

Abstract
Background and Purpose
The opioid system plays a crucial role in several physiological processes in the CNS and in the periphery. It has also been shown that selective opioid receptor agonists exert potent inhibitory action on pruritus and pain. In this study we examined whether two analogues of Salvinorin A, PR-37 and PR-38, exhibit antipruritic properties in mice.
Experimental Approach
To examine the antiscratch effect of PR-37 and PR-38 we used a mouse model of compound 48/80-induced pruritus. In order to elucidate the mechanism of action of tested compounds, specific antagonists of opioid and cannabinoid receptors were used. The effect of PR-37 on the CNS was assessed by measuring motor parameters and exploratory behaviours in mice.
Key Results
PR-37 and PR-38, jnjected s.c., significantly reduced the number of compound 48/80-induced scratching behaviours in mice in a dose- and time-dependent manner. PR-38 was also active when orally administered. The antiscratch activity of PR-37 was blocked by the selective κ opioid receptor antagonist, nor-binaltorphimine, and that of PR-38 by the selective μ opioid receptor antagonist, β-funaltrexamine.
Conclusion and Implications
In conclusion, a novel framework for the development of new antipruritic drugs derived from salvinorin A has been validated.
Table of Links
| TARGETS | |
|---|---|
| GPCRs | |
| DOR, δ-opioid receptor | |
| KOR, κ-opioid receptor | |
| MOR, μ-opioid receptor |
| LIGANDS | |
|---|---|
| AM 251 | NLTR, naltrindole |
| Butorphanol | NLX, naloxone |
| β-FNA, β-funaltrexamine | norBNI, nor-binaltorphimine |
| ICI 204 448 | Salvinorin A |
| Nalfurafine (TRK820) |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Pruritus is defined as a short- or long-term localized or generalized itch. There is no precise definition of itch, but it is generally referred to as an unpleasant cutaneous sensation, leading to a desire to scratch. Itch may be manifested by different sensations, such as tingling, crawling or irritation, but the common feature of itch-related sensations is their peripheral origin, in particular the skin (Bigliardi and Bigliardi-Qi, 2014). Pruritus negatively affects quality of life; it may lead to scratch-related skin damage, sleep disturbance and could even cause reactive depression (Benecke et al., 2013). Chronic pruritus may occur at any age; however, its prevalence is higher in older patients than in children and young adults (Benecke et al., 2013). Chronic pruritus may be caused by some diseases, such as atopic dermatitis, liver diseases or bile secretion disorders.
Clinical and molecular studies in patients affected by pruritus lead to identification of specific mediators and receptors involved in the development of itch, such as histamine 4 receptor, interleukin-31, together with opioid receptors, gastrin-releasing peptide receptor, neurokinin-1 receptor and transient receptor potential channel ion channels (Benecke et al., 2013; Bigliardi and Bigliardi-Qi, 2014). Regardless of the factor that triggers the sensation of itch, the first step in its induction is the interaction between the skin cells, for instance, keratinocytes, melanocytes, Merkel cells, mastocytes, endothelial cells and fibroblasts, and peripheral nonmyelinated sensory C-fibres (Bigliardi and Bigliardi-Qi, 2014). Neural signals are further transduced through the peripheral nerves, dorsal root ganglia and spinal cord to the higher centres in the brain (Bigliardi and Bigliardi-Qi, 2014). These signals can be modulated at each level of transduction to decrease the sensation of itch, as well as pain.
Three main types of opioid receptors, namely, μ-opioid receptor (MOR), κ-opioid receptor (KOR) and δ-opioid receptor (DOR), mediate the actions of opioid drugs and endogenous opioid peptides. They are located both in the CNS and the periphery, including neurons (located in the dermis, epidermis and around hair follicles) and immune cells (macrophages, neutrophils) (Bigliardi et al., 2009; Bigliardi and Bigliardi-Qi, 2014; Salaga et al., 2013; Sobczak et al., 2014). Opioid receptors mediate several biological functions and are attractive molecular targets in the development of new therapeutic agents. In the CNS, opioid agonists regulate, among others, pain perception and antinociception, mood, cognitive function and locomotor activity (Fichna et al., 2009a; Sobczak et al., 2014). On the cellular level, activation of opioid receptors results in inhibition of adenyl cyclase, increase in K+ conductance and activation of mitogen-activated protein kinases (Wang et al., 2005).
Recently, a broader attention has been given to KORs and their ligands, which may become a target in the treatment of pruritus. It is generally accepted that KOR signalling suppresses, while MOR signalling stimulates itch responses. However, a specific mechanism underlying this difference is not well understood. For example, pharmacological blockade of KOR by its selective antagonist nor-binaltorphimine (norBNI) was shown to induce scratching behaviour (Kamei and Nagase, 2001) Other studies found that centrally administered selective MOR agonists, such as [D-Ala2-N-MePhe4-Gly-ol]-enkephalin (DAMGO), produce facial scratching in mice (Kuraishi et al., 2000). Moreover, spinal analgesics acting on MOR, such as morphine, produce a dose-dependent itch in primates (Ko and Naughton, 2000). This side effect may lessen its potential in pain relief.
In this study, we characterized the antiscratch effect of the selective KOR agonist salvinorin A (SA), a natural diterpenoid isolated from the Mexican plant Salvia divinorum, and its two Michael acceptor-type analogues, methyl salvinorin B-2-O-malonate (PR-37) and 2-O-cinnamoylsalvinorin B (PR-38) (Fichna et al., 2009b; Polepally et al., 2013; Polepally et al., 2014). In the in vitro studies, these two analogs displayed high affinity at KOR with Ki values of 2.0 ± 0.9 and 9.6 ± 2.0 nM for PR-37 and PR-38, respectively (Polepally et al., 2013; 2014,), and potently inhibited adenylate cyclase in live HEK293 cells with EC50 values of 137 ± 15 and 7 ± 1 nM respectively (Polepally et al., 2013; 2014,). Of note, recently we showed that PR-38 has neither hallucinogenic nor anxiolytic or anxiogenic effects in mice (Salaga et al., 2014b). Here we performed similar behavioural tests to exclude the potential CNS-related effects of PR-37.
The results of this study showed that PR-37 and PR-38 potently attenuated pruritus in mice and confirmed the endogenous opioid system as a target for antipruritic drugs.
Methods
Animals
All animal care and experimental protocols were in accordance with the European Communities Council Directive 2010/63/EU and Polish legislation acts concerning animal experimentation. The experimental protocol was approved by the Local Ethics Committee at the Medical University of Lodz (#700/2012). Efforts were made to minimize animal suffering and to reduce the number of animals used. All in vivo experiments have been performed in accordance with ARRIVE guidelines (for details, see McGrath et al., 2010). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 490 animals were used in the experiments described here.
Swiss-Webster mice selectively bred towards high analgesia (HA) and low analgesia (LA) (Panocka et al., 1986) were obtained from the Institute of Genetics and Animal Breeding at the Polish Academy of Sciences. The selection process is briefly described below. Outbred mice were forced to swim for 3 min in water (20°C). Two minutes after completion of the swim, the latency of the nociceptive reflex on a hot plate (at 56°C) was measured. Males and females with the longest (50–60 s) and the shortest (<10 s) post-swim latencies of the hind paw flick or lick response were chosen as progenitors of the HA and the LA lines respectively. A similar procedure was repeated in each offspring generation, and only subjects displaying the longest and the shortest post-swim hotplate latencies were mated to maintain the HA and the LA lines respectively. HA mice were used to compare behavioural itch response between these two lines. In all other experiments described in this study, we used LA mice. Preliminary experiments revealed that the response of LA mice to a pruritus-inducing agent was similar to that of normal Swiss-Webster mice. In all experiments, we used male mice weighing 22–30 g (6–8 weeks of age). The animals were housed at a constant temperature (22°C) and maintained under a 12 h light/dark cycle (lights on 6:00 a.m.) in sawdust-lined plastic cages with access to chow pellets and tap water ad libitum.
Induction of pruritus and measurement of antiscratch activity
To measure the antiscratch effect, we used a model described by Kuraishi et al. (1995) with some modification. Briefly, each mouse was weighed and allowed to acclimatise for at least 1 h in an individual observation cage. Vehicle, SA, ICI 204 448, PR-37 and PR-38 were injected subcutaneously (s.c., 100 μL) into the flank before the s.c. injection of the pruritogen compound 48/80 (0.5 mg·mL−1, 100 μL) into the back of the neck (interscapular region). Opioid and cannabinoid antagonists were injected s.c. (100 μL) before agonists, into the opposite flank of the body. Oral administration of PR-37 and PR-38 was performed in the volume of 150 μL. After injection of the pruritogen compound, mice were returned to observation cages and the number of hind leg scratching movements directed to the neck was counted for 30 min. Mice were used only once; all experiments took place between 7 and 11 a.m.
Pharmacological treatments
SA was injected s.c. at 10 mg·kg−1, 45 min before compound 48/80. A reference antipruritic, ICI 204 448, was administered s.c. at a dose of 10 mg·kg−1, 45 min before 48/80 compound. In the dose–effect experiments, PR-37 and PR-38 were injected at doses of 1, 3, 5, 10 and 20 mg·kg−1 (s.c.) 45 min before the 48/80 compound. In the time-course experiments, PR-37 and PR-38 were administered at 10 mg·kg−1 (s.c.) 15, 45, 90 and 120 min before administration of 48/80 compound. DOR and cannabinoid receptor type 1 (CB1) antagonists were administered s.c. 30 min before PR-37 and PR-38 MOR and KOR antagonists as well as naloxone (NLX) were administered either 30 or 120 min before PR-37 and PR-38 (to ensure their selectivity). The antagonists were used at the following doses: naltrindole (NLTR, 10 mg·kg−1), N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (AM 251, 1 mg·kg−1), β-funaltrexamine (β-FNA, 1 mg·kg−1), norBNI (10 mg·kg−1) and NLX (1 mg·kg−1). Doses were selected based on the preliminary studies and published data (Chen et al., 2014; Salaga et al., 2014a,b,; Taylor et al., 2014). In experiments with oral administration, PR-37 and PR-38 were gavaged orally (p.o.) at a dose of 10 mg·kg−1, 45 min before the 48/80 compound. Control animals received vehicle (s.c. or p.o.) alone. In the measurements of spontaneous locomotor activity, morphine and SA were administered s,c, at 5 and 10 mg·kg−1, respectively, 30 min prior to the test. None of the antagonists influenced the observed parameters when given alone.
Measurement of locomotor activity
Locomotor activity was assessed as described previously by Salaga et al. (2014b). Briefly, measurement was made automatically in a Digiscan actimeter (Omnitech Electronics Inc., Columbus, OH, USA). The device measured horizontal displacements and vertical movements. Animals were placed individually in 20 × 20 × 30 cm compartments. The responses were expressed as the number of crossed infrared beams by mouse during six consecutive 10 min periods.
Elevated O-maze test
To identify differences in exploratory and anxiety-related behaviour, the elevated O-maze test was used, as described earlier by Salaga et al. (2014b). The maze consisted of a grey plastic runway (width 6 cm, outer diameter 46 cm, 50 cm above ground level), covered with black cardboard paper to prevent mice from slipping off. The maze consisted of two opposing open and two opposing sectors protected by inner and outer walls (height 10 cm). The maze was illuminated with 25 Lux. At the beginning of the experiment, animals were placed in one of the protected sectors and observed for 5 min. The following parameters were analysed: total time spent in the open and closed compartments, and total distance travelled in open and closed compartments.
Catwalk quantitative gait analysis test
The quantitative analysis of the gait was performed as described recently by Vandeputte et al. (2010) and Salaga et al. (2014b). The Catwalk™ is a video-based analysis system to measure gait parameters in voluntarily walking mice (Noldus, Wageningen, The Netherlands). The system measures various parameters of footfalls in a dynamic manner. When the animal crosses the walkway, the areas of contact between paws and the base are being illuminated. In this way the different paw contacts are visualized and multiple parameters are calculated. Here, in order to examine whether PR-37 affects the CNS-derived motor function, several selected parameters were analysed, including run duration, intensity of the paws (signal depends on the strength of contact between the paw and the glass plate), print width (width of the complete paw print), print length (length of the complete paw print), print area (surface area of the complete paw print), maximal area of print, swing (duration of the lack of contact with the runway in a step cycle), swing speed (speed of the paw during swing), stand time, stride length, step cycle and body speed. For data collection, three runs per animal were performed by placing the animal in front of the start zone of the catwalk runway. Analysis was performed on a minimum of four normal step sequence patterns in each uninterrupted run.
Data analysis
Statistical analyses were performed using PRISM 5.0 (GraphPad Software Inc., La Jolla, CA, USA). The data are expressed as means ± SEM. Student’s t-test was used to compare single treatment means with control means. anova followed by Newman–Keuls post hoc test was used for analysis of multiple treatment means. P values ≤ 0.05 were considered statistically significant.
Materials
All drugs and reagents, unless otherwise stated, were purchased from Sigma-Aldrich (Poznan, Poland). SA (purity: 99% by high-performance liquid chromatography) was isolated from S. divinorum leaves, purchased from The Sage Wisdom Salvia Shop (Malibu, CA, USA) by one of the authors (J.K.Z.). PR-37 and PR-38 were synthesized from SA at the Department of Pharmacognosy, University of Mississippi, USA, as described earlier (Polepally et al., 2013; 2014,). Nor-binaltorphimine dihydrochloride, β-FNA hydrochloride, NLTR hydrochloride and AM 251 were purchased from Tocris Bioscience (Ellisville, MO, USA). Drugs were dissolved in 5% DMSO in saline, which was used as vehicle in control experiments. The vehicles in the used concentrations had no effects on the observed parameters.
Results
Mice with hyperactivity of the endogenous opioid system are resistant to compound 48/80-induced pruritus
First, we compared the effect of 48/80 compound on behavioural itch response in LA and HA mice. LA mice displayed a significant susceptibility to compound 48/80-induced pruritus manifested by the high number of scratches per 30 min of observation (Figure 1). On the contrary, HA mice were highly resistant to compound 48/80-induced pruritus. LA mice treated with SA at the dose of 10 mg·kg−1 s.c. showed a lower number of scratches than vehicle-treated animals, but the difference did not reach statistical significance.
Figure 1.
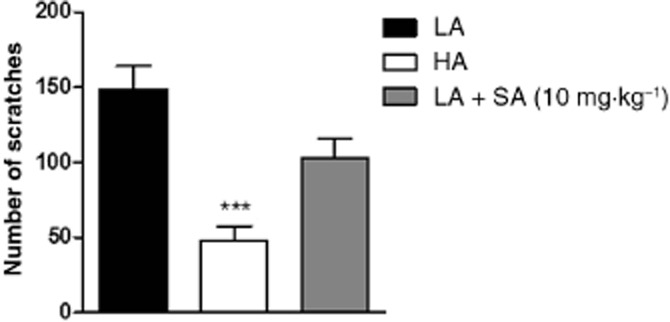
Pruritic effect of 48/80 compound in LA versus HA mice. Figure shows data for vehicle-treated LA and HA mice, and SA-treated LA mice (10 mg·kg−1, s.c.). Data represent mean ± SEM of n = 6–10 animals. ***P < 0.001, as compared with the LA group.
PR-37 and PR-38 protect against compound 48/80-induced pruritus in a dose- and time-dependent manner
To test the antiscratch effect of PR-37 and PR-38, we used a model of compound 48/80-induced pruritus. As shown in Figure 2A and B, both PR-37 and PR-38 reduced the number of compound 48/80-induced scratching behaviours in a dose-dependent manner. The effect of both compounds was statistically significant after administration at the dose of 10 and 20 mg·kg−1. Additionally, we used a well-known antipruritic, a peripherally restricted KOR agonist, ICI 204 448, as a reference compound. ICI 204 448 (10 mg·kg−1, s.c.) significantly reduced the number of compound 48/80-induced scratches and its effect was stronger than that of PR-37 and PR-38 (Figure 2C).
Figure 2.
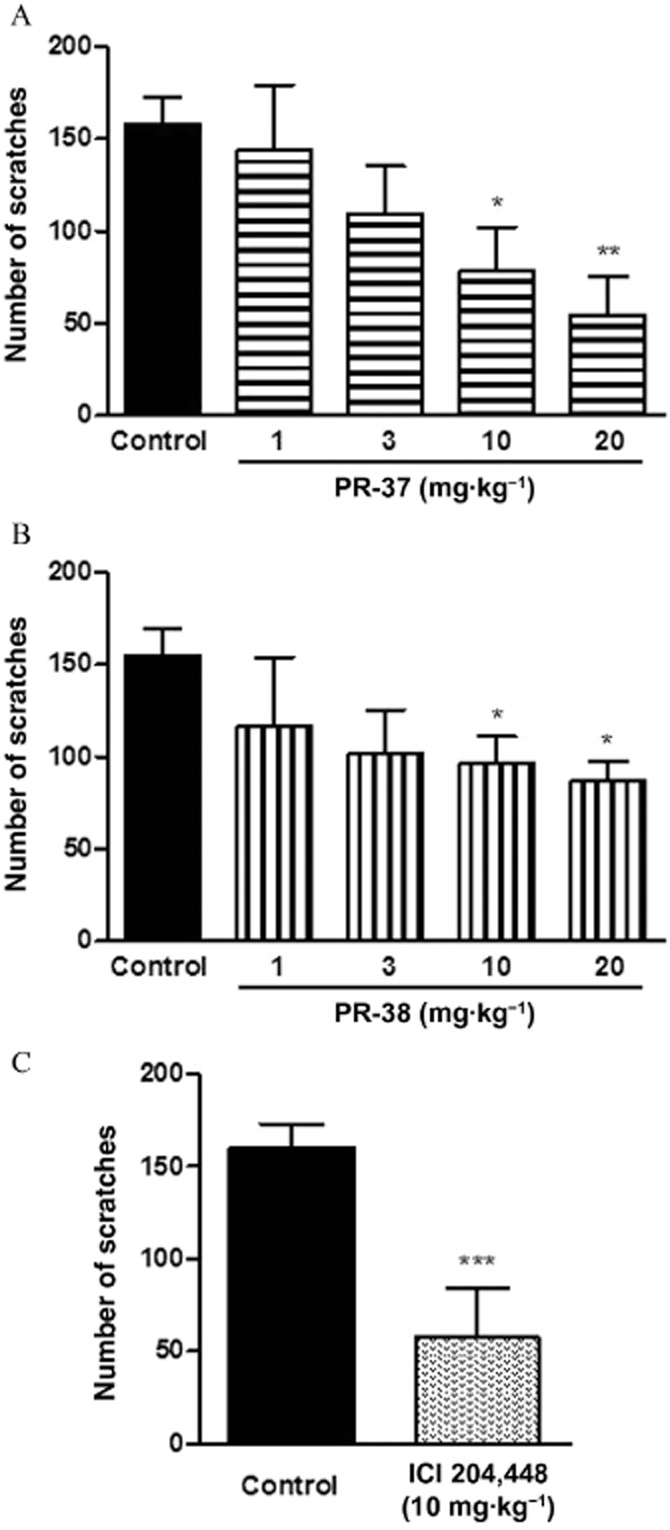
The effect of PR-37, PR-38 and ICI 204 448 on compound 48/80-induced pruritus in mice. (A) Effect of s.c. administration of PR-37 (1–20 mg·kg−1) on the number of scratches per 30 min. (B) Effect of s.c. administration of PR-38 (1–20 mg·kg−1) on the number of scratches per 30 min. (C) Effect of s.c. administration of a reference drug ICI 204 448 (10 mg·kg−1) on the number of scratches per 30 min. Data represent mean ± SEM of n = 6–10 animals. *P < 0.05, **P < 0.01, ***P < 0.001, as compared with the control group.
Encouraged by the above mentioned data, we examined the time-course of the antiscratch effect of PR-37 and PR-38. As shown in Figure 3A and B, the compounds (10 mg·kg−1, s.c.) exhibited a different duration of action in vivo. The antiscratch effect of PR-37 emerged 45 min after injection and was present until 90 min post-administration. The effect rapidly vanished between 90 and 120 min after administration. In the case of PR-38, a significant antiscratch activity was only present 45 min after administration, then it gradually faded between 45 and 120 min post-administration.
Figure 3.
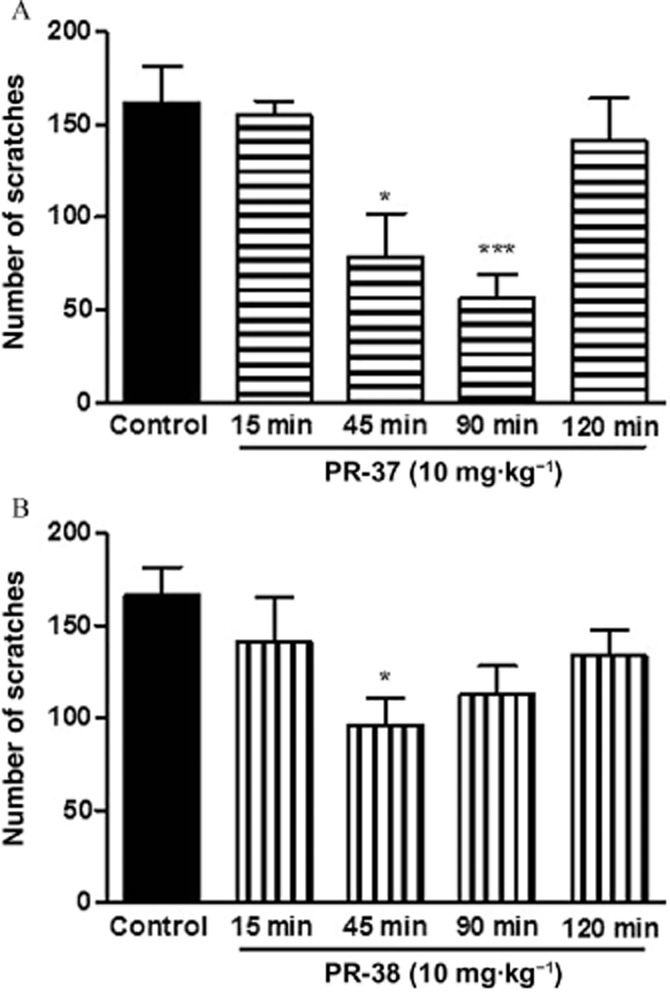
The time-course of the antiscratch activity of PR-37 and PR-38 after a single s.c. administration at the dose of 10 mg·kg−1 in mice. (A) Time-course of the effect of PR-37 on the number of scratches per 30 min. The strongest effect occurs 90 min post-administration (B) Time-course of the effect of PR-38 on the number of scratches per 30 min. The strongest effect occurs 45 min post-administration. Data represent mean ± SEM of n = 8–10 mice for each experimental group. *P < 0.05, ***P < 0.001, as compared with the control group.
PR-37 and PR-38 exhibit different mechanisms of action in vivo
To examine the mechanism of the antiscratch activity of PR-37 and PR-38, we used specific opioid and cannabinoid receptor antagonists. Although in the in vitro tests both compounds displayed affinity only for opioid receptors, in previous studies we observed that certain biological effects of PR-38 are blocked by the CB1 receptor antagonist AM 251, which is why it was used in this study (Salaga et al., 2014b,c,). The effect of both compounds was inhibited by the non-selective opioid receptor antagonist NLX; however, the difference did not reach statistical significance (Figures 4B and 5B). As shown, PR-37 (Figure 4) and PR-38 (Figure 5) exhibited different mechanisms of action in vivo. The antiscratch effect of PR-37 (10 mg·kg−1, s.c.) was blocked by the KOR antagonist norBNI (10 mg·kg−1, s.c.), whereas the effect of PR-38 (10 mg·kg−1, s.c.) was inhibited by the MOR antagonist β-FNA (10 mg·kg−1, s.c.). Neither NLTR nor AM 251 blocked the antiscratch activity of PR-37 and PR-38. Experiments with β-FNA and norBNI have been performed with two different time gaps between the antagonist and the test compound treatments to ensure their selectivity at MOR and KOR respectively. As shown in Figures 4B and 5B, extending the interval between treatments to 120 min did not change the outcomes of the experiment.
Figure 4.
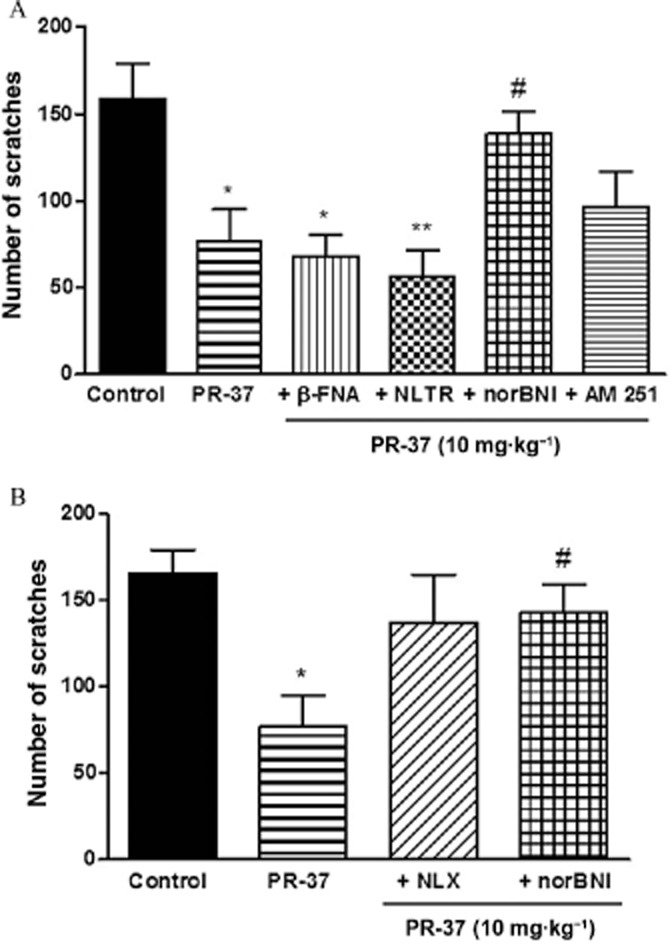
Antiscratch effect of s.c. administration (10 mg·kg−1) of PR-37 alone or in the presence of opioid and cannabinoid receptor antagonists. (A) Antiscratch effect of PR-37 is blocked by KOR antagonist norBNI (10 mg·kg−1, s.c.) administered 30 min prior to the test compound but not MOR, DOR and CB1 receptor antagonists (β-FNA, NLTR and AM 251 respectively). (B) Antiscratch effect of PR-37 is blocked by the non-selective opioid antagonist NLX (1 mg·kg−1, s.c.) and KOR antagonist norBNI (10 mg·kg−1, s.c.) administered 120 min prior to the test compound. Data represent mean ± SEM of n = 6–10 mice for each experimental group. *P < 0.05, **P < 0.01, as compared with the control group. #P < 0.05, as compared with PR-37.
Figure 5.
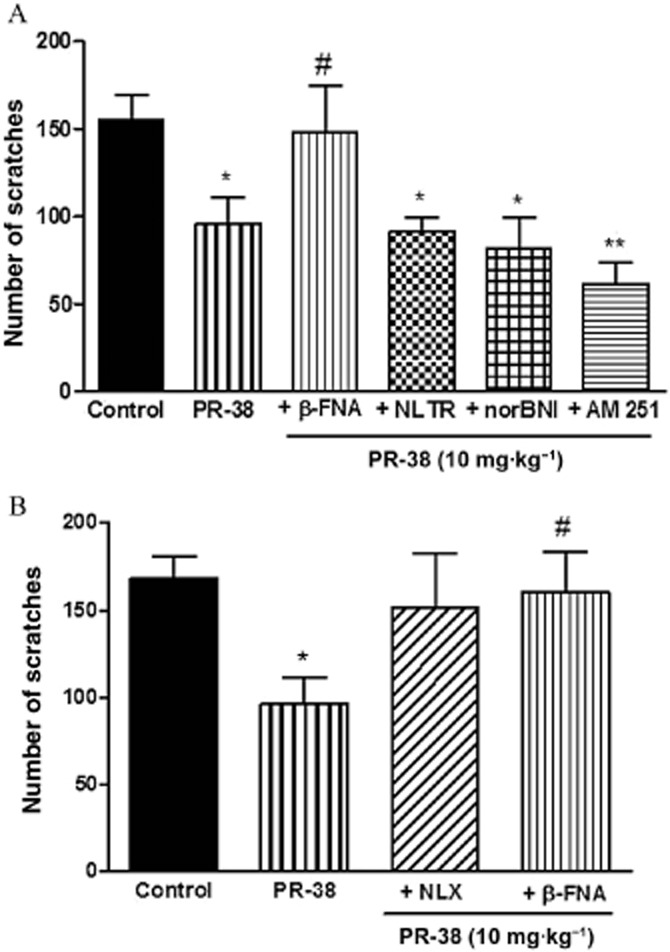
Antiscratch effect of s.c. administration (10 mg·kg−1) of PR-38 alone or in the presence of opioid and cannabinoid receptor antagonists. (A) Antiscratch effect of PR-38 is blocked by MOR antagonist β-FNA (1 mg·kg−1, s.c.) administered 30 min prior to the test compound, but not KOR, DOR and CB1 receptor antagonists (norBNI, NLTR and AM 251 respectively). (B) Antiscratch effect of PR-38 is blocked by the non-selective opioid receptor antagonist NLX (1 mg·kg−1, s.c.) and MOR antagonist β-FNA (1 mg·kg−1, s.c.) administered 120 min prior to the test compound. Data represent mean ± SEM of n = 6–10 mice for each experimental group. *P < 0.05, **P < 0.01, as compared with the control group. #P < 0.05, as compared with PR-37 or PR-38.
To further confirm the selectivity of β-FNA and norBNI at MOR and KOR, we examined whether they influence the effects elicited by morphine and SA on mouse locomotor activity. The hyperactivity induced by morphine was blocked by pretreatment with β-FNA (1 mg·kg−1, s.c.), but not norBNI (10 mg·kg−1, s.c.; Supporting Information Fig. S1A). The hypoactivity induced by treatment with SA was blocked by norBNI (10 mg·kg−1, s.c.), but not β-FNA (1 mg·kg−1, s.c.; Supporting Information Fig. S1B).
PR-38 exhibits antiscratch activity after p.o. administration
We also examined the antiscratch activity of PR-37 and PR-38 after p.o. administration. As shown in Figure 6A, PR-37 reduced the number of scratches, but the difference did not reach statistical significance but PR-38 significantly reduced the number of scratches after p.o. administration (Figure 6B).
Figure 6.
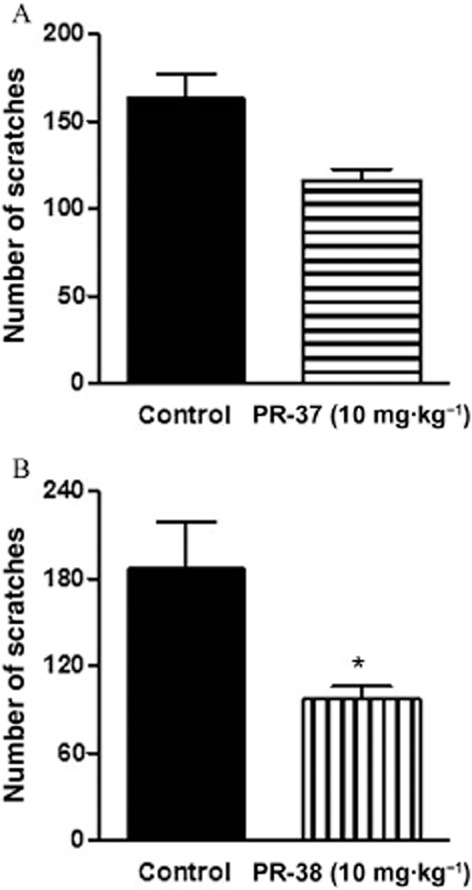
Antiscratch activity of PR-37 (A) and PR-38 (B) after oral administration (10 mg·kg−1) in mice. Data represent mean ± SEM of n = 6–8 mice for each experimental group. *P < 0.05, as compared with the control group.
PR-37 does not affect exploratory behaviour or anxiety levels in mice, but it impairs motor functions
SA, which is a parent compound of PR-37 and PR-38, is known for its CNS-related actions, such as hallucinogenic episodes. Previously, we have shown that PR-38 does not influence CNS-related behaviours after systemic administration (Salaga et al., 2014b). Here we investigated the effect of PR-37 in the CNS using appropriate mouse models. The influence of the i.p. administration of PR-37 on mouse locomotor activity was measured over six consecutive periods of 10 min each. The drug administered at a dose of 10 mg·kg−1 did not modify horizontal or vertical locomotor activity in any of the time periods of the test (Figure 7A and B).
Figure 7.

Effect of PR-37 on the CNS. (A) Effect of PR-37 (10 mg·kg−1, i.p.) on horizontal locomotor activity. (B) Effect of PR-37 (10 mg·kg−1, i.p.) on vertical locomotor activity. (C) Effect of PR-37 (10 mg·kg−1, i.p) on time spent in the opened and closed areas of elevated o-maze. (D–I) Catwalk automated gait analysis in mice after PR-38 administration (10 mg·kg−1, i.p.) Depicted are mean ratios ± SEM (8–10 mice per experimental group) of pawprint intensity (D), print width (E), print area (F), max area (G), swing (H) and swing speed (I), separately, for front and hind paws. **P < 0.01, ***P < 0.001, as compared with the control group.
To test the effect on exploratory behaviour and potential anxiogenic or anxiolytic activity of PR-37, an elevated O-maze test was used. No significant differences were found for measures of anxiety, such as the percentage of time spent in open and closed sectors after i.p. administration of PR-37 at a dose of 10 mg·kg−1 (Figure 7C).
We also analysed the gait parameters of mice after i.p. administration of PR-37 at a dose of 10 mg·kg−1. Eleven parameters of gait were measured for front and hind paws separately; four of them changed significantly after administration of PR-37 in comparison with control animals, including intensity of the paw prints (Figure 7D), print width (Figure 7E), print area (Figure 7F) and maximal area of print (Figure 7G). Other parameters, such as swing (Figure 7H), swing speed (Figure 7I), run duration, stand time, print length, stride length, step cycle and body speed (data not shown), were not changed after administration of PR-37.
Discussion
In this study, we showed that two derivatives of SA, PR-37 and PR-38, attenuate pruritus in mice by activation of opioid receptors. Of note, these two compounds exhibited different pharmacological features, such as bioavailability, as well as duration and mechanism of action. Furthermore, we have confirmed the endogenous opioid system as an attractive pharmacological target for antipruritic drugs.
To date, several studies suggested that itch sensation is closely associated with opioid receptor-dependent signalling. For example, Bergasa et al. (1993) found that plasma obtained from patients with pruritus administered into the medullary dorsal horn, which is the site of action of opiates in producing facial scratching, elicited facial scratching activity in monkeys, and this effect was abolished by the administration of the opioid receptor antagonist NLX (see Bigliardi et al., 2009; Benecke et al., 2013; Bigliardi and Bigliardi-Qi, 2014). In our study we add another line of evidence supporting the involvement of opioid receptors in the modulation of itch sensation by showing that mice bred selectively towards an increased activity of the endogenous opioid system (HA) are resistant to pruritus compared to mice exhibiting normal opioid system activity (LA).
There is a strong body of evidence based on animal and human studies supporting the hypothesis that the activation of KOR protects against pruritus (see Benecke et al., 2013; Bigliardi and Bigliardi-Qi, 2014; Ko, 2014). Moreover, some observations indicate the existence of a link between KOR and its endogenous agonists, dynorphins, and pruritic skin diseases. Immunohistochemical studies revealed down-regulation of KOR in the skin samples obtained from patients suffering from pruritic psoriasis (Tominaga et al., 2007). Animal experiments showed that the density of free nerve endings in the epidermis was higher in KOR knockout mice compared with the wild phenotype. In addition, scratching behaviour in KOR-deficient mice with dry skin dermatitis was reduced compared to wild-type animals (Bigliardi and Bigliardi-Qi, 2014). Of note, it has been shown that not only centrally acting but also peripherally restricted KOR agonists, such as ICI 204 448, inhibited chloroquine-induced pruritus in mice (Inan and Cowan, 2004). These observations fully justify further development of novel KOR ligands as novel antipruritic agents.
SA, the active component of S. divinorum, has recently been demonstrated to exhibit significant analgesic effects (Fichna et al., 2012). It has also been shown to reduce the number of itch responses in mice; however, the effect of SA was weaker than other KOR agonists, such as TRK820, which is probably due to the rapid degradation of SA in vivo (Wang et al., 2005). Moreover, the potential of SA to become a drug used in the clinical treatment of disorders dominated by pain and itch is strongly hampered by its adverse effects, as it rapidly crosses the blood–brain barrier and causes short-lived hallucinations. Therefore attempts to develop new analogues with similar activity have been undertaken. The fact that SA analogues can be synthesized in the simple two-step reaction from SA, which is readily available from accessible plant material, may provide a significant advantage over currently available semi-synthetic opioids, such as TRK820.
Here we found that PR-38, a novel SA analogue and a mixed KOR/MOR ligand, attenuated pruritus after peripheral administration, and this effect was blocked by the selective MOR antagonist β-FNA. Moreover, this study provides evidence for good bioavailability of PR-38, as we showed that this analogue maintains its antiscratch activity after oral administration. This is in line with our recent report, where we demonstrated that PR-38 is present in plasma at the detectable level after oral administration (Salaga et al., 2014b). Furthermore, the same study showed that PR-38 did not produce any CNS-related side effects, which is an unquestionable advantage over ‘classical’ opioids (Salaga et al., 2014b). Given both, a potent antipruritic effect and good bioavailability, as well as the lack of CNS-related side effects, PR-38 may become a scaffold for the design of novel compounds that could be used as supplemental agents to treat pruritus, also in patients receiving spinal opioid analgesics.
In our study we also showed that another SA analogue, PR-37, inhibited compound 48/80-induced pruritus in a dose-dependent manner. In the case of PR-37, this effect was blocked by the KOR antagonist norBNI. Of note, in vitro PR-37 also binds at DOR, but with low affinity (Ki = 4320 ± 680 nM). However, the antiscratch activity of PR-37 was not blocked by the DOR antagonist, which indicates that the interaction between PR-37 and DOR is negligible for its antiscratch effect (Polepally et al., 2013).
One of the main overall goals in our studies is to find a novel SA analogue lacking the CNS-related side effects, typical of the parent compound. It is widely known that SA has a strong, short-acting psychoactive effect and its actions in humans range from impaired motor activity, visual effects, memory impairment and unresponsiveness to feelings of anxiety/fear, distance from usual daily reality and paranoia (Ranganathan et al., 2012; MacLean et al., 2013). To determine the potential side effects of PR-37, we used the same set of behavioural experiments and dosage as previously described for PR-38 (Salaga et al., 2014b). We observed that the majority of parameters did not change after administration of PR-37, namely, the results obtained in the O-maze test suggest no anxiogenic and/or anxiolytic activity and no effects on spontaneous locomotor activity were found. However, we observed some significant disturbances in several gait parameters suggesting that PR-37 affects neuromotor functions of the CNS. These data show that PR-37 and PR-38 differ in their ability to affect CNS-related functions, in favour of the latter. A comparison of the structures of both SA analogues is thus recommended, as it may lead to the identification of certain moieties, which are responsible for either induction of psychopharmacological effects or determination of their ability to penetrate the blood–brain barrier. Higher efficiency of PR-37 than PR-38 in crossing the blood–brain barrier could also explain, at least partly, its longer and more potent antiscratch effect in vivo.
The majority of recent studies focus on MOR and KOR as targets for both pruritogenic and antipruritic compounds. While it is generally accepted that stimulation of KOR attenuates pruritus, it is still not clear whether pharmacological stimulation or blockade of MOR may have beneficial effects (Bigliardi and Bigliardi-Qi, 2014). Moreover, centrally administered MOR agonists were found to induce different scratching responses. For example, a synthetic MOR agonist, DAMGO, which is known for its higher efficacy at MOR than morphine, induced a more intense pruritus than morphine (Ko, 2014). Interestingly, it has been demonstrated that MOP1D, an isoform of MOR, is responsible for morphine-induced pruritus and that morphine-induced itch is separable from morphine-induced analgesia in mice (Liu et al., 2011).
As indicated by the in vitro and in vivo studies, PR-38 is a mixed MOR/KOR agonist with about fivefold lower efficacy at MOR. However, its antiscratch activity in vivo was blocked by the MOR, but not the KOR, antagonist. Previously, we have shown that the potent antinociceptive effect of PR-38 in mouse models of visceral pain were also mediated by MOR (Salaga et al., 2014b). Taking into account the data showing the involvement of central MORs in the induction of pruritus, we suggest that PR-38 does not activate the sites in the CNS due to a very weak penetration through the blood–brain barrier. However, this only partly explains the antipruritic activity of PR-38, in particular, when our observations also rule out the direct involvement of KOR. We hypothesize that there is a possible cross-talk between MOR and KOR in this process. If MOR-KOR heterodimers are formed, a signalling cascade causing an antipruritic effect may be evoked exclusively by activation of MOR, and in that way, the KOR antagonist will not block it. A more in-depth mechanistic study is needed to address this issue.
On the other hand, it may be hypothesized that PR-38 acts in vivo as a weak partial agonist or partial antagonist of MOR. It may be expected that such low-efficacy MOR ligands would antagonize scratching in contrast to high-efficacy MOR ligands, which would induce it. Indeed, it has been shown that a synthetic mixed KOR-agonist/MOR-partial antagonist butorphanol is highly effective in the treatment of intractable pruritus (Dawn and Yosipovitch, 2006; Lim et al., 2008). Moreover, some authors postulate that the itch sensation in the periphery and CNS is the result of an imbalance between MOR and KOR activities (Bigliardi and Bigliardi-Qi, 2014). Of interest, nalfurafine (TRK820), a mixed KOR agonist/MOR- and DOR-partial agonist, was effective against pruritus in animal studies (Togashi et al., 2002; Umeuchi et al., 2003; Wang et al., 2005; Inan et al., 2009). Clinical trials with nalfurafine are particularly encouraging, as they showed its effectiveness against haemodialysis-associated pruritus, both after intravenous and oral administration (Wikstrom et al., 2005; Kumagai et al., 2010; Inui, 2012; Phan et al., 2012; Benecke et al., 2013).
In conclusion, modification of the chemical structure of SA produced analogues displaying antiscratch activity but with a different pharmacological profile. One of these derivatives, PR-38, elicited a significant antiscratch effect in vivo, which was mediated by MOR, and retained its biological activity after oral administration. It seems likely that PR-38 will be further developed due to its significant effects on the pruritus combined with good bioavailability and the lack of psychotropic side effects. PR-38 may also be used as a scaffold for the further development of novel SA-derived agonists, partial agonists, and KOR/MOR dual agonists that do not cross the blood–brain barrier. On the other hand, based on the structure of PR-37, it may be seen what types of chemical modifications are needed to eliminate the potential CNS-related side effects of SA. These data may be used in the future for the design of novel SA derivatives with more desirable therapeutic profiles.
Acknowledgments
Supported by NIH Grant R01 DA017204 and the NIMH Psychoactive Drug Screening Program (PDSP-HHSN-271-2013-00017-C to BR), the Iuventus Plus program of the Polish Ministry of Science and Higher Education (#0107/IP1/2013/72 to JF) and the grants from the Medical University of Lodz (502-03/1-156-04/502-14-140 to MS and 503/1-156-04/503-01 to JF) and National Science Centre (#UMO-2013/11/N/NZ7/02354 to MS; #UMO-2013/11/B/NZ7/01301 and #UMO-2014/13/B/NZ4/01179 to JF).
Glossary
- β-FNA
β-funaltrexamine
- CB1
cannabinoid receptor type 1
- DAMGO
[D-Ala2-N-MePhe4-Gly-ol]-enkephalin
- DOR
δ-opioid receptor
- HA
high analgesia
- KOR
κ-opioid receptor
- LA
low analgesia
- MOR
μ-opioid receptor
- NLTR
naltrindole
- NLX
naloxone
- norBNI
nor-binaltorphimine
- SA
salvinorin A
- TRP
transient receptor potential channel
Author contributions
M. Salaga, M. Z., M. M., A. F., N. M., K. S. and J. C. D. R. performed the research. M. Salaga and J. F. designed the research study. J. K. Z., P. R. P., B. L. R., M. Sacharczuk and J. F. contributed essential tools and reagents. M. Salaga, J. C. D. R. and J. F. analysed the data. M. Sacharczuk critically reviewed the manuscript. M. Salaga and J. F. wrote the paper.
Conflict of interest
The authors have nothing to disclose.
Supporting Information
Figure S1 Confirmatory experiment demonstrating the selectivity of β-FNA and norBNI (administered 2 h prior to the respective agonist) at MOR and KOR receptors respectively. (A) Morphine-induced locomotor hyperactivity is blocked by pretreatment with β-FNA (1 mg·kg−1 s.c.) but not norBNI (10 mg·kg−1, s.c.). (B) The effect of SA on total spontaneous locomotor activity is blocked by norBNI (10 mg·kg−1, s.c.) but not β-FNA (1 mg·kg−1, s.c.). *P < 0.05, as compared with the control group. #P < 0.05, as compared with morphine or SA.
{kind=link}
References
- Alexander SP, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The concise guide to pharmacology 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benecke H, Lotts T, Stander S. Investigational drugs for pruritus. Expert Opin Investig Drugs. 2013;22:1167–1179. doi: 10.1517/13543784.2013.813932. [DOI] [PubMed] [Google Scholar]
- Bergasa NV, Thomas DA, Vergalla J, Turner ML, Jones EA. Plasma from patients with the pruritus of cholestasis induces opioid receptor-mediated scratching in monkeys. Life Sci. 1993;53:1253–1257. doi: 10.1016/0024-3205(93)90569-o. [DOI] [PubMed] [Google Scholar]
- Bigliardi PL, Bigliardi-Qi M. Peripheral opioids. In: Carstens E, Akiyama T, editors. Itch Mechanisms and Treatment. Boca Raton, FL: CRC Press; 2014. pp. 1–12. chapter 18. In: (eds). [Google Scholar]
- Bigliardi PL, Tobin DJ, Gaveriaux-Ruff C, Bigliardi-Qi M. Opioids and the skin – where do we stand? Exp Dermatol. 2009;18:424–430. doi: 10.1111/j.1600-0625.2009.00844.x. [DOI] [PubMed] [Google Scholar]
- Chen C, Fichna J, Laudon M, Storr M. Antinociceptive effects of novel melatonin receptor agonists in mouse models of abdominal pain. World J Gastroenterol. 2014;20:1298–1304. doi: 10.3748/wjg.v20.i5.1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawn AG, Yosipovitch G. Butorphanol for treatment of intractable pruritus. J Am Acad Dermatol. 2006;54:527–531. doi: 10.1016/j.jaad.2005.12.010. [DOI] [PubMed] [Google Scholar]
- Fichna J, Schicho R, Janecka A, Zjawiony JK, Storr M. Selective natural kappa opioid and cannabinoid receptor agonists with a potential role in the treatment of gastrointestinal dysfunction. Drug News Perspect. 2009a;22:383–392. doi: 10.1358/dnp.2009.22.7.1400219. [DOI] [PubMed] [Google Scholar]
- Fichna J, Schicho R, Andrews CN, Bashashati M, Klompus M, McKay DM, et al. Salvinorin A inhibits colonic transit and neurogenic ion transport in mice by activating kappa-opioid and cannabinoid receptors. Neurogastroenterol Motil. 2009b;21:1326–e128. doi: 10.1111/j.1365-2982.2009.01369.x. [DOI] [PubMed] [Google Scholar]
- Fichna J, Dicay M, Lewellyn K, Janecka A, Zjawiony JK, MacNaughton WK, et al. Salvinorin A has antiinflammatory and antinociceptive effects in experimental models of colitis in mice mediated by KOR and CB1 receptors. Inflamm Bowel Dis. 2012;18:1137–1145. doi: 10.1002/ibd.21873. [DOI] [PubMed] [Google Scholar]
- Inan S, Cowan A. Kappa opioid agonists suppress chloroquine-induced scratching in mice. Eur J Pharmacol. 2004;502:233–237. doi: 10.1016/j.ejphar.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Inan S, Dun NJ, Cowan A. Nalfurafine prevents 5′-guanidinonaltrindole- and compound 48/80-induced spinal c-fos expression and attenuates 5′-guanidinonaltrindole-elicited scratching behavior in mice. Neuroscience. 2009;163:23–33. doi: 10.1016/j.neuroscience.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inui S. Nalfurafine hydrochloride for the treatment of pruritus. Expert Opin Pharmacother. 2012;13:1507–1513. doi: 10.1517/14656566.2012.693164. [DOI] [PubMed] [Google Scholar]
- Kamei J, Nagase H. Norbinaltorphimine, a selective kappa-opioid receptor antagonist, induces an itch-associated response in mice. Eur J Pharmacol. 2001;418:141–145. doi: 10.1016/s0014-2999(01)00941-4. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko MC. Roles of central opioid receptor subtypes in regulating itch sensation. Peripheral opioids. In: Carstens E, Akiyama T, editors. Itch Mechanisms and Treatment. Boca Raton, FL: CRC Press; 2014. pp. 1–10. chapter 25. In: (eds). [PubMed] [Google Scholar]
- Ko MC, Naughton NN. An experimental itch model in monkeys: characterization of intrathecal morphine-induced scratching and antinociception. Anesthesiology. 2000;92:795–805. doi: 10.1097/00000542-200003000-00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai H, Ebata T, Takamori K, Muramatsu T, Nakamoto H, Suzuki H. Effect of a novel kappa-receptor agonist, nalfurafine hydrochloride, on severe itch in 337 haemodialysis patients: a phase III, randomized, double-blind, placebo-controlled study. Nephrol Dial Transplant. 2010;25:1251–1257. doi: 10.1093/ndt/gfp588. [DOI] [PubMed] [Google Scholar]
- Kuraishi Y, Nagasawa T, Hayashi K, Satoh M. Scratching behavior induced by pruritogenic but not algesiogenic agents in mice. Eur J Pharmacol. 1995;275:229–233. doi: 10.1016/0014-2999(94)00780-b. [DOI] [PubMed] [Google Scholar]
- Kuraishi Y, Yamaguchi T, Miyamoto T. Itch-scratch responses induced by opioids through central mu opioid receptors in mice. J Biomed Sci. 2000;7:248–252. doi: 10.1007/BF02255473. [DOI] [PubMed] [Google Scholar]
- Lim GJ, Ishiuji Y, Dawn A, Harrison B, Kim D, Atala A, et al. In vitro and in vivo characterization of a novel liposomal butorphanol formulation for treatment of pruritus. Acta Derm Venereol. 2008;88:327–330. doi: 10.2340/00015555-0480. [DOI] [PubMed] [Google Scholar]
- Liu XY, Liu ZC, Sun YG, Ross M, Kim S, Tsai FF, et al. Unidirectional cross-activation of GRPR by MOR1D uncouples itch and analgesia induced by opioids. Cell. 2011;147:447–458. doi: 10.1016/j.cell.2011.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean KA, Johnson MW, Reissig CJ, Prisinzano TE, Griffiths RR. Dose-related effects of salvinorin A in humans: dissociative, hallucinogenic, and memory effects. Psychopharmacology (Berl) 2013;226:381–392. doi: 10.1007/s00213-012-2912-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panocka I, Marek P, Sadowski B. Differentiation of neurochemical basis of stress-induced analgesia in mice by selective breeding. Brain Res. 1986;397:156–160. doi: 10.1016/0006-8993(86)91380-6. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan NQ, Lotts T, Antal A, Bernhard JD, Stander S. Systemic kappa opioid receptor agonists in the treatment of chronic pruritus: a literature review. Acta Derm Venereol. 2012;92:555–560. doi: 10.2340/00015555-1353. [DOI] [PubMed] [Google Scholar]
- Polepally PR, White K, Vardy E, Roth BL, Ferreira D, Zjawiony JK. Kappa-opioid receptor-selective dicarboxylic ester-derived salvinorin A ligands. Bioorg Med Chem Lett. 2013;23:2860–2862. doi: 10.1016/j.bmcl.2013.03.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polepally PR, Huben K, Vardy E, Setola V, Mosier PD, Roth BL, et al. Michael acceptor approach to the design of new salvinorin A-based high affinity ligands for the kappa-opioid receptor. Eur J Med Chem. 2014;85:818–829. doi: 10.1016/j.ejmech.2014.07.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan M, Schnakenberg A, Skosnik PD, Cohen BM, Pittman B, Sewell RA, et al. Dose-related behavioral, subjective, endocrine, and psychophysiological effects of the kappa opioid agonist salvinorin A in humans. Biol Psychiatry. 2012;72:871–879. doi: 10.1016/j.biopsych.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salaga M, Sobczak M, Fichna J. Inhibition of proteases as a novel therapeutic strategy in the treatment of metabolic, inflammatory and functional diseases of the gastrointestinal tract. Drug Discov Today. 2013;18:708–715. doi: 10.1016/j.drudis.2013.03.004. [DOI] [PubMed] [Google Scholar]
- Salaga M, Mokrowiecka A, Zakrzewski PK, Cygankiewicz A, Leishman E, Sobczak M, et al. Experimental colitis in mice is attenuated by changes in the levels of endocannabinoid metabolites induced by selective inhibition of fatty acid amide hydrolase (FAAH) J Crohns Colitis. 2014a;8:998–1009. doi: 10.1016/j.crohns.2014.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salaga M, Polepally PR, Sobczak M, Grzywacz D, Kamysz W, Sibaev A, et al. Novel orally available salvinorin A analog PR-38 inhibits gastrointestinal motility and reduces abdominal pain in mouse models mimicking irritable bowel syndrome. J Pharmacol Exp Ther. 2014b;350:69–78. doi: 10.1124/jpet.114.214239. [DOI] [PubMed] [Google Scholar]
- Salaga M, Polepally PR, Zakrzewski PK, Cygankiewicz A, Sobczak M, Kordek R, et al. Novel orally available salvinorin A analog PR-38 protects against experimental colitis and reduces abdominal pain in mice by interaction with opioid and cannabinoid receptors. Biochem Pharmacol. 2014c;92:618–626. doi: 10.1016/j.bcp.2014.09.018. [DOI] [PubMed] [Google Scholar]
- Sobczak M, Salaga M, Storr MA, Fichna J. Physiology, signaling, and pharmacology of opioid receptors and their ligands in the gastrointestinal tract: current concepts and future perspectives. J Gastroenterol. 2014;49:24–45. doi: 10.1007/s00535-013-0753-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Roberts KW, Pradhan AA, Akbari HA, Walwyn W, Lutfy K, et al. Anti-nociception mediated by a kappa opioid receptor agonist is blocked by a delta receptor agonist. Br J Pharmacol. 2014;172:691–703. doi: 10.1111/bph.12810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togashi Y, Umeuchi H, Okano K, Ando N, Yoshizawa Y, Honda T, et al. Antipruritic activity of the kappa-opioid receptor agonist, TRK-820. Eur J Pharmacol. 2002;435:259–264. doi: 10.1016/s0014-2999(01)01588-6. [DOI] [PubMed] [Google Scholar]
- Tominaga M, Ogawa H, Takamori K. Possible roles of epidermal opioid systems in pruritus of atopic dermatitis. J Invest Dermatol. 2007;127:2228–2235. doi: 10.1038/sj.jid.5700942. [DOI] [PubMed] [Google Scholar]
- Umeuchi H, Togashi Y, Honda T, Nakao K, Okano K, Tanaka T, et al. Involvement of central mu-opioid system in the scratching behavior in mice, and the suppression of it by the activation of kappa-opioid system. Eur J Pharmacol. 2003;477:29–35. doi: 10.1016/j.ejphar.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Vandeputte C, Taymans JM, Casteels C, Coun F, Ni Y, Van LK, et al. Automated quantitative gait analysis in animal models of movement disorders. BMC Neurosci. 2010;11:92. doi: 10.1186/1471-2202-11-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Tang K, Inan S, Siebert D, Holzgrabe U, Lee DY, et al. Comparison of pharmacological activities of three distinct kappa ligands (salvinorin A, TRK-820 and 3FLB) on kappa opioid receptors in vitro and their antipruritic and antinociceptive activities in vivo. J Pharmacol Exp Ther. 2005;312:220–230. doi: 10.1124/jpet.104.073668. [DOI] [PubMed] [Google Scholar]
- Wikstrom B, Gellert R, Ladefoged SD, Danda Y, Akai M, Ide K, et al. Kappa-opioid system in uremic pruritus: multicenter, randomized, double-blind, placebo-controlled clinical studies. J Am Soc Nephrol. 2005;16:3742–3747. doi: 10.1681/ASN.2005020152. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Confirmatory experiment demonstrating the selectivity of β-FNA and norBNI (administered 2 h prior to the respective agonist) at MOR and KOR receptors respectively. (A) Morphine-induced locomotor hyperactivity is blocked by pretreatment with β-FNA (1 mg·kg−1 s.c.) but not norBNI (10 mg·kg−1, s.c.). (B) The effect of SA on total spontaneous locomotor activity is blocked by norBNI (10 mg·kg−1, s.c.) but not β-FNA (1 mg·kg−1, s.c.). *P < 0.05, as compared with the control group. #P < 0.05, as compared with morphine or SA.