

Abstract
Background and Purpose
Nicotinic (ACh) receptor recovery from desensitization is modulated by PKC, but the PKC isozymes and the phosphorylation sites involved have not been identified. We investigated whether PKCε phosphorylation of α4β2 nAChRs regulates receptor recovery from desensitization.
Experimental Approach
Receptor recovery from desensitization was investigated by electrophysiological characterization of human α4β2 nAChRs. Phosphorylation of the α4 nAChR subunit was assessed by immunoblotting of mouse synaptosomes. Hypothermia induced by sazetidine-A and nicotine was measured in Prkce−/− and wild-type mice.
Key Results
Inhibiting PKCε impaired the magnitude of α4β2 nAChR recovery from desensitization. We identified five putative PKCε phosphorylation sites in the large intracellular loop of the α4 subunit, and mutating four sites to alanines also impaired recovery from desensitization. α4 nAChR subunit phosphorylation was reduced in synaptosomes from Prkce−/− mice. Sazetidine-A-induced hypothermia, which is mediated by α4β2 nAChR desensitization, was more severe and prolonged in Prkce−/− than in wild-type mice.
Conclusions and Implications
PKCε phosphorylates the α4 nAChR subunit and regulates recovery from receptor desensitization. This study illustrates the importance of phosphorylation in regulating α4β2 receptor function, and suggests that reducing phosphorylation prolongs receptor desensitization and decreases the number of receptors available for activation.
Tables of Links
| TARGETS | ||
|---|---|---|
| Ligand-gated ion channelsa | Ion channelsb | Enzymesc |
| α4 nicotinic AChR | TRPV1 | PKCε |
| β2 nicotinic AChR | PKA | |
| GABAA receptor |
| LIGANDS |
|---|
| ACh |
| ATP |
| Nicotine |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14
Alexander et al., 2013a,b,c,,).
Introduction
Neuronal nicotinic acetylcholine receptors (nAChRs) are members of a super-family of pentameric, cys-loop, ligand-gated ion channels that also include GABAA, 5-HT3 and glycine receptors (Zhang et al., 2013). There are 11 subunits (α2–7, α9–10, β2–4) in the mammalian nervous system that can assemble to form nAChRs. The α4β2 subtype is very widely expressed, has high affinity for nicotine and is important in mediating the reinforcing effects of nicotine (Picciotto et al., 1995; Pons et al., 2008; Albuquerque et al., 2009). The nAChRs mediate fast cholinergic neurotransmission and can exist in closed, open and desensitized states (Quick and Lester, 2002; Albuquerque et al., 2009). Brief application of agonist onto cells expressing α4β2 nAChRs causes desensitization that recovers quickly over milliseconds, but agonist exposure for several minutes induces a deep desensitization that recovers slowly over minutes (Paradiso and Steinbach, 2003). The greater the agonist concentration and the longer the exposure, the greater the desensitization (Paradiso and Steinbach, 2003). nAChR desensitization is thought to play an important role in mediating nicotine addiction by facilitating dopamine neuron firing in the ventral tegmental area and contributing to the salience of smoking-related cues (Picciotto et al., 2008).
Recovery from desensitization is regulated by PKC (Downing and Role, 1987; Eilers et al., 1997; Nishizaki and Sumikawa, 1998; Fenster et al., 1999). Human and rat α4β2 nAChR expressed in cell lines show an increased rate of recovery from deep desensitization when PKC is activated and impaired recovery when it is inhibited (Eilers et al., 1997; Fenster et al., 1999). The PKC family consists of nine serine-threonine kinases, each with distinct expression patterns and substrates in the brain (Olive and Messing, 2004). The α4 nAChR subunit contains a large intracellular loop between transmembrane domains 3 and 4 that contains several putative phosphorylation sites. Mutating a potential PKC phosphorylation site in this loop, S336, to alanine impairs receptor recovery from deep desensitization (Fenster et al., 1999), suggesting that PKC phosphorylation within this loop modulates the kinetics of desensitization recovery.
The PKC isozymes that phosphorylate and regulate the α4 subunit are not known. We recently reported that PKCε regulates nicotine consumption and reward, as PKCε knock-out mice (Prkce−/−) consume less nicotine and show less conditioned place preference for nicotine than wild-type mice (Lee and Messing, 2011). In this study, we sought to determine whether PKCε phosphorylates the α4 nAChR subunit and regulates α4β2 receptor function. We found that inhibiting PKCε impaired the recovery of α4β2 nAChRs from deep desensitization. We identified five putative PKCε phosphorylation sites in the large intracellular loop of the human α4 nAChR subunit, and mutating four of them to alanine impaired recovery to different degrees. Phosphorylation of at least one site was reduced in synaptosomes from Prkce−/− mice, and Prkce−/− mice showed prolonged hypothermia after administration of sazetidine-A, which desensitizes α4β2 nAChRs (Xiao et al., 2006). These results demonstrate that PKCε phosphorylates α4 nAChR subunits and regulates the recovery of α4β2 nAChRs from desensitization. Impaired receptor recovery from desensitization may reduce the pool of nicotinic receptors available for activation in Prkce−/− mice, and this may be one reason why the Prkce−/− mice show reduced nicotine consumption and reward.
Methods
Generation of HEKtsA201 cells stably transfected with analogue-specific PKCε
Generation of the ATP analogue-specific PKCε (as-PKCε) has been previously described (Qi et al., 2007). HEKtsA201 cells were transfected with 6 or 12 μg of as-PKCε cDNA with Lipofectamine 2000 according to the manufacturer’s instructions (Life Technologies, Grand Island, NY, USA). Pooled clones were selected 48 h later with 5 μg·mL−1 puromycin. Medium was replaced daily and the concentration of puromycin was reduced to 1 μg·mL−1 after 1 week.
Electrophysiological characterization of HEKtsA201 cells expressing α4β2 nAChRs
To inhibit as-PKCε, we incubated cells with an external solution containing 1 μM of 1Na-PP1 or vehicle for 30 min before initiating receptor desensitization and throughout the remainder of the experiment. The internal pipette solution contained (in mM): 140 CsCl, 4 NaCl, 4 MgCl2, 0.5 CaCl2, 10 HEPES, 5 EGTA, and 5 Mg2+-ATP (pH 7.3, adjusted with CsOH). The external solution contained (in mM): 140 NaCl, 5 KCl, 2 MgCl2, 2 CaCl2, 10 D-glucose and 10 HEPES (pH 7.3, adjusted with NaOH). Drugs were applied locally by pressure ejection at 10 psi (Picospritzer II, General Valve Corporation, Fairfield, NJ, USA) from a micropipette placed adjacent to the cell. Cells were voltage clamped at −70 mV and whole-cell currents were recorded using an Axopatch 700B patch amplifier (Axon Instruments, Sunnyvale, CA, USA), filtered at 2 kHz and digitized at 5 kHz with a Digidata 1322A interface and pClampex 9.2 (Axon Instruments). The series resistance was continuously monitored during each experiment and cells that showed more than 30% change in resistance were discarded. All recordings were obtained at room temperature.
nAChR desensitization experiments used a three-phase, conventional whole-cell patch-clamp protocol (Paradiso and Steinbach, 2003). Phase one consisted of three pulses of ACh (100 μM, 300 ms duration) applied at 2 min intervals to test response stability. If the responses showed more than 20% variation, the cell was discarded. Phase two consisted of a 60 s application of 100 μM ACh that induced deep desensitization. Phase three consisted of a washout of the ACh followed by ACh pulses of 300 ms duration applied over 23 min, which measured receptor recovery from desensitization. In most cases, a 60 s application of ACh was enough to induce complete desensitization; however, a small residual current occasionally remained. In those cases, the residual current was subtracted from peak current measurements taken during phase 3.
The response activation and deactivation times were calculated from the average of the responses obtained during the second and third pulse (100 μM ACh, 300 ms duration) of the stability test (phase 1), before initiating desensitization. The activation and deactivation times were calculated as the time from 10% to 90% of the peak of the averaged response (for activation) and from 90% to 10% (for deactivation) using Clampfit 10.4 (Molecular Devices, Sunnyvale, CA, USA).
Cell surface biotinylation of HEKtsA201 cells expressing α4β2 nAChRs
To assess receptor trafficking, cells were plated on 60 mm dishes and pre-incubated with 1 μM 1Na-PP1 or vehicle for 30 min before initiating desensitization (100 μM ACh for 60 s). Cells were quickly washed after the ACh application and then incubated with fresh medium containing 1 μM 1Na-PP1 or vehicle for 20 min. Cell surface proteins were biotinylated using sulfo-NHS-biotin and isolated using Immunopure Immobilized Monomeric Avidin beads (Thermo Fisher Scientific Inc., Rockford, IL, USA) according to the manufacturer’s instructions. Proteins were eluted with SDS loading buffer for immunoblotting.
Identification of PKCε phosphorylation sites
A cDNA encoding the large intracellular loop of the human α4 nACh receptor subunit (amino acids 328–600) was divided into five fragments and each was subcloned into pGEX-6P-2 (GE Healthcare Biosciences, Pittsburgh, PA, USA). Glutathione-S-transferase (GST) fusion proteins were produced in Escherichia coli BL21(DE3)pLysS cells (Life Technologies, Grand Island, NY, USA) and purified by affinity chromatography using glutathione-sepharose 4B (GE Healthcare Biosciences). PKCε phosphorylation was performed in buffer containing 20 mM HEPES, pH 7.4, 0.1 mM EGTA, 0.03% Triton X-100, 10 mM MgCl2, 1 mM 2-mercaptoethanol, 0.48 μg·μL−1 L-phosphatidylserine (Avanti Polar Lipids, Alabaster, AL, USA), 1 μM phorbol-12-myristate-13-acetate, 0.07 μM human recombinant PKCε (Life Technologies), 10 μCi [32P]-ATP and 1 μM of purified GST-α4 fragment. After incubation at 37°C for 3 h, 50 μL of 5× SDS sample buffer was added to stop the reaction. Proteins were separated by SDS-PAGE on 4–12% gradient gels (Life Technologies) and incorporated radioactivity was quantified by phosphorimaging (Typhoon 9410, GE Healthcare Biosciences). The primers used for subcloning the α4 loop were: TATGAGAATTCTCAACGTGCACCACCGCTCGCCACGC – fragment 1 forward, TATGACTCGAGGGGCTCCCCTTCTGGCTCGGGCC – fragment 1 reverse; TATGAGAATTCCCGAGCCAGAAGGGGAGCCCCCTG – fragment 2 forward, TATGACTCGAGCTGCTGAGGAGGGAGCTGGTCGGAGG – fragment 2 reverse; TATGAGAATTCAGCCCCTGGAAGCTGAGAAAGCCAGCC – fragment 3 forward, TATGACTCGAGGTCTGGGGGTGGGAGCTCAGCCG – fragment 3 reverse; TATGAGAATTCAGCTCCCACCCCCAGACCAGCCCTC – fragment 4 forward, TATGACTCGAGCACCGCCCGGGTCAGGGCCGG – fragment 4 reverse; TATGAGAATTCCGGTCAAGACCCGTAGCACCAAAGCGC – fragment 5 forward, TATGACTCGAGGCGGTCGATGACCATGGCCACGTACTTC – fragment 5 reverse; TATGAGAATTCTCAACGTGCACCACCGCTCGCCACGC – full loop forward, TATGACTCGAGGCGGTCGATGACCATGGCCACGTACTTC – full loop reverse. The nucleotides in italics were included to facilitate the digestion of the PCR product with the restriction enzymes EcoRI and XhoI.
For determination of phosphorylated residues by LC/MS/MS, a cold reaction was run with the full α4 loop using conditions identical to those described earlier but with 0.5 mM cold ATP, with and without PKCε. The bands were excised and phosphorylated residues were identified by the UT Southwestern Proteomics Core on an ABI 5000TM unit (https://proteomics.swmed.edu/wordpress; Dallas, TX, USA). Residues were numbered starting from the first methionine of the ATG start site.
Generation of mutant α4 nAChR subunits
Site-directed mutagenesis was performed using the Stratagene QuikChange II XL site-directed mutagenesis kit according to the manufacturer’s protocols (Agilent Technologies, Santa Clara, CA, USA). The vector pcDNA3.1-hygro (Life Technologies) was used as a template for the native and mutant cDNAs, and all mutants were confirmed with DNA sequencing. The primers used to generate the point mutations were: CTGGCCAAAGCCAGGGCCCTCAGCGTCCAGCAC – S465A forward, GTGCTGGACGCTGAGGGCCCTGGCTTTGGCCAG – S465A reverse; CAAAGCCAGGTCCCTCGCCGTCCAGCACATGTCC – S467A forward, GGACATGTGCTGGACGGCGAGGGACCTGGCTTTG – S467A reverse; CGGTCAAGACCCGCGCCACCAAAGCACCGCCC – S550A forward, GGGCGGTGCTTTGGTGGCGCGGGTCTTGACCG – S550A reverse; GTGTCCCCGAGTGCCGCCGTCAAGACCCGCAGC – T545A forward, GCTGCGGGTCTTGACGGCGGCACTCGGGGACAC – T545A reverse; CAAGACCCGCAGCGCCAAAGCACCGCCC – T551A forward and GGGCGGTGCTTTGGCGCTGCGGGTCTTG – T551A reverse.
Transient transfection of mutant α4β2 nAChRs for electrophysiological studies
The cDNAs for mutant α4, native β2 subunit and eGFP were transiently transfected into HEKtsA201 cells stably expressing as-PKCε (6 μg as-PKCε cDNA) at a 1:1:0.25 ratio with calcium chloride (2.5 M) and HEPES buffer (280 mM NaCl, 10 mM KCl, 12 mM glucose, 1.5 mM Na2HPO4, 50 mM HEPES acid at pH 7.0). DNA complexes were added drop-wise to the wells, and cells were incubated for 16 h in growth medium at 37°C in humidified 5% CO2:95% air. eGFP-positive cells were used for electrophysiological studies.
Synaptosome preparation
Synaptosomes were generated using a discontinuous sucrose gradient (Gray and Whittaker, 1962; Witzmann et al., 2005). Wild-type and Prkce−/− mouse brains were homogenized in buffer containing 0.32 mM sucrose with 20 mM HEPES, pH 7.4, and centrifuged at 1000× g for 10 min at 4°C to obtain the P1 pellet and S1 supernatant. The S1 fraction was centrifuged at 17 000× g for 15 min at 4°C to obtain the pellet P2 and supernatant S2. The P2 fraction was resuspended in homogenizing buffer and layered on top of a discontinuous sucrose density gradient, composed of a 1.2 M bottom fraction and a 0.8 M top fraction, and was centrifuged at 55 000× g for 90 min at 4°C. The synaptosome fraction was removed from the interface between the two gradients, diluted in three volumes of homogenizing buffer and centrifuged at 55 000× g for 20 min to obtain the P3 synaptosomal pellet that was used for immunoblotting. Halt protease and phosphatase inhibitor cocktails were added for all steps (Thermo Fisher Scientific Inc.).
Immunoblotting
PKCε, phosphorylated and total α4 subunit, β2 subunit and GAPDH were measured by Western blot analysis. PKCε immunoreactivity was detected using rabbit anti-PKCε SN134 antibody (Choi et al., 2002). GAPDH was detected with anti-GAPDH (D16H11, Cell Signaling, Danvers, MA, USA). The β2 subunit was detected with anti-β2 (sc-11372, Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA). Phospho- and non-phospho-α4 antibodies were generated against the human sequence KARSLSVQH (ProSci Inc., Poway, CA, USA), with serine 465 (corresponding to mouse S468) or serine 467 (mouse S470) as the phosphorylated residue. A HRP-conjugated anti-rabbit secondary antibody was used for all experiments (Jackson ImmunoResearch Laboratories Inc., West Grove, PA, USA). Band density was quantified using ImageJ (http://imagej.nih.gov/ij/). PKCε and β2 subunit bands were normalized to total protein loaded, which was measured by scanning an identical gel stained with Coomassie blue. For phospho-α4 assessment, pairs of gels were run in parallel, one for the phospho-antibody and one for the non-phospho-antibody. For every immunoblot, the nitrocellulose membrane was cut at approximately 50 kDa and probed for α4 and GAPDH separately. The phospho and non-phospho α4 bands were first normalized to GAPDH and then the ratio of phospho-α4 to non-phospho-α4 was calculated.
Animals
Hybrid C57BL/6J × 129S4/SvJae Prkce−/− and wild-type littermates were generated as previously described (Khasar et al., 1999; Lee and Messing, 2011). Male mice were a minimum of 8 weeks old and drug naïve for all studies. Mice were housed 2–4 per cage in a 12-12 h light-dark cycle room with lights on at 06:00 h and off at 18:00 h. Food and water were freely available. All drugs were dissolved in 0.9% physiological saline and injected at a volume of 10 μL·g−1. The pH of the nicotine and sazetidine-A solutions were 3.4 and 3.6, respectively, and were not adjusted. Nicotine concentrations are shown as the free base. Mice were killed by carbon dioxide after the in vivo experiments. A separate group of drug-naïve mice were killed by carbon dioxide to collect brain tissue for synaptosome preparation. All procedures were conducted under guidelines established by the Institutional Animal Care and Use Committee, the National Research Council Committee Guide for the Care and Use of Laboratory Animals (National Research Council Committee for the Update of the Guide for the Care and Use of Laboratory Animals, 2010). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Hypothermia
Body temperature was measured with an infrared thermometer (Braintree Scientific, Inc., Braintree, MA, USA) held a few millimetres from the belly of the mouse, which was shaved to facilitate temperature measurement. The mice were separated into individual cages maintained at room temperature for the duration of the experiment. A baseline body temperature was recorded before drug injection. Nicotine and sazetidine-A were injected s.c., and temperature was recorded every 5 min over 2 h. Fifteen wild-type and 14 Prkce−/− mice were used in the sazetidine-A experiment, and 13 mice per genotype were used in the nicotine experiment. A separate group of mice were injected with nicotine, n = 4 wild-type and n = 5 Prkce−/−, or sazetidine-A, n = 5 wild-type and n = 6 Prkce−/−, and the body temperature was measured at baseline and after 24 h. Mice were killed by carbon dioxide at the conclusion of the experiment.
Statistics
All statistics were calculated using Prism 6.0 (GraphPad Software, La Jolla, CA, USA). Data are presented as mean ± SEM values. In the studies of recovery from receptor desensitization, the plateau and rate constant for each recovery curve were calculated using non-linear regression and compared using an extra-sum-of-squares F test. The activation time, deactivation time, current density for the control versus 1Na-PP1 conditions and the levels of phosphorylated-α4 were analysed using Student’s t-test or Mann–Whitney U-test. The activation time, deactivation time and current density for the mutant α4β2 versus native receptors and surface β2 nAChR subunit levels were analysed using one-way anova followed by a Dunnett’s post hoc test or the Kruskal–Wallis test followed by a Dunn’s multiple comparison post hoc test. Hypothermia was analysed by anova with a repeated measure for time and genotype as a between-subjects factor, followed by a Bonferroni’s post hoc test. Cohen’s d effect size was calculated at http://www.uccs.edu/lbecker/effect-size.html, provided by the University of Colorado, Colorado Springs. Comparisons were considered statistically significant if P < 0.05.
Materials
Nicotine hydrogen tartrate salt was purchased from Sigma-Aldrich Inc. (Dallas, TX, USA). ACh chloride and sazetidine-A dihydrochloride were purchased from Tocris Bioscience, Inc., (R&D Systems, Inc., Minneapolis, MN, USA). 1-(1,1-Dimethylethyl)-3-(1-naphthalenyl)-1H-pyrazolo[3,4-d]pyrimidine-4-amine (1Na-PP1) was a gift from Dr. Kevan Shokat (University of California, San Francisco). The cDNAs for the human α4 and β2 nAChR subunits and HEKtsA201 cells stably expressing human α4β2 nAChRs (Kuryatov et al., 2005) were generously provided by Dr Jon Lindstrom (University of Pennsylvania).
Results
PKCε modulates α4β2 nAChR recovery from desensitization
To examine the effect of PKCε on α4β2 nAChR desensitization, we used HEKtsA201 cells that stably express human α4β2 nAChRs and transfected them to stably express an ATP as-PKCε, in which the gatekeeper methionine residue in the ATP binding pocket is exchanged for alanine, creating a silent mutation that preserves kinase activity but allows the kinase to be selectively inhibited by 1Na-PP1 (Bishop et al., 2000; Qi et al., 2007). This approach is advantageous as there are no drugs that selectively inhibit the kinase activity of PKCε. HEKtsA201-α4β2 nAChR cells express very low levels of endogenous PKCε, and stable transfection of 6 or 12 μg of cDNA encoding as-PKCε substantially increased PKCε immunoreactivity compared with non-transfected cells (Figure 1A), indicating that as-PKCε accounts for most of the PKCε in this cell line. The cell line transfected with 6 μg as-PKCε cDNA was used for all subsequent experiments.
Figure 1.
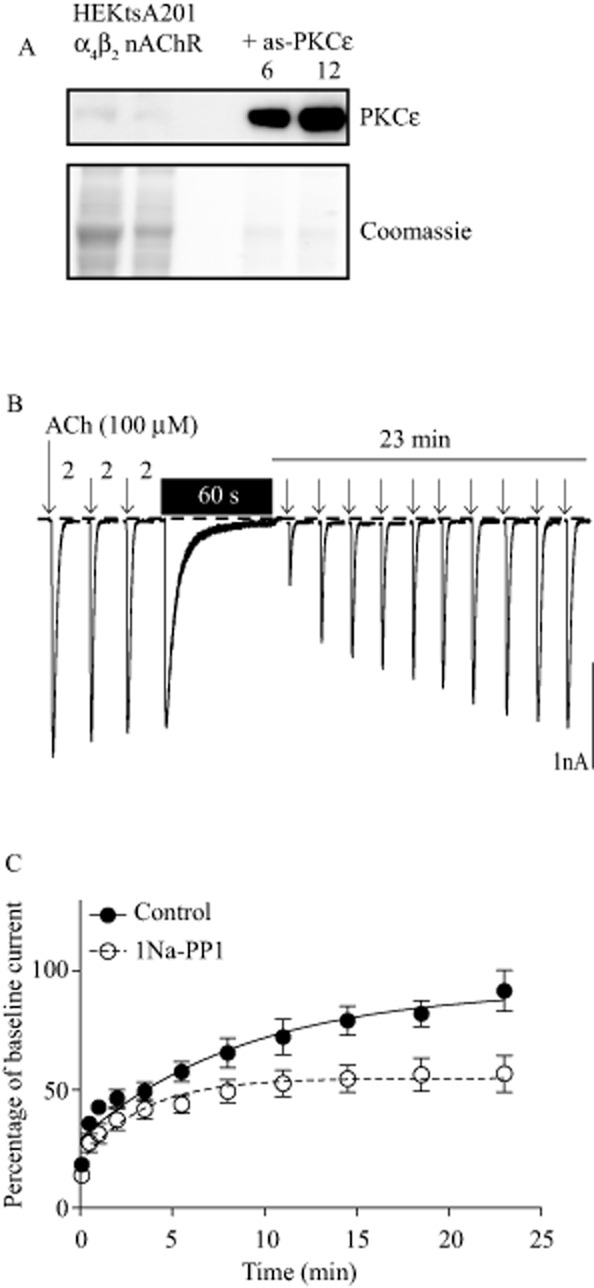
PKCε regulates α4β2 nAChR recovery from deep desensitization. (A) Upper panel, HEKtsA201 cells stably expressing human α4β2 nAChRs have very low levels of endogenous PKCε detected by immunoblotting (left two lanes). Stable transfection with 6 or 12 μg of as-PKCε cDNA resulted in much higher levels of PKCε immunoreactivity (right two lanes). Lower panel, Coomassie blue stain of a simultaneously run identical blot. Samples from as-PKCε-transfected cells were loaded at 20% of the amount of the protein used for native HEKtsA201-α4β2 samples. (B) The response of wild-type α4β2 nAChRs to 300 ms puffs of ACh (100 μM), denoted by the arrows. Baseline stability of the response was determined using three puffs of ACh spaced 2 min apart. A 60 s application of ACh (100 μM) was used to induce deep desensitization and was washed out immediately afterwards. Recovery from deep desensitization was assessed using repeated ACh puffs over 23 min. Time is not to scale. (C) Inhibition of as-PKCε by 1 μM of 1Na-PP1 impaired α4β2 nACh receptor recovery from deep desensitization (control n = 8, 1Na-PP1 n = 9 cells).
We used a whole-cell patch-clamp recording protocol (Paradiso and Steinbach, 2003) to measure nAChR recovery from deep desensitization (Figure 1B). Inhibition of PKCε activity with 1 μM 1Na-PP1 impaired the magnitude of receptor recovery from desensitization, with 1Na-PP1-treated cells recovering to 59% of the control cells [Figure 1C, control plateau = 92.7 ± 7.6%, 1Na-PP1 plateau = 54.7 ± 2.7%, F(1,173) = 12.00, P = 0.0007]. The rate of recovery between 1Na-PP1 and control-treated cells was not significantly different, although there was a trend towards a higher rate constant in 1Na-PP1-treated cells [control K = 0.11 ± 0.03, 1Na-PP1 K = 0.30 ± 0.10, F(1,173) = 3.313, P = 0.07]. Surface and intracellular β2 subunit immunoreactivity were not significantly different between control and 1Na-PP1-treated cells measured 20 min after washout of the 60 second application of ACh (surface control = 1.00 ± 0.04, 1Na-PP1 = 0.99 ± 0.02, t = 0.261, P = 0.80; intracellular control = 1.00 ± 0.04, 1Na-PP1 = 0.99 ± 0.04, t = 0.154, P = 0.88, control n = 6, 1Na-PP1 n = 7 experiments). We also examined the effect of 1Na-PP1 on current density and on receptor activation and deactivation. The current density was not significantly different between control and 1Na-PP1-treated cells (control = −89 ± 11, 1Na-PP1 = −103 ± 15 pA·pF−1, t = 0.731, P = 0.47). The activation time after a single 300 ms application of 100 μM ACh was also not significantly different between control and 1Na-PP1-treated cells (control = 77.0 ± 8.9 ms, 1Na-PP1 = 73.8 ± 9.2 ms, Mann–Whitney U = 35.50, P = 0.98). In addition, the response deactivation time was not significantly different between control and 1Na-PP1-treated cells (control = 453.1 ± 55.2 ms, 1Na-PP1 = 559.9 ± 52.7 ms, t = 1.399, P = 0.18, control n = 8 and 1Na-PP1 n = 9 cells). These data show that inhibiting PKCε reduces the extent of recovery from desensitization without altering the abundance of receptors at the cell surface or the kinetics of receptor activation and deactivation.
PKCε phosphorylation of α4β2 nAChRs in vitro
The intracellular loop between transmembrane regions three and four of the α4 subunit contains several potential PKC phosphorylation sites (Fenster et al., 1999). We investigated whether PKCε phosphorylates this loop in vitro. We truncated the human α4 nAChR loop into five fragments and incubated each fragment with PKCε and [32P]-ATP (Figure 2A). We found that PKCε phosphorylated fragments 3, 4 and 5 (Figure 2B), indicating that more than one PKCε phosphorylation site existed in the loop. We could not purify fragment 1, so it was not tested. To identify which residues were phosphorylated, we used LC/MS/MS. The full intracellular loop (amino acids 328–600) was cloned and used as the substrate in a non-radioactive in vitro kinase assay with or without PKCε. Amino acids that were phosphorylated in the presence of PKCε, but not in its absence, were identified as PKCε phosphorylation sites. There was 77% amino acid sequence coverage of the loop in the LC/MS/MS analysis and five amino acids were found to be phosphorylated by PKCε: S465, S467, T545, S550 and T551. The positions of the phosphorylated residues agreed with the pattern of phosphorylated fragments from our in vitro kinase assay (Figure 2B). With the exception of S550, these phosphorylated residues are conserved in mouse and rat α4 sequences (Figure 2C).
Figure 2.
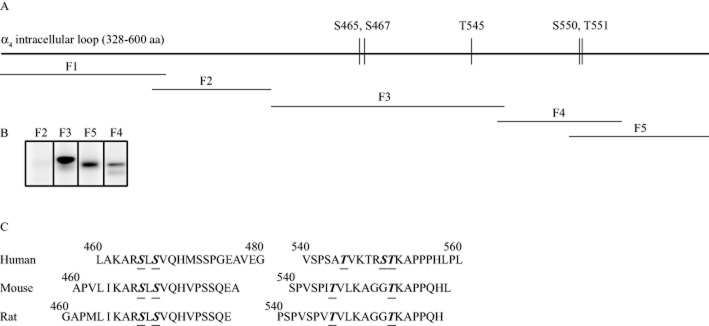
PKCε phosphorylates the large intracellular loop of the human α4 nAChR subunit in vitro. (A) Scheme of the large intracellular loop of the α4 subunit with the PKCε phosphorylation sites identified by LC/MS/MS. The GST-fusion fragments generated for the in vitro PKCε phosphorylation assay are shown underneath and labelled by fragment number, F1–F5. (B) The PKCε kinase assay showed 32P-labelling for fragments 3, 4 and 5. Fragment 1 could not be purified and was therefore not tested. (C) Alignment of amino acids 460–480 and 540–560 of the α4 subunits of human, mouse and rat. PKCε phosphorylation sites identified by MS are underlined.
Functional effect of α4 nACh receptor phosphorylation
To test the functional effect of phosphorylation at these residues, we mutated each site to alanine and transiently transfected each mutant α4 subunit with a native β2 subunit into HEKtsA201 cells expressing as-PKCε. All mutations except T545A resulted in receptors that had impaired recovery compared with native receptors (Figure 3A–E and Table 2009). The greatest deficit occurred with the S465A mutation, followed by the S467A, T551A and S550A mutations, with mutant receptors recovering to 61%, 71%, 75% and 81% of native receptors respectively (Figure 3, Table 2009). The rate of recovery from desensitization was also faster for α4(S465A)β2 receptors than for native receptors (native τ = 2.22 s, S465A τ = 0.76 s), whereas the other mutants showed a rate of recovery similar to native receptors (Table 2009).
Figure 3.

Mutation of the PKCε phosphorylation sites impaired α4β2 nAChR recovery from deep desensitization. (A–E) Recovery from desensitization induced by ACh (100 μM, 60 s) in native and mutant α4β2 nACh receptors containing PKCε phosphorylation site point mutations. (F) Mean current density (pA·pF−1) for the native and mutant receptors. Native α4β2 n = 14, S465A n = 12, S467A n = 9, T545A n = 9, S550A n = 13, T551A n = 9 cells. (G) Activation and (H) deactivation time for native and mutant receptors. *P < 0.05, **P < 0.01 compared with the native receptor. Native α4β2 n = 11, S465A n = 8, S467A n = 9, T545A n = 8, S550A n = 9, T551A n = 9 cells.
Table 1.
Plateau and K values for desensitization recovery curves for native and mutant α4β2 receptors
| Receptor | Plateau (%, mean ± SEM) | K (1 s−1, mean ± SEM) |
|---|---|---|
| Native α4β2 nACh | 99.49 ± 4.53 | 0.45 ± 0.09 |
| α4 S465A | 60.89 ± 2.07* [F(1,211) = 29.38, P < 0.0001] | 1.31 ± 0.31* [F(1,211) = 5.313, P = 0.02] |
| α4 S467A | 70.55 ± 2.28* [F(1,192) = 22.29, P < 0.0001] | 0.79 ± 0.21 [F(1,192) = 1.342, P = 0.25] |
| α4 T545A | 90.96 ± 3.16 [F(1,196) = 1.85, P = 0.18] | 0.35 ± 0.08 [F(1,196) = 0.356, P = 0.55] |
| α4 S550A | 80.45 ± 4.98* [F(1,194) = 4.98, P = 0.03] | 0.45 ± 0.14 [F(1,194) = 0.0006, P = 0.99] |
| α4 T551A | 74.77 ± 3.24* [F(1,192) = 9.05, P = 0.003] | 0.32 ± 0.07 [F(1,192) = 0.73, P = 0.39] |
Significantly different from native receptor.
In addition to recovery from desensitization, we measured current density and kinetics of activation and deactivation. The current density (pA·pF−1) of the T545A mutant was 90% greater than the current density of the native receptor, whereas all the current densities of the other mutants were not significantly different from the native receptor [Figure 3F, anova F(5,64) = 3.303, P = 0.01, Dunnett’s post hoc P < 0.05]. The current density of the S550A mutation was 49% smaller than the native receptor, but this did not reach statistical significance. Activation time was not significantly different between native and mutant receptors. The response deactivation time was 61–63% shorter for S467A, T545A and T551A mutants than for native receptors (Figure 3H, anova F(5,48) = 6.938, P < 0.0001, Dunnett’s post hoc P < 0.05), whereas deactivation times were similar for the S465A and S550A mutants and the native receptors.
PKCε phosphorylation of α4 subunits in vivo
To determine whether PKCε phosphorylates α4 nAChR subunits in vivo, we generated phospho-antibodies against S465 and S467, using the peptide KARSLSVQH. This sequence is identical in the human, mouse and rat α4 subunits. The S465 and S467 residues correspond to S468 and S470 in the mouse α4 nAChR subunit, but we refer to them here by the human sequence numbering for clarity. We first tested S465 as this mutation showed the greatest impairment in receptor recovery and this site has not been identified as a phosphorylation site. To enrich for α4β2 nAChRs from mouse brain tissue, we isolated the synaptosomal fraction from wild-type and Prkce−/− mouse brain. There was no significant difference in the level of S465 phospho-immunoreactivity in wild-type and Prkce−/− mouse synaptosomes (Figure 4A, t = 1.332, P = 0.21). We next tested S467 as it showed the second largest impairment in receptor recovery. S467 is also implicated in protein–protein interactions with the chaperone protein 14-3-3η (Jeanclos et al., 2001) and may be a PKA phosphorylation site (Jeanclos et al., 2001; Guo and Wecker, 2002; Pollock et al., 2007). However, it has not been identified as a PKC phosphorylation site or shown to be phosphorylated in vivo. The anti-phospho-S467 antibody was able to detect a strong band from native human α4β2 nACh receptors expressed in HEKtsA201 cells, but not from cells expressing the mutant α4(S467A)β2 receptor (Figure 4B). An antibody generated against the non-phosphorylated α4 sequence detected both the native and S467A receptors. When protein levels or exposure time was increased, the phospho-antibody detected non-specific bands in HEKts201 cells that did not express α4β2 nAChRs. Both antibodies detected bands in mouse brain synaptosomes (Figure 4C). We quantified the lower molecular weight band in the phospho-antibody immunoblot and found a small but significant 13% reduction in the phospho-α4 S467/non-phospho α4 S467 ratio in Prkce−/− synaptosomes compared with wild-type mouse synaptosomes (Figure 4D, t = 2.825, P = 0.02). The effect size for this comparison was large (Cohen’s d = 1.786). Our findings indicate that PKCε contributes to the phosphorylation of this residue in vivo.
Figure 4.

Phosphorylation of the α4 nAChR subunit in synaptosomes. Phospho-antibodies were raised to the PKCε phosphorylation sites S465 and S467 in the human α4 subunit, corresponding to S468 and S470 in mouse. (A) The level of phosphorylated S465 to non-phosphorylated S465 was not significantly different between wild-type and Prkce−/− mouse synaptosomes. (B) The S467 phospho-antibody detected a strong band from HEKtsA201 cells expressing native α4β2 nAChRs ‘S’ and did not detect a band from cells expressing the S467A mutation ‘A’. The non-phospho-α4 antibody detected bands from both native and S467A mutant receptors. Corresponding GAPDH levels are shown in the lower panel. The S467 phospho-antibody detected several non-specific bands in an overexposure from HEKtsA201 cells that do not express α4β2 nAChRs ‘E’. (C) The phospho-α4 S467 and non-phospho-α4 S467 antibodies detected bands in synaptosome preparations from wild-type and Prkce−/− mouse brains. The corresponding GAPDH blots are shown below each panel. The 60 kDa molecular weight band was quantified from the phospho-α4 S467 blots. Molecular weights are indicated on the side (kDa). (D) The level of phosphorylated α4 S467 to non-phosphorylated α4 S467 was 13% higher in synaptosomes from wild-type compared with Prkce−/− mouse brains. *P < 0.05 compared with wild-type mice, n = 6 mice per genotype.
Sazetidine-A- and nicotine-induced hypothermia in Prkce−/− mice
To determine if reduced PKCε phosphorylation of the α4 nAChR subunit is physiologically relevant, we measured hypothermia induced by sazetidine-A or nicotine in wild-type and Prkce−/− mice. Sazetidine-A and nicotine both cause acute hypothermia when administered to C57BL/6 mice (Rezvani et al., 2012). Sazetidine-A primarily desensitizes α4β2 nAChR with very little receptor activation (Xiao et al., 2006), while nicotine activates and desensitizes α4β2 nAChR, suggesting that both drugs induce hypothermia via α4β2 desensitization. We found that baseline skin temperatures in wild-type (34.3 ± 0.2°C) and Prkce−/− mice (34.3 ± 0.1°C) were identical (t = 0.156, P = 0.88). Injection of 2.0 mg·kg−1 sazetidine-A s.c. produced a greater drop in body temperature and delayed recovery from hypothermia in Prkce−/− mice compared with wild-type mice [Figure 5A, anova FgenotypeXtime (24,648) = 6.862, P < 0.001, Ftime (24,648) = 64.42, P < 0.0001, Fgenotype (1,648) = 5.653, P = 0.02]. Injection of 2.0 mg·kg−1 nicotine s.c. also produced a greater and more prolonged drop in body temperature in Prkce−/− mice compared with wild-type mice [Figure 5B, anova FgenotypeXtime (24,576) = 5.702, P < 0.001, Ftime (24,576) = 116.2, P < 0.0001, Fgenotype (1,576) = 8.507, P = 0.008]. Body temperature returned to baseline after 24 h and was not different between genotypes (Figure 5C and D, nicotine t = 0.019, P = 0.99, sazetidine-A t = 0.238, P = 0.82). These results indicate that absence of PKCε prolongs the hypothermic effect of sazetidine-A and nicotine in mice.
Figure 5.
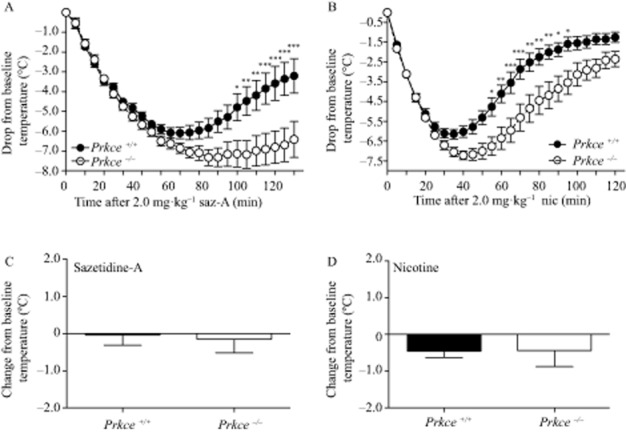
Prkce−/− mice show deeper and more prolonged hypothermia from acute sazetidine-A and nicotine. Drop in mouse skin temperature after injection of (A) 2.0 mg·kg−1 sazetidine-A s.c. in wild-type (n = 15) and Prkce−/− mice (n = 14), and (B) 2.0 mg·kg−1 nicotine s.c. in wild-type and Prkce−/− mice (n = 13 per genotype). Skin temperature 24 h after (C) 2.0 mg·kg−1 sazetidine-A (n = 5 wild-type and n = 6 Prkce−/− mice) and (D) 2.0 mg·kg−1 nicotine injections (n = 4 wild-type and n = 5 Prkce−/− mice). *P < 0.05, **P < 0.01, ***P < 0.001 compared with wild-type mice at the same time point.
Discussion and conclusion
Previous studies showed that activating PKC increases, whereas inhibiting PKC decreases, the recovery of α4β2 nAChR from deep desensitization (Eilers et al., 1997; Fenster et al., 1999). However, these studies did not identify specific PKC isozymes involved. In this study, we found that PKCε phosphorylates α4 nAChR subunits, and mutating the PKCε phosphorylation sites dramatically impaired α4β2 nAChR recovery from deep desensitization. Most of these phosphorylated residues are conserved between humans, mice and rats. We also found that phosphorylation of α4 at S467 is reduced in mice carrying a null mutation in Prkce, suggesting that PKCε phosphorylates the α4 subunit in vivo.
We used HEKtsA201 cells expressing human α4β2 nACh receptors to investigate the effect of inhibiting PKCε activity on receptor desensitization. These cells express both stoichiometries of the α4β2 receptor, α4(3)β2(2) and α4(2)β2(3) (Kuryatov et al., 2005). Our in vitro data do not distinguish between stoichiometries. In our electrophysiology studies, we transiently transfected HEKtsA201 cells with equal amounts of mutant α4 and native β2 cDNA, which likely results in a mixture of both mutant α4(3)β2(2) and α4(2)β2(3) nAChR. Determining the effect of phosphorylation on each stoichiometry would be of interest, as sazetidine-A has different effects depending on receptor stoichiometry (Zwart et al., 2008). The α4 subunit is also incorporated in numerous other nAChR subtypes, such as α4α5β2 and α4α6β2β3 receptors. Whether PKCε regulates desensitization of these subtypes is also of interest. In the Prkce−/− mice, PKCε phosphorylation is abolished for all subtypes of α4-containing nAChR. Altered phosphorylation on multiple receptor subtypes could contribute to the responses observed after nicotine and sazetidine-A injection in the Prkce−/− mice.
Pharmacological inhibition of PKCε reduced the magnitude of receptor recovery from desensitization without affecting the rate of recovery. We found no difference in cell surface or intracellular β2 subunit immunoreactivity when PKCε was inhibited, indicating that the reduced magnitude of recovery was not due to a lower number of receptors at the cell surface. We speculate that impaired PKCε phosphorylation causes α4β2 receptors to persist in an intermediate desensitized state, resulting in a lower magnitude, but unchanged rate of recovery. How inhibition of phosphorylation promotes this intermediate desensitized state warrants further investigation.
The large intracellular loop of the α4 subunit contains many putative phosphorylation sites and we identified five PKCε phosphorylation sites in the human α4 nAChR subunit: S465, S467, T545, S550 and T551. Our study is the first to identify S465, T545 and T551 as phosphorylation sites in vitro. Previous studies have suggested that S550 may be phosphorylated by PKC (Pollock et al., 2007), and S467 may be phosphorylated by PKA (Jeanclos et al., 2001; Guo and Wecker, 2002; Pollock et al., 2007). We found that mutation of the S465, S467, S550 and T551 residues to alanine impaired receptor recovery from desensitization. The presence of multiple PKCε phosphorylation sites raises the possibility that more than one residue is phosphorylated simultaneously. We did not assess the functional effect of multiple mutations, but speculate that mutating two or more phosphorylation sites might be additive. Previous work by Fenster et al. (1999) identified serine 336 of the rat α4 subunit as a potential PKC phosphorylation site (Fenster et al., 1999) and mutating S336 to alanine produced a receptor that also showed impaired recovery from desensitization (Pollock et al., 2007). S336 is also highly conserved between humans, rats and mice. Our mass spectrometry analysis covered 77% of the α4 intracellular loop sequence and S336 was included in the remaining 23% missed sequence; thus we could not identify S336 as a PKCε phosphorylation site in our study.
In addition to desensitization, PKCs may also be involved in regulating intracellular and surface α4 and β2 subunit protein levels, as treatment with phorbol esters, which activates all PKCs, increases intracellular and surface subunit levels in transfected SH-EP1 cells (Wecker et al., 2010). The identity of the PKC responsible for this effect is unknown. We found that acute inhibition of PKCε did not affect surface or intracellular β2 protein levels. The T545A PKCε phosphorylation mutant had greater current density than native receptors, suggesting that PKCε phosphorylation of this site may negatively regulate receptor expression. The S550A mutant showed a trend for reduction in current density, suggesting that this site may positively regulate receptor expression; however, it was not significantly different compared with the native receptor.
Increased receptor deactivation may dampen fast cholinergic neurotransmission by reducing receptor open time. Compared with the native α4β2 receptor, three of the PKCε phosphorylation site mutants, S467A, T545A and T551A, showed faster deactivation time, which suggests that impaired phosphorylation at these residues may dampen fast cholinergic neurotransmission. We did not see any difference in response deactivation time when PKCε was inhibited with 1Na-PP1, which may be due to the net effect of reducing PKCε phosphorylation at multiple residues.
A recent proteomics study of phosphorylated proteins in synaptosomes from adult C57BL/6 mice found that both S465 and S467 of the α4 nAChR subunit are phosphorylated in vivo (Trinidad et al., 2012). Our data suggest that S467 is phosphorylated by PKCε in vivo, but S465 may be phosphorylated by kinases other than PKCε. The S467A mutation resulted in both a large impairment in receptor desensitization recovery and increased response deactivation time. As previous data show that PKA phosphorylates the same site in vitro (Jeanclos et al., 2001; Guo and Wecker, 2002; Pollock et al., 2007), S467 may be phosphorylated by both kinases in vivo. S467 mediates the interaction between the α4 subunit and the chaperone protein 14-3-3η, and activation of PKA increases the interaction between α4 and 14-3-3η, which is associated with increased surface expression of the α4β2 nAChR in vitro (Jeanclos et al., 2001). Here, we find that S467 is also involved in receptor desensitization and deactivation.
To determine if reduced PKCε phosphorylation of the α4 nACh receptor subunit is physiologically relevant, we measured hypothermia induced by administration of either sazetidine-A or nicotine in Prkce−/− and wild-type mice because this response is thought to be mediated by α4β2 nAChR desensitization (Rezvani et al., 2012). Sazetidine-A was first identified as a desensitizer of α4β2 nAChR (Xiao et al., 2006), but was later discovered to have mixed effects depending on the stoichiometry of the α4β2 nAChR (Zwart et al., 2008). It is a very poor agonist at α4(3)β2(2) and a full agonist at α4(2)β2(3) (Zwart et al., 2008). Sazetidine-A mediated hypothermia in mice is prolonged by co-injection of di-hydro-β-erythroidine, a nAChR antagonist, which supports the conclusion that sazetidine-A mediates hypothermia via desensitization (Rezvani et al., 2012). Given that inhibition of PKCε impairs recovery of α4β2 nAChR from deep desensitization, we predicted that Prkce−/− would show a greater hypothermic response. Indeed, we found that compared with wild-type mice, Prkce−/− mice showed deeper and more prolonged hypothermia after administration of either drug. Our findings further support the conclusion that sazetidine-A- and nicotine-induced hypothermia is due to α4β2 nACh receptor desensitization, and indicate that PKCε regulates the response to both drugs. Although we did not measure sazetidine-A clearance in Prkce−/−- and wild-type mice, we have previously shown that nicotine clearance rates are similar in both genotypes (Lee and Messing, 2011), indicating that differences in nicotine-induced hypothermia are not due to altered nicotine clearance.
Desensitization of nAChR is thought to contribute to nicotine addiction (Picciotto et al., 2008); however, the impact of nAChR desensitization on nicotine consumption, reward and relapse is unclear. Although sazetidine-A reduces established nicotine consumption in rats (Levin et al., 2010; Rezvani et al., 2010; Johnson et al., 2012), it is also self-administered (Paterson et al., 2010), suggesting that sazetidine-A reduces nicotine consumption by substituting for nicotine. However, we previously found that Prkce−/− mice show reduced nicotine consumption and nicotine conditioned place preference (Lee and Messing, 2011), and here find that inhibiting PKCε prolongs recovery of α4β2 nACh receptors from deep desensitization. Based on these results, we hypothesize that impaired α4β2 nAChR recovery from deep desensitization reduces nicotine consumption and reward, and that modulating the phosphorylation status of α4β2 nAChR to prolong nAChR desensitization may be an additional strategy for drug development. Future studies to investigate whether altered phosphorylation of α4 nAChR subunits regulates nicotine consumption and reward in mice will be informative. Targeting PKCε activity would be one method to reduce α4β2 nAChR phosphorylation. The substrates of PKCε have not been fully elucidated. PKCε has been shown to phosphorylate additional receptors including GABAA receptors and TRPV1 receptors (Newton and Messing, 2010). Prkce−/− mice have normal home-cage behaviour, no significant differences in locomotor activity and no differences in saccharin consumption (Hodge et al., 1999), which suggests that inhibiting PKCε activity is unlikely to cause severe deficits in behaviour.
Our results are the first to demonstrate that PKCε phosphorylates α4β2 nAChR. We showed that the functional consequence of eliminating PKCε phosphorylation sites in the α4 subunit is impaired recovery from deep desensitization. This study illustrates the importance of phosphorylation on α4β2 nACh receptor function, and we suggest that targeting the phosphorylation status of nACh receptors may be useful for modulating receptor function and behaviour.
Acknowledgments
This work was supported by a Canadian Institute of Health Research post-doctoral fellowship to A. M. L., US PHS grant AA013588 to R. O. M. and by funds provided by the State of California for medical research on alcohol and substance abuse through University of California, San Francisco. We thank Dr. Jon Lindstrom and Dr. Kevan Shokat for generously providing reagents, Jackie Stecher for assistance with the animal breeding and genotyping and Ky Lee with assistance with molecular biology experiments.
Glossary
- 1Na-PP1
1-(1,1-dimethylethyl)-3-(1-naphthalenyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine
- as-PKCε
analogue-sensitive PKCε
- nAChR
nicotinic ACh receptor
- Prkce−/−
PKCε knock-out mice
Author contributions
A. M. L. designed and performed molecular biology and behavioural experiments and prepared the manuscript. D.-F. W. performed the electrophysiology experiments. J. D. and T. M. participated in molecular cloning experiments and generated the mutant receptors. D. W. performed the kinase assay. R. O. M. assisted in designing the experiments and co-authored the manuscript.
Conflicts of interest
No conflicts of interest to declare.
References
- Albuquerque EX, Pereira EF, Alkondon M, Rogers SW. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev. 2009;89:73–120. doi: 10.1152/physrev.00015.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: ligand-gated ion channels. Br J Pharmacol. 2013a;170:1582–1606. doi: 10.1111/bph.12446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, et al. The concise guide to pharmacology 2013/14: ion channels. Br J Pharmacol. 2013b;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: enzymes. Br J Pharmacol. 2013c;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, et al. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature. 2000;407:395–401. doi: 10.1038/35030148. [DOI] [PubMed] [Google Scholar]
- Choi DS, Wang D, Dadgar J, Chang WS, Messing RO. Conditional rescue of protein kinase C epsilon regulates ethanol preference and hypnotic sensitivity in adult mice. J Neurosci. 2002;22:9905–9911. doi: 10.1523/JNEUROSCI.22-22-09905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downing JE, Role LW. Activators of protein kinase C enhance acetylcholine receptor desensitization in sympathetic ganglion neurons. Proc Natl Acad Sci U S A. 1987;84:7739–7743. doi: 10.1073/pnas.84.21.7739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers H, Schaeffer E, Bickler PE, Forsayeth JR. Functional deactivation of the major neuronal nicotinic receptor caused by nicotine and a protein kinase C-dependent mechanism. Mol Pharmacol. 1997;52:1105–1112. [PubMed] [Google Scholar]
- Fenster CP, Beckman ML, Parker JC, Sheffield EB, Whitworth TL, Quick MW, et al. Regulation of alpha4beta2 nicotinic receptor desensitization by calcium and protein kinase C. Mol Pharmacol. 1999;55:432–443. [PubMed] [Google Scholar]
- Gray EG, Whittaker VP. The isolation of nerve endings from brain: an electron-microscopic study of cell fragments derived by homogenization and centrifugation. J Anat. 1962;96:79–88. [PMC free article] [PubMed] [Google Scholar]
- Guo X, Wecker L. Identification of three cAMP-dependent protein kinase (PKA) phosphorylation sites within the major intracellular domain of neuronal nicotinic receptor alpha4 subunits. J Neurochem. 2002;82:439–447. doi: 10.1046/j.1471-4159.2002.01027.x. [DOI] [PubMed] [Google Scholar]
- Hodge CW, Mehmert KK, Kelley SP, McMahon T, Haywood A, Olive MF, et al. Supersensitivity to allosteric GABA(A) receptor modulators and alcohol in mice lacking PKCepsilon. Nat Neurosci. 1999;2:997–1002. doi: 10.1038/14795. [DOI] [PubMed] [Google Scholar]
- Jeanclos EM, Lin L, Treuil MW, Rao J, DeCoster MA, Anand R. The chaperone protein 14-3-3eta interacts with the nicotinic acetylcholine receptor alpha 4 subunit. Evidence for a dynamic role in subunit stabilization. J Biol Chem. 2001;276:28281–28290. doi: 10.1074/jbc.M011549200. [DOI] [PubMed] [Google Scholar]
- Johnson JE, Slade S, Wells C, Petro A, Sexton H, Rezvani AH, et al. Assessing the effects of chronic sazetidine-A delivery on nicotine self-administration in both male and female rats. Psychopharmacology (Berl) 2012;222:269–276. doi: 10.1007/s00213-012-2642-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasar SG, Lin YH, Martin A, Dadgar J, McMahon T, Wang D, et al. A novel nociceptor signaling pathway revealed in protein kinase C epsilon mutant mice. Neuron. 1999;24:253–260. doi: 10.1016/s0896-6273(00)80837-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuryatov A, Luo J, Cooper J, Lindstrom J. Nicotine acts as a pharmacological chaperone to up-regulate human alpha4beta2 acetylcholine receptors. Mol Pharmacol. 2005;68:1839–1851. doi: 10.1124/mol.105.012419. [DOI] [PubMed] [Google Scholar]
- Lee AM, Messing RO. Protein kinase C epsilon modulates nicotine consumption and dopamine reward signals in the nucleus accumbens. Proc Natl Acad Sci U S A. 2011;108:16080–16085. doi: 10.1073/pnas.1106277108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin ED, Rezvani AH, Xiao Y, Slade S, Cauley M, Wells C, et al. Sazetidine-A, a selective alpha4beta2 nicotinic receptor desensitizing agent and partial agonist, reduces nicotine self-administration in rats. J Pharmacol Exp Ther. 2010;332:933–939. doi: 10.1124/jpet.109.162073. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Research Council Committee for the Update of the Guide for the Care and Use of Laboratory Animals. 2010. ). National Academies Press. [PubMed]
- Newton PM, Messing RO. The substrates and binding partners of protein kinase Cepsilon. Biochem J. 2010;427:189–196. doi: 10.1042/BJ20091302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizaki T, Sumikawa K. Effects of PKC and PKA phosphorylation on desensitization of nicotinic acetylcholine receptors. Brain Res. 1998;812:242–245. doi: 10.1016/s0006-8993(98)00836-1. [DOI] [PubMed] [Google Scholar]
- Olive MF, Messing RO. Protein kinase C isozymes and addiction. Mol Neurobiol. 2004;29:139–154. doi: 10.1385/mn:29:2:139. [DOI] [PubMed] [Google Scholar]
- Paradiso KG, Steinbach JH. Nicotine is highly effective at producing desensitization of rat alpha4beta2 neuronal nicotinic receptors. J Physiol. 2003;553:857–871. doi: 10.1113/jphysiol.2003.053447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson NE, Min W, Hackett A, Lowe D, Hanania T, Caldarone B, et al. The high-affinity nAChR partial agonists varenicline and sazetidine-A exhibit reinforcing properties in rats. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34:1455–1464. doi: 10.1016/j.pnpbp.2010.07.037. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picciotto MR, Zoli M, Léna C, Bessis A, Lallemand Y, Le Novère N, et al. Abnormal avoidance learning in mice lacking functional high-affinity nicotine receptor in the brain. Nature. 1995;374:65–67. doi: 10.1038/374065a0. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Addy NA, Mineur YS, Brunzell DH. It is not ‘either/or’: activation and desensitization of nicotinic acetylcholine receptors both contribute to behaviors related to nicotine addiction and mood. Prog Neurobiol. 2008;84:329–342. doi: 10.1016/j.pneurobio.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock VV, Pastoor TE, Wecker L. Cyclic AMP-dependent protein kinase (PKA) phosphorylates Ser362 and 467 and protein kinase C phosphorylates Ser550 within the M3/M4 cytoplasmic domain of human nicotinic receptor alpha4 subunits. J Neurochem. 2007;103:456–466. doi: 10.1111/j.1471-4159.2007.04853.x. [DOI] [PubMed] [Google Scholar]
- Pons S, Fattore L, Cossu G, Tolu S, Porcu E, McIntosh JM, et al. Crucial role of alpha4 and alpha6 nicotinic acetylcholine receptor subunits from ventral tegmental area in systemic nicotine self-administration. J Neurosci. 2008;28:12318–12327. doi: 10.1523/JNEUROSCI.3918-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi ZH, Song M, Wallace MJ, Wand D, Newton PM, McMahon T, et al. Protein kinase C epsilon regulates gamma-aminobutyrate type A receptor sensitivity to ethanol and benzodiazepines through phosphorylation of gamma2 subunits. J Biol Chem. 2007;282:33052–33063. doi: 10.1074/jbc.M707233200. [DOI] [PubMed] [Google Scholar]
- Quick MW, Lester RA. Desensitization of neuronal nicotinic receptors. J Neurobiol. 2002;53:457–478. doi: 10.1002/neu.10109. [DOI] [PubMed] [Google Scholar]
- Rezvani AH, Slade S, Wells C, Petro A, Lumeng L, Li TK, et al. Effects of sazetidine-A, a selective alpha4beta2 nicotinic acetylcholine receptor desensitizing agent on alcohol and nicotine self-administration in selectively bred alcohol-preferring (P) rats. Psychopharmacology (Berl) 2010;211:161–174. doi: 10.1007/s00213-010-1878-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezvani AH, Timofeeva O, Sexton HG, DeCuir D, Xiao Y, Gordon CJ, et al. Effects of sazetidine-A, a selective alpha4beta2* nicotinic receptor desensitizing agent, on body temperature regulation in mice and rats. Eur J Pharmacol. 2012;682:110–117. doi: 10.1016/j.ejphar.2012.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinidad JC, Barkan DT, Gulledge BF, Thalhammer A, Sali A, Schoepfer R, et al. Global identification and characterization of both O-GlcNAcylation and phosphorylation at the murine synapse. Mol Cell Proteomics. 2012;11:215–229. doi: 10.1074/mcp.O112.018366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wecker L, Pollock VV, Pacheco MA, Pastoor T. Nicotine-induced up regulation of alpha4beta2 neuronal nicotinic receptors is mediated by the protein kinase C-dependent phosphorylation of alpha4 subunits. Neuroscience. 2010;171:12–22. doi: 10.1016/j.neuroscience.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witzmann FA, Arnold RJ, Bai F, Hrncirova P, Kimpel MW, Mechref YS, et al. A proteomic survey of rat cerebral cortical synaptosomes. Proteomics. 2005;5:2177–2201. doi: 10.1002/pmic.200401102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Fan H, Musachio JL, Wei ZL, Chellappan SK, Kozikowski AP, et al. Sazetidine-A, a novel ligand that desensitizes alpha4beta2 nicotinic acetylcholine receptors without activating them. Mol Pharmacol. 2006;70:1454–1460. doi: 10.1124/mol.106.027318. [DOI] [PubMed] [Google Scholar]
- Zhang J, Xue F, Liu Y, Yang H, Wang X. The structural mechanism of the cys-loop receptor desensitization. Mol Neurobiol. 2013;48:97–108. doi: 10.1007/s12035-013-8420-z. [DOI] [PubMed] [Google Scholar]
- Zwart R, Carbone AL, Moroni M, Bermudez I, Mogg AJ, Folly EA, et al. Sazetidine-A is a potent and selective agonist at native and recombinant alpha 4 beta 2 nicotinic acetylcholine receptors. Mol Pharmacol. 2008;73:1838–1843. doi: 10.1124/mol.108.045104. [DOI] [PubMed] [Google Scholar]