

Abstract
Background and Purpose
Monoglyceride lipase (MGL) degrades 2-arachidonoyl glycerol (2-AG), an endogenous agonist of cannabinoid receptors (CB1/2). Because the CB1 receptor is involved in the control of gut function, we investigated the effects of pharmacological inhibition and genetic deletion of MGL on intestinal motility. Furthermore, we determined whether defective 2-AG degradation affects μ-opioid receptor (μ receptor) signalling, a parallel pathway regulating gut motility.
Experimental Approach
Gut motility was investigated by monitoring Evans Blue transit and colonic bead propulsion in response to MGL inhibition and CB1 receptor or μ receptor stimulation. Ileal contractility was investigated by electrical field stimulation. CB1 receptor expression in ileum and colon was assessed by immunohistochemical analyses.
Key Results
Pharmacological inhibition of MGL slowed down whole gut transit in a CB1 receptor-dependent manner. Conversely, genetic deletion of MGL did not affect gut transit despite increased 2-AG levels. Notably, MGL deficiency caused complete insensitivity to CB1 receptor agonist-mediated inhibition of whole gut transit and ileal contractility suggesting local desensitization of CB1 receptors. Accordingly, immunohistochemical analyses of myenteric ganglia of MGL-deficient mice revealed that CB1 receptors were trapped in endocytic vesicles. Finally, MGL-deficient mice displayed accelerated colonic propulsion and were hypersensitive to μ receptor agonist-mediated inhibition of colonic motility. This phenotype was reproduced by chronic pharmacological inhibition of MGL.
Conclusion and Implications
Constantly elevated 2-AG levels induce severe desensitization of intestinal CB1 receptors and increased sensitivity to μ receptor-mediated inhibition of colonic motility. These changes should be considered when cannabinoid-based drugs are used in the therapy of gastrointestinal diseases.
Tables of Links
| LIGANDS | |
|---|---|
| 2-AG | JZL184 |
| ACh | Loperamide |
| Arachidonic acid | Salvinorin A |
| Bethanechol | WIN55,212-2 |
| CP55,940 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14
Alexander et al., 2013a,b,).
Introduction
Cannabis has a long history as a traditional therapeutic agent for the treatment of gastrointestinal (GI) diseases. The therapeutic effects of cannabinoids include relief of nausea and vomiting, stimulation of appetite and normalization of gut motility (Izzo and Sharkey, 2010). Plant-derived and synthetic cannabinoids activate CB receptors, type 1 and/or type 2 (CB1 and CB2) (nomenclature follows Alexander et al., 2013a) with highest expression levels in neurons and immune cells respectively (Herkenham et al., 1990; Munro et al., 1993). A large body of evidence demonstrates that cannabinoids modulate GI function. They act as potent inhibitors of GI motility delaying gastric emptying and GI transit in animal models and humans (Duncan et al., 2005; Izzo and Sharkey, 2010). Rimonabant (also known as SR141716), an inverse agonist/antagonist of CB1 receptors, induces an increase in gut motility and causes diarrhoea in rodents (Colombo et al., 1998; Izzo et al., 1999; Aviello et al., 2008). Cannabinoids affect GI function primarily by activating CB1 receptors which are present on neurons of the enteric nervous system (ENS). CB1 receptor shows high expression levels in myenteric and submucosal plexus of the intestine (Coutts et al., 2002) where its activation results in the inhibition of ACh release from enteric neurons causing a reduction of intestinal peristaltic- and electrically-evoked contractions (Pertwee, 2001). Besides influencing the autonomously functioning ENS, cannabinoids may also regulate GI function by signals from the CNS through their central action or by modulating the activity of nerves of the brain–gut axis (Izzo et al., 2000). Although CB2 receptors have been shown to be expressed in the ENS (Duncan et al., 2008a), they are believed to play a minor role in the regulation of GI motility under normal conditions, but might become important in inflammatory states (Wright et al., 2008).
Cannabinoids mimic endogenously-produced signalling lipids termed endocannabinoids (ECs). The two best characterized ECs are derivatives of arachidonic acid, arachidonoyl ethanolamine (AEA) and 2-arachidonoyl glycerol (2-AG) (Devane et al., 1992; Mechoulam and Ben-Shabat, 1995; Sugiura et al., 1995). Both compounds activate CB receptors and thus may contribute to the physiological regulation of GI function. ECs act as retrograde inhibitors of synaptic transmission. They are produced on demand at postsynaptic terminals from phospholipid precursors and activate CB1 receptors located at presynaptic terminals. ECs are rapidly degraded by lipid hydrolases which terminate the EC action. AEA is hydrolyzed primarily by fatty acid amide hydrolase (FAAH) (Deutsch and Chin, 1993). FAAH-deficient mice exhibit normal gut motility (Capasso et al., 2005). Yet, pharmacological inhibition of FAAH has been shown to increase gut motility and to normalize endotoxin-induced hypermotility (Bashashati et al., 2012). 2-AG is hydrolyzed by monoglyceride lipase (MGL, also known as monoacylglycerol lipase) which is highly expressed in the GI tract of rodents (Duncan et al., 2008b). MGL is present in epithelial cells and enteric neurons, specifically in a subpopulation of excitatory motor neurons that innervate longitudinal muscles of the intestine (Duncan et al., 2008b). Previous studies demonstrated that treatment of mice with URB602, which acts as inhibitor of both MGL and FAAH (Vandevoorde et al., 2007), results in inhibition of gut motility (Duncan et al., 2008b). Very recently, Bashashati et al. (2015) showed that inhibition of 2-AG synthesis normalized intestinal motility in a mouse model of genetic constipation. Furthermore, pharmacological inhibition of MGL, using the specific inhibitor JZL184, is protective against trinitrobenzene sulfonic acid-induced colitis in mice (Alhouayek et al., 2011). A recent study indicates that JZL184 exerts gastroprotective effects against haemorrhages evoked by non-steroidal anti-inflammatory drugs (Kinsey et al., 2011). Together, the current knowledge obtained from genetically modified and pharmacologically treated mice suggests that ECs act as physiological regulators of gut motility and as gut protecting factors under pathophysiological conditions.
Based on these previous findings, we investigated whether genetic MGL deficiency may produce similar effects on gut motility as observed after acute pharmacological inhibition. In particular, we explored whether genetic MGL deficiency may cause local desensitization of CB1 receptors in the GI tract comparable with the brain (Schlosburg et al., 2010) and whether other systems that regulate intestinal motility, such as the opioid system, may be influenced by CB1 receptor desensitization. Our observations suggest that increased 2-AG levels in MGL-deficient mice (MGL-KO) or in animals chronically treated with an MGL inhibitor cause severe desensitization of intestinal CB1 receptors, which prevents 2-AG-mediated suppression of gut motility. Additionally, we observed that mice lacking MGL activity exhibit increased colonic motility and were hypersensitive to the μ-opioid receptor (μ receptor)-mediated inhibition of colonic motility. Thus, our experiments in MGL-KO mice revealed that MGL deficiency provokes functional antagonism through CB1 receptor desensitization in the intestine and changes in a parallel signalling pathway controlling GI motility.
Methods
Animals
Mice were maintained on a regular light-dark cycle (14 h light, 10 h dark) with a room temperature of 23 ± 1°C and kept ad libitum on a standard laboratory chow diet (4.5% w/w fat, Ssniff Spezialdiaeten, Soest, Germany). Animals used for experiments were 12–20 weeks of age. If not mentioned otherwise, male mice were used. MGL-KO mice were generated as described previously (Taschler et al., 2011). CB1-deficient (CB1-KO) mice were obtained from A. Zimmer (University of Bonn, Bonn, Germany) and generated as described (Zimmer et al., 1999). Animals were anaesthetized with IsoFlo/Isoflurane (Abbott, Animal Health, Queenborough, Kent, UK) and killed by cervical dislocation. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). Experimental procedures were approved by the Ethics committee of the University of Graz, the Austrian Federal Ministry of Science and Research (protocol number BMWF 66007/7-ll/3b/2013) and were conducted in accordance with the council of Europe Convention (ETS 123). Procedures were performed as humanely as possible to minimize suffering.
Tissue preparation
Tissues were surgically removed and washed with PBS. Homogenization was performed on ice in solution A (0.25 M sucrose, 1 mM EDTA, 20 μM dithiothreitol, 0.1% Triton X-100, 20 μg·mL−1 leupeptin, 2 μg·mL−1 antipain, 1 μg·mL−1 pepstatin, pH 7.0) using an Ultra Turrax (IKA, Staufen, Germany). 20 000×g infranatants were used for expression analyses.
Determination of whole gut transit
To determine whole gut transit, animals were kept in individual cages without bedding and were fasted overnight (14 h). The next day, mice were injected i.p. with carrier solution alone (0.9% NaCl, 5% Chremophor, 5% EtOH), WIN 55,212-2 (1 mg·kg−1), CP 55,940 (0.1 mg·kg−1), loperamide (5 mg·kg−1) or JZL184 (16 mg·kg−1). After 20 min, mice were given an oral gavage of 200 μL Evans Blue (0.9% NaCl, 5% Evans Blue, 5% gum arabica) and the time until the detection of Evans Blue in the faeces was recorded. For the determination of whole gut transit after chronic inhibition of MGL, C57bl/6J mice were injected daily with JZL184 (16 mg kg−1) for 7 consecutive days. Thereafter, whole gut transit was determined as described earlier. For the determination of upper GI transit and colonic transit, mice were killed after 3 and 6 h, and the distance moved by the marker was determined and expressed as percentage of small intestine and colon respectively. Concentrations of agonists and inhibitors used were similar to those used in previous studies (Capasso et al., 2005; Carai et al., 2006; Schlosburg et al., 2010; Taschler et al., 2011).
Determination of intestinal motility
The ileum was surgically excised from animals, washed with PBS and residual food pulp was carefully removed. Then, ileal segments (∼1 cm) were tied with a thread and incubated in a physiological solution at 37°C and 5% CO2. A tension of 1 mg was applied for each segment. First, basal contractions were monitored over 30 min. Electrical field stimulations (EFS) were carried out at 2, 4, 8 and 16 Hz (10 s stimulation duration, 0.5 ms pulse duration, 60 V). Then, increasing concentrations of bethanechol were added (10−10–10−5 M) to obtain maximal contractions. To investigate the effects of CB1 receptor or μ receptor activation, EFS and bethanechol-stimulated contractions were determined after 20 min pre-incubation with increasing concentrations of WIN 55,212-2 or loperamide (10−10–10−5 M). After each treatment, ileal segments were washed extensively.
Determination of colonic propulsion
Mice were fasted overnight (14 h). Before starting the experiment, mice were acclimatized to single cages without bedding for at least 1 h. Then, carrier solutions, WIN 55,212-2 (1 mg·kg−1) or loperamide (5 mg·kg−1) were injected i.p. After 20 min, a plastic bead (2 mm in diameter) was inserted 2 cm into the colon under mild anaesthesia using IsoFlo/Isoflurane (Abbott). Then, time until expulsion of the plastic bead was recorded.
Quantification of 2-AG
Tissues of overnight (14 h) fasted mice were homogenized and extracted twice with CHCl3/MeOH/H2O (2:1:0.6, v v−1v−1) containing 1-heptadecanoyl-rac-glycerol (C17:0-MG, Avanti Lipids, Alabaster, AL, USA) as an internal standard. The lipid-containing organic phase was dried and monoglycerides (MGs) were isolated by solid phase extraction using silica gel columns. Fractions were obtained by consecutive elution with 99/1 and 90/10 CHCl3/MeOH (v v−1). The latter fraction containing MGs was used for 2-AG quantification using an AQUITY-UPLC (Waters, Manchester, UK) equipped with a BEH-C18-column (2.1 × 150 mm, 1.7 μm; Waters) coupled to a SYNAPT™ G1 qTOF high-definition mass spectrometer (Waters) equipped with an electrospray ionization sources. Quantifier ions were MNa + (m/z 401) for 2-AG and MNa + (m/z 367) for C17:0-MG respectively. Concentrations of 0.5–100 pmol·μL−1 2-AG were used for the calibration curve together with C17:0-MG.
Immunofluorescence of CB1 receptors and lysosomal-associated membrane protein 1 (LAMP1) in mouse ileum and colon
Whole mount preparations of the ileum and the distal part of the colon were prepared for immunohistochemical detection of CB1 receptors and LAMP1 in myenteric ganglia as described previously (Li et al., 2013). Briefly, after incubation in PBS containing 4% donkey serum and 0.1% Triton X-100 to block non-specific binding, whole mounts were exposed to rabbit anti-CB1 receptor antibody (ab23703; Abcam, Cambridge, UK, diluted 1:100) and rat LAMP1 monoclonal antibody (1:400), clone 1D4B (Abnova, Taipei, Taiwan) (Poole et al., 2011) overnight at 4°C. To visualize immunoreactivity, the fluorophore-conjugated secondary antibodies anti-rabbit Alexa 488 (1:100; Life Technologies, Invitrogen, Vienna, Austria) and anti-rat Cy3 (1:600; Jackson ImmunoResearch, West Grove, PA, USA) were used. Specimens were examined under an Olympus IX 70 fluorescent microscope (Olympus, Vienna, Austria) and photographed with a Hamamatsu ORCA CCD camera (Hamamatsu Photonics, Herrsching am Ammersee, Germany) using Olympus xcellence® imaging software (Olympus). Only brightness and contrast of the images were adjusted using Corel Photoshop® (Corel Corporation, Ottawa, Ontario, Canada).
Western blotting analyses
Proteins of tissue homogenates were separated by 10% SDS-PAGE using Tris/glycine (TST, 0.8% SDS, 1.6 M glycine, 200 mM Tris/HCl, pH 8.5) as electrophoresis buffer. Then, proteins were transferred onto PVDF membranes (Carl Roth GmbH, Karlsruhe, Germany) using N-cyclohexyl-3-aminopropanesulfonic acid buffer. After non-specific binding had been blocked with 10% non-fat dry milk in TST, membranes were hybridized with respective antibodies for 1 h at room temperature. The membranes were then washed with TST and specific protein expression was detected using a HRP-conjugated antibody and visualized by enhanced chemiluminescence detection (ECL, GE Healthcare, Little Chalfont, UK). For detection of MGL, a polyclonal, in-house made rabbit anti-MGL serum was used.
Protein determination
Protein concentrations of tissue lysates were determined using the Bio-Rad protein assay kit according to the manufacturer’s instructions (Bio-Rad, Hercules, CA, USA), using BSA as a standard.
Statistical analyses
Values are presented as means ± SEM. Statistical significance was determined by Student’s unpaired t-test (two-tailed) for the comparison of single groups or treatments. Two-way anova followed by Bonferroni’s post hoc test was used for the analysis of multiple measurements. Group differences were considered statistically significant for genotypes: *P < 0.05, **P < 0.01 and ***P < 0.001; for treatments: #P < 0.05, ##P < 0.01 and ###P < 0.001.
Drugs and chemicals
The following were used in the experiments: Evans Blue (Sigma Aldrich, St. Louis, MO, USA), WIN 55,212-2 (Cayman Chemicals, Ann Arbor, MI, USA); CP 55,940 (Sigma Aldrich); bethanechol (Sigma Aldrich), loperamide (Sigma Aldrich), JZL184 (Cayman Chemicals).
Results
MGL deficiency causes accumulation of intestinal 2-AG and insensitivity to CB receptor agonist treatment in vivo
To investigate the contribution of MGL to intestinal 2-AG degradation, we first measured MGL expression in different segments of the intestinal tract and compared 2-AG levels of wild-type and MGL-KO mice. Western blotting analyses revealed that MGL was expressed in all the intestinal segments investigated, whereas no expression was detected in segments of MGL-ko mice. The highest expression of MGL was detected in the colon (Figure 1A). As shown in Figure 1B, MGL-KO mice exhibited increased 2-AG levels throughout the intestine in comparison with wild-type controls (1.6-, 1.9-, 2.3- and 2.7-fold in duodenum, jejunum, ileum and colon respectively).
Figure 1.
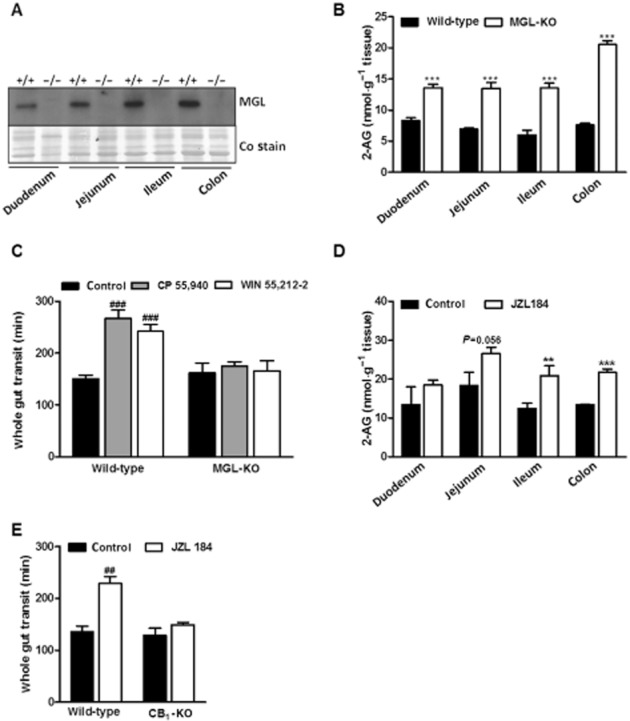
MGL deficiency causes 2-AG accumulation in the intestine and insensitivity to CB receptor agonist treatment. (A) MGL expression in different segments of the intestine. MGL expression was determined in preparations of duodenum, jejunum, ileum and colon of wild-type and MGL-KO mice using a rabbit polyclonal MGL antiserum. The membrane was stained with Coomassie blue (Co) as loading control. (B) 2-AG levels in different segments of the intestine of wild-type and MGL-KO mice. 2-AG was quantified by LC/MS measurement in tissue lipid extracts of overnight fasted mice using 1-heptadecanoyl-rac-glycerol as internal standard. Data are presented as means ± SEM (n = 6 per genotype). (C) Whole gut transit and effect of CB receptor agonists on gut transit in wild-type and MGL-KO mice. Animals were kept in single cages without bedding and fasted overnight. Subsequently, animals were injected i.p. with either carrier solution, CP 55,940 (0.1 mg·kg−1 mouse) or WIN 55,212-2 (1 mg·kg−1 mouse). After 20 min, mice were gavaged with Evans Blue and received free access to food. The time until the appearance of Evans Blue in the faeces was recorded. Data are presented as means ± SEM (n = 12 per genotype for carrier solution and CP 55,940; n = 6 per genotype for WIN 55,212-2). (D) 2-AG levels in different segments of the intestine of wild-type mice treated with JZL184. Data are presented as means ± SEM (n = 3–4 per group). (E) Whole gut transit and effect of JZL184 on gut transit in wild-type and CB1-KO mice. Animals were injected i.p. with either carrier solution or JZL184 (16 mg·kg−1 mouse). Then, Evans Blue was administered and its appearance recorded as in (C). Data are presented as means ± SEM (n = 5–6 per genotype). Statistical differences were determined using Student’s unpaired t-test or two-way anova followed by Bonferroni’s post hoc test, **P < 0.01 and ***P < 0.001 for comparison of genotypes; ##P < 0.01 and ###P < 0.001 for comparison of treatments.
To examine whether increased 2-AG levels in the intestine of MGL-KO mice caused alterations in whole gut transit, mice received a gavage containing Evans Blue and whole gut transit time of the marker was monitored. As shown in Figure 1C, we did not observe differences in whole gut transit between wild-type and MGL-KO mice. Next, we compared the sensitivity of wild-type and mutant mice to CB receptor agonists. In wild-type mice, the CB1/2 agonists CP 55,940 and WIN 55,212-2 decelerated gut transit 1.8- and 1.7-fold respectively. Notably, these compounds had no effect in MGL-KO mice (Figure 1C), indicating that increased 2-AG levels cause desensitization of CB receptors.
To compare the effect of acute MGL inhibition on whole gut transit, wild-type mice were treated with JZL184, a specific inhibitor of MGL (Long et al., 2009). Inhibition of MGL significantly increased 2-AG levels in ileum and colon 20 min after injection (1.7 and 1.6-fold, respectively) as compared with mice treated with carrier alone (Figure 1D). This treatment delayed whole gut transit 1.7-fold (Figure 1E), similar to that observed with CB receptor agonists (Figure 1C). To examine whether this effect was dependent on the CB1 receptor, we included CB1 receptor knockout (CB1-KO) mice in the experiment. In accordance with published data, this mouse model showed unchanged gut transit under basal conditions (Capasso et al., 2005; Carai et al., 2006). In contrast to wild-type mice, however, JZL184 did not affect whole gut transit in CB1-KO mice (Figure 1E). This experiment indicates that acute inhibitor-mediated inactivation of MGL decelerates whole gut transit via activation of CB1 receptors. Conversely, permanently elevated 2-AG levels in MGL-KO mice caused desensitization of CB1 receptors preventing hyperactivation of the intestinal EC system.
MGL deficiency causes CB receptor desensitization in the ileum
Gut motility is influenced by central and local signals. To investigate whether the insensitivity to CB receptor agonists is caused by desensitization of CB1 receptors expressed in the gut, we performed ex vivo experiments using ileal explants from small intestine. The sensitivity to CB receptor agonists was monitored by detecting contractions in EFS studies. We did not observe any differences in basal contraction number between wild-type and MGL-KO mice (wild-type 31.3 ± 2.45, MGL-KO 31.8 ± 1.25 contractions min-1; n = 6 per genotype). The addition of WIN 55,212-2 decreased EFS-induced contractions in a dose-dependent manner in wild-type mice (Figure 2A and C). Notably, WIN 55,212-2 had no effect on ileal segments of MGL-KO mice (Figure 2B and D), suggesting that the desensitization occurred in the small intestine. Bethanechol-stimulated contractions, a measure of the maximal contraction capacity (Jankovic et al., 1998), were not altered between genotypes (Supporting Information Fig. S1).
Figure 2.
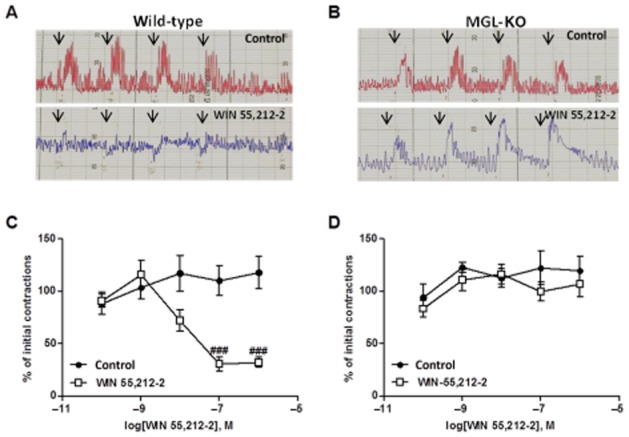
Ileal explants of MGL-KO mice are insensitive to CB receptor agonist treatment. Ileal segments of wild-type and MGL-KO mice were incubated in a physiological solution at 37°C and 5% CO2. Contractions were evoked using EFS. (A, B) Representative recordings of EFS-induced contractions of wild-type (A) and MGL-KO mice (B) treated with either control solution (red) or 10−7 M WIN 55,212-2 (blue). Arrows indicate the electrical stimuli at 2, 4, 8 and 16 Hz. (C, D) EFS-stimulated contractions (8 Hz) were determined in ileal explants of wild-type (C) and MGL-KO (D) after 20 min pre-incubation with the indicated concentrations of WIN 55,212-2. Data are presented as means ± SEM (n = 6 per genotype). Statistical differences were determined using two-way anova followed by Bonferroni’s post hoc test, ###P < 0.001.
MGL-deficiency causes accumulation of CB1 receptors in intracellular vesicles
The CB1 receptor is highly expressed in myenteric ganglia (Duncan et al., 2005). In order to investigate changes in intestinal CB1 receptor expression and localization, we performed immunohistochemical analyses. The staining of ganglia in the ileum and colon revealed different distribution patterns of CB1 receptors in whole mount preparations of wild-type and MGL-KO mice. In wild-type mice, CB1 receptors were uniformly distributed with only some cells showing vesicular CB1 receptor localization. In contrast, MGL-KO preparations showed strong granular appearance of the receptor close to neuronal nuclei (Figure 3A). Co-labelling with the late endosomal/lysosomal marker LAMP1 revealed a partial colocalization of the receptor suggesting that CB1 receptors accumulate in the endocytic vesicles (Figure 3B). The CB1 receptor antibody did not stain brain sections from CB1-KO mice confirming its specificity (Supporting Information Fig. S2).
Figure 3.
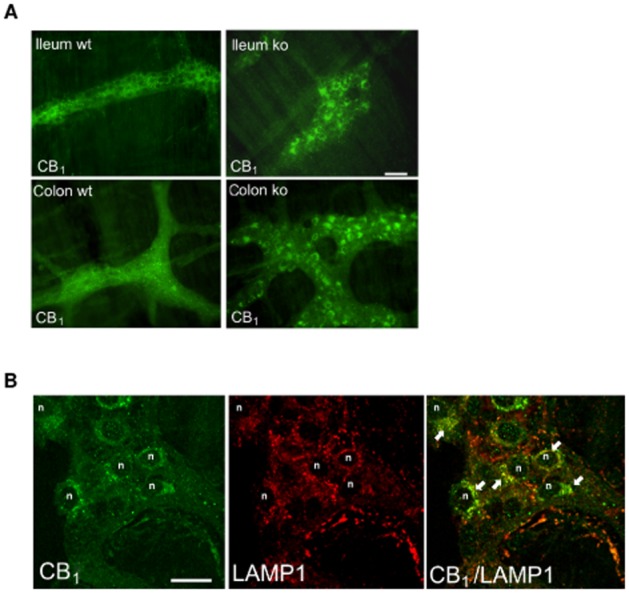
Immunohistochemical analyses of CB1 receptor expression in myenteric ganglia of MGL-KO and wild-type (wt) mice. (A) Immunofluorescence images of CB1 receptors show strong granular appearance of the receptor tightly packed and close to neuronal nuclei of myenteric ganglia from MGL-KO ileum and colon (ileum ko,and colon ko). Images are representative of immunostainings in four different MGL-KO and wild-type mice. Scale bar: 50 μm. (B) CB1 receptors were co-labelled with the lysosomal marker LAMP1 to show internalized CB1 receptors in neurons of myenteric ganglia from MGL-KO mice. Confocal images of a myenteric ganglion from the colon show granular perinuclear distribution of CB1 receptors (some nuclei of neurons are indicated with n). CB1 receptors partially colocalize with LAMP1 (yellow colour through merged images; CB1/LAMP1 colocalization marked by arrows). Scale bar: 20 μm.
MGL-KO mice show increased sensitivity to μ receptor agonist treatment
To investigate whether MGL deficiency influences other signalling pathways, we treated mice with loperamide, a selective μ receptor agonist, which inhibits gut motility (De Luca and Coupar, 1996). In accordance with published data (Carai et al., 2006), loperamide decelerated whole gut transit in wild-type mice 4.4-fold (Figure 4A). In comparison with wild-type mice, MGL-KO animals exhibited a more than 4 h delay in gut transit (7.7-fold compared with carrier treated mice), indicating that MGL-KO mice are hypersensitive to μ receptor stimulation (Figure 4A). To examine whether this increased sensitivity is caused by desensitization of CB1 receptors, we compared the effect of μ receptor stimulation on gut transit in wild-type and CB1-KO mice. Although CB1-KO mice showed a trend towards delayed transit, we did not observe a significant difference between groups (Figure 4B). This observation suggests that a CB1 receptor-independent mechanism causes μ receptor hypersensitivity in MGL-KO mice.
Figure 4.
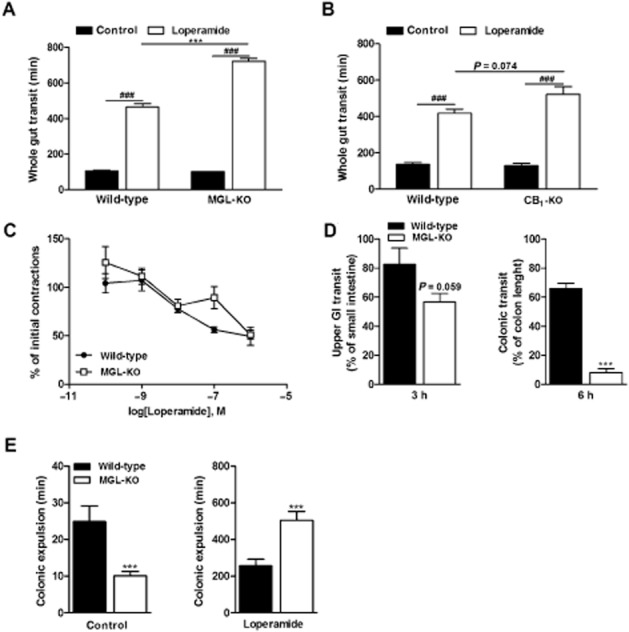
MGL-KO mice are hypersensitive to μ receptor agonist-induced constipation. (A) Effect of loperamide on whole gut transit in wild-type and MGL-KO mice (n = 10 per genotype). Animals were kept in single cages without bedding and fasted overnight. Loperamide (5 mg·kg−1 mouse) or carrier solution were injected i.p. After 20 min, mice were gavaged with Evans Blue and received free access to food and water. The time period until the appearance of Evans Blue in the faeces was recorded. (B) Effect of loperamide on whole gut transit of wild-type and CB1-KO mice (n = 5–6 per genotype). The experiment was performed exactly as in (A). (C) Effect of loperamide on the contractility of ileal segments obtained from wild-type and MGL-KO mice. Explants were incubated in a physiological solution at 37°C and 5% CO2. Contractions were evoked using EFS (8 Hz). EFS-stimulated contractions were determined 20 min after pre-incubation with the indicated concentrations of loperamide (n = 3 per genotype). (D) Upper GI transit and colonic transit in loperamide-treated wild-type and MGL-KO mice. Animals were treated with loperamide and Evans Blue as in (A). After 3 and 6 h, mice were killed and the progression of the marker in the GI tract was determined. Progression of the dye was expressed as % of small intestine (left panel) or colon length (right panel). (E) Effect of loperamide on the colonic propulsion of plastic beads in wild-type and MGL-KO mice. Animals (n = 10 per genotype) were treated with loperamide and Evans Blue as in (A). After 20 min, a 2 mm plastic bead was inserted 2 cm into the colon under mild anaesthesia. Subsequently, the time periods until the appearance of the bead were monitored. Data are presented as means ± SEM. Statistical differences were determined using Student’s unpaired t-test or two-way anova followed by Bonferroni’s post hoc test, ***P < 0.001 for comparison of genotypes; ###P < 0.001 for comparison of treatments.
To investigate changes in gut motility in more detail, we performed EFS studies in ileal explants of wild-type and MGL-KO mice in the presence of increasing concentrations of loperamide. However, ileal contractility was inhibited in explants of both genotypes to a similar extent (Figure 4C), indicating that μ receptor sensitivity in the ileum was unchanged. Bethanechol-stimulated contractions were not different between genotypes (Supporting Information Fig. S3).
To elucidate whether loperamide affects upper or lower gut transit, mice received a gavage containing Evans Blue and were killed after 3 and 6 h. As shown in Figure 4D (left panel), upper GI transit was not significantly different between wild-type and MGL-KO mice, whereas colonic transit was strongly delayed in MGL-KO mice (Figure 4D, right panel).
MGL-KO mice show accelerated colonic propulsion and are hypersensitive to loperamide-mediated inhibition of colonic motility
To confirm the changes in colonic μ receptor sensitivity in MGL-KO mice, we monitored the colonic propulsion of plastic beads in vivo in the absence or presence of loperamide. In contrast to our observations for whole gut transit, we observed a clear difference between genotypes in the absence of loperamide. Colonic propulsion in MGL-KO mice was accelerated by 50% as compared with wild-type controls (Figure 4E, left panel). Loperamide strongly decelerated bead expulsion in both genotypes. Yet, expulsion in MGL-KO animals was delayed twofold in comparison with wild-type mice (Figure 4E, right panel). These findings suggest that the decelerated gut transit in loperamide-treated MGL-KO mice is primarily caused by inhibition of colonic motility.
Chronic inhibition of MGL leads to pharmacological tolerance and hypersensitivity to μ receptor agonist treatment
Chronic inhibition of MGL has been shown to cause CB1 receptor desensitization in the brain (Schlosburg et al., 2010). To investigate whether repeated inhibitor treatment leads to intestinal desensitization, C57Bl/6J mice received a daily injection of JZL184 for 7 days. After chronic treatment, we did not observe differences in whole gut transit between inhibitor and carrier-treated animal groups (Figure 5A). Furthermore, this treatment sensitized mice to loperamide-mediated inhibition of gut transit. Thus, chronic MGL inhibition elicits CB1 receptor desensitization and hypersensitivity to μ receptor agonists mimicking the intestinal phenotype of MGL-KO mice.
Figure 5.
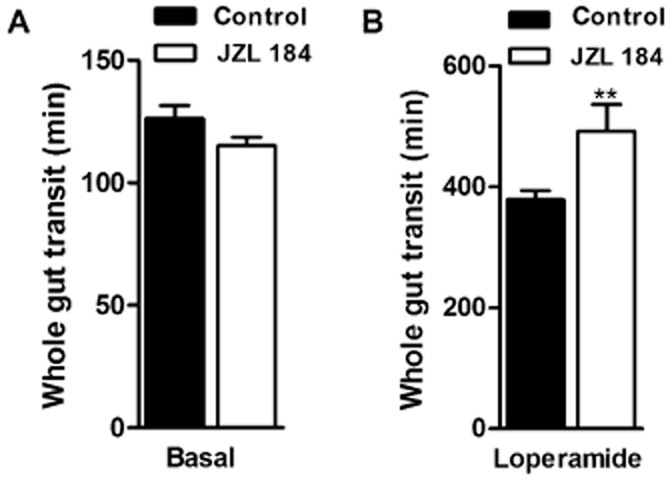
Chronic inhibition of MGL leads to pharmacological tolerance and hypersensitivity to μ receptor agonist treatments. (A, B) Whole gut transit of mice chronically treated with JZL184 (16 mg·kg−1) for 7 consecutive days. On day 7, animals were fasted overnight and kept in single cages without bedding. Subsequently, animals received an i.p. injection of JZL184 (n = 8, 16 mg·kg−1) (A) or loperamide (n = 8, 5 mg·kg−1) (B). Control mice were treated with carrier solution. Twenty minutes after injection, mice were gavaged with Evans Blue and received free access to food. The time until the appearance of Evans Blue in the faeces was recorded. Data are presented as means ± SEM. Statistical differences were determined using Student’s unpaired t-test, **P < 0.01.
Discussion and conclusions
In the current study, we investigated the effects of acute and chronic pharmacological MGL inhibition on intestinal motility. Furthermore, we used MGL-KO mice to explore possible desensitizing effects on CB1 receptors through long-term increase of 2-AG. In accordance with published observations (Duncan et al., 2008b), we found that acute inhibition of MGL delays gut transit via activation of CB1 receptors. MGL-KO mice exhibited chronically increased 2-AG levels in the small and large intestine. Therefore, we speculated that these mice also display a delay in GI transit. In contrast to our expectations, whole gut transit was unaltered and colonic propulsion was even accelerated. Notably, gut transit of MGL-KO mice was completely insensitive to CB1 receptor agonist treatment. Accordingly, ex vivo experiments using ileal explants confirmed severe intestinal desensitization of this signalling pathway. Our observations therefore indicate that chronically elevated 2-AG levels cause CB1 receptor desensitization which counteracts 2-AG-mediated inhibition of intestinal motility.
We then performed immunohistochemistry in enteric neurons to investigate potential effects of chronically elevated 2-AG levels on CB1 receptors. Our immunohistochemical data suggest that increased 2-AG levels cause internalization of CB1 receptors in enteric neurons. In general, ligand-induced internalization of GPCRs is an important mechanism for the termination of signal transduction leading to desensitization. The endocytic trafficking towards lysosomes is the main mechanism of receptor internalization which is followed either by recycling of the receptors back to the plasma membrane or degradation within lysosomes (Hanyaloglu and von Zastrow, 2008). The mechanism of CB1 receptor internalization is still not fully understood. Published studies suggest that CB1 receptor internalization and desensitization are dependent on phosphorylation events and the recruitment of β-arrestin 2 (Jin et al., 1999; Daigle et al., 2008). Our observations indicate that CB1 receptors of the myenteric plexus are trapped in endocytic vesicles. Partial colocalization with the lysosomal protein LAMP1 further suggests that internalized receptors undergo lysosomal degradation. In accordance with our observations, previous studies have already shown that long-term pharmacological and genetic MGL inactivation reduces CB1 receptor sensitivity in the brain, which is associated with a reduction of cannabimimetic effects of CB1 receptor agonists on pain sensitivity (Chanda et al., 2010; Schlosburg et al., 2010), locomotor activity and energy metabolism (Taschler et al., 2011). To our knowledge, this is the first report showing that elevated 2-AG levels cause peripheral desensitization of CB1 receptors.
Like cannabinoids, opioids have been used for the traditional treatment of GI dysfunction and diarrhoea for centuries. Opioids and cannabinoids are known to share pharmacological effects in the GI tract, including inhibition of gastric emptying, intestinal transit and secretion (Holzer, 2009; Izzo and Sharkey, 2010). In analogy to our findings, chronic treatment with opioid receptor agonists provokes drug tolerance and receptor desensitization (Williams et al., 2013). To investigate whether MGL deficiency affects other signalling pathways that inhibit GI motility, we treated mice with loperamide, which acts as a peripherally restricted agonist of μ receptors (Manara and Bianchetti, 1985; De Luca and Coupar, 1996). Notably, MGL-KO mice responded with increased sensitivity to loperamide treatment which was manifested in a greater inhibition of colonic motility as compared with wild-type. The increase in μ receptor sensitivity seems to be independent of CB1 receptor signalling, as CB1-KO mice did not show changes in loperamide sensitivity. This observation confirms data of a previous study showing that intestinal μ receptor sensitivity of CB1-KO mice is similar to wild-type mice (Carai et al., 2006). These authors concluded that cannabinoid and opioid signalling pathways act in parallel but independently of each other, although other studies showed a physical interaction between CB1 and opioid receptors (Rios et al., 2006; Canals and Milligan, 2008; Hojo et al., 2008). Some drugs, such as salvinorin A, have been shown to act via the cannabinoid as well as the opioid pathway in the regulation of gut motility, suggesting a close interplay between these two systems (Fichna et al., 2009). The mechanism of how MGL deficiency causes hypersensitivity to loperamide agonists remains elusive. Interestingly, however, another study suggests that 2-AG and AEA are able to alter cholinergic contractions in human colon independently of CB1 receptors (Smid et al., 2007). Thus, it is reasonable to assume that the increase in colonic μ receptor sensitivity in MGL-KO mice was caused by CB1 receptor-independent pathways.
In conclusion, our data show an important role for MGL in the regulation of intestinal 2-AG levels and motility. Contrary to acute treatment with MGL inhibitors, constantly elevated 2-AG levels in response to genetic MGL deletion or long-term pharmacological inhibition provoke complex changes in signalling processes including severe desensitization of intestinal CB1 receptors and hypersensitivity to μ receptor agonists. Our findings on the cross-talk between cannabinoid and opioid signalling pathways, as well as the desensitization processes are of high importance for a future cannabinoid -based therapy. Fine tuning of drug dosages of CB1 receptor agonists or MGL inhibitors is most likely essential for the long-term use of these drugs. Furthermore, increased opioid sensitivity in response to long-term MGL inhibition may be useful in the understanding and treatment of opioid tolerance and therefore warrants further research.
Acknowledgments
This research was supported by grants of the Austrian Science Funds (FWF), projects P21296 (R. Z.), P22771 (R. S.), P25633 (R. S.) and by the DK Molecular Enzymology (W901-B05 DK). The authors gratefully acknowledge support from NAWI Graz. We thank M. Kollroser for help with MS analysis.
Glossary
- 2-AG
2-arachidonoyl glycerol
- AEA
arachidonoyl ethanolamine
- CB
cannabinoid
- EC
endocannabinoid
- EFS
electrical field stimulation
- ENS
enteric nervous system
- FAAH
fatty acid amide hydrolase
- GI
gastrointestinal
- LAMP1
lysosomal-associated membrane protein 1
- MGL
monoglyceride lipase
Author contributions
U. T., T. O. E., F. P. W. R., G. F. G., H. W. and R. S. performed the experiments. U. T., R. S., A. L., M. S. and R. Z. designed the experiments and wrote the manuscript. All authors contributed intellectually to this work, reviewed and edited the manuscript.
Conflict of interest
The authors declare no conflict of interest.
Supporting Information
Figure S1 Bethanechol-stimulated contractions of ileal segments of wild-type and MGL-ko mice after WIN 55,212-2 treatment. (A, B) Bethanechol-stimulated contractions were determined in ileal segments of wild-type (A) and MGL-ko (B) after 20 min pre-incubation with indicated concentrations of WIN 55,212-2. Data are presented as means ± SEM. (n = 6/genotype). Statistical differences were determined two-way anova followed by Bonferroni’s post hoc test.
Figure S2 Immunohistochemistry of CB1 receptor expression in the brain. Images show immunofluorescence staining of the hippocampus of wild-type and CB1-ko mice using a CB1 receptor-specific antibody (dilution: 1/100). Immunofluorescence staining was performed as described in the Methods section.
Figure S3 Bethanechol-stimulated contractions of wild-type and MGL-ko mice after loperamide treatment. Bethanechol-stimulated contractions were determined in ileal segments of wild-type and MGL-ko mice after 20 min pre-incubation with the indicated concentrations of loperamide. Data are presented as means ± SEM. (n = 3/genotype). Statistical differences were determined by two-way anova followed by Bonferroni’s post hoc test.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: enzymes. Br J Pharmacol. 2013b;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhouayek M, Lambert DM, Delzenne NM, Cani PD, Muccioli GG. Increasing endogenous 2-arachidonoylglycerol levels counteracts colitis and related systemic inflammation. FASEB J. 2011;25:2711–2721. doi: 10.1096/fj.10-176602. [DOI] [PubMed] [Google Scholar]
- Aviello G, Romano B, Izzo AA. Cannabinoids and gastrointestinal motility: animal and human studies. Eur Rev Med Pharmacol Sci. 2008;12(Suppl. 1):81–93. [PubMed] [Google Scholar]
- Bashashati M, Storr MA, Nikas SP, Wood JT, Godlewski G, Liu J, et al. Inhibiting fatty acid amide hydrolase normalizes endotoxin-induced enhanced gastrointestinal motility in mice. Br J Pharmacol. 2012;165:1556–1571. doi: 10.1111/j.1476-5381.2011.01644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashashati M, Nasser Y, Keenan C, Ho W, Piscitelli F, Nalli M, et al. Inhibiting endocannabinoid biosynthesis: a novel approach to the treatment of constipation. Br J Pharmacol. 2015;172:3099–3111. doi: 10.1111/bph.13114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canals M, Milligan G. Constitutive activity of the cannabinoid CB1 receptor regulates the function of co-expressed Mu opioid receptors. J Biol Chem. 2008;283:11424–11434. doi: 10.1074/jbc.M710300200. [DOI] [PubMed] [Google Scholar]
- Capasso R, Matias I, Lutz B, Borrelli F, Capasso F, Marsicano G, et al. Fatty acid amide hydrolase controls mouse intestinal motility in vivo. Gastroenterology. 2005;129:941–951. doi: 10.1053/j.gastro.2005.06.018. [DOI] [PubMed] [Google Scholar]
- Carai MA, Colombo G, Gessa GL, Yalamanchili R, Basavarajappa BS, Basavarajppa BS, et al. Investigation on the relationship between cannabinoid CB1 and opioid receptors in gastrointestinal motility in mice. Br J Pharmacol. 2006;148:1043–1050. doi: 10.1038/sj.bjp.0706824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda PK, Gao Y, Mark L, Btesh J, Strassle BW, Lu P, et al. Monoacylglycerol lipase activity is a critical modulator of the tone and integrity of the endocannabinoid system. Mol Pharmacol. 2010;78:996–1003. doi: 10.1124/mol.110.068304. [DOI] [PubMed] [Google Scholar]
- Colombo G, Agabio R, Lobina C, Reali R, Gessa GL. Cannabinoid modulation of intestinal propulsion in mice. Eur J Pharmacol. 1998;344:67–69. doi: 10.1016/s0014-2999(97)01555-0. [DOI] [PubMed] [Google Scholar]
- Coutts AA, Irving AJ, Mackie K, Pertwee RG, Anavi-Goffer S. Localisation of cannabinoid CB(1) receptor immunoreactivity in the guinea pig and rat myenteric plexus. J Comp Neurol. 2002;448:410–422. doi: 10.1002/cne.10270. [DOI] [PubMed] [Google Scholar]
- Daigle TL, Kwok ML, Mackie K. Regulation of CB1 cannabinoid receptor internalization by a promiscuous phosphorylation-dependent mechanism. J Neurochem. 2008;106:70–82. doi: 10.1111/j.1471-4159.2008.05336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Luca A, Coupar IM. Insights into opioid action in the intestinal tract. Pharmacol Ther. 1996;69:103–115. doi: 10.1016/0163-7258(95)02053-5. [DOI] [PubMed] [Google Scholar]
- Deutsch DG, Chin SA. Enzymatic synthesis and degradation of anandamide, a cannabinoid receptor agonist. Biochem Pharmacol. 1993;46:791–796. doi: 10.1016/0006-2952(93)90486-g. [DOI] [PubMed] [Google Scholar]
- Devane WA, Breuer A, Sheskin T, Jarbe TU, Eisen MS, Mechoulam R. A novel probe for the cannabinoid receptor. J Med Chem. 1992;35:2065–2069. doi: 10.1021/jm00089a018. [DOI] [PubMed] [Google Scholar]
- Duncan M, Davison JS, Sharkey KA. Review article: endocannabinoids and their receptors in the enteric nervous system. Aliment Pharmacol Ther. 2005;22:667–683. doi: 10.1111/j.1365-2036.2005.02648.x. [DOI] [PubMed] [Google Scholar]
- Duncan M, Mouihate A, Mackie K, Keenan CM, Buckley NE, Davison JS, et al. Cannabinoid CB2 receptors in the enteric nervous system modulate gastrointestinal contractility in lipopolysaccharide-treated rats. Am J Physiol Gastrointest Liver Physiol. 2008a;295:G78–G87. doi: 10.1152/ajpgi.90285.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan M, Thomas AD, Cluny NL, Patel A, Patel KD, Lutz B, et al. Distribution and function of monoacylglycerol lipase in the gastrointestinal tract. Am J Physiol Gastrointest Liver Physiol. 2008b;295:G1255–G1265. doi: 10.1152/ajpgi.90500.2008. [DOI] [PubMed] [Google Scholar]
- Fichna J, Schicho R, Andrews CN, Bashashati M, Klompus M, McKay DM, et al. Salvinorin A inhibits colonic transit and neurogenic ion transport in mice by activating kappa-opioid and cannabinoid receptors. Neurogastroenterol Motil. 2009;21:1326–e128. doi: 10.1111/j.1365-2982.2009.01369.x. [DOI] [PubMed] [Google Scholar]
- Hanyaloglu AC, von Zastrow M. Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu Rev Pharmacol Toxicol. 2008;48:537–568. doi: 10.1146/annurev.pharmtox.48.113006.094830. [DOI] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, de Costa BR, et al. Cannabinoid receptor localization in brain. Proc Natl Acad Sci U S A. 1990;87:1932–1936. doi: 10.1073/pnas.87.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hojo M, Sudo Y, Ando Y, Minami K, Takada M, Matsubara T, et al. μ-opioid receptor forms a functional heterodimer with cannabinoid CB 1 receptor: electrophysiological and FRET assay analysis. J Pharmacol Sci. 2008;319:308–319. doi: 10.1254/jphs.08244fp. [DOI] [PubMed] [Google Scholar]
- Holzer P. Opioid receptors in the gastrointestinal tract. Regul Pept. 2009;155:11–17. doi: 10.1016/j.regpep.2009.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo AA, Sharkey KA. Cannabinoids and the gut: new developments and emerging concepts. Pharmacol Ther. 2010;126:21–38. doi: 10.1016/j.pharmthera.2009.12.005. [DOI] [PubMed] [Google Scholar]
- Izzo AA, Mascolo N, Borrelli F, Capasso F. Defaecation, intestinal fluid accumulation and motility in rodents: implications of cannabinoid CB1 receptors. Naunyn Schmiedebergs Arch Pharmacol. 1999;359:65–70. doi: 10.1007/pl00005325. [DOI] [PubMed] [Google Scholar]
- Izzo AA, Pinto L, Borrelli F, Capasso R, Mascolo N, Capasso F. Central and peripheral cannabinoid modulation of gastrointestinal transit in physiological states or during the diarrhoea induced by croton oil. Br J Pharmacol. 2000;129:1627–1632. doi: 10.1038/sj.bjp.0703265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankovic S, Kouvelas D, Mirtsou-Fidani V. Time course of isolated rat fundus response to muscarinic agonists: a measure of intrinsic efficacy. Physiol Res. 1998 [PubMed] [Google Scholar]
- Jin W, Brown S, Roche JP, Hsieh C, Celver JP, Kovoor A, et al. Distinct domains of the CB1 cannabinoid receptor mediate desensitization and internalization. J Neurosci. 1999;19:3773–3780. doi: 10.1523/JNEUROSCI.19-10-03773.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Nomura DK, O’Neal ST, Long JZ, Mahadevan A, Cravatt BF, et al. Inhibition of monoacylglycerol lipase attenuates nonsteroidal anti-inflammatory drug-induced gastric hemorrhages in mice. J Pharmacol Exp Ther. 2011;338:795–802. doi: 10.1124/jpet.110.175778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K, Fichna J, Schicho R, Saur D, Bashashati M, Mackie K, et al. A role for O-1602 and G protein-coupled receptor GPR55 in the control of colonic motility in mice. Neuropharmacology. 2013;71:255–263. doi: 10.1016/j.neuropharm.2013.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, et al. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol. 2009;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manara L, Bianchetti A. The central and peripheral influences of opioids on gastrointestinal propulsion. Annu Rev Pharmacol Toxicol. 1985;25:249–273. doi: 10.1146/annurev.pa.25.040185.001341. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl. Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG. Cannabinoids and the gastrointestinal tract. Gut. 2001;48:859–867. doi: 10.1136/gut.48.6.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole DP, Pelayo J-C, Scherrer G, Evans CJ, Kieffer BL, Bunnett NW. Localization and regulation of fluorescently labeled delta opioid receptor, expressed in enteric neurons of mice. Gastroenterology. 2011;141:982–991. doi: 10.1053/j.gastro.2011.05.042. e1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios C, Gomes I, Devi LA. mu opioid and CB1 cannabinoid receptor interactions: reciprocal inhibition of receptor signaling and neuritogenesis. Br J Pharmacol. 2006;148:387–395. doi: 10.1038/sj.bjp.0706757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlosburg JE, Blankman JL, Long JZ, Nomura DK, Pan B, Kinsey SG, et al. Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat Neurosci. 2010;13:1113–1119. doi: 10.1038/nn.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smid SD, Bjorklund CK, Svensson KM, Heigis S, Revesz A. The endocannabinoids anandamide and 2-arachidonoylglycerol inhibit cholinergic contractility in the human colon. Eur J Pharmacol. 2007;575:168–176. doi: 10.1016/j.ejphar.2007.07.036. [DOI] [PubMed] [Google Scholar]
- Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, et al. 2-arachidonoylglycerol: a possible endogenous cbr ligand in brain. Biochem Biophys Res Commun. 1995;215:89–97. doi: 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]
- Taschler U, Radner FP, Heier C, Schreiber R, Schweiger M, Schoiswohl G, et al. Monoglyceride lipase deficiency in mice impairs lipolysis and attenuates diet-induced insulin resistance. J Biol Chem. 2011;286:17467–17477. doi: 10.1074/jbc.M110.215434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandevoorde S, Jonsson KO, Labar G, Persson E, Lambert DM, Fowler CJ. Lack of selectivity of URB602 for 2-oleoylglycerol compared to anandamide hydrolysis in vitro. Br J Pharmacol. 2007;150:186–191. doi: 10.1038/sj.bjp.0706971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JT, Ingram SL, Henderson G, Chavkin C, Zastrow M, von Schulz S, et al. Regulation of μ-opioid receptors: desensitization, phosphorylation, internalization, and tolerance. Pharmacol Rev. 2013;65:223–254. doi: 10.1124/pr.112.005942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright KL, Duncan M, Sharkey KA. Cannabinoid CB2 receptors in the gastrointestinal tract: a regulatory system in states of inflammation. Br J Pharmacol. 2008;153:263–270. doi: 10.1038/sj.bjp.0707486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer AM, Hohmann AG, Herkenham M, Bonner TI. Increased mortality, hypoactivity, and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proc Natl Acad Sci U S A. 1999;96:5780–5785. doi: 10.1073/pnas.96.10.5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Bethanechol-stimulated contractions of ileal segments of wild-type and MGL-ko mice after WIN 55,212-2 treatment. (A, B) Bethanechol-stimulated contractions were determined in ileal segments of wild-type (A) and MGL-ko (B) after 20 min pre-incubation with indicated concentrations of WIN 55,212-2. Data are presented as means ± SEM. (n = 6/genotype). Statistical differences were determined two-way anova followed by Bonferroni’s post hoc test.
Figure S2 Immunohistochemistry of CB1 receptor expression in the brain. Images show immunofluorescence staining of the hippocampus of wild-type and CB1-ko mice using a CB1 receptor-specific antibody (dilution: 1/100). Immunofluorescence staining was performed as described in the Methods section.
Figure S3 Bethanechol-stimulated contractions of wild-type and MGL-ko mice after loperamide treatment. Bethanechol-stimulated contractions were determined in ileal segments of wild-type and MGL-ko mice after 20 min pre-incubation with the indicated concentrations of loperamide. Data are presented as means ± SEM. (n = 3/genotype). Statistical differences were determined by two-way anova followed by Bonferroni’s post hoc test.