

Abstract Abstract
Respiratory and limb muscle dysfunction is emerging as an important pathophysiological abnormality in pulmonary arterial hypertension (PAH). Muscle abnormalities appear to occur frequently and promote dyspnea, fatigue, and exercise limitation in patients with PAH. Preliminary data suggest that targeted muscle training may be of benefit, although further evidence is required to consolidate these findings into specific recommendations for exercise training in patients with PAH. This article reviews the current evidence on prevalence, risk factors, and implications of respiratory and limb muscle dysfunction in patients with PAH. It also reviews the impact of exercise rehabilitation on morphologic, metabolic, and functional muscle profile and outcomes in PAH. Future research priorities are highlighted.
Keywords: pulmonary arterial hypertension, respiratory muscles, skeletal muscle, rehabilitation
Pulmonary arterial hypertension (PAH), especially the idiopathic subtype (IPAH), represents the type of pulmonary hypertension (PH) in which the most important advances in the understanding and disease-specific treatment have been achieved in the recent years. Pathogenetically, all forms of PAH, regardless of underlying etiology, share a common pulmonary vasculopathy characterized by proliferative and obstructing remodeling, together with vasoconstriction and the development of in situ thrombosis of the distal pulmonary arterioles in particular.1 The consequent increase in pulmonary vascular resistance raises the pulmonary arterial pressure and right ventricular (RV) afterload and leads to RV hypertrophy and dilatation and eventually to RV failure and death. Traditionally, cardiopulmonary abnormalities have been considered the primary limiting factors in PAH. However, although often unrecognized in clinical practice, muscle dysfunction is increasingly appreciated as being present as a potential contributor to several manifestations of PAH.2 Pathophysiologically, muscle dysfunction promotes dyspnea, fatigue, and impaired exercise capacity, which are cardinal and prognostic features in patients with PAH.1
Manifestations beyond the primary organ-target have long been recognized in many chronic cardiopulmonary diseases, such as chronic obstructive pulmonary disease (COPD)3,4 and chronic heart failure (CHF).5 Thus, despite the debate as to whether they are truly systemic in nature or their systemic manifestations represent mere consequences of the primary pathophysiological process, many chronic diseases are increasingly regarded as conditions with a significant systemic component.6 Importantly, significant outcomes such as functional capacity, quality of life, and survival are affected by the interplay between the primary and systemic effects that occur in chronic cardiopulmonary disease.7-9 In PAH, muscle dysfunction seems to constitute a manifestation beyond the pulmonary vasculature, and therefore PAH appears to follow the paradigm of other chronic cardiopulmonary diseases.
This article reviews the current evidence on prevalence, risk factors, and implications of respiratory and limb muscle dysfunction in patients with PAH. It also reviews the impact of exercise rehabilitation on muscle structure and function and outcomes in PAH. Future research priorities are highlighted.
Risk factors
A conceptual model of the factors and mechanisms that lead to muscle dysfunction in PAH and their implications is delineated in Figure 1. Although evidence of a causal relationship is still missing, both local and systemic mechanisms could potentially trigger and aggravate muscle dysfunction in PAH. First, cardiac output in PAH is impaired and commonly fails to accommodate increases in oxygen demand by the skeletal muscles during exercise. Combined with the frequently occurring chronic arterial hypoxemia, this may exert a deleterious effect on the overall muscle function.10 Hypoxia has been described to inhibit skeletal muscle differentiation through accelerating degradation of MyoD, a myogenic transcription factor of myoblast differentiation,11 and it induces a shift from aerobic to anaerobic muscle metabolism. Second, inflammation, which is thought to play an important role in the pulmonary vascular remodeling and the pathogenesis of PAH, may induce skeletal muscle impairment. Elevated levels of circulating proinflammatory cytokines, such as tumor necrosis factor α (TNF-α), interleukin 1 (IL-1), and IL-6, that have been demonstrated in patients with PAH and animal models of PH12 may initiate injury of striated muscle, impair contractile protein function, stimulate proteolysis,13-15 reduce skeletal muscle force,16 and alter skeletal muscle metabolism.17 Third, the increased sympathetic tone in PAH18 may increase peripheral vascular resistance and reduce the perfusion of the skeletal muscles. Fourth, muscle function in PAH may also be aggravated by the inhibition of hormonal/anabolic pathways, such as the insulin-signaling pathway. Insulin resistance has been described in PAH,19 and its potential role in the pathogenesis of PAH is currently under investigation.20 Notably, insulin signaling is impaired by chronic inflammation and cytokines such as TNF-α and IL-6.21
Figure 1.

Conceptual model of the factors and mechanisms that lead to muscle dysfunction in pulmonary arterial hypertension and their implications. SNA: sympathetic nerve activity.
Ultimately, patients with PAH often adopt a sedentary lifestyle.22,23 Physical inactivity, in turn, establishes a spiral between muscle disuse and muscle impairment.24 Muscle disuse is associated with decreases in myofibrillar protein synthesis; increases in proteolysis, inflammation, and muscle fibrosis via alteration in the expression of various genes24 (such as upregulation of the proteolytic genes Muscle Ring Finger 1 [MuRF-1] and Muscle Atrophy F-box [MAFbx]/atrogin-125); and activation of molecular mechanisms, such as the classical and alternative nuclear factor-κB signaling pathways.26
Respiratory muscle dysfunction
The first evidence of significant inspiratory and expiratory muscle dysfunction in patients with PAH was provided by Meyer et al.27 in 2005. In a prospective study of 37 patients with IPAH in World Health Organization (WHO) functional classes II–IV, both maximal inspiratory pressure (MIP) and maximal expiratory pressure (MEP) were reduced by 25% compared with those in healthy controls. This magnitude of reduction in MIP is similar to the 28% MIP reduction in patients with congestive left ventricular failure.28 In addition, there was a close correlation between MIP and MEP, signifying a parallel reduction in inspiratory and expiratory pressures. In 2008, Kabitz et al.29 confirmed substantially impaired respiratory muscle function in a sample of patients with precapillary PH (25 with IPAH and 6 with chronic thromboembolic PH). These patients demonstrated a 20% decrease in mouth MIP and MEP and sniff nasal pressure. The reduction in inspiratory muscle strength was even more prominent with the use of nonvolitional tests: twitch mouth and transdiaphragmatic pressures were reduced by 34%. Of note, nonvolitional tests, although more complex to perform, are considered more accurate and provide a definitive diagnosis of respiratory muscle weakness.2,30 This suggests that patients with precapillary PH may actually have more severely impaired inspiratory muscle strength than chronic left ventricular failure patients.28
The function of the diaphragm, the primary inspiratory muscle, is also impaired. The maximal and submaximal diaphragmatic isometric force–generating capacities in PH models and 2 PH patients were reduced by 30%31,32 and 50%, respectively.32 However, isotonic contractile properties are more physiologically relevant properties of diaphragm function in vivo, as the diaphragm does not perform maximum isometric contractions in vivo but rather shortens against submaximal loads.33 Isotonic contractile properties of the diaphragm were also abnormal in PH mice and associated with 40% slower maximal shortening velocity and 60% lower peak power.31 These values are normalized to the cross-sectional area (CSA) of the muscle fibers, which suggests that the intrinsic capacity of diaphragm fibers to generate force is impaired.31 Clinical evidence of impaired isotonic contractile properties of the diaphragm in PAH patients is suggested by the 20% decrease in maximal sniff nasal pressure (a volitional dynamic maneuver that involves diaphragm shortening) reported by Kabitz et al.29 and a 30% decrease in maximal voluntary ventilation in PAH subjects compared to healthy controls.34
Muscle atrophy
As in all skeletal muscles, depression of the force-generating capacity of the diaphragm can result from atrophy (decrease in fiber size or CSA and/or contractile dysfunction, defined as muscle force normalized for CSA). Both these abnormalities have been shown to underlie diaphragmatic muscle weakness in patients with PAH and experimental PH.31,32 In particular, the fiber CSA has been found to be significantly decreased in 6 patients with PH (75%) compared to that in healthy controls,32 but also in PH animal models (40% in PH rats32 and 25% in PH mice31). De Man et al.32 found increased expression of the E3 ligases MAFbx/atrogin-1 and MuRF-1, which are considered key markers of activation of the ubiquitin-proteasome proteolytic pathway in the muscles. This indicates that the reduction in fiber CSA (irrespective of fiber type) is most likely due to elevated degradation of myofibrillar proteins rather than changes in protein synthesis. The concept that protein degradation accounts for the diaphragmatic contractile dysfunction in PAH was opposed by Ahn et al.,31 who did not find any association between contractile impairment and decreases in myofibrillar proteins but suggested that posttranslational modifications affecting the phosphorylation or thiol oxidation status of myofibrillar proteins were the likely cause of isometric and isotonic contractile impairments in PAH.31
Impaired contractility
Manders et al.35 provided further evidence of intrinsic myopathy. At a single-fiber level, they demonstrated that diaphragmatic isometric weakness in PH rats is at least partly caused by diaphragm-specific sarcomeric dysfunction due to decrease in the force generated per cross bridge, mainly in type II fibers. Type II fibers also showed reduced Ca2+ sensitivity, which could contribute to diaphragm weakness at submaximal activation.35 Kanj et al.36 observed a progressive increase in calcium accumulation in diaphragmatic tissues in animal models of PH. This could possibly be due to an increase in the muscle sarcoplasm permeability to calcium and/or an increase in the amount of calcium binding to troponin during the excitation-contraction coupling. Whether this actually affects diaphragmatic contractility remains unknown. Nevertheless, this observation leads to the speculation that PH may trigger a compensatory response in the diaphragm to increase its uptake of calcium in response to the reduced sensitivity of its muscle fibers to calcium.
Capillarity and oxidative capacity
Capillary density and fiber oxidative enzyme activity of the diaphragm were preserved despite atrophy.32 This excludes the possibility that the diaphragmatic atrophy in PAH represents an adaptive mechanism with the aim of reducing oxygen diffusion distance and preventing tissue hypoxia in the context of low cardiac output.32
The role of central drive
The minute ventilation in precapillary PH is known to be increased both at rest and during exercise.34 In line with this, ventilatory drive (P0.1; mouth occlusion pressure after 0.1 s of inspiration) showed trends toward higher values (although without reaching statistical significance),27,29 whereas respiratory capacity (P0.1/PImax) either tended to increase29 or increased27 relative to that in controls. Increased P0.1/PImax suggests inadequate muscle effort in response to central drive and argues against a central role in the respiratory muscle impairment in PAH. Of course, a chronically increased central drive could contribute to respiratory muscle weakness via an increase in workload,27 which the respiratory musculature is insufficiently equipped to tolerate. However, Kabitz et al.29 found no difference in the effective inspiratory impedance (P0.1/VT/ti, a marker of the load imposed on the inspiratory muscles) between patients with precapillary PH and controls, indicating that the load imposed on the inspiratory muscles was not increased. Collectively, these findings suggest that the impairment of respiratory muscle function in patients with PH is unlikely to be accounted for by the changes in the breathing pattern. Rather, the commonly occurring increased central drive in PAH could be a response to respiratory muscle weakness per se.
Limb muscle dysfunction
Bauer et al.37 were the first to report on occurrence of limb muscle dysfunction in PAH. In a prospective study, 24 patients with IPAH in WHO functional class II or III exhibited 30% reduction in isometric forearm muscle strength (hand grip) compared to healthy controls. Subsequently, it has been shown that IPAH patients have also lower strength of the quadriceps muscles compared to healthy controls, demonstrated by both maximal voluntary contraction and nonvolitional magnetic stimulation of the femoral nerve.38
Alterations in the muscle fiber type
Skeletal striated muscle fibers are broadly divided into myosin type I (or slow-twitch oxidative) fibers and type II (or fast-twitch glycolytic) fibers. Type I fibers present higher mitochondrial density and oxidative capacity and, therefore, increased aerobic activity and resistance to fatigue that allow for more repetitive and extended contractions over a longer time compared to type II fibers. Type II fibers are divided further into type IIa and type IIx fibers, of which the latter are the “classic” fast-twitch fibers that excel at producing quick, powerful bursts of speed but also have a much faster rate of fatigue, as they rely completely on anaerobic metabolism. Two recent studies on the molecular mechanisms mediating muscle dysfunction in PAH suggest that skeletal muscle–type changes occur in PAH. Biopsies of vastus lateralis by Mainguy et al.38 and Batt et al.39 in 10 IPAH patients and 12 PAH patients, respectively, displayed a decreased type I/type II muscle fiber ratio. Analysis of the fiber types suggested that either a smaller CSA in the type I fibers39 or an increase in the proportion of type IIx fibers38 could be responsible for this shift in fiber-type balance. The proportion of type I fibers negatively correlated with disease duration.38
Muscle atrophy
PAH patients tend to have lower thigh muscle CSA, as measured by computed tomography (CT),38,39 although this is not in line with findings of other studies,40 where no significant difference in thigh muscle area was observed. This discordance seems unlikely to be accounted by hemodynamic severity of the disease, as the hemodynamic profiles of the study populations were similar, and raises questions about the sensitivity of the CT in detecting small differences in muscle mass. It is possible that differences in thigh muscle area may be more significant for patients with advanced muscle depletion, as is found in COPD patients.41 Perhaps measurement of muscle mass by magnetic resonance imaging would help to resolve this issue.
Enzyme analysis in biopsied vastus lateralis muscle of patients with PAH suggests engagement of cellular signaling networks stimulating ubiquitin-proteasome-mediated proteolysis of muscle, with concurrent depression of networks mediating muscle hypertrophy.39 Also, a study by Wüst et al.42 showed a reduction in muscle fiber CSA in the plantaris muscle of a monocrotaline rat model of PH with RV failure. Of note, the reduction in CSA was more pronounced in the glycolytic fibers, although there was no shift in the fiber-type distribution.42
Impaired contractility
Patients with PAH demonstrated increased phosphorylation of ryanodine receptor 1 receptors, suggesting altered sarcoplasmic reticulum Ca2+ sequestration, which may impair excitation-contraction coupling in the PAH muscle.39
Reduced oxidative capacity
In PAH patients, discriminating enzyme ratios suggested a relatively higher potential for anaerobic than for aerobic energy metabolism (lower oxidative enzyme activities) in IPAH patients,38 and also, protein levels of a positive regulator of mitochondrial fusion (Mitofusin 2) were significantly lower.39 Mitochondrial volume density and oxidative capacity were reduced in PH rats.42
Reduced capillarity
Decreased capillarity around type I muscle fibers has been shown in IPAH patients.38 Evidence of substantial impairments of limb muscle microcirculation in patients with PAH has also been provided by Dimopoulos et al.43 By employing near-infrared spectroscopy with vascular occlusion, they obtained tissue oxygen saturation (defined as the hemoglobin saturation [%], an index of tissue oxygen) of the thenar muscle in PAH patients, healthy subjects, and patients with CHF. PAH patients showed substantially decreased tissue oxygen saturation, slower reactive hyperemia time (suggestive of endothelium dysfunction), and increased peripheral systemic vasoconstriction compared to the other groups. A reduced capillary-to-fiber ratio in limb muscles has also been demonstrated in experimental PH.42
Endothelial dysfunction
The skeletal muscle microcirculation controls nutrient and oxygen delivery to muscle fibers. Although pulmonary endothelial dysfunction and remodeling of small pulmonary arteries have long been recognized as key elements in the pathophysiology of PAH, the role of the peripheral systemic microcirculation has been less investigated. By measuring postocclusion brachial artery dilation (a nitric oxide–mediated, endothelial-dependent vasoreactive response), recent studies showed that a systemic component of endothelial dysfunction might exist in many PAH subclasses, including idiopathic, scleroderma-associated, and chronic thromboembolic PH.44-46 Peripheral endothelial dysfunction in adults with PAH measured by this method was associated with disease severity, as measured by New York Heart Association (NYHA) classification, systolic pulmonary pressure, 6-minute walk distance (6MWD), and oxygen desaturation on exercise,44 and was correlated with pulmonary vascular response to inhaled iloprost in IPAH patients.45 These abnormalities seem to persist despite the use of drugs such as sildenafil and are inversely related to response to inhaled prostacyclin analogs.47 Peripheral endothelial dysfunction also correlated directly with cardiac index and inversely with tricuspid regurgitation and RV myocardial performance index in a pediatric population of IPAH subjects.48 These findings suggest that peripheral endothelial dysfunction may serve as a clinical tool for the evaluation of pulmonary vascular disease and progression of pulmonary vasoreactivity in PAH.
Finally, macroscopic changes in the systemic arterial circulation (i.e., brachial artery diameter) have been associated with pulmonary vascular dysfunction and abnormalities in RV perfusion (greater RV mass and RV end-diastolic volume, as well as lower RV ejection fraction).49 Whether this signifies a pathophysiological link between the pulmonary and peripheral circulation in PAH remains to be confirmed. These findings suggest that a global vasculopathy possibly occurs in PAH.
Respiratory, limb, or global myopathy?
A parallel reduction in the strength of both limb and respiratory muscles may suggest a generalized systemic myopathic syndrome in patients with PAH, but study findings are discordant. On one hand, Bauer et al.37 reported a strong correlation between impaired maximal isometric forearm muscle strength and reduced MIP and MEP in IPAH patients. Coexistence of reduced resistance and strength of the quadriceps muscle and overall respiratory muscle impairment in PAH patients has also been reported.40
Conversely, in animal models and PAH patients with demonstrated respiratory muscle impairment, the force-generating capacity of the extensor digitorum longus and quadriceps, respectively, was preserved, and no morphologic changes were identified.32,35 The maximal tension and Ca2+ sensitivity of force generation of fibers from the extensor digitorum longus were also normal in animal models of PH.35 These findings suggest that selective diaphragmatic weakness results from a local process rather than a systemic process such as inflammation or hypoxia; if such a systemic etiology is at play, then alterations in the respiratory and limb skeletal muscles would be expected to share a high degree of similarity and, to a certain extent, to develop simultaneously.32
Further evidence against the concept of systemic myopathy in PAH is suggested by the finding that the reduction in muscle fiber CSA in the plantaris muscle of a monocrotaline rat model of PH was more prominent in the type II fibers.42 The preferential CSA loss in glycolytic fibers, rather than a decline in all fiber types, renders systemic factors per se, such as hypoxia and inflammation, unlikely explanations for the phenomenon of muscle dysfunction in PAH. Also, there was capillary loss and consequent reduction in capillary-to-fiber-area ratio that was restricted to muscle regions with a high oxidative capacity (there are two distinct regions of the plantaris muscle: [1] a deep region with more oxidative type I and IIa fibers and a more extensive capillary network and [2] a superficial region containing predominantly glycolytic, type IIx, and type IIb fibers and a spare capillary network), but it was not dependent on the fiber type around which capillaries were expressed. Also, mitochondria oxidative capacity was low independently of regional capillarity (even in muscle regions without capillary loss).42 These observations suggest that local/autocrine factors influenced by the metabolic surrounding of the fiber, rather than the fiber type or systemic hypoxia, are responsible for the skeletal muscle capillary remodeling in PAH.42 It appears to be a delicate regional interplay between the requirement for oxygen delivery (oxidative capacity) and the peak potential for oxygen supply (reflected in capillarity) that is resolved differentially in different muscle regions. It is also possible that structural remodeling of capillaries in skeletal muscle is more detrimental to muscle regions with high oxidative capacity, the mechanism for which is currently unknown.42
Physiological and clinical implications of muscle dysfunction in PAH
An association between muscle strength and quality of life in PAH has been recently presented in preliminary results of an ongoing trial by Garfield et al.50 In 12 patients with IPAH, quality of life, measured by the total St. George’s respiratory questionnaire score, correlated significantly with quadriceps maximal volitional capacity–to–body mass index ratio, fat-free mass index measured by bioelectrical impedance, and ultrasound CSA of the rectus femoris.
The reported abnormalities in mitochondrial volume and activity and in fiber size and type in skeletal muscle in PAH patients would be expected to induce a reduction in aerobic capacity and muscle performance. Also, reductions in capillarity around each fiber may limit the potential for oxygen delivery and metabolite removal in skeletal muscle, especially during exercise. Strong correlation between maximal isometric forearm muscle strength and 6MWD has been found by Bauer et al.37 However, there was no correlation between forearm muscle strength and systolic pulmonary artery pressure.37 Of note, walking distances, rather than hemodynamics, have a strong, independent association with functional status and survival in patients with PAH.1,51 Similar findings were produced by Breda et al.40 and Mainguy et al.,38 who showed an independent association between quadriceps muscle strength,38 overall function (total work) and morphology (type I fiber area),40 oxidative capacity (oxidative enzyme levels and type I fiber capillarity),38 and exercise capacity (maximal oxygen consumption [Vo2max] and anaerobic threshold) in PAH patients but no association with resting pulmonary hemodynamics.38,40 The correlation between muscle performance and exercise tolerance could be explained by the influence of muscle function on the perception of leg effort during exercise, which is the main limiting symptom in a significant proportion of patients with IPAH.38 In any case, this observation suggests that limb muscle dysfunction in PAH may be clinically relevant to PAH.
The implications of the respiratory muscle impairment per se in PAH are under study. A decrease in respiratory muscle strength and function would be expected to lead to impaired ventilatory capacity. However, patients with PAH typically manifest resting and exercise hypocapnia, suggesting that increased, out-of-proportion ventilation, rather than ventilatory impairment, occurs in PAH. Alternatively, it could be hypothesized that ventilatory capacity is maintained only at the expense of higher energy expenditure by the dysfunctional respiratory muscles. In other words, the respiratory muscles in PAH operate on the elevated portion of the relationship between ventilatory requirement and capacity. This shift in the balance between ventilatory capacity and demand is considered one of the main causative mechanisms of dyspnea in chronic cardiopulmonary disease.52
The pioneer studies by Meyer et al.27 and Kabitz et al.29 attempted to draw associations between respiratory muscle function and physiological and clinical variables. In the former study,27 inspiratory and expiratory muscle weakness was independent of pulmonary hemodynamics, 6MWD, and ventilatory inefficiency (estimated from the slope VE/Vco2 [minute ventilation–to–carbon dioxide production ratio]). These findings might have been confounded by the small patient population size.2 Kabitz et al.29 were able to demonstrate that the reduced MIP and sniff transdiaphragmatic pressure were significantly associated with 6MWD. More recent findings40 also reported a significant correlation between inspiratory weakness (reduced MIP) and impaired exercise capacity (6MWD and Vo2max) in PAH patients. These findings are supported by evidence in CHF patients relating reduced MIP to worse functional class, maximal oxygen consumption, exercise capacity, and prognosis.28,53
Evidence of muscle dysfunction fired the “muscle hypothesis” for PAH,2 which suggests the occurrence of the muscle metaboreflex (MM), a process in which reflex sympathetic nervous system activation further degrades muscle function (Fig. 2). Activated by accumulation of metabolites in the fatiguing respiratory and/or limb muscles, the MM augments chemoreceptor gain and ergoreceptor drive, which, mediated supraspinally via chemically sensitive afferent fibers, leads to increases in central sympathetic nerve activity outflow.54 Theoretically, this could account for the persistently increased sympathetic drive18 in patients with PAH. Importantly, the increased sympathetic drive may provoke hyperventilation and dyspnea,2 and increase in the muscle sympathetic nerve activity promotes vasoconstriction, perfusion limitation, and redistribution of blood flow.54 From this perspective, the fundamental goal of the MM may be the protection (redirection) of oxygen delivery to the (fatiguing) muscles that initiated the MM or, simply, to the most “vital” muscles. This may become essential during physiological states in which there is competition for cardiac output, such as moderate-to-severe exercise. Alternatively, it may become essential in states of functional weakening of the respiratory and/or limb muscles. Acute fatigue of the inspiratory muscles has been shown to cause reflex reduction in resting leg blood flow in healthy subjects but also in patients with COPD and CHF (respiratory MM).55 Of course, regional muscle vasoconstriction promotes fatigue and impairs exercise capacity. Finally (via feedback), the MM may intensify effort perceptions and cause reflex inhibition of the motor output. Activation of the MM occurs in health during heavy sustained exercise;54 however, factors such as hypoxia and inherent muscle dysfunction lower the threshold for its activation and perpetuation in disease.56 Accordingly, the MM has been implicated in CHF,57 but it is also increasingly considered in a range of conditions from COPD58 to muscular atrophy.59 Whether the MM is also relevant to PAH remains unexplored and theoretical.
Figure 2.

The muscle hypothesis. Muscle work and accumulation of products of muscle metabolism during exercise lead to augmented chemoreceptor gain and ergoreceptor drive in the impaired skeletal musculature. This leads to enhanced transmission to the CNS through sensitive, afferent unmyelinated nerve fibers. The CNS responds with increased sympathetic efferent discharge to the peripheral muscles (MSNA), which causes vasoconstriction and limitation of blood flow and oxygen transportation to the exercising skeletal muscles, thus promoting fatigue and premature cessation of exercise. Finally, enhanced sympathetic drive promotes hyperventilation and breathlessness. MSNA: muscle sympathetic nerve activity; CNS: central nervous system.
The role of exercise rehabilitation
Previously, because of the presumed risk of sudden cardiac death with exercise, experts used to recommend significant limitations in physical activities and, certainly, avoidance of exercise, including pulmonary rehabilitation programs.60 However, the advent of multiple-targeted medical therapies has promoted a trend of improved prognosis and function and has fired an interest in the role of exercise training in PAH, which is currently being investigated by a number of clinical trials worldwide.61
Several small-scale studies have already demonstrated that closely supervised and monitored low-level exercise training programs may have potential benefits in the management of stable PAH patients by improving exercise capacity (6MWD, peak oxygen consumption, peak workload), functional status, and quality of life, with an acceptably low risk of adverse events.62-64 In the only randomized control study to date, Mereles et al.65 found that a 15-week exercise and respiratory training program induced a major improvement in 6MWD (96 m; 22%) in patients with severe precapillary PH receiving stable disease-targeted medication. The mean difference between these patients and matched control subjects who did not receive training was 111 m. This improvement in exercise capacity was greater than any medical intervention had achieved previously in randomized controlled trials of mono- or combination therapy.65
The first findings on the effect of exercise training on limb muscle function and morphology in IPAH were reported by de Man et al.66 In that study, 19 stable IPAH patients in NYHA classes 2 or 3 attended a 12-week outpatient exercise program consisted of cycling and quadriceps muscle training. The end of the program observed a significant increase (89%) in the overall endurance capacity but no improvement in maximal exercise capacity. The same phenomenon was seen in quadriceps function: a large improvement in quadriceps endurance (34% increase) and a smaller (13%) but significant increase in quadriceps strength were found.66 Importantly, the exercise-induced increase in endurance capacity was associated with morphologic changes in quadriceps muscle compatible with improved aerobic capacity. Histological analyses in 12 patients revealed increased quadriceps capillarization and oxidative enzyme activity (especially of the type I muscle fibers). No hypertrophy (increase in CSA) or fiber-type switch was shown in this study, although this could have been confounded by the relative short exercise period at a low intensity.66 However, exercise-induced fiber-type switch was demonstrated in a later study of 5 IPAH patients who underwent a 12-week program of endurance and both arm and quadriceps strength training.63 At the end of the program, not only did the type I fiber surface and the capillaries/fiber ratio tend to increase but the proportion of type IIx fibers also significantly decreased. The latter correlated strongly with the relative decrease in carbon dioxide production for any given workload observed in these patients, suggesting that this less fatigable muscle profile may have resulted in a higher anaerobic threshold.63
Kabitz et al.67 were the first to study the effect of combined respiratory and limb muscle training. Seven stable patients with PAH in WHO functional classes II–IV underwent 3 weeks of in-hospital complex whole-body exercise and multimodal respiratory training, which was continued at home for another 12 weeks. Exercise capacity (6MWD) and respiratory muscle function (assessed by twitch mouth pressure during nonvolitional supramaximal magnetic phrenic nerve stimulation) increased significantly in the period between baseline and the final assessment at the end of the program. Unfortunately, the limb muscle function was not assessed in this study. Whether the sole application of respiratory muscle training in PH patients without whole-body training components (e.g., cycling, walking) also would be beneficial remains to be shown. Also unknown is whether the combination of the two training modalities might lead to further benefits compared to singular modalities.
In a prospective study by Grünig et al.,68 a cohort of 183 patients with PH (PAH, chronic thromboembolic PH, and PH due to respiratory or left heart diseases) obtained significant benefits from 3 weeks of hospital-based exercise training followed by 15 weeks of home-based training. After 3 and 15 weeks, patients significantly improved their 6MWD, scores of quality of life, WHO functional class, peak oxygen consumption, oxygen pulse, heart rate, and systolic pulmonary artery pressure at rest and maximal workload compared to baseline. The improvement in 6MWD was similar in patients with different PH forms and functional classes. Even in severely affected patients (WHO functional class IV), exercise training was highly effective.68
Data from patients with systolic heart failure provide insight into the mechanisms of the exercise-induced improvement of muscle function that could be relevant to PAH.64 Although patients with PAH are much younger and the disease duration is much shorter,38 the two patient groups seem to share some similar cardiopulmonary, hemodynamic, and muscle derangements. Patients with CHF, even those with the most severe disease, who often suffer from pulmonary vascular hypertension, clearly benefit from cardiac rehabilitation. Well-documented physiological benefits of exercise training in patients with CHF include reduced inflammatory cytokines,69 improvement of endothelial function,70 reduction in sympathetic neural activation,71 increased skeletal muscle oxidative capacity,72 and amelioration of limb muscle wasting.73 Whether similar adaptations occur in PAH remains to be proven. In addition, exercise training may induce morphologic muscle changes. Although skeletal muscle fiber-type proportion is largely genetically determined,74 Mainguy et al.63 reported exercise-induced fiber-type shifting from type IIx to IIa and a trend to increased type I fiber surface in subjects with IPAH, as previously described in healthy subjects.75 This less fatigable muscle profile may result in a higher anaerobic threshold. Exercise may also attenuate chemoreceptor gain and ergoreceptor drive implicated in the “muscle hypothesis” by improving muscle aerobic efficiency and reducing the rate of accumulation of muscle metabolites during physical activity. Other adaptations, such as neural, neuroendocrine, and metabolic changes, may also occur, but these possibilities remain completely unexplored. Finally, findings by Mainguy et al.63 suggest that improvement in cardiac function is unlikely to be responsible for the exercise-induced clinical benefits in PAH patients, as no trend in isotime oxygen uptake, heart rate, or oxygen pulse was observed after training. However, Grünig et al.68 showed that exercise training may improve central hemodynamics (systolic pulmonary artery pressure at rest) and cardiac function (peak oxygen consumption, oxygen pulse, and heart rate) in PAH patients.
In summary, emerging evidence suggests that in a disease with severe functional impairment, exercise training may serve as a safe, feasible, and beneficial adjunct therapy. Current international guidelines by the American Thoracic Society and the European Respiratory Society support exercise training within the context of pulmonary rehabilitation for PAH.60 Recommended forms of exercise include light or moderate aerobic and light resistive training.60 The potential benefits of exercise training in patients with PAH are presented in Figure 3.
Figure 3.
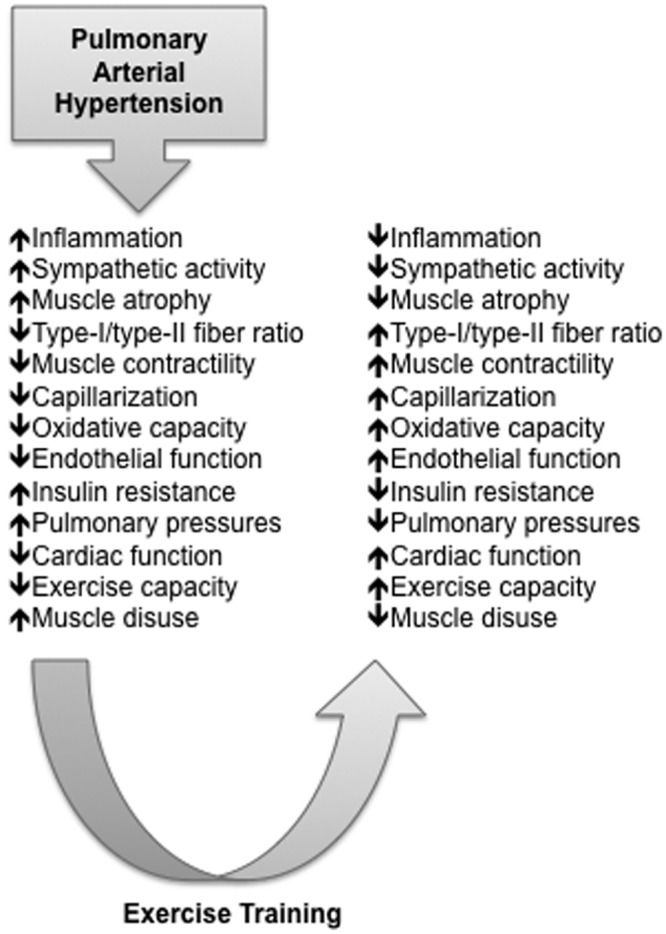
Potential impact of exercise training in patients with pulmonary arterial hypertension.
Future directions
The phenomenon of muscle dysfunction in PAH calls for a wider appreciation and deeper understanding. The pathogenesis, molecular basis, and extent of muscle dysfunction should be further explored. Larger, appropriately designed studies are needed to establish whether both respiratory and limb muscles are mutually affected. Also, whether the demonstrated muscle defects represent consequences of systemic abnormalities stemming from the primary pathobiology of PAH or constitute manifestations of a primary myopathic process remains to be explored. Likewise, the physiological and clinical correlations of the muscle dysfunction (innocent bystander or active mediator of the disease process?) and the relevance of the MM should be further explored. For the latter, additional experiments including microneurographic measurements, ventilatory responses to hypoxia and hypercapnia, and measurements of ventilatory variables at exercise will be necessary.2 Finally, studies are needed to explore whether PAH-specific treatment influences aspects of skeletal muscle function, morphology, enzyme activities, and vascularization. Ultimately, all aspects of muscle alterations in PAH should be considered and interpreted within the frame of disease heterogeneity, duration, and severity.
The origin and mechanisms of peripheral endothelial dysfunction in PAH and its impact on skeletal muscle function are yet to be investigated. The role of inflammation, oxidative stress, impaired balance between vasoactive mediators and vasoconstrictors, and the possible effect of PAH-specific therapy on peripheral endothelial dysfunction should be explored. It also remains unknown to what extent, if any, peripheral endothelial dysfunction is related to the abnormalities in the pulmonary microcirculation.
Further trials are required to better establish the potential value of targeted muscle training in improving exercise capacity in PAH. Such trials should include larger populations and should be randomized by design. Until then, the observations on the beneficial effect of exercise on muscle dysfunction in PAH should continue to be considered exploratory. Likewise, correlations between changes in muscle characteristics and exercise capacity in studies to date do not necessarily imply causal relationships. Furthermore, until the evidence becomes conclusive on whether a systemic myopathy occurs in PAH, future trials should include assessment of the effects of combined respiratory and limb muscle training on muscle characteristics, exercise capacity, and other clinical outcomes in PAH.
Clinical trials are required to determine the optimal timing for initiation, modality (strength vs. aerobic exercise), duration, intensity, and framework (hospital vs. community vs. home) of exercise training, as well as factors that can predict which patients can benefit most from it. Finally, it remains to be conclusively investigated whether training affects central hemodynamic parameters. Ultimately, an area of further fruitful exploration could be the possible effect of training on RV remodeling and function, both of which are of prognostic significance in PAH.76,77 Ultimately, future trials in exercise training in PAH will ideally examine the effects of targeted multimodal exercise on all three major systems implicated in the pathophysiology of dyspnea, fatigue, and exercise limitation: respiratory, cardiovascular, and muscle. Besides, as with other chronic cardiopulmonary diseases, exercise limitation in PAH is most likely multifactorial and, as such, is not limited by any single component of the oxygen transport/utilization process but rather by their collective quantitative interaction(s).78
Conclusion
Emerging evidence suggests that adverse alterations of respiratory and peripheral skeletal muscle morphology and function occur in PAH. These alterations may be clinically relevant, as they are associated with functional limitations. Ultimately, muscle dysfunction offers a potentially fruitful field for therapeutic interventions, as it seems partially reversible by exercise training. However, more trials are needed to consolidate these findings into specific recommendations for exercise rehabilitation in patients with PAH.
Source of Support: Nil.
Conflict of Interest: None declared.
References
- 1.Galiè N, Hoeper MM, Humbert M, Torbicki A, Vachiéry JL, Barberà JA, Beghetti M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 2009;30(20):2493–2537. [DOI] [PubMed]
- 2.Naeije R. Breathing more with weaker respiratory muscles in pulmonary arterial hypertension. Eur Respir J 2005;25(1):6–8. [DOI] [PubMed]
- 3.Garcia-Aymerich J, Serra I, Gómez FP, Farrero E, Balcells E, Rodriguez DA, de Batlle J, et al. Physical activity and clinical and functional status in COPD. Chest 2009;136(1):62–70. [DOI] [PubMed]
- 4.Agustí A. Systemic effects of chronic obstructive pulmonary disease: what we know and what we don’t know (but should). Proc Am Thorac Soc 2007;4(7):522–525. [DOI] [PubMed]
- 5.Yndestad A, Damås JK, Øie E, Ueland T, Gullestad L, Aukrust P. Systemic inflammation in heart failure—the whys and wherefores. Heart Fail Rev 2006;11(1):83–92. [DOI] [PubMed]
- 6.Huertas A, Palange P. COPD: a multifactorial systemic disease. Ther Adv Respir Dis 2011;5(3):217–224. [DOI] [PubMed]
- 7.Barnes PJ, Celli BR. Systemic manifestations and comorbidities of COPD. Eur Respir J 2009;33(5):1165–1185. [DOI] [PubMed]
- 8.Vijayan VK. Morbidities associated with obstructive sleep apnea. Expert Rev Respir Med 2012;6(5):557–566. [DOI] [PubMed]
- 9.Sprunger DB, Olson AL, Huie TJ, Fernandez-Perez ER, Fischer A, Solomon JJ, Brown KK, Swigris JJ. Pulmonary fibrosis is associated with an elevated risk of thromboembolic disease. Eur Respir J 2012;39(1):125–132. [DOI] [PMC free article] [PubMed]
- 10.Fowler RM, Gain KR, Gabbay E. Exercise intolerance in pulmonary arterial hypertension. Pulm Med 2012;2012:359204. doi:10.1155/2012/359204. [DOI] [PMC free article] [PubMed]
- 11.Di Carlo A, De Mori R, Martelli F, Pompilio G, Capogrossi MC, Germani A. Hypoxia inhibits myogenic differentiation through accelerated MyoD degradation. J Biol Chem 2004;279(16):16332–16338. [DOI] [PubMed]
- 12.Groth A, Vrugt B, Brock M, Speich R, Ulrich S, Huber LC. Inflammatory cytokines in pulmonary hypertension. Respir Res 2014;15:47. doi:10.1186/1465-9921-15-47. [DOI] [PMC free article] [PubMed]
- 13.Janssen SP, Gayan-Ramirez G, Van den Bergh A, Herijgers P, Maes K, Verbeken E, Decramer M. Interleukin-6 causes myocardial failure and skeletal muscle atrophy in rats. Circulation 2005;111(8):996–1005. [DOI] [PubMed]
- 14.Reid MB, Lännergren J, Westerblad H. Respiratory and limb muscle weakness induced by tumor necrosis factor-α: involvement of muscle myofilaments. Am J Respir Crit Care Med 2002;166(4):479–484. [DOI] [PubMed]
- 15.Li YP, Chen Y, John J, Moylan J, Jin B, Mann DL, Reid MB. TNF-α acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J 2005;19(3):362–370. [DOI] [PMC free article] [PubMed]
- 16.Adams V, Mangner N, Gasch A, Krohne C, Gielen S, Hirner S, Thierse HJ, et al. Induction of MuRF1 is essential for TNF-α-induced loss of muscle function in mice. J Mol Biol 2008;384(1):48–59. [DOI] [PubMed]
- 17.Remels A, Gosker H, Verhees K, Langen R, Schols A. TNF-α-induced NF-κB activation stimulates skeletal muscle glycolytic metabolism through activation of HIF-1α. Endocrinology 2015;156(5):1770–1781. [DOI] [PubMed]
- 18.Velez-Roa S, Ciarka A, Najem B, Vachiéry JL, Naeije R, van de Borne P. Increased sympathetic nerve activity in pulmonary artery hypertension. Circulation 2004;110(10):1308–1312. [DOI] [PubMed]
- 19.Zamanian RT, Hansmann G, Snook S, Lilienfeld D, Rappaport KM, Reaven GM, Rabinovitch M, Doyle RL. Insulin resistance in pulmonary arterial hypertension. Eur Respir J 2009;33(2):318–324. [DOI] [PMC free article] [PubMed]
- 20.Naderi N, Boobejame P, Bakhshandeh H, Amin A, Taghavi S, Maleki M. Insulin resistance in pulmonary arterial hypertension, is it a novel disease modifier? Res Cardiovasc Med 2014;3(3):e19710. doi:10.5812/cardiovascmed.19710. [DOI] [PMC free article] [PubMed]
- 21.Wei Y, Chen K, Whaley-Connell AT, Stump CS, Ibdah JA, Sowers JR. Skeletal muscle insulin resistance: role of inflammatory cytokines and reactive oxygen species. Am J Physiol Regul Integr Comp Physiol 2008;294(3):R673–R680. [DOI] [PubMed]
- 22.Pugh ME, Buchowski MS, Robbins IM, Newman JH, Hemnes AR. Physical activity limitation as measured by accelerometry in pulmonary arterial hypertension. Chest 2012;142(6):1391–1398. [DOI] [PMC free article] [PubMed]
- 23.Mainguy V, Provencher S, Maltais F, Malenfant S, Saey D. Assessment of daily life physical activities in pulmonary arterial hypertension. PloS ONE 2011;6(11):e27993. doi:10.1371/journal.pone.0027993. [DOI] [PMC free article] [PubMed]
- 24.Jackman RW, Kandarian SC. The molecular basis of skeletal muscle atrophy. Am J Physiol Cell Physiol 2004;287(4):C834–C843. [DOI] [PubMed]
- 25.Bodine SC. Disuse-induced muscle wasting. Int J Biochem Cell Biol 2013;45(10):2200–2208. [DOI] [PMC free article] [PubMed]
- 26.Li H, Malhotra S, Kumar A. Nuclear factor-kappa B signaling in skeletal muscle atrophy. J Mol Med 2008;86(10):1113–1126. [DOI] [PMC free article] [PubMed]
- 27.Meyer FJ, Lossnitzer D, Kristen AV, Schoene AM, Kübler W, Katus HA, Borst MM. Respiratory muscle dysfunction in idiopathic pulmonary arterial hypertension. Eur Respir J 2005;25(1):125–130. [DOI] [PubMed]
- 28.Meyer FJ, Borst MM, Zugck C, Kirschke A, Schellberg D, Kübler W, Haass M. Respiratory muscle dysfunction in congestive heart failure: clinical correlation and prognostic significance. Circulation 2001;103(17):2153–2158. [DOI] [PubMed]
- 29.Kabitz HJ, Schwoerer A, Bremer HC, Sonntag F, Walterspacher S, Walker D, Schaefer V, et al. Impairment of respiratory muscle function in pulmonary hypertension. Clin Sci 2008;114(2):165–171. [DOI] [PubMed]
- 30.American Thoracic Society/European Respiratory Society. ATS/ERS statement on respiratory muscle testing. Am J Respir Crit Care Med 2002;166(4):518–624. [DOI] [PubMed]
- 31.Ahn B, Empinado HM, Al-Rajhi M, Judge AR, Ferreira LF. Diaphragm atrophy and contractile dysfunction in a murine model of pulmonary hypertension. PloS ONE 2013;8(4):e62702. doi:10.1371/journal.pone.0062702. [DOI] [PMC free article] [PubMed]
- 32.de Man FS, van Hees HW, Handoko ML, Niessen HW, Schalij I, Humbert M, Dorfmüller P, et al. Diaphragm muscle fiber weakness in pulmonary hypertension. Am J Respir Crit Care Med 2011;183(10):1411–1418. [DOI] [PubMed]
- 33.van Hees HW, van der Heijden HF, Hafmans T, Ennen L, Heunks LM, Verheugt FW, Dekhuijzen PN. Impaired isotonic contractility and structural abnormalities in the diaphragm of congestive heart failure rats. Int J Cardiol 2008;128(3):326–335. [DOI] [PubMed]
- 34.Sun XG, Hansen JE, Oudiz RJ, Wasserman K. Exercise pathophysiology in patients with primary pulmonary hypertension. Circulation 2001;104(4):429–435. [DOI] [PubMed]
- 35.Manders E, de Man FS, Handoko ML, Westerhof N, van Hees HW, Stienen GJ, Vonk-Noordegraaf A, Ottenheijm CA. Diaphragm weakness in pulmonary arterial hypertension: role of sarcomeric dysfunction. Am J Physiol Lung Cell Mol Physiol 2012;303(12):L1070–L1078. [DOI] [PubMed]
- 36.Kanj NA, Nasser MG, Medawar WA, Al Tayeh AU, Khoury MY, Nassar CF. Reversal of impaired calcium homeostasis in the rat diaphragm subjected to monocrotaline-induced pulmonary hypertension. Toxicol Lett 1999;105(3):177–182. [DOI] [PubMed]
- 37.Bauer R, Dehnert C, Schoene P, Filusch A, Bartsch P, Borst MM, Katus HA, Meyer FJ. Skeletal muscle dysfunction in patients with idiopathic pulmonary arterial hypertension. Respir Med 2007;101(11):2366–2369. [DOI] [PubMed]
- 38.Mainguy V, Maltais F, Saey D, Gagnon P, Martel S, Simon M, Provencher S. Peripheral muscle dysfunction in idiopathic pulmonary arterial hypertension. Thorax 2010;65(2):113–117. [DOI] [PubMed]
- 39.Batt J, Ahmed SS, Correa J, Bain A, Granton J. Skeletal muscle dysfunction in idiopathic pulmonary arterial hypertension. Am J Respir Cell Mol Biol 2014;50(1):74–86. [DOI] [PubMed]
- 40.Breda AP, Pereira de Albuquerque AL, Jardim C, Morinaga LK, Suesada MM, Fernandes CJ, Dias B, Lourenço RB, Salge JM, Souza R. Skeletal muscle abnormalities in pulmonary arterial hypertension. PloS ONE 2014;9(12):e114101. doi:10.1371/journal.pone.0114101. [DOI] [PMC free article] [PubMed]
- 41.Debigaré R, Marquis K, Côté CH, Tremblay RR, Michaud A, LeBlanc P, Maltais F. Catabolic/anabolic balance and muscle wasting in patients with COPD. Chest 2003;124(1):83–89. [DOI] [PubMed]
- 42.Wüst RC, Myers DS, Stones R, Benoist D, Robinson PA, Boyle JP, Peers C, White E, Rossiter HB. Regional skeletal muscle remodeling and mitochondrial dysfunction in right ventricular heart failure. Am J Physiol Heart Circ Physiol 2012;302(2):H402–H411. [DOI] [PMC free article] [PubMed]
- 43.Dimopoulos S, Tzanis G, Manetos C, Tasoulis A, Mpouchla A, Tseliou E, Vasileiadis I, Diakos N, Terrovitis J, Nanas S. Peripheral muscle microcirculatory alterations in patients with pulmonary arterial hypertension: a pilot study. Respir Care 2013;58(12):2134–2141. [DOI] [PubMed]
- 44.Peled N, Bendayan D, Shitrit D, Fox B, Yehoshua L, Kramer MR. Peripheral endothelial dysfunction in patients with pulmonary arterial hypertension. Respir Med 2008;102(12):1791–1796. [DOI] [PubMed]
- 45.Wolff B, Lodziewski S, Bollmann T, Opitz CF, Ewert R. Impaired peripheral endothelial function in severe idiopathic pulmonary hypertension correlates with the pulmonary vascular response to inhaled iloprost. Am Heart J 2007;153(6):1088.e1–1088.e7. [DOI] [PubMed]
- 46.Peled N, Shitrit D, Fox BD, Shlomi D, Amital A, Bendayan D, Kramer MR. Peripheral arterial stiffness and endothelial dysfunction in idiopathic and scleroderma associated pulmonary arterial hypertension. J Rheumatol 2009;36(5):970–975. [DOI] [PubMed]
- 47.Gabrielli LA, Castro PF, Godoy I, Mellado R, Bourge RC, Alcaino H, Chiong M, et al. Systemic oxidative stress and endothelial dysfunction is associated with an attenuated acute vascular response to inhaled prostanoid in pulmonary artery hypertension patients. J Card Fail 2011;17(12):1012–1017. [DOI] [PubMed]
- 48.Friedman D, Szmuszkovicz J, Rabai M, Detterich JA, Menteer J, Wood JC. Systemic endothelial dysfunction in children with idiopathic pulmonary arterial hypertension correlates with disease severity. J Heart Lung Transplant 2012;31(6):642–647. [DOI] [PMC free article] [PubMed]
- 49.Dibble CT, Shimbo D, Barr RG, Bagiella E, Chahal H, Ventetuolo CE, Herrington DM, Lima JA, Bluemke DA, Kawut SM. Brachial artery diameter and the right ventricle: the Multi-Ethnic Study of Atherosclerosis–Right Ventricle Study. Chest 2012;142(6):1399–1405. [DOI] [PMC free article] [PubMed]
- 50.Garfield BE, Parfitt L, Harries C, Dimopoulos K, Gatzoulis M, Kemp P, Polkey MI, Wort SJ. S144. Quality of life in idiopathic pulmonary arterial hypertension is associated with quadriceps function and size. Thorax 2014;69(suppl. 2):A76–A77.
- 51.Miyamoto S, Nagaya N, Satoh T, Kyotani S, Sakamaki F, Fujita M, Nakanishi N, Miyatake K. Clinical correlates and prognostic significance of six-minute walk test in patients with primary pulmonary hypertension: comparison with cardiopulmonary exercise testing. Am J Respir Crit Care Med 2000;161(2):487–492. [DOI] [PubMed]
- 52.Vogiatzis I, Zakynthinos S. Factors limiting exercise tolerance in chronic lung diseases. Compr Physiol 2012;2(3):1779–1817. [DOI] [PubMed]
- 53.Chua TP, Anker SD, Harrington D, Coats AJ. Inspiratory muscle strength is a determinant of maximum oxygen consumption in chronic heart failure. Br Heart J 1995;74(4):381–385. [DOI] [PMC free article] [PubMed]
- 54.Boushel R. Muscle metaboreflex control of the circulation during exercise. Acta Physiol 2010;199(4):367–383. [DOI] [PubMed]
- 55.Sheel AW, Derchak PA, Morgan BJ, Pegelow DF, Jacques AJ, Dempsey JA. Fatiguing inspiratory muscle work causes reflex reduction in resting leg blood flow in humans. J Physiol 2001;537(1):277–289. [DOI] [PMC free article] [PubMed]
- 56.Amann M, Pegelow DF, Jacques AJ, Dempsey JA. Inspiratory muscle work in acute hypoxia influences locomotor muscle fatigue and exercise performance of healthy humans. Am J Physiol Regul Integr Comp Physiol 2007;293(5):R2036–R2045. [DOI] [PubMed]
- 57.Clark AL, Poole-Wilson PA, Coats AJ. Exercise limitation in chronic heart failure: central role of the periphery. J Am Coll Cardiol 1996;28(5):1092–1102. [DOI] [PubMed]
- 58.Chiappa GR, Vieira PJ, Umpierre D, Correa AP, Berton DC, Ribeiro JP, Neder JA. Inspiratory resistance decreases limb blood flow in COPD patients with heart failure. Eur Respir J 2014;43(5):1507–1510. [DOI] [PubMed]
- 59.Smith SA, Downey RM, Williamson JW, Mizuno M. Autonomic dysfunction in muscular dystrophy: a theoretical framework for muscle reflex involvement. Front Physiol 2014;5:47. doi:10.3389/fphys.2014.00047. [DOI] [PMC free article] [PubMed]
- 60.Spruit MA, Singh SJ, Garvey C, ZuWallack R, Nici L, Rochester C, Hill K, et al. An official American Thoracic Society/European Respiratory Society statement: key concepts and advances in pulmonary rehabilitation. Am J Respir Crit Care Med 2013;188(8):e13–e64. [DOI] [PubMed]
- 61.Babu AS, Padmakumar R, Maiya AG. A review of ongoing trials in exercise based rehabilitation for pulmonary arterial hypertension. Indian J Med Res 2013;137(5):900–906. [PMC free article] [PubMed]
- 62.Grünig E, Ehlken N, Ghofrani A, Staehler G, Meyer FJ, Juenger J, Opitz CF, et al. Effect of exercise and respiratory training on clinical progression and survival in patients with severe chronic pulmonary hypertension. Respiration 2011;81(5):394–401. [DOI] [PubMed]
- 63.Mainguy V, Maltais F, Saey D, Gagnon P, Martel S, Simon M, Provencher S. Effects of a rehabilitation program on skeletal muscle function in idiopathic pulmonary arterial hypertension. J Cardiopulm Rehabil Prev 2010;30(5):319–323. [DOI] [PubMed]
- 64.Zafrir B. Exercise training and rehabilitation in pulmonary arterial hypertension: rationale and current data evaluation. J Cardiopulm Rehabil Prev 2013;33(5):263–273. [DOI] [PubMed]
- 65.Mereles D, Ehlken N, Kreuscher S, Ghofrani S, Hoeper MM, Halank M, Meyer FJ, et al. Exercise and respiratory training improve exercise capacity and quality of life in patients with severe chronic pulmonary hypertension. Circulation 2006;114(14):1482–1489. [DOI] [PubMed]
- 66.de Man FS, Handoko ML, Groepenhoff H, van ’t Hul AJ, Abbink J, Koppers RJ, Grotjohan HP, et al. Effects of exercise training in patients with idiopathic pulmonary arterial hypertension. Eur Respir J 2009;34(3):669–675. [DOI] [PubMed]
- 67.Kabitz HJ, Bremer HC, Schwoerer A, Sonntag F, Walterspacher S, Walker DJ, Ehlken N, Staehler G, Windisch W, Grünig E. The combination of exercise and respiratory training improves respiratory muscle function in pulmonary hypertension. Lung 2014;192(2):321–328. [DOI] [PubMed]
- 68.Grünig E, Lichtblau M, Ehlken N, Ghofrani HA, Reichenberger F, Staehler G, Halank M, et al. Safety and efficacy of exercise training in various forms of pulmonary hypertension. Eur Respir J 2012;40(1):84–92. [DOI] [PubMed]
- 69.Gielen S, Adams V, Möbius-Winkler S, Linke A, Erbs S, Yu J, Kempf W, Schubert A, Schuler G, Hambrecht R. Anti-inflammatory effects of exercise training in the skeletal muscle of patients with chronic heart failure. J Am Coll Cardiol 2003;42(5):861–868. [DOI] [PubMed]
- 70.Linke A, Schoene N, Gielen S, Hofer J, Erbs S, Schuler G, Hambrecht R. Endothelial dysfunction in patients with chronic heart failure: systemic effects of lower-limb exercise training. J Am Coll Cardiol 2001;37(2):392–397. [DOI] [PubMed]
- 71.Roveda F, Middlekauff HR, Rondon MU, Reis SF, Souza M, Nastari L, Barretto AC, Krieger EM, Negrão CE. The effects of exercise training on sympathetic neural activation in advanced heart failure: a randomized controlled trial. J Am Coll Cardiol 2003;42(5):854–860. [DOI] [PubMed]
- 72.Gielen S, Adams V, Linke A, Erbs S, Möbius-Winkler S, Schubert A, Schuler G, Hambrecht R. Exercise training in chronic heart failure: correlation between reduced local inflammation and improved oxidative capacity in the skeletal muscle. Eur J Cardiovasc Prev Rehabil 2005;12(4):393–400. [DOI] [PubMed]
- 73.Gielen S, Laughlin MH, O’Conner C, Duncker DJ. Exercise training in patients with heart disease: review of beneficial effects and clinical recommendations. Prog Cardiovasc Dis 2015;57(4):347–355. [DOI] [PubMed]
- 74.Simoneau JA, Bouchard C. Genetic determinism of fiber type proportion in human skeletal muscle. FASEB J 1995;9(11):1091–1095. [DOI] [PubMed]
- 75.Andersen P, Henriksson J. Training induced changes in the subgroups of human type II skeletal muscle fibres. Acta Physiol Scand 1977;99(1):123–125. [DOI] [PubMed]
- 76.Fukuda Y, Tanaka H, Motoji Y, Ryo K, Sawa T, Imanishi J, Miyoshi T, et al. Utility of combining assessment of right ventricular function and right atrial remodeling as a prognostic factor for patients with pulmonary hypertension. Int J Cardiovasc Imaging 2014;30(7):1269–1277. [DOI] [PubMed]
- 77.Vonk Noordegraaf A, Galiè N. The role of the right ventricle in pulmonary arterial hypertension. Eur Respir Rev 2011;20(122):243–253. [DOI] [PMC free article] [PubMed]
- 78.Ross RM. ATS/ACCP statement on cardiopulmonary exercise testing. Am J Respir Crit Care Med 2003;167(10):1451; author reply 1451. [DOI] [PubMed]