

Abstract
Background and Purpose
Myeloid differentiation 2 (MD-2) recognizes LPS, which is required for TLR4 activation, and represents an attractive therapeutic target for severe inflammatory disorders. We previously found that a chalcone derivative, L6H21, could inhibit LPS-induced overexpression of TNF-α and IL-6 in macrophages. Here, we performed a series of biochemical experiments to investigate whether L6H21 specifically targets MD-2 and inhibits the interaction and signalling transduction of LPS-TLR4/MD-2.
Experimental Approach
The binding affinity of L6H21 to MD-2 protein was analysed using computer docking, surface plasmon resonance analysis, elisa, fluorescence measurements and flow cytometric analysis. The effects of L6H21 on MAPK and NF-κB signalling were determined using EMSA, fluorescence staining, Western blotting and immunoprecipitation. The anti-inflammatory effects of L6H21 were confirmed using elisa and RT-qPCR in vitro. The anti-inflammatory effects of L6H21 were also evaluated in septic C57BL/6 mice.
Key Results
Compound L6H21 inserted into the hydrophobic region of the MD-2 pocket, forming hydrogen bonds with Arg90 and Tyr102 in the MD-2 pocket. In vitro, L6H21 subsequently suppressed MAPK phosphorylation, NF-κB activation and cytokine expression in macrophages stimulated by LPS. In vivo, L6H21 pretreatment improved survival, prevented lung injury, decreased serum and hepatic cytokine levels in mice subjected to LPS. In addition, mice with MD-2 gene knockout were universally protected from the effects of LPS-induced septic shock.
Conclusions and Implications
Overall, this work demonstrated that the new chalcone derivative, L6H21, is a potential candidate for the treatment of sepsis. More importantly, the data confirmed that MD-2 is an important therapeutic target for inflammatory disorders.
Tables of Links
| LIGANDS | |
|---|---|
| Auranofin | IL-6 |
| Curcumin | IL-10 |
| E5564 (eritoran) | IL-12 |
| IFN | LPS |
| IL-1β | TNF-α |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14
Introduction
Sepsis remains the most common cause of death in intensive care units in the world. Endotoxin (lipopolysaccharide from Gram-negative bacteria, or LPS) has been implicated as a major cause of sepsis (Esteban et al., 2013; Oblak and Jerala, 2015). The receptor capable of recognizing LPS is mainly Toll-like receptor 4 (TLR4) (Park and Lee, 2013; Tan and Kagan, 2014). TLR signalling involves a family of adaptor proteins which recruit downstream protein kinases to activate transcription factors such as MAPKs, NF-κB and IFN-regulatory factors (Park and Lee, 2013). However, LPS does not bind to TLR4 directly. A small protein, myeloid differentiation 2 (MD-2), participates in the LPS–TLR4 complex by associating with the TLR4 extracellular domain (Duan et al., 2014). Recognition of LPS by TLR4 requires MD-2 since most of the lipid chains of LPS interact with a hydrophobic pocket in MD-2 (Duan et al., 2014). Engagement of LPS with the TLR4/MD-2 complex triggers dimerization of two TLR4/MD-2 complexes, which leads to the recruitment of two major adaptor molecules, MyD88 and TRIF, and the activation of intracellular signalling pathways (i.e. MAPKs and NF-κB) for the production of pro-inflammatory cytokines (Park and Lee, 2013). It has been demonstrated that MD-2 is essential for correct intracellular distribution and LPS recognition of TLR4, and MD-2−/− mice do not respond to LPS and survive endotoxic shock (Nagai et al., 2002). Therefore, MD-2 is proposed as a potential target for a therapy that neutralizes the toxic effects of endotoxin.
Although the studies on anti-sepsis drugs have been researched extensively, few have reached the market. The current knowledge on the structure and function of the TLR4/MD-2 complex has opened up the possibility of the development of new drug targets to prevent sepsis and other diseases associated with this signalling molecule. A series of LPS-like TLR4/MD-2 antagonists, such as fatty acid chain-containing CRX-526, E5531 and E5564 (eritoran), which directly target the MD-2 protein, have been evaluated in clinical and pre-clinical studies (Park et al., 2012). Unfortunately, the phase III study of eritoran completed in 2011 failed due to no significant difference between the eritoran treated group and the placebo group (Barochia et al., 2011; Park et al., 2012). In addition, some small-molecule TLR4/MD-2 inhibitors have been developed for the treatment of sepsis (Park et al., 2012). Due to the seemingly higher importance of TLR4, more attention has been paid to TLR4 than MD-2 in the past decades. However, clinical trials of the TLR4 inhibitor, TAK-242, also failed as it did not suppress the cytokine surge in patients with severe sepsis and related respiratory or cardiovascular disease (Rice et al., 2010).
From recent studies, it has been deduced that some natural and synthetic chemicals unrelated to the structure of bacterial lipids or fatty acids are MD-2 inhibitors (Figure 1A) (Park et al., 2012). These small molecules, such as, JSH (Roh et al., 2011), curcumin (Gradisar et al., 2007), xanthohumol (Peluso et al., 2010) and CAPE (Kim et al., 2013), bind directly to the MD-2 pocket and block the ability of LPS to recognize TLR4/MD-2, causing inflammatory signalling inactivation and prevention of septic shock, even though their specificities remain to be defined. As shown in Figure 1A, the structures of the MD-2 inhibitors share the same chalcone skeleton. Thus, naturally occurring chalcone may have potential as a substance with the ability to target MD-2. Previously, our group synthesized and evaluated a series of chalcone derivatives as anti-inflammatory agents in LPS-stimulated macrophages and mouse models (Wu et al., 2011). Among these synthetic chalcones, (E)-2,3-dimethoxy-4′-methoxychalcone (L6H21) showed excellent anti-inflammatory activity (Figure 1B) (Wu et al., 2011). Thus, it is hypothesized that the new chalcone derivative L6H21 may target MD-2 and serve as an anti-sepsis candidate. In this study, we demonstrated that L6H21 has anti-inflammatory effects, identified its mechanisms as a new MD-2 inhibitor and further validated that this small-molecule MD-2 inhibitor has the potential to be developed as an anti-sepsis drug.
Figure 1.
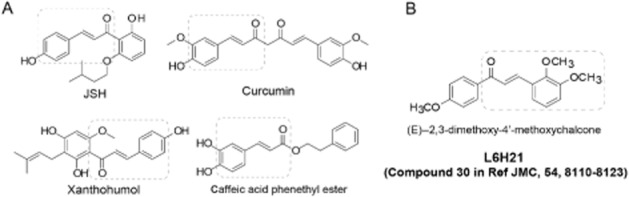
Structures of the current MD-2 inhibitors and compound L6H21.
Methods
Cell lines and reagents
Mouse RAW 264.7 macrophages were purchased from ATCC (Manassas, VA, USA). Recombinant human MD-2 (rhMD-2) protein and rhTLR4 were purchased from R&D (Minneapolis, MN, USA). Mutated rhMD-2 was obtained using the methods described in the Supporting Information. Curcumin, LPS (from Salmonella typhosa), TLR2 agonist Pam3CK and LPS labelled with FITC (LPS-FITC) (from E. coli 055:B5) were purchased from Sigma Chemical Co. (St. Louis, MO, USA). The TLR2 inhibitor CU-CPT22 was purchased from R&D Systems, Inc. (Minneapolis, MN, USA). Anti-p-ERK, anti-ERK, anti-p-p38, anti-p38, anti-p-JNK, anti-JNK, anti-β-actin and anti-I-κB were from Cell Signaling (Danvers, MA, USA). Anti-MD-2 antibody was purchased from eBioscience (San Diego, CA, USA). L6H21 was synthesized and structurally identified using MS and 1H NMR analyses, as described in our previous paper (Wu et al., 2011). Before being use in the biological experiments, L6H21 was recrystallized from CHCl3/EtOH, with an HPLC purity of >99%. In in vitro experiments, L6H21 was used in DMSO solution with DMSO as a vehicle control. In the in vivo study, L6H21 was firstly dissolved in water with macrogol 15 hydroxystearate (a non-ionic solubilizer for injection from BASF) in water. The concentration of L6H21 and solubilizer were 2 mg·mL−1 and 8% in water solution respectively. For the vehicle, the solubilizer was prepared at 8% in water.
Animals
Male C57BL/6 mice weighing 18–22 g were obtained from the Wenzhou Medical University Animal Centre. Male MD2−/− mice (B6.129P2-Ly96 KO) with C57BL/6 background were provided by Riken BioResource Center of Japan (Tsukuba, Ibaraki, Japan). In this study, 170 C57BL/6 mice and 20 MD2−/− mice were used. All animal care and experimental procedures complied with the ‘The Detailed Rules and Regulations of Medical Animal Experiments Administration and Implementation’ (Order No. 1998-55, Ministry of Public Health, China) and ‘Ordinance in Experimental Animal Management’ (Order No. 1998-02, Ministry of Science and Technology, China) and were approved by the Wenzhou Medical University Animal Policy and Welfare Committee (Approval Document Nos. wydw2013-0167 and wydw2014-0062). The results of all studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Preparation of mouse peritoneal macrophages
Mouse primary peritoneal macrophages (MPMs) were prepared and cultured from C57BL/6 mice and MD2−/− mice, respectively, using the method described in our previous paper (Pan et al., 2013).
LPS-induced septic mortality in C57BL/6 mice
Male C57BL/6 mice weighing 18–22 g were randomly divided into four groups (n = 10 per group). Mice were injected with 200 μL of LPS (at 20 mg·kg−1 in 0.9% saline, i.v. through the tail vein) 15 min before (for treatment) or after (for prevention) an injection of L6H21 (at 10 mg·kg−1, i.v. through the tail vein) respectively. Mice in both vehicle control group and LPS alone group received 100 μL of vehicle, and mice in the vehicle control group also received 200 μL of saline. After the LPS injection, body weight change and mortality were recorded for 7 days.
LPS-induced inflammatory responses in C57BL/6 mice
Male C57BL/6 mice weighing 18–22 g were randomly divided into six groups (n = 10 per group). Mice in the LPS + L6H21 group were injected with 200 μL of LPS (at 20 mg·kg−1, i.v. through the tail vein) 15 min after being injected with L6H21 (at 10 mg·kg−1, i.v. through the tail vein). Mice in both vehicle control group and LPS alone group received 100 μL of vehicle, and mice in the vehicle control group also received 200 μL of saline. Two or eight hours after the LPS injection, the mice were anaesthetized and killed. The blood samples were collected from the right ventricle using a heparin-containing syringe with a needle. The serum TNF-α and IL-6 levels were determined using elisa. Both lung and liver were harvested. Liver tissues were homogenized for RNA extraction and real-time qPCR analysis.
LPS-induced septic mortality in MD2−/− mice
Male MD2−/− mice weighing 18–22 g were given an i.v. injection of LPS (20 mg·kg−1). As a comparison, the wide-type male C57BL/6 mice were also i.v. injected with LPS at the same dosage. Their survival was recorded for 7 days (n = 10 in each group).
Docking of L6H21 to the MD-2 structural model
Docking simulation of L6H21 with MD-2 protein (PDB ID: 2E56) was carried out with Tripos molecular modelling packages, Sybyl-2.0 (Tripos, St. Louis, MO, USA). The ligand–receptor complex was subjected to energy minimization using the Tripos force field and electrostatic charges of Gasteiger–Hückel with their energies minimized using the protocol previously indicated (Roh et al., 2011). The ligand-binding groove on MD-2 was kept rigid, whereas all torsible bonds of L6H21 were free, allowing flexible docking to yield more than 100 structures. The final docked conformations were then clustered within the tolerance of 1 Å root-mean-square deviation.
Surface plasmon resonance (SPR) analysis
The binding affinity of L6H21 to recombinant human MD-2 (rhMD-2, or rhMD-2 mutations) or rhTLR4 was determined using a ProteOn XPR36 Protein Interaction Assay system (Bio-Rad Laboratories, Hercules, CA, USA) with an HTE sensor chip (ProteOn™, #176–5033). Briefly, the rhMD-2 or rhTLR4 protein (in acetate acid buffer pH 5.5) was loaded onto the sensors and then 10 mM NiSO4 was added for activation of the chip. Different concentrations of L6H21 (at 64, 32, 16, 8, 4, 2, 1 and 0 μM) were prepared with running buffer (PBS, 0.1% SDS, 5% DMSO). Sensor and sample plate were placed on the instrument. The interactions were determined according to the manufacturer’s instructions at a flow rate of 30 μL·min−1 for 120 s during the association phase followed by 120 s for the dissociation phase at 25°C. The data were analysed with ProteOn manager software. Binding kinetic parameters including the KD values were calculated by global fitting of the kinetic data from various concentrations of L6H21 using a 1:1 Langmuir binding model.
Elisa
LPS binding to MD-2 was determined in a cell-free assay, anti-human MD-2 antibody (eBioscience) was coated in a 96-well plate overnight at 4°C in 10 mM Tris–HCl buffer (pH 7.5). The plate was washed with PBST and blocked with 3% BSA for 1.5 h at room temperature. rhMD2 (4 μg·mL−1) in 10 mM Tris–HCl buffer (pH 7.5) was added to the pre-coated plate and incubated for 1.5 h at room temperature. After being washed with PBST, biotin-labelled LPS (Biotin-LPS, InvivoGen, San Diego, CA, USA) was incubated for 1 h at room temperature with or without the presence of L6H21 (0.1 or 1.0 μM). After further washing, streptavidin-conjugated horseradish peroxidase (Beyotime, Shanghai, China) was added for 1 h at room temperature. The horseradish peroxidase activity was determined in an M5 microplate reader at 450 nm after the addition of TMB substrate solution (eBioscience).
Fluorescence measurements
Fluorescence measurements were performed using an M5 microplate reader (Molecular Devices, Sunnyvale, CA, USA). All measurements were done at 25°C in a 1 cm path-length quartz cuvette. Briefly, 1,1′-bis(anilino)-4,4′-bis(naphthalene)-8,8′-disulfonate (bis-ANS, 5 μM) and rhMD-2 protein (5 nM) were mixed in PBS (pH 7.4) and incubated for 15 min to reach stable relative fluorescence units (RFUs) emitted at 430–590 nm under the excitation at 385 nm. Non-fluorescent L6H21 at 2.5, 5, 10, 20 or 30 μM was then incubated for 5 min followed by measurement of RFUs emitted at 430–590 nm.
Flow cytometric analysis
Cellular binding of fluorescein isothiocyanate-labelled LPS (LPS-FITC, from E. coli 055:B5; Sigma, St. Louis, MO, USA) was measured as described in the previous studies (Roh et al., 2011). Briefly, HUVEC304 cells (1 × 105) were incubated with LPS-FITC (50 μg·mL−1) for 30 min with or without the presence of L6H21. After being washed, the cells bound to LPS-FITC were determined and counted using flow cytometry.
EMSA
Cells were pretreated with L6H21 (2.5, 5 or 10 μM) or vehicle control (DMSO) for 30 min and then incubated with LPS (0.5 μg·mL−1) for 1 h. The binding reaction was detected by incubating 4 μg of nuclear extract prepared through a nucleoprotein and plasmosin extraction kit (KeyGEN, Nanjing, China), followed by a 20 min incubation at room temperature with biotinylated NF-κB probe (KeyGEN). Products were electrophoresed and dried gels were analysed by BeyoECL Plus Reagent with a gel imaging system (Bio-Rad Laboratories).
Assay of cellular NF-κB p-65 translocation
The cells were immunofluorescence-labelled according to the manufacturer’s instruction using a Cellular NF-κB p65 Translocation Kit (Beyotime Biotech, Nantong, China). P65 protein and nuclei fluoresce as red and blue, respectively, and can be simultaneously viewed by fluorescence microscope (200×, Nikon, Tokyo, Japan) at an excitation wavelength of 350 nm for DAPI and 540 nm for cyanine 3 (Cy3). To create a two-colour image, the red and blue images were overlaid.
Western blotting
Cell protein samples (50 μg) were subjected to 10% SDS-PAGE and transferred onto a PVDF membrane (Bio-Rad Laboratories). After being blocked in blocking buffer (5% milk in tris-buffered saline containing 0.05% Tween 20) for 1.5 h at room temperature, membranes were incubated with different primary antibodies overnight at 4°C. The membranes then were washed in TBS-T and reacted with secondary horseradish peroxidase-conjugated antibody (Santa Cruz, CA, USA; 1:5000) for 1–2 h at room temperature. Blots were then visualized using enhanced chemiluminescence reagents (Bio-Rad Laboratories). The density of the immunoreactive bands was analysed using Image J software (NIH, Bethesda, MD, USA).
Immunoprecipitation
TLR4 was co-precipitated with MD2 from macrophages to detect the association of TLR4 with MD2. RAW264.7 macrophages were treated with LPS for 15 min after L6H21 pretreatment. Cell extracts were incubated with anti-MD2 antibody for 1 h and then precipitated with protein G-Sepharose beads at 4°C overnight. The TLR4 level in the beads was further detected by immunoblot using an anti-TLR4 antibody.
RNA extraction and real-time quantitative PCR assay
Total RNA was isolated from cells or tissues (50–100 mg) using TRIZOL (Invitrogen, Carlsbad, CA, USA). Reverse transcription and quantitative PCR (RT-qPCR) were performed using M-MLV Platinum RT-qPCR Kit (Invitrogen). Real-time quantitative PCR was carried out using the Eppendorf Realplex4 instrument (Eppendorf, Hamburg, Germany). Primers of genes including TNF-α, IL-6, IL-1β, IL-12, IL-10, COX-2 and β-actin were synthesized from Invitrogen (Invitrogen, Shanghai, China). The primer sequences used were shown in Supporting Information Table S1. The relative amount of each gene was normalized to the amount of β-actin.
Determination of cytokine levels
For the determination of cytokine levels in cell medium or mouse plasma, cytokine-specific elisa kits (eBioscience) were used according to the manufacturer’s instructions. The total amount of TNF-α or IL-6 in the cell medium was normalized to the total amount of protein in the viable cell pellets. Experiments were performed in triplicate.
Lung histopathology
Lung tissues were fixed in 4% paraformaldehyde solution, embedded in paraffin and sectioned at 5 μm. After dehydration, sections were stained with haematoxylin and eosin (H&E). To evaluate the histopathological damage, each image of sections was obtained using a light microscope (400× amplification; Nikon).
Statistical analysis
Data are presented as means ± SDs. The statistical significance between groups was obtained by Student’s t-test or anova multiple comparisons in GraphPad Pro 5.0 (GraphPad, San Diego, CA, USA). Differences were considered to be significant when P < 0.05.
Results
L6H21 is a specific MD-2 inhibitor and directly binds to MD-2 protein, blocking the formation of the LPS-TLR4/MD-2 complex
The direct interaction of L6H21 and MD-2 was examined by fluorescence spectroscopy using Bis-ANS, a fluorescent probe, to map the lipid-binding sites on several proteins, including MD-2 (Pastukhov and Ropson, 2003; Mancek-Keber and Jerala, 2006). As shown in Figure 2A, the fluorescence values of bis-ANS were markedly enhanced upon binding to cell-free rhMD-2 protein, while incubation with L6H21 decreased the fluorescence intensity of bis-ANS in a dose-dependent manner, suggesting that L6H21 binds rhMD-2 and displaces bis-ANS from rhMD-2. Next, we evaluated the binding of L6H21 with MD-2 by SPR experiments, which showed that L6H21 binds MD-2 protein in a dose-dependent manner with a high affinity and low KD value of 33.3 μM (Figure 2B). In addition, the SPR assay showed that L6H21 interacted with recombinant human TLR4 proteins (Supporting Information Fig. S1).
Figure 2.

L6H21 is an MD-2 inhibitor. (A) Fluorescence measurements showed that L6H21 dose-dependently inhibited the binding of bis-ANS and rhMD-2. (B) SPR analysis showed the direct interaction between L6H21 and rhMD-2. (C) Flow cytometry analysis showed that L6H21 decreased the levels of LPS-FITC on the cell surface in a dose-dependent manner. (D) Immunoprecipitation assay showed that L6H21 pretreatment (1, 5 and 10 μM) significantly reduced the formation of the TLR4–MD2 complex induced by 0.5 μg·mL−1 LPS. (E) As determined by elisa, L6H21 dose-dependently inhibited the binding of biotin-LPS to rhMD-2. (F and G) RAW264.7 macrophages were pretreated with vehicle control (DMSO), L6H21 (5 or 10 μM) or TLR2 inhibitor CU-CPT22 (5 or 10 μM) for 2 h followed by incubation with Pam3CK (0.1 μg·mL−1) for 12 h. The protein level of TNF-α (F) and IL-6 (G) in the culture medium was measured by elisa and normalized to the total amount of protein respectively. Data are mean values (±SEM) of at least three separate experiments (**P < 0.01).
We then tested the effect of L6H21 on the LPS–MD2 and TLR4–MD2 interactions. At the cellular level, HUECV304 cells, which express both TLR4 and MD-2, were incubated with FITC-labelled LPS (FITC-LPS) and then subjected to flow cytometry analysis. Figure 2C shows that FITC-LPS alone binds to the cell surface with an MFI of 17.4, while treatment with L6H21 dose-dependently reduced FITC-LPS binding to the receptor on the cell surface. To demonstrate the effect of L6H21 on the formation of the TLR4/MD-2 complex, immunoprecipitation experiments were performed. As shown in Figure 2D, the co-precipitation of TLR4/MD-2 profoundly increased in the LPS alone group, while treatment with L6H21 significantly inhibited the LPS-induced TLR4/MD-2 complex in a dose-dependent manner. At the molecular level, a biotin-streptavidin-based elisa system was established for the determination of the LPS–rhMD-2 interaction. Figure 2E shows that biotin-labelled LPS (biotin-LPS) bound to rhMD-2 in the plates, while co-incubation with L6H21 significantly blocked the interaction of biotin-LPS and rhMD-2. This also suggests that the binding site for L6H21 on MD-2 may overlap with the binding site for LPS.
We further investigated whether L6H21 specifically targets MD-2 and blocks the MD-2/TLR4 pathway. MD-2 is an assistant protein of TLR4, which plays no role in TLR2 signalling. Thus, RAW264.7 macrophages were pretreated with L6H21 for 2 h followed by incubation with Pam3CK, a specific activator of TLR2, for 12 h, and then the inflammatory cytokine levels in the culture medium were determined by elisa. As shown in Figure 2F and G, unlike a specific TLR2 inhibitor CU-CPT22, L6H21 did not inhibit the overexpression of TNF-α and IL-6 induced by Pam3CK and TLR2 activation, suggesting that L6H21 does not target the TLR2 and common downstream signalling of TLR2/TLR4. Thus, all these data prove that L6H21 binds directly to MD-2, and specifically antagonizes the LPS-TLR4/MD-2 interactions.
L6H21 acts on the Arg90 and Tyr102 residues in the MD-2 protein pocket
To investigate the underlying structural mechanism of L6H21 binding to the MD-2 protein, we performed a molecular simulation of the L6H21–MD2 complex using docking software. As shown in Figure 3A, L6H21 was fitted into the binding pocket of MD-2, displaying close contact with hydrophobic residues in the most energetically favourable simulation. The whole molecule of L6H21 was buried inside the lipid-binding pocket, indicating overlap and competition between L6H21 and LPS. The computer-assistant prediction showed that several binding conformations of L6H21 may exist in the MD-2 pocket, among which, two confirmations with the highest docking score may be more likely to form hydrogen bonds with Arg90 and Tyr102 respectively (Figure 3A). Thus, in order to validate the important role of Arg90 and Tyr102 in L6H21 binding to MD-2, two new MD-2 proteins with an R90A or Y102A mutation were prepared respectively. The SPR assay indicated that L6H21 no longer binds to MD-2 with an R90A mutation (Figure 3B); the elisa method showed that L6H21 did not inhibit the binding of biotin-LPS with MD-2R90A (Figure 3C). Similar results were observed with the MD-2Y102A mutation (Figure 3D and E). These results demonstrate a possible binding mechanism for L6H21 with an MD-2 protein.
Figure 3.
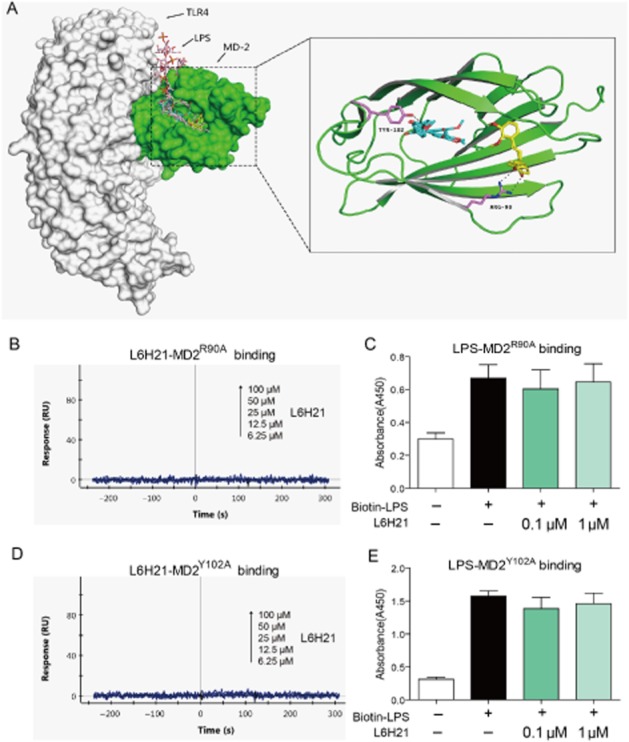
L6H21 binds MD2 via acting on Arg90 and Tyr102. (A) Molecular docking of L6H21 with MD-2 protein was carried out with the programme Tripos molecular modelling packages Sybyl-x.v1.1.083. (B) SPR analysis showed no interaction between L6H21 and rhMD-2R90A. (C) As determined by elisa, L6H21 did not inhibit the binding of Biotin-LPS to rhMD-2R90A. (D) SPR analysis showed no interaction between L6H21 and rhMD-2Y102A. (E) As determined by elisa, L6H21 did not inhibit the binding of biotin-LPS to rhMD-2Y102A. Data are mean values (±SEM) of at least three separate experiments.
L6H21 inhibits LPS-induced MAPK and NF-κB activation in macrophages
MAPKs consist of three kinases: ERK, p38 and JNK. Figure 4A shows that treatment with L6H21 dose-dependently prevented LPS-induced phosphorylation of ERK, p-38 and JNK in mouse primary macrophages (MPMs). We next evaluated the degradation of IκB, which is required for NF-κB p65 subunit transclocation and NF-κB activation. As shown in Figure 4B, after exposure to LPS, MPM cells markedly increased the degradation of IκB. However, treatment with L6H21 dose-dependently reversed the LPS-induced IκB degradation. Figure 4C further shows that LPS accelerated the NF-κB p65 translocation from the cytoplasm to nuclei, whereas this was significantly reduced in the L6H21-pretreated cells. We also analysed the DNA-binding transcriptional activity of NF-κB. An EMSA assay revealed that LPS stimulation for 1 h increased the activity of NF-κB, and pretreatment with L6H21 markedly reduced this increased NF-κB activity in a dose-dependent manner (Figure 4D).
Figure 4.

L6H21 inhibited LPS-induced MAPK phosphorylation and NF-κB activation. (A and B) MPMs were pretreated with vehicle control (DMSO) or L6H21 (2.5, 5 or 10 μM) for 2 h followed by incubation with LPS (0.5 μg·mL−1) for 30 min. The protein levels of p-ERK, ERK, p-p38, p38, p-JNK, JNK, I-κB, β-actin and GAPDH were examined by Western blot respectively. Values below the Western blots represent the mean optical density ratio in three independent experiments, and the column figure data (A) were normalized to β-actin and then expressed versus total level of MAPKs (*P < 0.05, **P < 0.01). (C) MPMs were pretreated with vehicle control (DMSO) or L6H21 (2, 2.5 or 10 μM) for 2 h, and then stimulated with LPS (0.5 μg·mL−1) for 1 h. Nuclear extracts were analysed for NF-κB activity by EMSA. (D) MPMs were pretreated with L6H21 (10 μM) or vehicle control (DMSO) for 2 h, and then stimulated with LPS (0.5 μg·mL−1) for 1 h. The cells were incubated with p65 antibody and then Cy3 fluorescein-conjugated secondary antibody, and the nuclei were stained with DAPI. The images (200×) were obtained by fluorescence microscopy and overlay. Similar results were obtained in three independent experiments.
L6H21 suppresses LPS-induced inflammatory cytokine expression
MPMs were pretreated with L6H21 for 2 h and then incubated with LPS for 22 h. Figure 5A shows that the protein levels of TNF-α and IL-6 in the culture medium were markedly increased upon exposure to LPS, while treatment with L6H21 dose-dependently suppressed the release of these two cytokines, with IC50 values <10 μM. Similar results were also observed in the cultured RAW264.7 macrophages (Figure 5B). In addition, pretreatment with 10 μM L6H21 significantly attenuated LPS-induced mRNA expression of inflammatory genes, that is, TNF-α, IL-6, IL-1β, IL-12 and COX-2 (Figure 5C).
Figure 5.

L6H21 inhibited LPS-induced inflammatory cytokine expression in macrophages. (A and B) MPMs (A) or RAW 264.7 macrophages (B) were pretreated with vehicle control (DMSO) or L6H21 (2, 2.5 or 10 μM) for 2 h followed by incubation with LPS (0.5 μg·mL−1) for 22 h. The protein level of TNF-α and IL-6 in the culture medium was measured by elisa and normalized to the total amount of protein respectively. Each bar represents mean ± SD of three to five independent experiments (*P < 0.05, **P < 0.01). (C) RAW 264.7 cells were pretreated with vehicle control (DMSO) or L6H21 (2, 2.5, or 10 μM) for 2 h followed by incubation with LPS (0.5 μg·mL−1) for 6 h. The mRNA levels of inflammatory cytokines were quantified by real-time qPCR with β-actin mRNA as the internal control. Each bar represents mean ± SD of three to five independent experiments (*P < 0.05, **P < 0.01, ***P < 0.001).
L6H21 increased mouse survival in LPS-induced septic models
As shown in Figure 6A, all animals treated with LPS alone died within 36 h as a result of septic shock. In animals receiving L6H21 at 10 mg·kg−1 either 15 min before LPS injection (for preventive effect) or 15 min after LPS injection (for therapeutic effect), the survival rates were significantly increased compared with that of the control group (60% survivals in the ‘prevention’ group and 50% survivals in the ‘treatment’ group, P < 0.01 in both groups vs. LPS alone group). Meanwhile, the body weight of L6H21-treated mice decreased during the 0–48 h after LPS but recovered slowly 48 h after LPS treatment (Figure 6B).
Figure 6.
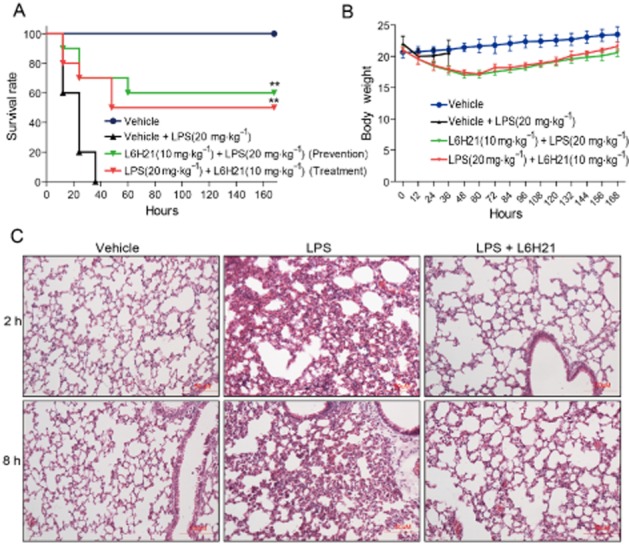
L6H21 enhanced survival and improved lung injury in LPS-induced septic mice. (A and B) C57BL/6 mice (n = 10 per group) were injected i.v. with 10 mg·kg−1 L6H21 15 min before or after i.v. injection of 20 mg·kg−1 LPS respectively. Survival rates (A) and body weight (B) were recorded for 7 days after LPS injection (** P < 0.01 vs. LPS-treated group). (C) Mice were treated with L6H21 (10 mg·kg−1, i.v.) 15 min before LPS (10 mg·kg−1, i.v.) administration. Mice were killed at 2 h or 8 h after LPS injection. Lung histopathological analysis was performed using H&E staining as described in the Methods section (n = 10 per group).
Effects of L6H21 on lung histopathology in mice after LPS administration
The lungs are the organs most sensitive to LPS-induced septic injury. Two or eight hours after injection with LPS (10 mg·kg−1, i.v.), mice were killed and the lung tissues were harvested for pathological analysis. As shown in Figure 6C, histological changes in the LPS-treated mouse lung, such as inflammatory cell infiltration, perivascular oedema and marked alveolar haemorrhage, were observed by H&E staining. These pathological changes were markedly improved by L6H21 pretreatment at 10 mg·kg−1, in both the 2 h and the 8 h treatment groups (Figure 6C).
L6H21 decreased inflammatory gene expression in LPS-induced septic mouse models
We also measured TNF-α and IL-6 levels in the blood and tissues of mice. As shown in Figure 7A and B, serum TNF-α and IL-6 levels were significantly increased at both 2 and 8 h after LPS injection, while pretreatment with L6H21 markedly prevented these increases. In addition, it was observed that cytokine levels after 2 h were significantly higher than that after 8 h of LPS injection, which is consistent with our previous report. Inflammatory gene expression in liver tissues was determined by real-time qPCR assay; the expressions of pro-inflammatory TNF-α, IL-1β and IL-6 genes in the liver were significantly increased by LPS administration at either the 2 h (Figure 7C) or 8 h (Figure 7D) time points, while these changes were significantly reversed after treatment with L6H21. With regard to the anti-inflammatory cytokine IL-10, its expression was slightly increased by 2 or 8 h after the LPS challenge via a feedback pathway, whereas it was significantly increased by L6H21 treatment (Figure 7C and D).
Figure 7.

L6H21 inhibited LPS-induced inflammatory cytokine production in mice. Mice were treated with L6H21 (10 mg·kg−1, i.v.) 15 min before LPS (10 mg·kg−1, i.v.) administration. Mice were killed and the blood and livers were collected at 2 or 8 h after LPS injection (n = 10 per group). (A and B) Serum level of TNF-α (A) and IL-6 (B) was detected by elisa. (C and D) The mRNA levels of hepatic inflammatory cytokines were determined by real-time qPCR at 2 h (C) or 8 h (D) after LPS treatment. Cytokine expression is normalized to β-actin and reported as the ratio to DMSO control.
MD-2 deficiency attenuates LPS-induced inflammatory cytokine production in macrophage and septic shock in mice
To validate the critical role played by MD-2 in the LPS response and endotoxic shock, MD-2−/− mice and primary macrophages from MD-2−/− mice were used for further investigations. LPS responsiveness in MD-2−/− MPMs was tested by measuring cytokine production in the culture medium. In contrast to wild-type (WT) MPMs, MD2-knockout MPMs produced undetectable amounts of TNF-α and IL-6 upon LPS stimulation (Figure 8A). However, there were no differences in TNF-α and IL-6 secretion in MD2-knockout and WT MPMs, when stimulated with the TLR2 agonist Pam3CK, respectively, indicating that an MD-2 deficiency does not affect the TLR2-mediated inflammatory response (Figure 8B). Subsequently, mice were challenged with LPS (20 mg·kg−1, i.v. injection). WT mice all died within 7 days after LPS administration, whereas MD-2−/− mice all survived the LPS challenge (Figure 8C). These data further validate the importance of MD-2 as a therapeutic target in LPS-induced inflammation and sepsis.
Figure 8.

MD-2 deficiency abolished LPS-induced inflammatory response. (A and B) MPMs were extracted from WT mice and MD2−/− mice, respectively, using the methods described in the Methods section. WT MPMs and MD2−/− MPMs were treated with LPS (0.5 μg·mL−1) for 22 h (A), or treated with Pam3CK (0.1 μg·mL−1) for 12 h (B) respectively. The protein level of TNF-α and IL-6 in the culture medium was measured by elisa and normalized to the total amount of protein respectively. Each bar represents mean ± SD of four independent experiments (**P < 0.01). (C) C57BL/6 mice and MD2−/− mice (n = 10 per group) were i.v. injected with 20 mg·kg−1 LPS respectively. Survival rates were recorded for 7 days after LPS injection (***P < 0.001). (D) Illustration of the protection provided by L6H21, via targeting MD-2, against LPS-induced inflammation and sepsis.
Discussion
In a previous study, we identified a new chalcone derivative L6H21 that inhibited LPS-induced inflammatory cytokine production in RAW264.7 macrophages (Wu et al., 2011). Further experiments with L6H21 identified MD-2, not TLR4, as its molecular target. In this study, we evaluated the anti-inflammatory profiles of L6H21 induced by targeting MD-2 both in vitro and in vivo.
MD-2 directly recognizes the lipid A domain of LPS for TLR4 activity and is a more desirable pharmacological target than TLR4 for the intervention of inflammatory conditions due to infection (Park and Lee, 2013). Neutralizing therapies against inflammatory mediators, such as TNF-α or even CD14 and TLR4, have failed in clinical trials (Reinhart et al., 2004; Dobesh and Olsen, 2014), and thus it is important to ascertain the clinical efficacy of agents targeting MD-2. MD-2 inhibitors are divided into two categories: lipid-like antagonists and non-lipid compounds. Recently, the phase III study of eritoran, used for treating severe sepsis, was performed; however, the results showed no significant difference between the eritoran treated groups and the placebo groups (Opal et al., 2013). Although there are other lipid A analogues in clinical trials for treating sepsis, so far, they have not shown better therapeutic efficacy than eritoran (Duan et al., 2014).
Recently, several natural and synthetic chemicals (Figure 1A), unrelated to the structure of LPS or lipid A, have also been reported as MD-2 inhibitors that attenuate LPS-induced sepsis and inflammatory injury both in vitro and in vivo (Park et al., 2012). These small molecules specifically bind to the MD-2 protein pocket and may prevent the issue of eritoran non-specifically binding to plasma lipoproteins with the loss of its antagonistic activity (Park et al., 2012). In addition, these small molecules are not rapidly metabolized in the circulation (Wasan et al., 2003; Yao et al., 2004). So far, more than eight small-molecule MD-2 inhibitors have been identified. Although they showed excellent anti-inflammatory and anti-sepsis actions, none of the studies have entered clinical trials. Due to its structural similarity to these compounds, we presumed that L6H21 may be a new MD-2 inhibitor. Thus, we designed a series of experiments to prove this hypothesis. At the molecular level, fluorescence measurements (Figure 2A) and SPR analysis (Figure 2B) validated that L6H21 binds directly to the recombinant MD-2 protein; elisa (Figure 2E) further confirmed that L6H21 directly affected the binding between LPS and rhMD-2. At the cellular level, flow cytometry (Figure 2C) and immunoprecipitation (Figure 2D) demonstrated the inhibitory effects of L6H21 on LPS–MD-2 interactions and MD–2-TLR4 complex formation respectively. Additionally, we found that L6H21 did not interact with the recombinant TLR4 protein nor directly inhibit the phosphorylation activity of downstream kinases JNK, ERK, p38 and IKK-β using the cell-free recombinant kinases (Supporting Information Fig. S1). Finally, L6H21 did not affect TLR2-mediated cytokine expression (Figure 2F). From these data, we clearly identified L6H21 as a novel and specific MD-2 inhibitor and showed that it is able to block the LPS-TLR4/MD-2 interactions and signalling transduction.
After validating that there is direct interaction between L6H21 and MD-2, we tried to determine the binding mechanism of L6H21 in the MD-2 pocket. Although several non-lipid compounds have been reported to bind with the MD-2 protein and their binding sites proposed, the X-ray diffraction-based structural information and exact binding mechanism are still unclear, which may be due to the difficulty in obtaining the MD-2 inhibitor co-crystal. The mechanisms of the MD-2-binding action of non-lipid compounds, predicted by computer-assistant simulation, are either (1) covalent interaction with the Cys133 residue of MD-2 or (2) linear alignment from the mouth to the bottom inside the pocket (Park et al., 2012). The Cys133 inside the MD-2 pocket is positioned at a distance of about 3 Å from the R4-alkyl chain of eritoran bound to the same pocket (Kim et al., 2007) and is considered a molecular target in the inhibition of LPS-induced inflammatory responses. Small-molecule inhibitors with α,β-unsaturated ketones are thought to form a covalent bond with Cys133 via a Michael-type reaction. Auranofin (Jeon et al., 2003), JTT-705 (Mancek-Keber et al., 2009) and CAPE (Kim et al., 2013) have been predicted to covalently modify the Cys133 residue, several of which showed an irreversible inhibition against MD-2; however, it is not always the outcome. The α,β-unsaturated ketone-containing curcumin interacts with MD-2 via a non-covalent mechanism. This concept is supported by studies involving its removal from the complex bound to MD-2 by chloroform extraction and through its ability to inhibit LPS from binding to the mutant MD-2 Cys133Phe in the same manner as the wild-type MD-2 (Gradisar et al., 2007). In addition, the Tyr102, Ser120, Lys122, Phe126, Gly123 and Lys130 residues were predicted, by molecular docking, to play a possible role in the small-molecule inhibitor and MD-2 interaction (Park et al., 2012). Here, after failing to get the co-crystal of L6H21-MD2, we predicted the binding mechanism of L6H21-MD2 using docking software. The docking studies suggested that the binding site for L6H21 on MD-2 may overlaps with the binding site for LPS (Figure 3A), which is consistent with the experimental data at the molecular and cellular levels. Using simulation and molecular dynamics, we were able to show that two residues, Arg90 and Tyr102, are more susceptible to form hydrogen bonds with L6H21 (Figure 3A). We further validated the contributions of these two residues by SPR and elisa assay using recombinant MD-2 site-directed mutations. Interestingly, mutating one of these two residues completely prevented the binding of L6H21 to rhMD-2 (Figure 3B–E), confirming their importance in the L6H21–MD2 interaction. The Tyr102 residue was predicted to be involved in the binding of isoxanthohumol to the MD-2 pocket (Peluso et al., 2010), while this is the first time that Arg90 has been shown to be an important molecular target for inhibiting MD-2. Although these results need further validation by structural biology methods, they already provide important information for the design of new MD-2 inhibitors and anti-inflammatory agents.
Conclusion
In conclusion, we first defined that MD-2 is a molecular target of the chalcone derivative L6H21 for its inhibition of the TLR4-mediated inflammatory response and septic injury both in vitro and in vivo. A schematic for the protection of L6H21 from LPS-induced sepsis is illustrated in Figure 8D. Compound L6H21 competitively displaces LPS from MD-2 and is fitted into the hydrophobic region of the MD-2 pocket. Arg90 and Tyr102 in the MD-2 protein contribute to the binding of L6H21 and MD-2 via the formation of a hydrogen bond with L6H21. Subsequently, L6H21 suppresses MAPK phosphorylation, NF-κB activation and cytokine expression in LPS-stimulated macrophages. In vivo, L6H21 treatment improved survival, protected the lungs from injury and decreased serum and hepatic cytokine levels in mice subjected to a lethal dose of LPS. This study proves that the new chalcone derivative, L6H21, can be used as a potential candidate in the treatment of sepsis and should be considered for further studies. More importantly, the data involving pharmacological inhibition combined with gene knockout confirmed that MD-2 is an important therapeutic target for inflammatory disorders.
Acknowledgments
This study was supported by the Natural Science Foundation of China (21272179 to G. L., 81472307 to Y. W., 81200572 to J. W. and 81371028 to W. W.), Natural Science Foundation of Zhejiang Province (LY14C050004 to X. L. and LQ14H310003 to Y. Z.), the Key Construction Academic Subject of Zhejiang Province (Traditional Chinese Medicine, 2012-XK-A28 to Z. L.).
Glossary
- L6H21
(E)-2,3-dimethoxy-4′-methoxychalcone
- MD-2
myeloid differentiation 2
- MPMs
mouse primary macrophages
- rhMD-2
recombinant human MD-2
- SPR
surface plasmon resonance
- TLR
toll like receptor
Author contributions
X. S., G. C., L. J., Z. W. and Q. F. performed the research. G. L. and Y. W. designed the research study. X. L. and J. W. contributed essential reagents or tools. Y. Z., W. W. and Y. W. analysed the data. G. L. and Y. W. wrote the paper.
Conflicts of interest statement
All authors declare no competing financial interest.
Supporting Information
Figure S1 L6H21 does not directly bind to TLR4 protein and inhibit the cell-free kinase activity. (A) SPR analysis showed no interaction between L6H21 and rhTLR4. (B) Kinase activity assay showed that L6H21 does not inhibit the kinase activity of recombinant kinases including JNK2, ERK1, p38a, p38b and IKK-β.
Figure S2 L6H21 treatment exhibited no significant toxicity in vitro. The primary macrophages (A), primary liver cells (B), and the cardiomyocytes (C) were treated with different doses of L6H21 (μM) for 24 h. The proliferation of the cells was determined through an MTT assay. The in vitro experiment were performed in triplicate (n = 3), and the data are expressed as mean ± SEM using GraphPad Prism 5.0 (GraphPad, San Diego, CA, USA). Student’s t-test was used to analyse the differences between the sets of data. A P value of <0.05 was considered significant.
Figure S3 L6H21 treatment exhibited no significant toxicity in vivo. (A) C56BL/6 mice were treated with L6H21 (1 or 2 g·kg−1) or 1% CMC-Na (Con) by gavage. After treatment, body weights were recorded daily for 15 days. (B–F) C56BL/6 mice were treated with L6H21 (100 mg·kg−1) or 1% CMC-Na (Con) by gavage once a day for 70 days. Body weights were recorded weekly (B).70 days after treatment, mice were anaesthetized with diethyl ether and killed. Creatinine levels in urine (C) and in serum (D), alanine transaminase (ALT) levels in serum (E), and cyanomethaemoglobin levels in serum (F) were determined by the automatic biochemical analyser. Data are presented as mean ± SEM, n = 6.
Figure S4 L6H21 administration induced no significant histological abnormality in vivo. C56BL/6 mice were treated with L6H21 (100 mg·kg−1) or 1% CMC-Na (Con) by gavage once a day for 70 days. 70 days after treatment, mice were anaesthetized with diethyl ether and killed. Hearts, livers and kidneys were harvested and performed H&E assay for histopathological evaluation. The specimens were observed under a microscope (400×, Nikon, Tokyo, Japan), and representative figures are shown.
Table S1 Primer sequences for real-time quantitative PCR.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: catalytic receptors. Br J Pharmacol. 2013a;170:1676–1705. doi: 10.1111/bph.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: enzymes. Br J Pharmacol. 2013b;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barochia A, Solomon S, Cui X, Natanson C, Eichacker PQ. Eritoran tetrasodium (E5564) treatment for sepsis: review of preclinical and clinical studies. Expert Opin Drug Metab Toxicol. 2011;7:479–494. doi: 10.1517/17425255.2011.558190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobesh PP, Olsen KM. Statins role in the prevention and treatment of sepsis. Pharmacol Res. 2014;88C:31–40. doi: 10.1016/j.phrs.2014.04.010. [DOI] [PubMed] [Google Scholar]
- Duan G, Zhu J, Xu J, Liu Y. Targeting myeloid differentiation 2 for treatment of sepsis. Front Biosci (Landmark Ed) 2014;19:904–915. doi: 10.2741/4256. [DOI] [PubMed] [Google Scholar]
- Esteban E, Ferrer R, Alsina L, Artigas A. Immunomodulation in sepsis: the role of endotoxin removal by polymyxin B-immobilized cartridge. Mediators Inflamm. 2013;2013:507539. doi: 10.1155/2013/507539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradisar H, Keber MM, Pristovsek P, Jerala R. MD-2 as the target of curcumin in the inhibition of response to LPS. J Leukoc Biol. 2007;82:968–974. doi: 10.1189/jlb.1206727. [DOI] [PubMed] [Google Scholar]
- Jeon KI, Byun MS, Jue DM. Gold compound auranofin inhibits IkappaB kinase (IKK) by modifying Cys-179 of IKKbeta subunit. Exp Mol Med. 2003;35:61–66. doi: 10.1038/emm.2003.9. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HM, Park BS, Kim JI, Kim SE, Lee J, Oh SC, et al. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell. 2007;130:906–917. doi: 10.1016/j.cell.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Kim SY, Koo JE, Seo YJ, Tyagi N, Jeong E, Choi J, et al. Suppression of Toll-like receptor 4 activation by caffeic acid phenethyl ester is mediated by interference of LPS binding to MD2. Br J Pharmacol. 2013;168:1933–1945. doi: 10.1111/bph.12091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancek-Keber M, Jerala R. Structural similarity between the hydrophobic fluorescent probe and lipid A as a ligand of MD-2. FASEB J. 2006;20:1836–1842. doi: 10.1096/fj.06-5862com. [DOI] [PubMed] [Google Scholar]
- Mancek-Keber M, Gradisar H, Inigo Pestana M, Martinez de Tejada G, Jerala R. Free thiol group of MD-2 as the target for inhibition of the lipopolysaccharide-induced cell activation. J Biol Chem. 2009;284:19493–19500. doi: 10.1074/jbc.M109.003756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai Y, Akashi S, Nagafuku M, Ogata M, Iwakura Y, Akira S, et al. Essential role of MD-2 in LPS responsiveness and TLR4 distribution. Nat Immunol. 2002;3:667–672. doi: 10.1038/ni809. [DOI] [PubMed] [Google Scholar]
- Oblak A, Jerala R. The molecular mechanism of species-specific recognition of lipopolysaccharides by the MD-2/TLR4 receptor complex. Mol Immunol. 2015;63:134–142. doi: 10.1016/j.molimm.2014.06.034. [DOI] [PubMed] [Google Scholar]
- Opal SM, Laterre PF, Francois B, LaRosa SP, Angus DC, Mira JP, et al. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA. 2013;309:1154–1162. doi: 10.1001/jama.2013.2194. [DOI] [PubMed] [Google Scholar]
- Pan Y, Zhu G, Wang Y, Cai L, Cai Y, Hu J, et al. Attenuation of high-glucose-induced inflammatory response by a novel curcumin derivative B06 contributes to its protection from diabetic pathogenic changes in rat kidney and heart. J Nutr Biochem. 2013;24:146–155. doi: 10.1016/j.jnutbio.2012.03.012. [DOI] [PubMed] [Google Scholar]
- Park BS, Lee JO. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp Mol Med. 2013;45:e66. doi: 10.1038/emm.2013.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SH, Kim ND, Jung JK, Lee CK, Han SB, Kim Y. Myeloid differentiation 2 as a therapeutic target of inflammatory disorders. Pharmacol Ther. 2012;133:291–298. doi: 10.1016/j.pharmthera.2011.11.001. [DOI] [PubMed] [Google Scholar]
- Pastukhov AV, Ropson IJ. Fluorescent dyes as probes to study lipid-binding proteins. Proteins. 2003;53:607–615. doi: 10.1002/prot.10401. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peluso MR, Miranda CL, Hobbs DJ, Proteau RR, Stevens JF. Xanthohumol and related prenylated flavonoids inhibit inflammatory cytokine production in LPS-activated THP-1 monocytes: structure-activity relationships and in silico binding to myeloid differentiation protein-2 (MD-2) Planta Med. 2010;76:1536–1543. doi: 10.1055/s-0029-1241013. [DOI] [PubMed] [Google Scholar]
- Reinhart K, Gluck T, Ligtenberg J, Tschaikowsky K, Bruining A, Bakker J, et al. CD14 receptor occupancy in severe sepsis: results of a phase I clinical trial with a recombinant chimeric CD14 monoclonal antibody (IC14) Crit Care Med. 2004;32:1100–1108. doi: 10.1097/01.ccm.0000124870.42312.c4. [DOI] [PubMed] [Google Scholar]
- Rice TW, Wheeler AP, Bernard GR, Vincent JL, Angus DC, Aikawa N, et al. A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis. Crit Care Med. 2010;38:1685–1694. doi: 10.1097/CCM.0b013e3181e7c5c9. [DOI] [PubMed] [Google Scholar]
- Roh E, Lee HS, Kwak JA, Hong JT, Nam SY, Jung SH, et al. MD-2 as the target of nonlipid chalcone in the inhibition of endotoxin LPS-induced TLR4 activity. J Infect Dis. 2011;203:1012–1020. doi: 10.1093/infdis/jiq155. [DOI] [PubMed] [Google Scholar]
- Tan Y, Kagan JC. A cross-disciplinary perspective on the innate immune responses to bacterial lipopolysaccharide. Mol Cell. 2014;54:212–223. doi: 10.1016/j.molcel.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasan KM, Sivak O, Cote RA, MacInnes AI, Boulanger KD, Lynn M, et al. Association of the endotoxin antagonist E5564 with high-density lipoproteins in vitro: dependence on low-density and triglyceride-rich lipoprotein concentrations. Antimicrob Agents Chemother. 2003;47:2796–2803. doi: 10.1128/AAC.47.9.2796-2803.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Li J, Cai Y, Pan Y, Ye F, Zhang Y, et al. Evaluation and discovery of novel synthetic chalcone derivatives as anti-inflammatory agents. J Med Chem. 2011;54:8110–8123. doi: 10.1021/jm200946h. [DOI] [PubMed] [Google Scholar]
- Yao LH, Jiang YM, Shi J, Tomas-Barberan FA, Datta N, Singanusong R, et al. Flavonoids in food and their health benefits. Plant Foods Hum Nutr. 2004;59:113–122. doi: 10.1007/s11130-004-0049-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 L6H21 does not directly bind to TLR4 protein and inhibit the cell-free kinase activity. (A) SPR analysis showed no interaction between L6H21 and rhTLR4. (B) Kinase activity assay showed that L6H21 does not inhibit the kinase activity of recombinant kinases including JNK2, ERK1, p38a, p38b and IKK-β.
Figure S2 L6H21 treatment exhibited no significant toxicity in vitro. The primary macrophages (A), primary liver cells (B), and the cardiomyocytes (C) were treated with different doses of L6H21 (μM) for 24 h. The proliferation of the cells was determined through an MTT assay. The in vitro experiment were performed in triplicate (n = 3), and the data are expressed as mean ± SEM using GraphPad Prism 5.0 (GraphPad, San Diego, CA, USA). Student’s t-test was used to analyse the differences between the sets of data. A P value of <0.05 was considered significant.
Figure S3 L6H21 treatment exhibited no significant toxicity in vivo. (A) C56BL/6 mice were treated with L6H21 (1 or 2 g·kg−1) or 1% CMC-Na (Con) by gavage. After treatment, body weights were recorded daily for 15 days. (B–F) C56BL/6 mice were treated with L6H21 (100 mg·kg−1) or 1% CMC-Na (Con) by gavage once a day for 70 days. Body weights were recorded weekly (B).70 days after treatment, mice were anaesthetized with diethyl ether and killed. Creatinine levels in urine (C) and in serum (D), alanine transaminase (ALT) levels in serum (E), and cyanomethaemoglobin levels in serum (F) were determined by the automatic biochemical analyser. Data are presented as mean ± SEM, n = 6.
Figure S4 L6H21 administration induced no significant histological abnormality in vivo. C56BL/6 mice were treated with L6H21 (100 mg·kg−1) or 1% CMC-Na (Con) by gavage once a day for 70 days. 70 days after treatment, mice were anaesthetized with diethyl ether and killed. Hearts, livers and kidneys were harvested and performed H&E assay for histopathological evaluation. The specimens were observed under a microscope (400×, Nikon, Tokyo, Japan), and representative figures are shown.
Table S1 Primer sequences for real-time quantitative PCR.