

Abstract
Background and Purpose
In pigs, ivabradine reduces infarct size even when given only at reperfusion and in the absence of heart rate reduction. The mechanism of this non-heart rate-related cardioprotection is unknown. Hence, in the present study we assessed the pleiotropic action of ivabradine in more detail.
Experimental Approach
Anaesthetized mice were pretreated with ivabradine (1.7 mg·kg−1 i.v.) or placebo (control) before a cycle of coronary occlusion/reperfusion (30/120 min ± left atrial pacing). Infarct size was determined. Isolated ventricular cardiomyocytes were exposed to simulated ischaemia/reperfusion (60/5 min) in the absence and presence of ivabradine, viability was then quantified and intra- and extracellular reactive oxygen species (ROS) formation was detected. Mitochondria were isolated from mouse hearts and exposed to simulated ischaemia/reperfusion (6/3 min) in glutamate/malate- and ADP-containing buffer in the absence and presence of ivabradine respectively. Mitochondrial respiration, extramitochondrial ROS, mitochondrial ATP production and calcium retention capacity (CRC) were assessed.
Key Results
Ivabradine decreased infarct size even with atrial pacing. Cardiomyocyte viability after simulated ischaemia/reperfusion was better preserved with ivabradin, the accumulation of intra- and extracellular ROS decreased in parallel. Mitochondrial complex I respiration was not different without/with ivabradine, but ivabradine significantly inhibited the accumulation of extramitochondrial ROS, increased mitochondrial ATP production and increased CRC.
Conclusion and Implications
Ivabradine reduces infarct size independently of a reduction in heart rate and improves ventricular cardiomyocyte viability, possibly by reducing mitochondrial ROS formation, increasing ATP production and CRC.
Tables of Links
| LIGANDS | |
|---|---|
| ADP | H2O2 |
| Ascorbate | Ivabradine |
| ATP | Malate |
| Glutamate |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14
Introduction
Myocardial oxygen consumption per cardiac cycle is fairly constant, and it increases proportionately with an increase in heart rate. In contrast, the duration of diastole, during which coronary blood flow occurs, decreases overproportionately with an increase in heart rate. As a consequence, both increased myocardial oxygen extraction and metabolic coronary vasodilation cooperate to match an increased oxygen supply to the increased oxygen consumption with increased heart rate. Such matching is increasingly difficult at higher heart rate, even in normal myocardium (Heusch, 2008a).
In coronary artery disease, adjacent myocardial areas supplied by normal and by atherosclerotic coronary arteries co-exist, and metabolic vasodilatation in normal myocardium together with increased extravascular compression in poststenotic myocardium act together to decrease the driving pressure gradient for collateral blood flow (Heusch and Yoshimoto, 1983; Custodis et al., 2010) and myocardial ischaemia ensues. Blockade of β-adrenoceptors attenuates increases in heart rate during exercise, stress and pain, and thereby attenuates myocardial ischaemia (Matsuzaki et al., 1984). However, β-blockade also impairs ventricular function and augments α-adrenoceptor-mediated coronary vasoconstrictor tone that contributes to myocardial ischaemia (Seitelberger et al., 1988; Heusch, 1990; Heusch et al., 2000). In contrast, more selective bradycardic agents limit the increases in heart rate and attenuate myocardial ischaemia without such detrimental effects (Guth et al., 1987; Heusch, 2008a).
Ivabradine is a selective bradycardic agent that inhibits the If current in the sinus node (DiFrancesco and Camm, 2004). Ivabradine has been shown to reduce heart rate and attenuate myocardial ischaemia in various experimental studies (Vilaine et al., 2003; Monnet et al., 2004; Heusch et al., 2008; Ceconi et al., 2009). In clinical settings of myocardial ischaemia, however, the effect of ivabradine is currently contentious. In patients with chronic stable angina, electrocardiographic alterations and clinical symptoms were attenuated by ivabradine (Borer et al., 2003; Tardif et al., 2005; 2009,; Lopez-Bescos et al., 2007; Ruzyllo et al., 2007). In a retrospective subgroup analysis of the BEAUTIFUL trial, the benefit from ivabradine in terms of reduced mortality and hospitalization for acute myocardial infarction in patients with anginal symptoms, left ventricular dysfunction and heart rate >70 per min at baseline was much greater than expected from the modest placebo-corrected heart rate reduction (Fox et al., 2009), suggesting a protective effect of ivabradine beyond that induced by heart rate reduction alone (Heusch, 2009). In contrast, in the recent SIGNIFY trial in patients with stable coronary artery disease and without left ventricular dysfunction, the ivabradine dose was higher, and it failed to reduce outcome in terms of mortality from cardiovascular causes and myocardial infarction. In patients with significant angina, ivabradine even increased cardiovascular mortality and non-fatal myocardial infarction (Fox et al., 2014). These findings suggest that heart rate might not be the appropriate target for cardioprotection by ivabradine in patients with myocardial ischaemia but without left ventricular dysfunction. In line with the notion of pleiotropic cardioprotection, ivabradine still reduced infarct size in a pig model of acute reperfused myocardial infarction even when heart rate was held constant by atrial pacing and even when ivabradine was only given at reperfusion (Heusch et al., 2008). The effectiveness of ivabradine at reperfusion when a burst of reactive oxygen species (ROS) is released (Bolli et al., 1988) indicated that it might attenuate ROS formation (Heusch, 2008b), and indeed in dyslipidaemic mice with ivabradine-induced heart rate reduction, an attenuated ROS formation in vascular tissue was reported (Custodis et al., 2008; Kröller-Schön et al., 2011).
In the present study, we assessed the pleiotropic action of ivabradine in more detail. We confirmed that ivabradine reduces infarct size in the absence of heart rate lowering in a different species (mouse). In mouse isolated ventricular cardiomyocytes, ivabradine provided better preservation of viability and reduced ROS formation during simulated ischaemia/reperfusion. Finally, experiments in mitochondria identified this organelle as a source of ROS formation that was sensitive to ivabradine. Because aging is an established confounder of cardioprotection (Boengler et al., 2009; Ferdinandy et al., 2014), we also demonstrated that ivabradine protected cardiomyocytes of aged mice from the effects of ischaemia/reperfusion.
Methods
Ischaemia/reperfusion in mice in vivo
The experimental protocols were approved by Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen, Recklinghausen, Germany (Az: 84-02.04.2013) and comply with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath et al., 2010).
Male mice (aged 20 ± 3 weeks; weighing 28 ± 2 g) were anaesthetized with pentobarbital (80 mg·kg−1 i.p.), and anaesthesia was maintained with additional pentobarbital as needed. Mice were intubated and ventilated with a respirator (Hugo Sachs Elektronik, March, Germany). Heart rate was recorded from a surface ECG using needle electrodes inserted s.c. into both forelimbs and into one hindlimb. The signal was enhanced, digitized (Picoscope2203, Pico Technology, Cambridgeshire, UK) and recorded over 60–90 s. Heart rate was determined from RR intervals over 8 s in a stable steady state. After measuring baseline heart rate, mice received ivabradine (1.7 mg·kg−1 i.v.) or saline (200 μL i.v.) as placebo (n = 6 each). This dose reduced the heart rate by about 20%, and this heart rate reduction is known to reduce infarct size in a pig model of myocardial ischaemia and reperfusion (Heusch et al., 2008). Heart rate was reduced by ∼70 min−1 within 5 min after ivabradine administration and remained stable during the experiment. An additional group of mice, which were initially treated with ivabradine to decrease heart rate, were paced back to their baseline heart rate and then maintained at constant heart rate by left atrial pacing (impulse generator, PCGU1000, Velleman, Gavere, Belgium: pulse width 1 ms, pulse amplitude 0–5 V) (n = 6). Mice were then subjected to 30 min occlusion and 120 min reperfusion of the left anterior descending coronary artery. Next, mice were killed by cervical dislocation, and Evans blue (5% v/v) was injected after re-occlusion of the left anterior descending coronary artery to delineate the area at risk (AAR). The hearts were removed and flushed with saline, left ventricles were excised, frozen for 30 min, sectioned into five transverse slices and immersed in buffer containing triphenyltetrazolium chloride (1% w/v) for 5 min at 37°C. The Evans blue-stained AAR and the triphenyltetrazolium chloride-negative stained regions of the myocardium were quantified using planimetry by Corel Photo-Paint (Corel Inc., Ottawa, Canada). Infarct size was expressed as a percentage of the AAR.
Isolation of cardiomyocytes
In separate studies, mice were killed by cervical dislocation and their hearts rapidly removed. Ventricular cardiomyocytes were isolated from young (aged 23 ± 3 weeks; weighing 28 ± 1 g) and old (aged 131 ± 6 weeks; weighing 34 ± 5 g) mice using a procedure described previously with some modifications (Li et al., 2004). Briefly, after the heart had been removed, the aorta was directly cannulated. Hearts were flushed with 4°C Tryode’s solution containing (in mmol·L−1) 113 NaCl, 4.7 KCl, 0.6 KH2PO4, 0.6 Na2HPO4, 1.2 MgSO4, 12 NaHCO3, 10 KHCO3, 10 HEPES, 30 taurine, 5.5 glucose, 10 2,3-butanedione monoxime and perfused in a Langendorff apparatus for 7 min at constant flow of 3 mL·min−1 with 37°C Tryode’s solution. Perfusion was then switched to Tryode’s solution with liberase (75 μg·mL−1) and trypsin (0.01% w/v) for 90 s. Atria were removed and discarded, ventricles were sectioned and cells were dispersed and resuspended in Tryode’s solution containing bovine calf serum (10% v/v) and CaCl2 (12.5 μmol·L−1). Isolated myocytes were purified from myocardial tissue by filtering through a nylon filter (200 μm mesh, Millipore, Billerica, MA, USA); CaCl2 was titrated at 20°C in five steps (each 10 min duration) to a final concentration of 1 mmol·L−1.
Simulated ischaemia/reperfusion injury in isolated cardiomyocytes
Isolated cardiomyocytes were assigned to a time control group exposed to normoxic buffer and to groups that were exposed to simulated ischaemia/reperfusion in the absence or presence of ivabradine. Final concentrations of 1 and 3 μmol·L−1 ivabradine were used, respectively; these concentrations correspond to the minimal and maximal doses that were previously used in different in vitro experiments (Bucchi et al., 2006; El Chemaly et al., 2007). Ischaemia (60 min) was induced by pelleting the cardiomyocytes in hypoxic buffer with pH adjusted to 6.5 and an osmolarity of 310 mOsm·L−1 containing (in mmol·L−1) 119 NaCl, 12 KCl, 0.5 MgCl2, 0.9 CaCl2, 5 HEPES, 20 Na lactate and sealing with a layer of mineral oil. Simulated reperfusion was induced by introducing normoxic buffer with pH adjusted to 7.4 and an osmolarity of 250 mOsm·L−1 containing (in mmol·L−1) 88 NaCl, 5.4 KCl, 1.2 Na2HPO4, 0.5 MgCl2, 1 CaCl2, 2.5 creatine, 20 HEPES, 5 taurine and 15 glucose for 5 min at room temperature. Normoxic buffer (in mmol·L−1: 125 NaCl, 5.4 KCl, 1.2 Na2HPO4, 0.5 MgCl2, 1 CaCl2, 2.5 creatine, 20 HEPES, 5 taurine, 15 glucose; 310 mOsm·L−1) was added to the cardiomyocytes assigned to the corresponding control group. Ivabradine (final concentration 1 or 3 μmol·L−1, respectively) or saline (control) was added before and during simulated ischaemia/reperfusion (n = 7 heart preparations). The timing of ivabradine addition (3 μmol·L−1) varied: cardiomyocytes were either treated for 30 min before and continued during simulated ischaemia/reperfusion or ivabradine was administered 10 min before and continued during reperfusion (n = 7 heart preparations). To determine the effect of ivabradine on cardiomyocytes of old mice, ivabradine (3 μmol·L−1) or saline was added 30 min before and during simulated ischaemia/reperfusion, or ivabradine was administered 10 min before and continued during reperfusion (n = 7 heart preparations).
Cardiomyocyte viability
Cardiomyocyte viability was determined at baseline, after 65 min normoxia (time control) and after simulated ischaemia/reperfusion by assessing trypan blue (0.5% v/v) exclusion and morphological criteria. Images of cardiomyocyte suspensions were recorded at 100× magnification (Leica DMLB, Wetzlar, Germany) for subsequent offline analysis. Viability was quantified as the fraction of rod-shaped, unstained cells over all cells, quantified in >200 cells per preparation and expressed as a percentage. Preparations with a yield of viable cardiomyocytes at baseline of less than 60% for young hearts or less than 30% for old hearts were discarded.
ROS during simulated ischaemia/reperfusion of isolated cardiomyocytes
Cardiomyocytes were loaded with the redox probe 5-(and-6)-chloromethyl-2′,7′-dichloro-dihydrofluorescein diacetate (CM-H2DCFDA; final concentration 5 μmol·L−1, Molecular Probes, Eugene, OR, USA) (Zhang et al., 2008) before and after 60 min simulated ischaemia (without/with 3 μmol·L−1 ivabradine). In the time control group, normoxic buffer (without/with 3 μmol·L−1 ivabradine) was added (n = 8 hearts). After cleavage by intracellular esterases, CM-H2DCFDA is retained in the intracellular compartment and oxidized to the highly fluorescent chloromethyl-2′,7′-dichlorofluorescein (CM-DCF) (Ku et al., 2013). CM-DCF fluorescence intensity was detected at 100× magnification (DMLB, Leica) and excitation and emission wavelengths of 480 and 520 nm respectively. In subsequent offline analysis, the background signal intensity was subtracted and fluorescence quantified in >20 cells per image as arbitrary units (a.u.). Only viable cells were included in this quantification. Viability was defined by an elongated shape, visible striations and lack of parietal blebs. It was impossible to quantify the fluorescence intensity during reperfusion as the fluorescence signal leaked into the extracellular buffer.
The Amplex Red Hydrogen Peroxide Assay (Invitrogen, Darmstadt, Germany) was used to detect peroxides as a marker for ROS in the extracellular space at baseline and after simulated ischaemia/reperfusion respectively. Amplex Red reacts in 1:1 stoichiometry with peroxides under catalysis by HRP and produces highly fluorescent resorufin. A total of 50 μmol·L−1 Amplex UltraRed and 2 U·mL−1 HRP were added to the buffers (control, simulated ischaemia and reperfusion respectively) (Schroder et al., 2012). Isolated cardiomyocytes were resuspended in 800 μL buffer without/with ivabradine (3 μmol·L−1) added before and during simulated ischaemia/reperfusion or only 10 min before and during reperfusion (each n = 8 heart preparations). After simulated ischaemia/reperfusion, the supernatant was collected and the yield of resorufin determined using a 96-well black plate and a Cary Eclipse fluorescence spectrophotometer (Varian, Mulgrave, Victoria, Australia) at 540 nm extinction and 580 nm emission wavelength respectively. Cardiomyocyte protein concentration was determined after cell lysis with 1 mmol·L−1 Tris/2% (w/v) SDS lysis buffer using a commercial kit (Lowry method, Bio-Rad, Hercules, CA, USA) with BSA as standard. ROS in the cardiomyocyte supernatant was then quantified in comparison with a H2O2 standard curve; the background fluorescence was subtracted, normalized for protein concentration (100 μg) of the cardiomyocytes and expressed as extracellular ROS concentration (μmol per 100 μg protein).
All experiments were performed in the absence and presence of 50 U·mL−1 catalase. The accumulation of ROS was determined as the catalase-sensitive component of the Amplex Red oxidation. To exclude an interference of buffer substances or ivabradine (3 μmol·L−1) with the Amplex Red detection assay, H2O2 standards (0; 0.025; 0.05; 0.1; 0.25; 0.5; 0.75 μmol·L−1) were added to each buffer.
Isolation of mitochondria
Mice (aged 20 ± 3 weeks; weighing 28 ± 3 g) were killed by cervical dislocation and their hearts rapidly removed, placed in ice-cold isolation buffer containing (in mmol·L−1) 250 sucrose, 10 HEPES, 1 EGTA, pH 7.4 with 0.5% w/v BSA, minced thoroughly using scissors and then homogenized with a tissue homogenizer (Ultra-Turrax, IKA, Staufen, Germany) using two 10 s treatments at a shaft rotation rate of 6500 rpm. This homogenate was further homogenized with proteinase type XXIV (8 IU·mg−1 tissue weight) using a Teflon pestle. The homogenate was centrifuged at 700× g for 10 min at 4°C. The supernatant was collected and centrifuged at 14 000× g for 10 min. The resulting pellet was resuspended in isolation buffer without BSA and centrifuged at 10 000× g for 5 min at 4°C. This procedure was repeated, and the pellet was resuspended in isolation buffer. The protein concentration of the isolated pellet was determined using a protein assay (Lowry method, Bio-Rad) in comparison with a BSA standard (Thermo Scientific, Waltham, MA, USA).
Mitochondrial oxygen consumption under normoxia and simulated ischaemia/reperfusion
Oxygen uptake of mitochondrial proteins was measured with a Clark-type electrode (Strathkelvin, Glasgow, UK) at 30°C during magnetic stirring in incubation buffer containing (in mmol·L−1) 125 KCl, 10 MOPS, 2 MgCl2, 5 KH2PO4, 0.2 EGTA with glutamate (5 mmol·L−1) and malate (5 mmol·L−1) as substrates for complex I and in the presence of 2 μmol·L−1 free Ca2+ (Heusch et al., 2011). Extramitochondrial Ca2+ was adjusted using Ca2+/EGTA buffers (Schild et al., 2003). For the calculation of the concentration of free calcium, the maxchelator programme (maxchelator.stanford.edu, Stanford, CA, USA) was used. The oxygen electrode was calibrated using a solubility coefficient of 237 nmol O2 mL−1 at 30°C.
For the measurement of ADP-stimulated respiration at baseline, 50 μg mitochondrial proteins were added to 0.5 mL incubation buffer supplemented without/with 3 mmol·L−1 ivabradine. Two minutes after incubation, 1 mmol·L−1 ADP was added and stimulated respiration (state 3) measured over 2–3 min (n = 34 heart preparations).
For the measurement of ADP-stimulated respiration after simulated ischaemia/reperfusion, the buffer (without glutamate and malate) was made hypoxic by the introduction of purified argon until the oxygen concentration was <15 nmol O2 mL−1. Mitochondrial proteins (200 μg) were added to 0.25 mL hypoxic buffer supplemented without/with 3 μmol·L−1 ivabradine, the maximal concentration that was used during simulated ischaemia/reperfusion of isolated cardiomyocytes. After 6 min, air-saturated incubation buffer (0.25 mL), again supplemented without/with 3 μmol·L−1 ivabradine, was added to achieve re-oxygenation for 3 min. After simulated ischaemia/reperfusion or the respective normoxic time control (9 min), glutamate (5 mmol·L−1) and malate (5 mmol·L−1) were given as substrates for complex I, mitochondria were stimulated with ADP and respiration was measured over 2–3 min. Finally, mitochondria were used to either measure complex IV respiration and maximal uncoupled oxygen uptake, or extramitochondrial ROS concentration, or ATP production.
In a subset of eight mitochondrial preparations, after measurement of ADP-stimulated respiration during normoxia (baseline and time control) and simulated ischaemia/reperfusion, respectively, complex IV respiration was determined by adding N,N,N,N′-tetramethyl-p-phenylenediamine (300 μmol·L−1) plus ascorbate (3 mmol·L−1), which donates electrons to cytochrome oxidase via the reduction of cytochrome c. Maximal uncoupled oxygen uptake was measured in the presence of 30 nmol·L−1 carbonyl cyanide-p-trifluoromethoxyphenylhydrazone.
Mitochondrial ROS after normoxia and simulated ischaemia/reperfusion
In a subset of 11 mitochondrial preparations, after measurement of ADP-stimulated respiration during normoxia (baseline and time control) and simulated ischaemia/reperfusion, respectively, the incubation buffer containing mitochondria was removed from the respiration chamber, mitochondrial protein concentration adjusted to 1 μg per 10 μL and immediately supplemented with 50 μmol·L−1 Amplex UltraRed and 2 U·mL−1 HRP (Schroder et al., 2012). The supernatant was collected after 30 min of incubation in the dark. Extramitochondrial ROS concentration was detected fluorometrically and quantified as described earlier.
Mitochondrial ATP production under normoxia and simulated ischaemia/reperfusion
In a subset of 15 mitochondrial preparations, after measurement of ADP-stimulated respiration during normoxia (baseline and time control) and simulated ischaemia/reperfusion, respectively, the incubation buffer with the mitochondria was taken from the respiration chamber, mitochondrial protein concentration adjusted to 1 μg per 10 μL and immediately supplemented with ATP assay mix (diluted 1:5). Mitochondrial ATP production after each measurement of respiration was determined immediately and compared with ATP standards using a 96-well white plate and a Cary Eclipse spectrophotometer (Varian) at 560 nm emission wavelength (Boengler et al., 2012).
Mitochondrial calcium retention capacity (CRC) under normoxia and simulated ischaemia/reperfusion
CRC was used as an indicator of mitochondrial permeability transition pore opening. In a subset of eight mitochondrial preparations, after measurement of ADP-stimulated respiration during normoxia (baseline and time control) and simulated ischaemia/reperfusion, respectively, the incubation buffer (without EGTA) containing mitochondria was removed from the respiration chamber. Mitochondrial protein concentration was adjusted to 1 μg per 10 μL and immediately supplemented with calcium green-5N (0.5 μmol·L−1, Invitrogen, Carlsbad, CA, USA), which was used as an indicator to detect extramitochondrial calcium spectrophotometrically (Cary Eclipse, Varian) at excitation and emission wavelengths of 500 and 530 nm respectively. Pulses of 5 nmol CaCl2 were added every minute until calcium was no longer taken up by the mitochondria and a rapid increase in calcium green fluorescence was detected. CRC was expressed as maximal mitochondrial calcium uptake (Heusch et al., 2011; Gedik et al., 2013).
Statistics
Data are mean ± SEM. AAR and infarct size were analysed by one-way anova followed by Fisher’s post hoc test. Heart rate, cardiomyocyte viability, intra-, extracellular and mitochondrial ROS, mitochondrial respiration, ATP production and CRC were analysed by two-way anova for repeated measurements. When a significant difference was detected, anova was followed by Fisher’s post hoc tests (SigmaStat 2.03, SPSS Inc., Chicago, IL, USA).
Materials
Chemicals were of the highest quality available, and all solutions were freshly prepared using MilliQ® Water (Merck Millipore, Darmstadt, Germany) or high-quality analytical-grade organic solvents and, where appropriate, sterilized before use. Unless otherwise indicated, materials were obtained from Sigma-Aldrich (Deisenhofen, Germany) or purchased from vendors as indicated. Ivabradine was provided by Servier (Suresnes, France). C57Bl6/J mice were purchased from Charles River Laboratories (Kisslegg, Germany).
Results
Myocardial ischaemia/reperfusion in vivo
Baseline heart rate was well matched between groups. After ivabradine, heart rate was reduced by 18 ± 1%, and atrial pacing abolished this reduction of heart rate. Heart rate was constant during ischaemia and reperfusion in all groups (Figure 1A). The AAR was not significantly different between groups (Figure 1B). Ivabradine reduced infarct size, and this benefit remained with atrial pacing (Figure 1C).
Figure 1.

Heart rate-independent infarct size reduction by ivabradine in mice in vivo. Heart rate was reduced by ivabradine; this reduction was abolished by atrial pacing. Heart rate did not change during ischaemia and reperfusion in all groups respectively. *P < 0.01 ivabradine versus placebo and versus ivabradine with atrial pacing, using two-way repeated measures anova followed by Fisher’s post hoc test. (A) AAR was comparable between groups (B), whereas infarct size was reduced with or without heart rate reduction by ivabradine (C). Data represent mean ± SEM of n = 6 independent hearts. *P < 0.01 versus placebo using one-way anova followed by Fisher’s post hoc test.
Cardiomyocyte viability with simulated ischaemia/reperfusion
The viability of ventricular cardiomyocytes from young mice was comparable at baseline, and it remained relatively stable with 65 min normoxia (Figure 2A and B). Simulated ischaemia/reperfusion reduced cardiomyocyte viability, whereas it was better preserved with ivabradine at both 1 and 3 μmol·L−1. Surprisingly, administration of ivabradine just before and during reperfusion provided greater protection than ivabradine administration throughout (Figure 2A and B). The maintenance of cardiomyocyte viability was also better during normoxia with ivabradine than without (Figure 2A and B).
Figure 2.

Cardiomyocyte viability during simulated ischaemia/reperfusion (I/R) was better preserved by ivabradine, irrespectively of its concentration [1 (A) vs. 3 μmol·L−1 (B, C)], and mouse age [(A, B) young vs. (C) old mice]. Data represent mean ± SEM of n = 7 different heart preparations. *P < 0.05 using two-way repeated measures anova followed by Fisher’s post hoc tests.
The viability of ventricular cardiomyocytes from old mice at baseline was much less than that of young mice (56 ± 1% vs. 74 ± 1%; P < 0.01). The viability of old cardiomyocytes remained relatively stable during normoxia. Simulated ischaemia/reperfusion also reduced the viability of old cardiomyocytes, and 3 μmol·L−1 ivabradine attenuated the loss of viability. The extent of protection during simulated ischaemia/reperfusion was not different between young and old cardiomyocytes, whereas the better maintenance of viability during normoxia was not seen in old cardiomyocytes, and the greater magnitude of protection with ivabradine just before and during reperfusion in young mouse cardiomyocytes was also not seen in those of old mice (Figure 2C).
ROS during simulated ischaemia/reperfusion
Intracellular ROS-related fluorescence was comparable at baseline without and with ivabradine. Simulated ischaemia increased intracellular ROS-related fluorescence, and ivabradine prevented this increase (Figure 3A and B). Under normoxia, there was no detectable increase in intracellular ROS-related fluorescence over 40 min (without/with ivabradine: 5.2 ± 0.4/5.3 ± 0.5 a.u.).
Figure 3.

Invabradine reduces the intracellular ROS concentration in preparations of isolated cardiomyocytes. (A) Immunofluorescence images of isolated cardiomyocytes, incubated without/with ivabradine and with CM-DCF for ROS detection. (B) Detection of intracellular ROS-related fluorescence with CM-DCF. Ivabradine reduced intracellular ROS-related fluorescence of isolated cardiomyocytes during simulated ischaemia (I) (n = 8). Data represent mean ± SEM. §P < 0.05 without (−) ivabradine versus with (+) ivabradine, two-way repeated measures anova followed by Fisher’s post hoc tests.
There was no difference in the standard curves between the buffer without/with ivabradine excluding its interference with the extracellular ROS detection by Amplex Red (slope without/with ivabradine 2.58/2.61; intercept without/with ivabradine 0.040/0.004; n = 6; Figure 4A). These data also indicate that ivabradine is not a direct scavenger of ROS.
Figure 4.
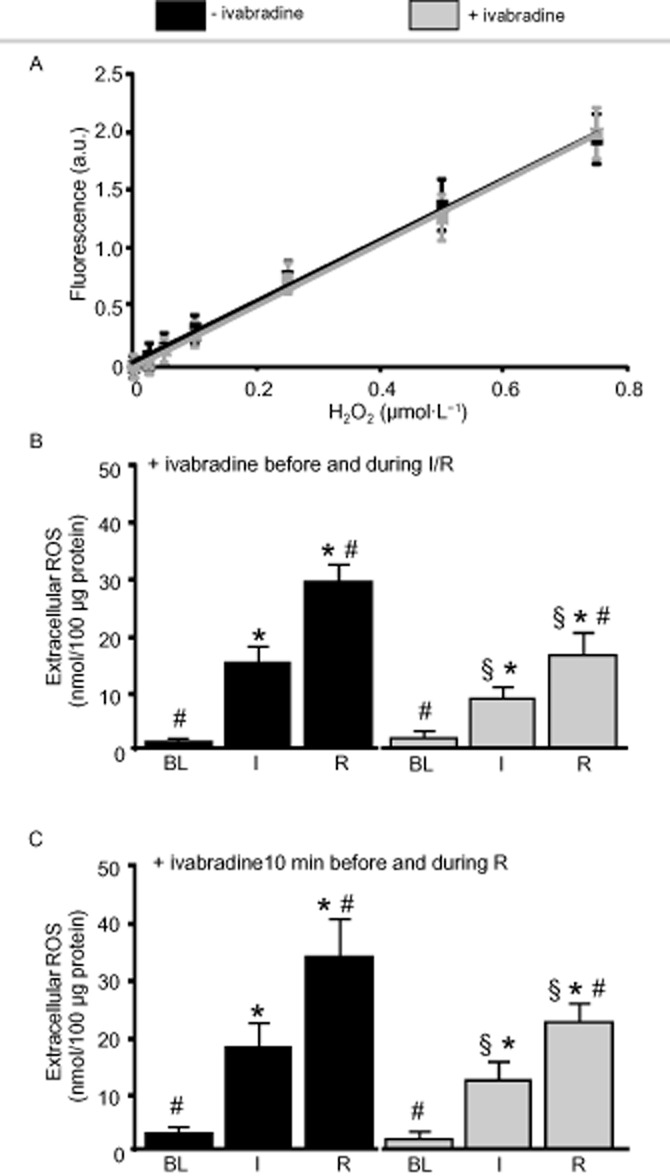
Ivabradine reduces the extracellular ROS concentration in preparations of isolated cardiomyocytes; detected with Amplex Red. (A) There was no difference in the standard curves between the buffer without/with ivabradine, excluding its interference with the ROS detection and direct scavenging of ROS by ivabradine. (B) Ivabradine reduced the extracellular ROS concentration when given before and during ischaemia (I) and reperfusion (R) (n = 8), (C) or given 10 min before and during reperfusion (n = 8). Data represent mean ± SEM. §P < 0.05 without (−) ivabradine versus with (+) ivabradine. *P < 0.05 versus baseline (BL). #P < 0.05 versus I two-way repeated measures anova followed by Fisher’s post hoc tests.
ROS in the extracellular space at baseline was comparable without and with ivabradine. The increase in the extracellular ROS concentration during simulated ischaemia/reperfusion was attenuated with ivabradine (Figure 4B). The increase in the extracellular ROS concentration was also attenuated when ivabradine was given no earlier than 10 min before the end of ischaemia and during reperfusion (Figure 4C).
Mitochondrial respiration, ROS, ATP production and CRC
ADP-stimulated complex I respiration was reduced after simulated ischaemia/reperfusion compared with baseline and the time control, but was not different without and with ivabradine (Figure 5A). Complex IV respiration and maximal uncoupled oxygen uptake (Figure 5B) were also not different without and with ivabradine. The extramitochondrial ROS concentration was increased after simulated ischaemia/reperfusion compared with baseline and the time control. Ivabradine reduced the extramitochondrial ROS concentration at baseline, during the time control and after simulated ischaemia/reperfusion respectively (Figure 5C). Mitochondrial ATP production was decreased after simulated ischaemia/reperfusion compared with baseline and the time control. Ivabradine improved ATP production at baseline, during the time control and after simulated ischaemia/reperfusion respectively (Figure 5D). Mitochondrial CRC (Figure 5E) was decreased after simulated ischaemia/reperfusion compared with baseline and the time control. Ivabradine improved CRC after simulated ischaemia/reperfusion.
Figure 5.

Mitochondrial respiration, ROS, ATP production and CRC after simulated ischaemia/reperfusion (I/R). (A) ADP-stimulated complex I respiration (n = 42) was reduced after I/R compared with baseline (BL) and time control, but was not different without and with ivabradine. (B) Complex IV respiration and maximal uncoupled oxygen uptake (n = 8) were also not different without and with ivabradine. (C) The extramitochondrial ROS concentration (n = 11) was increased after I/R compared with BL and the time control. Ivabradine reduced the extramitochondrial ROS concentration at BL, during time control and after simulated I/R respectively. (D) Mitochondrial ATP production (n = 15) was decreased after simulated I/R compared with BL and the time control. Ivabradine improved ATP production at BL, during the time control and after simulated I/R respectively. (E) Mitochondrial CRC (n = 8) was decreased after I/R compared with BL and the time control. Ivabradine improved CRC after simulated I/R. Data are mean ± SEM. §P < 0.05 without (−) ivabradine versus with (+) ivabradine. *P < 0.05 versus BL. #P < 0.05 versus time control using two-way repeated measures anova followed by Fisher’s post hoc tests.
Discussion
Consistent with data previously reported from anaesthetized pigs (Heusch et al., 2008), data from the present study confirm the heart rate-independent reduction of infarct size by ivabradine in mice. We emphasize that our focus on such pleiotropic protection by ivabradine is not meant to detract from its established protective effect mediated through heart rate reduction in studies where there was left ventricular dysfunction and/or heart failure (Fox et al., 2009; Swedberg et al., 2010). In fact, although heart rate reduction is beneficial in myocardial ischaemia and heart failure, it comes at the price of negative inotropism and augmented α-adrenoceptor-mediated coronary vasoconstriction when achieved through blockade of β-adrenoceptors (Seitelberger et al., 1988; Heusch, 1990; Heusch et al., 2000) and is associated with other serious side effects when achieved by calcium antagonism (Heusch, 1992) or digitalis (Castagno et al., 2012; Dobre et al., 2014). In contrast, ivabradine provides cardioprotection, which goes beyond that mediated by heart rate reduction per se. In patients with stable coronary artery disease and left ventricular dysfunction, heart rate reduction by ivabradine improves patients’ outcome (Fox et al., 2009). In patients with stable coronary artery disease without left ventricular dysfunction, there was also an ivabradine-mediated heart rate reduction accompanied by a reduction in symptoms. However, the heart rate reduction with ivabradine did not result in an improved outcome for the patients (Fox et al., 2014). Thus, the clinical outcome in settings of myocardial ischaemia per se may not be heart rate-dependent. Instead, protection may arise from the pleiotropic actions of ivabradine and a negative outcome may arise from arrhythmias induced by high doses of ivabradine.
The heart rate-independent cardioprotection mediated by ivabradine is provided through an effect at the ventricular level, as shown in our isolated murine cardiomyocyte preparations, and is independent of contraction frequency, as these isolated cardiomyocytes are non-contractile. Whereas ivabradine is relatively selective in comparison with β-blockers, calcium antagonists and digitalis in reducing heart rate by its inhibition of If current in the sinus node (Thollon et al., 1997; DiFrancesco and Camm, 2004), there are also ivabradine targets in ventricular cardiomyocytes. The If channels are members of the hyperpolarization-activated cyclic nucleotide gated (HCN) channel family, and HCN4 is the main isoform expressed in the sinus node of the heart (Bucchi et al., 2006; 2013,). However, HCN2 and 4 have also been demonstrated in ventricular myocardium of mice (Herrmann et al., 2011; Hofmann et al., 2012) and humans, particularly in failing human hearts (Cerbai et al., 2001; Stillitano et al., 2008). Whether or not heart rate-independent cardioprotection mediated by ivabradine was provided by HCN inhibition in ventricular cardiomyocytes or some other action is unclear from our study and deserves further investigation.
In our present study, the cardioprotective action of ivabradine was associated with reduced ROS formation, when detected intracellularly or extracellularly, from isolated cardiomyocytes or, more specifically, isolated mitochondria. Extramitochondrial ROS was also reduced at baseline; however, extracellular ROS was not. We can only speculate that this could be due to the scavenging of ROS within the cytosol. In fact, reduced ROS formation has been suggested before as a mechanism of ivabradine’s cardioprotection, when it reduced infarct size after it had been administered just during reperfusion (Heusch, 2009) when a burst of ROS formation is known to occur (Bolli et al., 1988). Here, we were able to confirm this effect when ivabradine was given to isolated cardiomyocytes just shortly before and during simulated reperfusion. Somewhat surprisingly, in this model of isolated cardiomyocytes, the protective effect of ivabradine was even greater when given shortly before reperfusion than when given before and during simulated ischaemia/reperfusion. However, this time-dependent benefit was not observed in isolated cardiomyocytes of old mice. Aging increases the ROS formation in cardiomyocytes in general (Boengler et al., 2009) and is possibly the reason for such reduced sensitivity to ivabradine. However, aging per se did not affect the protective effect of ivabradine in the absence of ischaemia/reperfusion. Decreased ROS formation by ivabradine has been demonstrated secondary to NADPH oxidase inhibition (Custodis et al., 2008) and prevention of eNOS uncoupling (Kröller-Schön et al., 2011) in vascular tissue of apolipoprotein E knockout mice. In both these vascular preparations, reduced ROS could be attributed to the reduced shear stress at reduced heart rate. Clearly, in our non-contractile ventricular cardiomyocytes a reduced heart rate was not evident, and the demonstrable decrease in ROS was not secondary to heart rate reduction. Our data show a marked reduction in ROS formation with no change in complex I or complex IV respiration, in line with a better coupling of respiration to ATP production, as well as an inhibition of the mitochondrial permeability transition pore opening, as reflected by increased CRC. A quantitative stoichiometric relationship between ROS formation, ATP production, CRC and cellular survival is certainly complex and not known in detail. In cardiomyocytes, the most important ROS generating source are mitochondria (Santos et al., 2011). However, our data do not specify the specific mitochondrial source of ROS and do not exclude sites of ROS formation other than the mitochondria where ivabradine might also act. In mitochondria ROS are generated at complexes I and III of the electron transport chain. Beside ROS generation, the ROS catabolism determines the final ROS content; there is a close link between the redox potential, which is significantly reduced during hypoxia, and the antioxidant defence. In cardiomyocytes, ROS are also generated by NADPH oxidases, xanthine oxidase or NOS. Also ROS generation and metabolism vary – especially during ischaemia/reperfusion (Santos et al., 2011). Also in our set-up, the detected ROS formation is a result of ROS generation and ROS catabolism. Thus, it is possible that the reduction of ROS is a secondary response or a consequence of other cardioprotective signals. The fact that ivabradine also reduced mitochondrial ROS formation and increased the viability of isolated cardiomyocytes under control conditions indicates that the protective effect is not restricted to ROS formation during ischaemia/reperfusion. Although our study identified mitochondria as a target of ivabradine’s protective effect, this does by no means exclude other targets, such as NADPH oxidase or eNOS uncoupling. Further biochemical and/or electrophysiological studies are necessary to clarify the target structure/s of ivabradine; for example, the verification of HCN proteins within the mitochondrial membranes using immunoblotting and/or the recording of ion channel currents using patch clamp techniques.
Taken together, ivabradine reduces infarct size and provides protection beyond that mediated by a reduction in heart rate. Ivabradine improves ventricular cardiomyocyte viability when given before and during simulated ischaemia/reperfusion – even when given shortly before reperfusion. This benefit appears to be mediated by reduced mitochondrial ROS formation, increased ATP production and increased CRC.
Study limitations
Here we used freshly isolated adult cardiomyocytes. The enzymatic digestion during separation stresses the cells and may therefore induce an activation of different stress signals that could influence the ivabradine-induced protection.
The protective effect of ivabradine on cardiomyocyte viability was evident in young and old mice and therefore independent of ageing. The underlying mechanism, notably the reduction in ROS formation during simulated ischaemia/reperfusion, as well as the identification of mitochondria as a source of ROS formation was only proven in isolated ventricular cardiomyocytes of young mice or their mitochondria respectively.
The translation of our findings into the clinical situation is difficult, not only because the drug doses in our current study were higher than in the clinical setting but even more so because the clinical trial data in patients with myocardial ischaemia are currently contentious (Fox et al., 2014). In reperfused acute myocardial infarction, a bolus of i.v. ivabradine during reperfusion reduced heart rate without unwanted side effects, but did not reduce the release of biomarkers that reflect myocardial damage (Steg et al., 2013), most likely because it was given too late to reduce reperfusion injury (Ibanez et al., 2015).
Acknowledgments
The technical assistance of Christin Roeskes and Jelena Löblein is acknowledged.
Glossary
- a.u
arbitrary units
- AAR
area at risk
- CM-DCF
chloromethyl-2′,7′-dichlorofluorescein
- CM-H2DCFDA
5-(and-6)-chloromethyl-2′,7′-dichloro-dihydrofluorescein diacetate
- HCN
hyperpolarization-activated cyclic nucleotide gated
- ROS
reactive oxygen species
Author contributions
P. K. and N. G. performed the experiments and analysed the data. P. K. and G. H. designed the study. P. W. and B. F. contributed essential reagents or tools. P. K., N. G., P. W., B. F., N. K. and G. H. wrote the paper.
Conflict of interest
G. H. serves as a consultant to Servier. P. W. and B. F. were supported by a grant from Servier. B. F. has been a principal investigator on the BEAUTIFUL and SIGNIFY studies run by Servier. B. F. has been on Servier’s Australian advisory board.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, et al. The concise guide to pharmacology 2013/14: ion channels. Br J Pharmacol. 2013b;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: enzymes. Br J Pharmacol. 2013c;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boengler K, Schulz R, Heusch G. Loss of cardioprotection with ageing. Cardiovasc Res. 2009;83:247–261. doi: 10.1093/cvr/cvp033. [DOI] [PubMed] [Google Scholar]
- Boengler K, Ruiz-Meana M, Gent S, Ungefug E, Soetkamp D, Miro-Casas E, et al. Mitochondrial connexin 43 impacts on respiratory complex I activity and mitochondrial oxygen consumption. J Cell Mol Med. 2012;16:1649–1655. doi: 10.1111/j.1582-4934.2011.01516.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolli R, Patel BS, Jeroudi MO, Lai EK, McCay PB. Demonstration of free radical generation in ‘stunned’ myocardium of intact dogs with the use of the spin trap α-phenyl N-tert-butyl nitrone. J Clin Invest. 1988;82:476–485. doi: 10.1172/JCI113621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borer JS, Fox K, Jaillon P, Lerebours G. Antianginal and antiischemic effects of ivabradine, an If inhibitor, in stable angina. Circulation. 2003;107:817–823. doi: 10.1161/01.cir.0000048143.25023.87. [DOI] [PubMed] [Google Scholar]
- Bucchi A, Tognati A, Milanesi R, Baruscotti M, DiFrancesco D. Properties of ivabradine-induced block of HCN1 and HCN4 pacemaker channels. J Physiol. 2006;572:335–346. doi: 10.1113/jphysiol.2005.100776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucchi A, Baruscotti M, Nardini M, Barbuti A, Micheloni S, Bolognesi M, et al. Identification of the molecular site of ivabradine binding to HCN4 channels. PLoS ONE. 2013;8:e53132. doi: 10.1371/journal.pone.0053132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castagno D, Petrie MC, Claggett B, McMurray J. Should we SHIFT our thinking about digoxin? Observations on ivabradine and heart rate reduction in heart failure. Eur Heart J. 2012;33:1137–1141. doi: 10.1093/eurheartj/ehs004. [DOI] [PubMed] [Google Scholar]
- Ceconi C, Cargnoni A, Francolini G, Parinello G, Ferrari R. Heart rate reduction with ivabradine improves energy metabolism and mechanical function of isolated ischaemic rabbit heart. Cardiovasc Res. 2009;84:72–82. doi: 10.1093/cvr/cvp158. [DOI] [PubMed] [Google Scholar]
- Cerbai E, Sartiani L, DePaoli P, Pino R, Maccherini M, Bizzarri F, et al. The properties of the pacemaker current If in human ventricular myocytes are modulated by cardiac disease. J Mol Cell Cardiol. 2001;33:441–448. doi: 10.1006/jmcc.2000.1316. [DOI] [PubMed] [Google Scholar]
- Custodis F, Baumhäkel M, Schlimmer N, List F, Gensch C, Böhm M, et al. Heart rate reduction by ivabradine reduces oxidative stress, improves endothelial function, and prevents atherosclerosis in apolipoprotein E deficient mice. Circulation. 2008;117:2377–2387. doi: 10.1161/CIRCULATIONAHA.107.746537. [DOI] [PubMed] [Google Scholar]
- Custodis F, Schirmer SH, Baumhäkel M, Heusch G, Böhm M, Laufs U. Vascular pathophysiology in response to increased heart rate. J Am Coll Cardiol. 2010;56:1973–1983. doi: 10.1016/j.jacc.2010.09.014. [DOI] [PubMed] [Google Scholar]
- DiFrancesco D, Camm JA. Heart rate lowering by specific and selective If current inhibition with ivabradine. A new therapeutic perspective in cardiovascular disease. Drugs. 2004;64:1757–1765. doi: 10.2165/00003495-200464160-00003. [DOI] [PubMed] [Google Scholar]
- Dobre D, Borer JS, Fox K, Swedberg K, Adams KF, Cleland JG, et al. Heart rate: a prognostic factor and therapeutic target in chronic heart failure. The distinct roles of drugs with heart rate-lowering properties. Eur J Heart Fail. 2014;16:76–85. doi: 10.1093/eurjhf/hft129. [DOI] [PubMed] [Google Scholar]
- El Chemaly A, Magaud C, Patri S, Jayle C, Guinamard R, Bois P. The heart rate-lowering agent ivabradine inhibits the pacemaker current I(f) in human atrial myocytes. J Cardiovasc Electrophysiol. 2007;18:1190–1196. doi: 10.1111/j.1540-8167.2007.00955.x. [DOI] [PubMed] [Google Scholar]
- Ferdinandy P, Hausenloy DJ, Heusch G, Baxter GF, Schulz R. Interaction of risk factors, comorbidities and comedications with ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning, and remote conditioning. Pharmacol Rev. 2014;66:1142–1174. doi: 10.1124/pr.113.008300. [DOI] [PubMed] [Google Scholar]
- Fox K, Ford I, Steg PG, Tendera M, Robertson M, Ferrari R. Relationship between ivabradine treatment and cardiovascular outcomes in patients with stable coronary artery disease and left ventricular systolic dysfunction with limiting angina: a subgroup analysis of the randomized, controlled BEAUTIFUL trial. Eur Heart J. 2009;30:2337–2345. doi: 10.1093/eurheartj/ehp358. [DOI] [PubMed] [Google Scholar]
- Fox K, Ford I, Steg PG, Tardif JC, Tendera M, Ferrari R. Ivabradine in stable coronary artery disease without clinical heart failure. N Engl J Med. 2014;371:1091–1099. doi: 10.1056/NEJMoa1406430. [DOI] [PubMed] [Google Scholar]
- Gedik N, Heusch G, Skyschally A. Infarct size reduction by cyclosporine A at reperfusion involves inhibition of the mitochondrial permeability transition pore but does not improve mitochondrial respiration. Arch Med Sci. 2013;9:968–975. doi: 10.5114/aoms.2013.38175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guth BD, Heusch G, Seitelberger R, Ross J., Jr Elimination of exercise-induced regional myocardial dysfunction by a bradycardic agent in dogs with chronic coronary stenosis. Circulation. 1987;75:661–669. doi: 10.1161/01.cir.75.3.661. [DOI] [PubMed] [Google Scholar]
- Herrmann S, Layh B, Ludwig A. Novel insights into the distribution of cardiac HCN channels: an expression study in the mouse heart. J Mol Cell Cardiol. 2011;51:997–1006. doi: 10.1016/j.yjmcc.2011.09.005. [DOI] [PubMed] [Google Scholar]
- Heusch G. α-Adrenergic mechanisms in myocardial ischemia. Circulation. 1990;81:1–13. doi: 10.1161/01.cir.81.1.1. [DOI] [PubMed] [Google Scholar]
- Heusch G. Myocardial stunning: a role for calcium antagonists during ischaemia? Cardiovasc Res. 1992;26:14–19. doi: 10.1093/cvr/26.1.14. [DOI] [PubMed] [Google Scholar]
- Heusch G. Heart rate in the pathophysiology of coronary blood flow and myocardial ischaemia: benefit from selective bradycardic agents. Br J Pharmacol. 2008a;153:1589–1601. doi: 10.1038/sj.bjp.0707673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heusch G. Pleiotropic action(s) of the bradycardic agent ivabradine: cardiovascular protection beyond heart rate reduction. Br J Pharmacol. 2008b;155:970–971. doi: 10.1038/bjp.2008.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heusch G. A BEAUTIFUL lesson – ivabradine protects from ischaemia, but not from heart failure: through heart rate reduction or more? Eur Heart J. 2009;30:2300–2301. doi: 10.1093/eurheartj/ehp360. [DOI] [PubMed] [Google Scholar]
- Heusch G, Yoshimoto N. Effects of heart rate and perfusion pressure on segmental coronary resistances and collateral perfusion. Pflügers Arch. 1983;397:284–289. doi: 10.1007/BF00580262. [DOI] [PubMed] [Google Scholar]
- Heusch G, Baumgart D, Camici P, Chilian W, Gregorini L, Hess O, et al. α-Adrenergic coronary vasoconstriction and myocardial ischemia in humans. Circulation. 2000;101:689–694. doi: 10.1161/01.cir.101.6.689. [DOI] [PubMed] [Google Scholar]
- Heusch G, Skyschally A, Gres P, van Caster P, Schilawa D, Schulz R. Improvement of regional myocardial blood flow and function and reduction of infarct size with ivabradine: protection beyond heart rate reduction. Eur Heart J. 2008;29:2265–2275. doi: 10.1093/eurheartj/ehn337. [DOI] [PubMed] [Google Scholar]
- Heusch G, Musiolik J, Gedik N, Skyschally A. Mitochondrial STAT3 activation and cardioprotection by ischemic postconditioning in pigs with regional myocardial ischemia/reperfusion. Circ Res. 2011;109:1302–1308. doi: 10.1161/CIRCRESAHA.111.255604. [DOI] [PubMed] [Google Scholar]
- Hofmann F, Fabritz L, Stieber J, Schmitt J, Kirchhof P, Ludwig A, et al. Ventricular HCN channels decrease the repolarization reserve in the hypertrophic heart. Cardiovasc Res. 2012;95:317–326. doi: 10.1093/cvr/cvs184. [DOI] [PubMed] [Google Scholar]
- Ibanez B, Heusch G, Ovize M, Van de Werf F. Evolving therapies for myocardial ischemia/reperfusion injury. J Am Coll Cardiol. 2015;65:1454–1471. doi: 10.1016/j.jacc.2015.02.032. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kröller-Schön S, Schulz E, Wenzel P, Kleschyov AL, Hortmann M, Torzewski M, et al. Differential effects of heart rate reduction with ivabradine in two models of endothelial dysfunction and oxidative stress. Basic Res Cardiol. 2011;106:1147–1158. doi: 10.1007/s00395-011-0227-3. [DOI] [PubMed] [Google Scholar]
- Ku HC, Chen WP, Su MJ. DPP4 deficiency exerts protective effect against H2O2 induced oxidative stress in isolated cardiomyocytes. PLoS ONE. 2013;8:e54518. doi: 10.1371/journal.pone.0054518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Heinzel FR, Boengler K, Schulz R, Heusch G. Role of connexin 43 in ischemic preconditioning does not involve intercellular communications through gap junctions. J Mol Cell Cardiol. 2004;36:161–163. doi: 10.1016/j.yjmcc.2003.10.019. [DOI] [PubMed] [Google Scholar]
- Lopez-Bescos L, Filipova S, Martos R. Long-term safety and efficacy of ivabradine in patients with chronic stable angina. Cardiology. 2007;108:387–396. doi: 10.1159/000108387. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Patritti J, Tajimi T, Miller M, Kemper WS, Ross J., Jr Effects of β-blockade on regional myocardial flow and function during exercise. Am J Physiol. 1984;247:H52–H60. doi: 10.1152/ajpheart.1984.247.1.H52. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monnet X, Colin P, Ghaleh B, Hittinger L, Giudicelli J-F, Berdeaux A. Heart rate reduction during exercise-induced myocardial ischaemia and stunning. Eur Heart J. 2004;25:579–586. doi: 10.1016/j.ehj.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzyllo W, Tendera M, Ford I, Fox KM. Antianginal efficacy and safety of ivabradine compared with amlodipine in patients with stable effort angina pectoris: a 3-month randomised, double-blind, multicentre, noninferiority trial. Drugs. 2007;67:393–405. doi: 10.2165/00003495-200767030-00005. [DOI] [PubMed] [Google Scholar]
- Santos CX, Anilkumar N, Zhang M, Brewer AC, Shah AM. Redox signaling in cardiac myocytes. Free Radic Biol Med. 2011;50:777–793. doi: 10.1016/j.freeradbiomed.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schild L, Reinheckel T, Reiser M, Horn TFW, Wolf G, Augustin W. Nitric oxide produced in rat liver mitochondria causes oxidative stress and impairment of respiration after transient hypoxia. FASEB J. 2003;17:2194–2201. doi: 10.1096/fj.02-1170com. [DOI] [PubMed] [Google Scholar]
- Schroder K, Zhang M, Benkhoff S, Mieth A, Pliquett R, Kosowski J, et al. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ Res. 2012;110:1217–1225. doi: 10.1161/CIRCRESAHA.112.267054. [DOI] [PubMed] [Google Scholar]
- Seitelberger R, Guth BD, Heusch G, Lee JD, Katayama K, Ross J., Jr Intracoronary α2-adrenergic receptor blockade attenuates ischemia in conscious dogs during exercise. Circ Res. 1988;62:436–442. doi: 10.1161/01.res.62.3.436. [DOI] [PubMed] [Google Scholar]
- Steg P, Lopez-de-Sa E, Schiele F, Hamon M, Meinertz T, Goicolea J, et al. Safety of intravenous ivabradine in acute ST-segment elevation myocardial infarction patients treated with primary percutaneous coronary intervention: a randomized, placebo-controlled, double-blind, pilot study. Eur Heart J Acute Cardiovasc Care. 2013;2:270–279. doi: 10.1177/2048872613489305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stillitano F, Lonardo G, Zicha S, Varro A, Cerbai E, Mugelli A, et al. Molecular basis of funny current If in normal and failing human heart. J Mol Cell Cardiol. 2008;45:289–299. doi: 10.1016/j.yjmcc.2008.04.013. [DOI] [PubMed] [Google Scholar]
- Swedberg K, Komajda M, Böhm M, Borer JS, Ford I, Dubost-Brama A, et al. Ivabradine and outcomes in chronic heart failure (SHIFT): a randomised placebo-controlled study. Lancet. 2010;376:875–885. doi: 10.1016/S0140-6736(10)61198-1. [DOI] [PubMed] [Google Scholar]
- Tardif J-C, Ford I, Tendera M, Bourassa MG, Fox K. Efficacy of ivabradine, a new selective If inhibitor, compared with atenolol in patients with chronic stable angina. Eur Heart J. 2005;26:2529–2536. doi: 10.1093/eurheartj/ehi586. [DOI] [PubMed] [Google Scholar]
- Tardif JC, Ponikowski P, Kahan T. Efficacy of the I(f) current inhibitor ivabradine in patients with chronic stable angina receiving beta-blocker therapy: a 4-month, randomized, placebo-controlled trial. Eur Heart J. 2009;30:540–548. doi: 10.1093/eurheartj/ehn571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thollon C, Bidouard J-P, Cambarrat C, Lesage L, Reure H, Delescluse I, et al. Stereospecific in vitro and in vivo effects of the new sinus node inhibitor (+)-S 16257. Eur J Pharmacol. 1997;339:43–51. doi: 10.1016/s0014-2999(97)01364-2. [DOI] [PubMed] [Google Scholar]
- Vilaine J-P, Bidouard J-P, Lesage L, Reure H, Péglion J-L. Anti-ischemic effects of ivabradine, a selective heart rate-reducing agent, in exercise-induced myocardial ischemia in pigs. J Cardiovasc Pharmacol. 2003;42:688–696. doi: 10.1097/00005344-200311000-00016. [DOI] [PubMed] [Google Scholar]
- Zhang XQ, Yamada S, Barry WH. Ranolazine inhibits an oxidative stress-induced increase in myocyte sodium and calcium loading during simulated-demand ischemia. J Cardiovasc Pharmacol. 2008;51:443–449. doi: 10.1097/FJC.0b013e318168e711. [DOI] [PubMed] [Google Scholar]