; Singh et al., 2012). Together, our data suggest that the lipid peroxides-coupled COX-2/TGF-β1 signalling might account for the exaggerated nephrotoxicity caused by the combined CSA/Indo regimen.
Since endothelin ETA receptors enhance vascular and mesangial cell proliferation, renal fibrosis and extracellular matrix production (Leask, 2009; Neuhofer and Pittrow, 2009), we asked if these receptors mediate the renal CSA–Indo interaction. Pharmacological data showed that selective blockade of ETA receptors by atrasentan alleviated most of nephrotoxic manifestations caused by CSA/Indo administration, including biochemical and structural indices of renal dysfunction, lipid peroxidation and reciprocal changes in COX-2 (decreases) and TGF-β1 levels (increases). Conceivably, these findings favour a pivotal role for the ETA receptor as an upstream effector whose activation would trigger a cascade of molecular and cellular changes that incite the CSA/Indo-evoked renal abnormalities. In a similar paradigm, selective ETA receptor antagonism has been shown to protect against diabetic glomerulopathy by reducing mRNA levels of extracellular matrix components and growth factors including TGF-β1 (Nakamura et al., 1995).
The presumption that ETA receptors positively correlate with the exaggerated nephrotoxicity in CSA/Indo-treated rats might be challenged by the observation that the immunohistochemical signal of ETA receptors was significantly reduced in glomerular and tubular tissues of these rats. While these findings might apparently argue against a causal relationship between ETA receptors and the deteriorated renal profile in this rat model, it is possible that the reduced abundance of renal ETA receptors might constitute an adaptive mechanism to counterbalance the expected rises in renovascular tone caused by concurrent increases in renal ET-1 and lipid peroxidation, on the one hand, and decreases in COX-2 expression and vasodilator prostaglandin production, on the other hand. The paradigm of a compensatory ETA receptor down-regulation in the face of elevated levels of vasoconstrictors and oxidative stress has been documented by others (Rajagopalan et al., 1997; Hink and Münzel, 2006). More importantly, we have recently shown that unlike its current effect in renal glomerular and tubular tissues, the combined CSA/Indo regimen significantly increased the renal arteriolar ETA receptor expression (El-Mas et al., 2015). Although the reason for the contrasting, region-dependent effect of the CSA/Indo regimen on ETA receptor density in vascular (increases, El-Mas et al., 2015) and non-vascular renal tissues (decreases, this study) is not clear, the contribution of arteriolar ETA receptor up-regulation to the exaggerated nephrotoxicity in CSA/Indo-treated rats cannot be ruled out.
It is imperative to comment on the role of renal ET-1 in the developed nephrotoxicity. Examination of the individual and combined effects of CSA and Indo showed that changes in renal ET-1 (Figure 5) were not consistently related to ETA receptor expression (Figure 7) or biochemical (Figure 1) or structural indices of nephrotoxicity (Figures 3 and 4). Specifically, smaller rises in renal ET-1 were demonstrated in CSA/Indo-treated rats compared with rats receiving CSA alone. These findings do not favour a role for renal ET-1 as such in the aggravated nephrotoxicity in CSA/Indo-treated rats and might infer a more prominent role for ETA receptors or downstream inflammatory and oxidative pathways in the evoked nephrotoxicity. With that in mind, a potential contribution of ET-1 in this context cannot be overlooked. Indeed, evidence suggests that even with low concentrations of ET-1, which produce either no or minimal vasoconstriction on their own, substantial increases in responses to other vasoconstrictor agents might occur (Henrion and Laher, 1993; Millette et al., 2003).
In summary, the current functional and histopathological data provide the first experimental evidence that implicate the enhanced renal oxidative damage due to excessive generation of lipid peroxides in the worsened renal profile in rats co-treated with CSA and Indo. The detrimental renal effect of lipid peroxidation seems to be exacerbated by the concomitant directional opposite changes in ET-1/TGF-β1 signalling (facilitation) and COX-2 expression (suppression). Despite the reduced ETA receptor expression in kidneys of CSA/Indo-treated rats, pharmacological antagonist studies showed that the residual ETA receptor sites remain conducive to the developed nephrotoxicity and predisposing cellular mechanisms. Clinically, the study emphasizes the additive nephrotoxic consequences of the CSA/Indo regimen and highlights a potential renoprotective effect for selective ETA receptor antagonism against renal dysfunction caused by this particular regimen or probably by other similar insults.
Acknowledgments
This work was supported by the ALEX-REP Grant Fund from Alexandria University, Egypt (Grant No. HLTH-13-01). The authors thank Dr. Reda S. Saad, Adjunct Professor, Department of Pathology, Western University, Ontario, Canada, and Hanan Mostafa Menisi, Department of Pathology, Faculty of Medicine, Alexandria University, for their help with the histopathology.
Glossary
- BUN
blood urea nitrogen
- CSA
cyclosporine
- DAB
diaminobenzidine
- ET-1
endothelin-1
- Indo
indomethacin
- MDA
malondialdehyde
- NSAIDs
non-steroidal anti-inflammatory drugs
- SOD
superoxide dismutase
Author contributions
M. W. H. was responsible for the experimental design, data analysis and presentation, and manuscript preparation. H. M. E.-G. was responsible for the experimental design, data analysis and presentation, and manuscript preparation. R. M. A. performed the experiments and data analysis. M. M. E.-M. was responsible for research idea, experimental design, data analysis and presentation, manuscript preparation, and work overseeing.
Conflict of interest
There is no conflict of interest to declare.
References
- Abassi Z, Brodsky S, Gealekman O, Rubinstein I, Hoffman A, Winaver J. Intrarenal expression and distribution of cyclooxygenase isoforms in rats with experimental heart failure. Am J Physiol Renal Physiol. 2001;280:F43–F53. doi: 10.1152/ajprenal.2001.280.1.F43. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: enzymes. Br J Pharmacol. 2013b;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman RD, Perez GO, Sfakianakis GN. Interaction of cyclosporine A and nonsteroidal anti-inflammatory drugs on renal function in patients with rheumatoid arthritis. Am J Med. 1992;93:396–402. doi: 10.1016/0002-9343(92)90169-c. [DOI] [PubMed] [Google Scholar]
- Amudha G, Josephine A, Sudhahar V, Varalakshmi P. Protective effect of lipoic acid on oxidative and peroxidative damage in cyclosporine A-induced renal toxicity. Int Immunopharmacol. 2007;7:1442–1449. doi: 10.1016/j.intimp.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Anjaneyulu M, Tirkey N, Chopra K. Attenuation of cyclosporine-induced renal dysfunction by catechin: possible antioxidant mechanism. Ren Fail. 2003;25:691–707. doi: 10.1081/jdi-120024285. [DOI] [PubMed] [Google Scholar]
- de Arriba G, Calvino M, Benito S, Parra T. Cyclosporine A-induced apoptosis in renal tubular cells is related to oxidative damage and mitochondrial fission. Toxicol Lett. 2013;218:30–38. doi: 10.1016/j.toxlet.2013.01.007. [DOI] [PubMed] [Google Scholar]
- Cauduro RL, Costa C, Lhulier F, Garcia RG, Cabral RD, Gonçalves LF, et al. Endothelin-1 plasma levels and hypertension in cyclosporine-treated renal transplant patients. Clin Transplant. 2005;19:470–474. doi: 10.1111/j.1399-0012.2005.00357.x. [DOI] [PubMed] [Google Scholar]
- Chatterjee PK, Cuzzocrea S, Brown PA, Zacharowski K, Stewart KN, Mota-Filipe H, et al. Tempol, a membrane-permeable radical scavenger, reduces oxidant stress-mediated renal dysfunction and injury in the rat. Kidney Int. 2000;58:658–673. doi: 10.1046/j.1523-1755.2000.00212.x. [DOI] [PubMed] [Google Scholar]
- Chen GF, Sun Z. Effects of chronic cold exposure on the endothelin system. J Appl Physiol. 2006;100:1719–1726. doi: 10.1152/japplphysiol.01407.2005. [DOI] [PubMed] [Google Scholar]
- Compton SJ, Jones CG. Mechanism of dye response and interference in the Bradford protein assay. Anal Biochem. 1985;151:369–374. doi: 10.1016/0003-2697(85)90190-3. [DOI] [PubMed] [Google Scholar]
- Dai Y, Palade P, Wang X, Mercanti F, Ding Z, Dai D, et al. High fat diet causes renal fibrosis in LDLr-null mice through MAPK-NF-κB pathway mediated by Ox-LDL. J Cardiovasc Pharmacol. 2014;63:158–166. doi: 10.1097/FJC.0000000000000035. [DOI] [PubMed] [Google Scholar]
- Deray G, Le Hoang P, Aupetit B, Achour A, Rottembourg J, Baumelou A. Enhancement of cyclosporine A nephrotoxicity by diclofenac. Clin Nephrol. 1987;27:213–214. [PubMed] [Google Scholar]
- Drury RAB, Wallington EA. Carleton’s Histological Techniques. 5th edn. New York: Oxford University Press; 1980. [Google Scholar]
- Duru M, Nacar A, Yönden Z, Kuvandik G, Helvaci MR, Koç A, et al. Protective effects of N-acetylcysteine on cyclosporine-A-induced nephrotoxicity. Ren Fail. 2008;30:453–459. doi: 10.1080/08860220801985942. [DOI] [PubMed] [Google Scholar]
- El-Mas MM, Afify EA, Omar AG, Mohy El-Din MM, Sharabi FM. Testosterone depletion contributes to cyclosporine-induced chronic impairment of acetylcholine renovascular relaxations. Eur J Pharmacol. 2003;468:217–224. doi: 10.1016/s0014-2999(03)01720-5. [DOI] [PubMed] [Google Scholar]
- El-Mas MM, Mohy El-Din MM, El-gowilly SM, Sharabi FM. Regional and endothelial differences in the cyclosporine attenuation of adenosine receptor-mediated vasorelaxations. J Cardiovasc Pharmacol. 2004a;43:562–573. doi: 10.1097/00005344-200404000-00012. [DOI] [PubMed] [Google Scholar]
- El-Mas MM, Mohy El-Din MM, El-Gowilly SM, Sharabi FM. Relative roles of endothelial relaxing factors in cyclosporine-induced impairment of cholinergic and beta-adrenergic renal vasodilations. Eur J Pharmacol. 2004b;487:149–158. doi: 10.1016/j.ejphar.2004.01.025. [DOI] [PubMed] [Google Scholar]
- El-Mas MM, Zhang J, Abdel-Rahman AA. Upregulation of vascular inducible nitric oxide synthase mediates the hypotensive effect of ethanol in conscious female rats. J Appl Physiol. 2006;100:1011–1018. doi: 10.1152/japplphysiol.01058.2005. [DOI] [PubMed] [Google Scholar]
- El-Mas MM, Sharabi FM, El-gowilly SM, Mohy El-Din MM. Inhibition of nitric oxide-guanylate cyclase-dependent and -independent signaling contributes to impairment of β-adrenergic vasorelaxations by cyclosporine. Biochem Pharmacol. 2007;73:359–367. doi: 10.1016/j.bcp.2006.10.015. [DOI] [PubMed] [Google Scholar]
- El-Mas MM, El-Gowelli HM, Abd-Elrahman KS, Saad EI, Abdel-Galil AG, Abdel-Rahman AA. Pioglitazone abrogates cyclosporine-evoked hypertension via rectifying abnormalities in vascular endothelial function. Biochem Pharmacol. 2011;81:526–533. doi: 10.1016/j.bcp.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Mas MM, Mohy El-Din MM, Helmy MM, Omar AG. Redox imbalances incite the hypertensive, baroreflex, and autonomic effects of cyclosporine in rats. Eur J Pharmacol. 2012;694:82–88. doi: 10.1016/j.ejphar.2012.08.021. [DOI] [PubMed] [Google Scholar]
- El-Mas MM, Helmy MW, Ali RM, El-Gowelli HM. Celecoxib, but not indomethacin, ameliorates the hypertensive and perivascular fibrotic actions of cyclosporine in rats: role of endothelin signaling. Toxicol Appl Pharmacol. 2015;284:1–7. doi: 10.1016/j.taap.2015.01.018. [DOI] [PubMed] [Google Scholar]
- Gao X, Yang T, Liu M, Peleli M, Zollbrecht C, Weitzberg E, et al. NADPH oxidase in the renal microvasculature is a primary target for blood pressure-lowering effects by inorganic nitrate and nitrite. Hypertension. 2015;65:161–170. doi: 10.1161/HYPERTENSIONAHA.114.04222. [DOI] [PubMed] [Google Scholar]
- Gerloni V, Cimaz R, Gattinara M, Arnoldi C, Pontikaki I, Fantini F. Efficacy and safety profile of cyclosporin A in the treatment of juvenile chronic (idiopathic) arthritis. Results of a 10-year prospective study. Rheumatology (Oxford) 2001;40:907–913. doi: 10.1093/rheumatology/40.8.907. [DOI] [PubMed] [Google Scholar]
- Gökçe A, Oktar S, Yönden Z, Aydin M, Ilhan S, Ozkan OV, et al. Protective effect of caffeic acid phenethyl ester on cyclosporine A-induced nephrotoxicity in rats. Ren Fail. 2009;31:843–847. doi: 10.3109/08860220903137517. [DOI] [PubMed] [Google Scholar]
- Hardy G, Stanke-Labesque F, Deveaux G, Devillier P, Sessa C, Bessard G. Cyclosporine A and cremophor EL induce contractions of human saphenous vein: involvement of thromboxane A2 receptor-dependent pathway. J Cardiovasc Pharmacol. 2000;36:693–698. doi: 10.1097/00005344-200012000-00002. [DOI] [PubMed] [Google Scholar]
- Harris RC, Breyer MD. Physiological regulation of cyclooxygenase- 2 in the kidney. Am J Physiol Renal Physiol. 2001;281:F1–F11. doi: 10.1152/ajprenal.2001.281.1.F1. [DOI] [PubMed] [Google Scholar]
- Helmy MM, El-Gowelli HM. Montelukast abrogates rhabdomyolysis-induced acute renal failure via rectifying detrimental changes in antioxidant profile and systemic cytokines and apoptotic factors production. Eur J Pharmacol. 2012;683:294–300. doi: 10.1016/j.ejphar.2012.03.018. [DOI] [PubMed] [Google Scholar]
- Henrion D, Laher I. Potentiation of norepinephrine-induced contractions by endothelin-1 in the rabbit aorta. Hypertension. 1993;22:78–83. doi: 10.1161/01.hyp.22.1.78. [DOI] [PubMed] [Google Scholar]
- Hink U, Münzel T. COX-2, another important player in the nitric oxide-endothelin cross-talk: good news for COX-2 inhibitors? Circ Res. 2006;98:1344–1346. doi: 10.1161/01.RES.0000228471.38761.93. [DOI] [PubMed] [Google Scholar]
- Ishikawa A, Homma Y. Beneficial effect of ubiquinol, the reduced form of coenzyme Q10, on cyclosporine nephrotoxicity. Int Braz J Urol. 2012;38:230–234. doi: 10.1590/s1677-55382012000200011. [DOI] [PubMed] [Google Scholar]
- Kaczmarek E, Górna A, Majewski P. Techniques of image analysis for quantitative immunohistochemistry. Rocz Akad Med Bialymst. 2004;49(Suppl. 1):155–158. [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koca O, Gökçe AM, Öztürk MI, Ercan F, Yurdakul N, Karaman MI. Effects of intensive cell phone (Philips Genic 900) use on the rat kidney tissue. Urol J. 2013;10:886–891. [PubMed] [Google Scholar]
- Leask A. Signaling in fibrosis: targeting the TGF beta, endothelin-1 and CCN2 axis in scleroderma. Front Biosci (Elite Ed) 2009;1:115–122. doi: 10.2741/E12. [DOI] [PubMed] [Google Scholar]
- Liu F, Mih JD, Shea BS, Kho AT, Sharif AS, Tager AM, et al. Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J Cell Biol. 2010;190:693–706. doi: 10.1083/jcb.201004082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher TF, Barton M. Endothelins and endothelin receptor antagonists: therapeutic considerations for a novel class of cardiovascular drugs. Circulation. 2000;102:2434–2440. doi: 10.1161/01.cir.102.19.2434. [DOI] [PubMed] [Google Scholar]
- Marklund S, Marklund G. Involvement of the superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur J Biochem. 1974;47:469–474. doi: 10.1111/j.1432-1033.1974.tb03714.x. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura T, Suzuki T, Aizawa K, Sawaki D, Munemasa Y, Ishida J, et al. Regulation of transforming growth factor-beta-dependent cyclooxygenase-2 expression in fibroblasts. J Biol Chem. 2009;284:35861–35871. doi: 10.1074/jbc.M109.014639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihara M, Uchiyama M. Determination of malonaldehyde precursor in tissues by thiobarbituric acid test. Anal Biochem. 1978;86:271–278. doi: 10.1016/0003-2697(78)90342-1. [DOI] [PubMed] [Google Scholar]
- Miller LW. Cardiovascular toxicities of immunosuppressive agents. Am J Transplant. 2002;2:807–818. doi: 10.1034/j.1600-6143.2002.20902.x. [DOI] [PubMed] [Google Scholar]
- Millette E, de Champlain J, Lamontagne D. Contribution of endogenous endothelin in the enhanced coronary constriction in DOCA-salt hypertensive rats. J Hypertens. 2003;21:115–123. doi: 10.1097/00004872-200301000-00021. [DOI] [PubMed] [Google Scholar]
- Musson RE, Cobbaert CM, Smit NP. Molecular diagnostics of calcineurin-related pathologies. Clin Chem. 2012;58:511–522. doi: 10.1373/clinchem.2011.167296. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Ebihara I, Fukui M, Tomino Y, Koide H. Effect of a specific endothelin receptor A antagonist on mRNA level for extracellular matrix components and growth factors in diabetic glomeruli. Diabetes. 1995;44:895–899. doi: 10.2337/diab.44.8.895. [DOI] [PubMed] [Google Scholar]
- Nasr MA, El-gowilly SM, El-Mas MM. Comparable renovascular protective effects of moxonidine and simvastatin in rats exposed to cigarette smoke. Vascul Pharmacol. 2010;53:53–60. doi: 10.1016/j.vph.2010.03.006. [DOI] [PubMed] [Google Scholar]
- Nasser SA, Elmallah AI, Sabra R, Khedr MM, El-Din MM, El-Mas MM. Blockade of endothelin ETA, but not thromboxane, receptors offsets the cyclosporine-evoked hypertension and interrelated baroreflex and vascular dysfunctions. Eur J Pharmacol. 2014;727:52–59. doi: 10.1016/j.ejphar.2014.01.034. [DOI] [PubMed] [Google Scholar]
- Neuhofer W, Pittrow D. Endothelin receptor selectivity in chronic kidney disease: rationale and review of recent evidence. Eur J Clin Invest. 2009;39(Suppl. 2):50–67. doi: 10.1111/j.1365-2362.2009.02121.x. [DOI] [PubMed] [Google Scholar]
- Papachristou E, Papadimitropoulos A, Kotsantis P, Goumenos DS, Katsoris PG, Vlachojannis JG. Cyclosporine induces endothelin-1 mRNA synthesis and nitric oxide production in human proximal tubular epithelial cell cultures. Ren Fail. 2009;31:372–376. doi: 10.1080/08860220902882022. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perazella MA, Tray K. Selective cyclooxygenase-2 inhibitors: a pattern of nephrotoxicity similar to traditional non-steroidal anti-inflammatory drugs. Am J Med. 2001;111:64–67. doi: 10.1016/s0002-9343(01)00757-4. [DOI] [PubMed] [Google Scholar]
- Rajagopalan S, Laursen JB, Borthayre A, Kurz S, Keiser J, Haleen S, et al. Role for endothelin-1 in angiotensin II-mediated hypertension. Hypertension. 1997;30:29–34. doi: 10.1161/01.hyp.30.1.29. [DOI] [PubMed] [Google Scholar]
- Richardson RJ, Murphy SD. Effect of glutathione depletion on tissue deposition of methylmercury in rats. Toxicol Appl Pharmacol. 1975;31:505–519. doi: 10.1016/0041-008x(75)90274-4. [DOI] [PubMed] [Google Scholar]
- Rosenberger C, Griethe W, Gruber G, Wiesener M, Frei U, Bachmann S, et al. Cellular responses to hypoxia after renal segmental infarction. Kidney Int. 2003;64:874–886. doi: 10.1046/j.1523-1755.2003.00159.x. [DOI] [PubMed] [Google Scholar]
- Singh M, Chaudhry P, Parent S, Asselin E. Ubiquitin-proteasomal degradation of COX-2 in TGF-β stimulated human endometrial cells is mediated through endoplasmic reticulum mannosidase I. Endocrinology. 2012;153:426–437. doi: 10.1210/en.2011-1438. [DOI] [PubMed] [Google Scholar]
- Tedesco D, Haragsim L. Cyclosporine: a review. J Transplant. 2012;2012:230386. doi: 10.1155/2012/230386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner L, Yin M, Glaser KJ, Woollard JA, Carrascal CA, Korsmo MJ, et al. Noninvasive in vivo assessment of renal tissue elasticity during graded renal ischemia using MR elastography. Invest Radiol. 2011;46:509–514. doi: 10.1097/RLI.0b013e3182183a95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting PH, Barnard N, Neilsch A, Simpson JG, Burke MD. Interactions between cyclosporin A, indomethacin and 16,16-dimethyl prostaglandin E2: effects on renal, hepatic and gastrointestinal toxicity in the rat. Br J Exp Pathol. 1987;68:777–786. [PMC free article] [PubMed] [Google Scholar]
- Williamson HE. Interaction of cyclosporine and indomethacin in the rat. Res Commun Chem Pathol Pharmacol. 1988;61:141–144. [PubMed] [Google Scholar]
- Wolf G. Renal injury due to renin-angiotensin-aldosterone system activation of the transforming growth factor-beta pathway. Kidney Int. 2006;70:1914–1919. doi: 10.1038/sj.ki.5001846. [DOI] [PubMed] [Google Scholar]
- Wong YF, Zhou H, Wang JR, Xie Y, Xu HX, Liu L. Anti-inflammatory and analgesic effects and molecular mechanisms of JCICM-6, a purified extract derived from an anti-arthritic Chinese herbal formula. Phytomedicine. 2008;15:416–426. doi: 10.1016/j.phymed.2008.02.008. [DOI] [PubMed] [Google Scholar]
- Yılmaz N, Ilhan S, Nazıroğlu M, Oktar S, Nacar A, Arıca V, et al. Ceftriaxone ameliorates cyclosporine A-induced oxidative nephrotoxicity in rat. Cell Biochem Funct. 2011;29:102–107. doi: 10.1002/cbf.1727. [DOI] [PubMed] [Google Scholar]