

Abstract Abstract
Ranolazine, a late inward sodium current and fatty acid oxidation inhibitor, may improve right ventricular (RV) function in pulmonary arterial hypertension (PAH); however, the safety and efficacy of ranolazine in humans with PAH is unknown. Therefore, we sought to (1) determine whether ranolazine is safe and well tolerated in PAH and (2) explore ranolazine’s effect on symptoms, exercise capacity, RV structure and function, and hemodynamic characteristics. We therefore conducted a 3-month, prospective, open-label pilot study involving patients with symptomatic PAH (n = 11) and echocardiographic evidence of RV dysfunction. We evaluated the safety and tolerability of ranolazine and compared symptoms, exercise capacity, exercise bicycle echocardiographic parameters, and invasive hemodynamic parameters between baseline and 3 months of ranolazine therapy using paired t tests. Of the 11 patients enrolled, one discontinued ranolazine therapy due to a drug-drug interaction after 3 days of therapy. All 10 of the remaining patients continued therapy for 3 months, and 8 (80%) of 10 completed all study tests. After 3 months, ranolazine administration was safe and associated with improvement in functional class (P = 0.0013), reduction in RV size (P = 0.015), improved RV function (improvement in RV strain during exercise at 3 months; P = 0.037), and a trend toward improved exercise time and exercise watts on bicycle echocardiography (P = 0.06 and 0.01, respectively). Ranolazine was not associated with improvement in invasive hemodynamic parameters. In conclusion, in a pilot study involving PAH, ranolazine therapy was safe and well tolerated, and it resulted in improvement in symptoms and echocardiographic parameters of RV structure and function but did not alter invasive hemodynamic parameters.
ClinicalTrials.gov Identifier: NCT01174173.
Keywords: pulmonary arterial hypertension, right ventricle, speckle-tracking echocardiography, hemodynamics
Pulmonary arterial hypertension (PAH) is a progressive disease of the pulmonary vasculature leading to increased pulmonary vascular resistance, elevated pulmonary artery (PA) pressures, and ultimately right ventricular (RV) dysfunction and failure.1,2 Despite significant progress in the treatment of World Health Organization (WHO) group 1 PAH with pulmonary vasodilator therapies, 5-year mortality rates remain high.3,4 Furthermore, abnormalities in echocardiographic RV parameters have been associated with adverse outcomes in PAH, which may be related to the multifactorial pathophysiology of progressive RV dysfunction in PAH, which includes (1) RV ischemia due to altered right coronary artery blood flow in the setting of near equalization of aortic and RV systolic pressure in systole and increased RV diastolic pressure;5 (2) metabolic abnormalities in the RV;6,7 and (3) the inability of the RV, a thin-walled structure accustomed to low PA pressures, to withstand the elevated afterload associated with PAH.8 To date, no PAH-specific therapies have specifically and directly targeted RV ischemia and the altered RV metabolic substrate present in PAH. Ranolazine is an approved therapy for the treatment of chronic stable angina that is safe and well tolerated in patients with coronary artery disease. Recent evidence suggests that ranolazine (1) reduces calcium overload of failing myocytes through inhibition of the inward late sodium current (INa) and (2) inhibition of fatty acid oxidation.9,10 By ameliorating calcium overload, ranolazine may reduce RV diastolic tension and improve myocardial blood flow during diastole in the ischemic RV, thereby improving RV performance and contractility. This latter effect may increase stroke volume and cardiac output, which could translate into benefits in exercise capacity. Furthermore, inhibition of fatty acid oxidation, which is upregulated in RV hypertrophy, may improve RV workload and hemodynamics. In a rat model of PAH induced by monocrotaline, treatment with ranolazine attenuated the development of RV hypertrophy and RV systolic dysfunction, further supporting the potential therapeutic role of ranolazine in PAH.11 We therefore investigated the feasibility and safety of the use of ranolazine in a prospective pilot study in patients with WHO group 1 PAH for a 3-month period with comprehensive assessment of symptoms, functional parameters, echocardiographic findings, and invasive right heart catheterization results. We hypothesized that ranolazine would be safe and well tolerated and would improve symptoms, exercise capacity, and parameters of RV structure and function.
Methods
Study population
During the period 2010–2012, we screened all adult patients with WHO group I PAH who were being cared for at the Northwestern University Pulmonary Hypertension Program for entry into our single-center, open-label, single-arm pilot study of ranolazine. Patients were screened for eligibility during a routine pulmonary hypertension clinic visit using the following inclusion and exclusion criteria.
Inclusion criteria included (1) fulfillment of the contemporary diagnostic criteria for WHO group 1 PAH, including mean PA pressure ≥25 mmHg, pulmonary capillary wedge pressure (PCWP) ≤15 mmHg, and no other causes of pulmonary hypertension or PAH (i.e., no significant left heart disease, lung disease, chronic thromboembolic disease, or miscellaneous causes of PAH, such as sarcoidosis); (2) WHO functional class II or III; (3) evidence of RV dysfunction as defined by RV fractional area change (RVFAC) <35% or tricuspid annular plane systolic excursion (TAPSE) <1.6 cm in accordance with the American Society of Echocardiography guidelines12 or evidence of interventricular septal flattening, indicative of RV pressure and/or volume overload; and (4) stable doses of pulmonary vasodilator therapy (prostacyclins, endothelin receptor antagonists, and/or phosphodiesterase inhibitors) with no new therapy initiation or dose escalation >50% in the 4 weeks before randomization.
Exclusion criteria included (1) history of epicardial coronary artery disease (defined as any coronary stenosis >50%; history of percutaneous coronary intervention or coronary artery bypass grafting; or abnormal noninvasive stress test results suggestive of myocardial ischemia in a coronary distribution); (2) WHO functional class IV symptoms; (3) history of complex ventricular arrhythmias requiring anti-arrhythmic medications; (4) corrected QT interval >500 milliseconds or history of QT prolongation (including congenital long QT syndrome) or receiving other QT-prolonging drugs; (5) history of hepatic dysfunction, including participants with WHO group I PAH due to portopulmonary hypertension; (6) current use of strong CYP3A inhibitors, which included bosentan; (7) severe or end-stage renal disease (estimated glomerular filtration rate <30 mL/min/1.73 m2); and (9) women who were pregnant or lactating. In addition, participants were excluded if they were physically unable to undergo the study procedures, including the 6-minute walk test, exercise echocardiographic examination, and right heart catheterization. All study participants gave written, informed consent, and the institutional review board at Northwestern University approved the study.
Study protocol
Study participants initiated ranolazine therapy at a dosage of 1,000 mg twice daily at the baseline study visit and continued to receive the study drug for 3 months. Dosage reduction to 500 mg twice daily was allowed if the patient was experiencing adverse effects. Adherence to study medication was documented by pill counts done by the study coordinator at follow-up visits. Follow-up visits occurred at months 1, 2, and 3. Serious adverse events during follow-up were defined as a fatal or serious deterioration in health resulting in death, risk of immediate death, admission to the hospital and hospitalization >24 hours, disability or incapacitation, or intervention to prevent a life-threatening condition.
Study procedures included a clinic visit with physical examination, questionnaires, electrocardiographic examination, 6-minute walk test, exercise echocardiographic examination, and invasive hemodynamic assessment at baseline and at the 3-month visit. As stated above, clinic visits, with assessment of WHO functional class and determination of adverse events, were also performed at 1 and 2 months after initiation of ranolazine.
Assessment of symptoms and functional status
Study participants completed a quality of life and functional status questionnaire (Kansas City Cardiomyopathy Questionnaire [KCCQ])13 at baseline and at 3 months. All participants also underwent a standardized 6-minute walk test at baseline and at 3 months.
Exercise stress echocardiography
All study participants underwent comprehensive 2-dimensional (2D) echocardiographic examination with Doppler and tissue Doppler imaging (TDI) at rest and at peak bicycle exercise using a commercially available exercise ergometer (MPI; Medical Positioning, Kansas City, MO) at both the baseline and 3-month study visits. No medications were adjusted or held before exercise testing. A commercially available ultrasound system with harmonic imaging (Vivid 7; GE Healthcare, Waukesha, WI) was used for all echocardiographic examinations. Cardiac structure and function were quantified at rest and at peak stress as recommended by the American Society of Echocardiography.12,14,15 Specifically, RV size, RVFAC, TAPSE, tricuspid regurgitation gradient, and RV strain (based on speckle-tracking analysis) were analyzed at rest and at peak stress during the baseline visit and the 3-month visit. Exercise capacity was recorded as both total exercise time and peak watts achieved. All echocardiographic measurements were made by an experienced research sonographer (blinded to clinical data and study outcomes). ProSolv 4.0 echocardiographic analysis software (FujiFilm, Indianapolis, IN) and EchoPAC (GE Healthcare, Waukesha, WI) for speckle-tracking analysis were used for echocardiographic quantitation, and all measurements were verified by a physician experienced in echocardiography. The 2D images of the RV were obtained at frame rates of 50–70 frames per second to optimize speckle-tracking analyses, which were performed after isolating the highest-quality cardiac cycle; the endocardial and epicardial borders were traced at end systole in each view. Computerized speckle-tracking analysis was performed and border tracings manually adjusted to optimize tracking. Strain values were converted to absolute values for ease of display. Lower absolute strain values correlated with worse RV function.16
Cardiac catheterization
All study participants underwent right heart catheterization at baseline as a requirement for entry into the study, and follow-up right heart catheterization was performed at 3 months. Right heart and PA catheterization were performed from the right internal jugular vein approach using standard Seldinger technique under fluoroscopic guidance. Study participants underwent recording of invasive hemodynamic assessments (mean right atrial pressure; systolic, diastolic, and mean PA pressure [mPAP]; and PCWP) using a fluid-filled, 6F PA catheter (Edwards Lifesciences, Irvine, CA) and a properly zeroed pressure transducer. Pressure recordings were analyzed off line using a WITT Hemodynamic Workstation (Philips Medical Systems, Andover, MA) at a 50 mm/s paper speed with adjustment of pressure (mmHg) scale as needed. All hemodynamic pressure measurements were made at end expiration and in duplicate using a standardized measurement protocol by a physician blinded to all clinical data. Cardiac output was calculated using the thermodilution method, and pulmonary vascular resistance (PVR) was calculated as the transpulmonary gradient (mPAP – PCWP) divided by cardiac output.17
Statistical analysis
Baseline demographic, clinical, functional status, echocardiographic, and invasive hemodynamic assessment data were summarized for the entire study sample. Continuous data with a normal distribution were displayed as mean ± standard deviation, and categorical data were displayed as counts and percentages. Next, differences in KCCQ summary score, WHO functional class, 6-minute walk distance, echocardiographic parameters, stress echocardiography variables, and invasive hemodynamic assessments were compared between baseline and 3 months of ranolazine therapy using paired t tests. A two-sided P value <0.05 was considered statistically significant. Because the pilot study did not have any a priori statistical hypotheses for the primary end point, no sample size or formal statistical power analyses were conducted for this exploratory study. All analyses were performed using Stata version 10.1 (StataCorp, College Station, TX).
Results
Baseline clinical and echocardiographic characteristics of the study patients
We prospectively enrolled 11 patients with WHO group 1 PAH with baseline clinical characteristics displayed in Table 1. Mean age was 48 ± 13 years, and 73% of patients were women. Nearly half of the study participants were nonwhite. The etiology of PAH in the majority of patients (n = 9; 73%) was connective tissue disease, and the majority of those (7 [78%] of 9) had systemic sclerosis (SSc). Four patients had evidence of interstitial lung disease, and 1 patient had chronic obstructive pulmonary disease (lung disease severity was graded clinically as mild in all of these patients). None of the study participants had epicardial coronary artery disease (as required by the enrollment criteria); 7 of 11 patients underwent coronary angiography, which showed no significant coronary stenosis, and 4 of 11 patients underwent noninvasive stress testing, which showed no evidence of myocardial ischemia.
Table 1.
Baseline demographic and clinical characteristics
| Characteristic | All patients (n = 11) |
|---|---|
| Age, mean ± SD, years | 48 ± 13 |
| Female sex | 8 (73) |
| Race | |
| White | 5 (45.5) |
| Black | 5 (45.5) |
| Other | 1 (9) |
| Etiology of PAH | |
| Idiopathic | 1 (9) |
| HIV associated | 2 (18) |
| Connective tissue disease associated | 8 (73) |
| Comorbidity | |
| Systemic hypertension | 4 (36) |
| Dyslipidemia | 2 (18) |
| Diabetes mellitus | 1 (9) |
| Coronary artery disease | 0 (0) |
| Chronic kidney disease | 1 (9) |
| Obesity | 5 (46) |
| Current tobacco use | 2 (18) |
| Past tobacco use | 3 (27) |
| Interstitial lung disease | 4 (36) |
| Chronic obstructive pulmonary disease | 1 (9) |
| Obstructive sleep apnea | 1 (9) |
| Venous thromboembolism | 3 (27) |
| WHO functional class | |
| Class II | 7 (64) |
| Class III | 4 (37) |
| Kansas City Cardiomyopathy Questionnaire summary score, mean ± SD | 60.3 ± 19.7 |
| Medication | |
| Phosphodiesterase inhibitor (sildenafil) | 10 (91) |
| Endothelin receptor antagonist (ambrisentan) | 4 (37) |
| Prostacyclin (treprostinil) | 2 (18) |
| Warfarin | 3 (27) |
| Statin | 2 (18) |
| Digitalis | 2 (18) |
| Diuretic | 7 (64) |
| Calcium-channel blocker | 3 (27) |
| Physical examination findings, mean ± SD | |
| Body mass index | 29 ± 7 |
| Heart rate, bpm | 96 ± 19 |
| Systolic blood pressure, mmHg | 117 ± 17 |
| Diastolic blood pressure, mmHg | 76 ± 14 |
Data are no. (%) of patients, unless otherwise indicated. Body mass index was defined as weight in kilograms divided by the square of height in meters. HIV: human immunodeficiency virus; PAH: pulmonary arterial hypertension; SD: standard deviation; WHO: World Health Organization.
All patients were receiving stable pulmonary vasodilator therapy without dose adjustment over follow-up; as shown in Table 1, almost all patients (91%) were receiving a phosphodiesterase-5 inhibitor. All study participants had either WHO functional class II or III symptoms at baseline. Heart rate and systemic blood pressure were within the normal range for the majority of patients.
Table 2 summarizes the echocardiographic characteristics of the study cohort at baseline. The majority of participants had RV dilation, as demonstrated by increased RV basal diameter (4.9 ± 0.8 cm), RV length (8.6 ± 1.1 cm), and RV end-diastolic area (38 ± 12 cm2). Furthermore, RV/LV maximum diameter ratio (measured in the apical 4-chamber view) was markedly increased at 1.4 ± 0.3 (normal value, < 0.67). RVFAC was reduced at 29% ± 11%, as was TAPSE (1.6 ± 0.5 cm), consistent with the enrollment criteria requiring evidence of RV dysfunction. Absolute RV global longitudinal strain was also reduced at 15.8% ± 5.2%.
Table 2.
Baseline echocardiographic examination findings and invasive hemodynamic parameters
| Characteristic | All patients (n = 11) |
|---|---|
| Echocardiographic parameter | |
| LV end-diastolic diameter, cm | 4.2 ± 0.5 |
| LV end-systolic diameter, cm | 2.6 ± 0.5 |
| Septal wall thickness, cm | 0.9 ± 0.1 |
| Posterior wall thickness, cm | 0.9 ± 0.1 |
| LV mass index, g/m2 | 63 ± 12 |
| LV end-diastolic volume index, mL/m2 | 35.8 ± 6.0 |
| LV end-systolic volume index, mL/m2 | 13.1 ± 2.5 |
| LV ejection fraction, % | 63 ± 5 |
| Pericardial effusion, no. (%) of patients | |
| Trace | 2 (18) |
| Mild | 6 (55) |
| Moderate | 3 (27) |
| Left atrial volume index, mL/m2 | 22.1 ± 3.5 |
| E, cm/s | 78 ± 29 |
| A, cm/s | 84 ± 17 |
| E/A ratio | 1.0 ± 0.4 |
| E/e′ ratio, lateral annulus | 6.8 ± 1.9 |
| RV basal diameter,a cm | 4.9 ± 0.8 |
| RV maximal diameter,a cm | 5.8 ± 1.0 |
| RV length,a cm | 8.6 ± 1.1 |
| RV wall thickness, cm | 0.6 ± 0.1 |
| RV end-diastolic area, cm2 | 38 ± 12 |
| RV end-systolic area, cm2 | 28 ± 11 |
| Right atrial area, cm2 | 25 ± 8 |
| Ratio of RV maximal diameter to LV maximal diametera | 1.4 ± 0.3 |
| RV fractional area change, % | 29 ± 11 |
| Tricuspid annular plane systolic excursion, cm | 1.6 ± 0.5 |
| RV tissue Doppler s′ velocity, cm/s | 12.4 ± 3.0 |
| Absolute RV global longitudinal strain, % | 15.8 ± 5.2 |
| Pulmonary artery systolic pressure, mmHg | 69 ± 33 |
| Right atrial pressure, mmHg | 8 ± 4 |
| Cardiac index, L/min/m2 | 3.0 ± 0.8 |
| Invasive hemodynamic parameter | |
| Right atrial pressure, mmHg | 9.5 ± 8.5 |
| Pulmonary artery systolic pressure, mmHg | 73 ± 21 |
| Pulmonary artery diastolic pressure, mmHg | 31 ± 10 |
| Mean pulmonary arterial pressure, mmHg | 48 ± 14 |
| Pulmonary capillary wedge pressure, mmHg | 13 ± 2.5 |
| Cardiac output, L/min | 5.4 ± 1.8 |
| Cardiac index, L/min/m2 | 2.9 ± 0.9 |
| Pulmonary arterial saturation, % | 65 ± 9 |
| Pulmonary vascular resistance, Wood units | 7.4 ± 4.0 |
Data are expressed as mean value (±SD) unless otherwise indicated. Baseline echocardiographic and invasive hemodynamic parameter data were measured for all 11 study participants, because all participants enrolled in the study completed baseline cardiac testing. A: late mitral inflow velocity; E: early mitral inflow velocity; e′: early diastolic mitral annular velocity; LV: left ventricle; RV: right ventricle; s′: peak systolic velocity.
Measured in the apical 4-chamber view
Effects of ranolazine on symptoms and exercise capacity
All study participants who continued to receive the study drug for the 3-month period (n = 10) completed symptom assessment and 6-minute walk testing. WHO functional class improved over the duration of the study. Figure 1 demonstrates improvement in functional class over the course of the monthly follow-up visits. Of the study participants, 36% reported advanced functional class III symptoms at baseline with improvement in all patients to functional class I or II symptoms by 3 months of follow-up (P = 0.0013). KCCQ summary score did not change significantly after 3 months of ranolazine (baseline vs. 3-month ranolazine, 60.3 ± 19.7 vs. 64.2 ± 17.5; P = 0.37). Figure 2 displays changes in parameters of exercise tolerance. The 6-minute walk test distance demonstrated a trend toward improvement from 383 ± 60 m to 419 ± 80 m, although this change did not reach statistical significance (P = 0.09).
Figure 1.
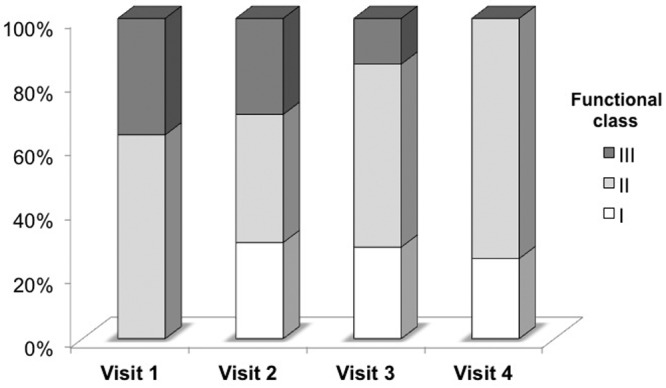
Effect of ranolazine on functional class in pulmonary arterial hypertension. Monthly changes in World Health Organization functional class from baseline to after 3 months of ranolazine therapy.
Figure 2.

Effect of ranolazine on functional status in pulmonary arterial hypertension. Changes in functional parameters including mean World Health Organization functional class, 6-minute walk distance, exercise capacity, and exercise time from baseline to after 3 months of ranolazine therapy.
Of the 10 patients who received ranolazine for the 3-month period, 2 refused to undergo the end-of-study (3-month) bicycle stress echocardiographic examination or invasive hemodynamic testing. In the remaining 8 patients, exercise tolerance, as measured by exercise time (s) and exercise capacity (W), improved (Fig. 2); however, these changes also did not reach statistical significance (P = 0.06 and 0.11, respectively).
Effects of ranolazine on cardiac structure and function
Ranolazine had no effect on left-sided cardiac parameters, including LV volumes, LV dimensions, LV systolic or diastolic function, and LA size. We found that resting values of RV size (RV end-diastolic area, RV end-systolic area, and RV/LV maximal diameter ratio) all improved significantly after 3 months of ranolazine (Fig. 3). Resting values of RV systolic function (TAPSE and RVFAC) also improved after 3 months of ranolazine. On speckle-tracking echocardiographic analysis of longitudinal RV systolic function, we found the difference between resting and peak exercise global longitudinal RV peak systolic strain was quite different when baseline was compared with 3 months of ranolazine administration (Fig. 4). At baseline (n = 8), absolute RV longitudinal strain decreased by 1.4 percentage-units during exercise, indicative of worsening of RV function with exercise. However, after 3 months of ranolazine therapy, absolute RV longitudinal strain increased by 1.0 percentage-unit (P = 0.037), indicating a significant improvement in the ability of the RV to compensate for exercise-induced increases in RV afterload.
Figure 3.

Effect of ranolazine on right ventricular structure and function in pulmonary arterial hypertension. LV: left ventricle; RV: right ventricle; TAPSE: tricuspid annular plane systolic excursion.
Figure 4.
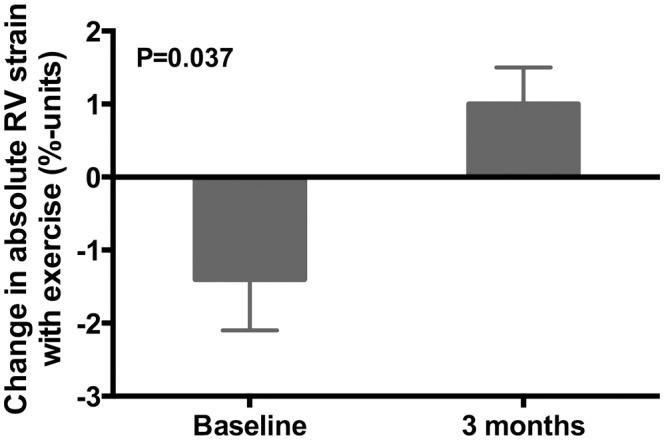
Effect of ranolazine on exercise-induced changes in right ventricular (RV) mechanics. Bar graphs represent change in absolute RV free-wall strain values from rest to peak exercise, with comparison of baseline data with data obtained after 3 months of ranolazine therapy.
Table 3 summarizes the invasive hemodynamic data obtained at baseline and after 3 months of ranolazine therapy in the 8 patients who underwent repeat cardiac catheterization at the end of the study period. We found no statistically significant differences in invasive hemodynamic data between baseline and 3 months of therapy with ranolazine. Consistent with the improvement in RV systolic function demonstrated on echocardiographic examination, invasively determined cardiac index increased from 2.54 L/min/m2 at baseline to 2.87 L/min/m2 after 3 months of ranolazine therapy, but this change did not reach statistical significance (P = 0.23). Similarly, there was a reduction in pulmonary vascular resistance from 8.6 to 7.0 Wood units after 3 months of ranolazine therapy, but this change also did not attain statistical significance (P = 0.27). There were no significant changes in systemic blood pressure, heart rate, weight, or PA saturation over the 3-month period (P > 0.05 for all comparisons by paired t test).
Table 3.
Invasive hemodynamic parameters at baseline versus after 3 months of ranolazine therapy
| Invasive hemodynamic parameter | Baseline (n = 8) |
After 3 months (n = 8) |
P |
|---|---|---|---|
| Right atrial pressure, mmHg | 9 ± 4 | 13 ± 3 | 0.41 |
| Mean pulmonary artery pressure, mmHg | 51 ± 6 | 48 ± 6 | 0.66 |
| Pulmonary capillary wedge pressure, mmHg | 13 ± 1 | 12 ± 1 | 0.32 |
| Cardiac index, L/min/m2 | 2.54 ± 0.26 | 2.87 ± 0.25 | 0.23 |
| Pulmonary vascular resistance, Wood units | 8.6 ± 1.4 | 7.0 ± 1.1 | 0.27 |
Data are mean value ± standard error. Baseline hemodynamic data differ from data given in Table 2, because the data in this table are only for those participants who underwent both baseline and 3-month invasive hemodynamic testing.
Safety and tolerability of ranolazine
Given the possibility that ranolazine (an inhibitor of CYP3A isoenzymes) may theoretically increase plasma concentrations of CYP3A4 substrates, no patients receiving bosentan were enrolled in the study, because bosentan is a CYP3A4 substrate. In addition, none of the study patients were withdrawn from bosentan therapy for the purposes of enrollment into the study. Of the 11 enrolled patients, 1 patient reported symptoms of dizziness, leg weakness, and hand tremors; review of her medications revealed that she had been taking darifenacin (a CYP3A4 substrate), a competitive muscarinic receptor antagonist (for the treatment of urinary incontinence), but had forgotten to report this medication during the baseline visit. Because of the possibility of an interaction between ranolazine and darifenacin, ranolazine was discontinued (with complete resolution of neurologic symptoms), and the patient was withdrawn from the study.
Of the remaining 10 patients, 8 (80%) tolerated the 1,000-mg twice-daily dose of ranolazine throughout the duration of the 3-month study period; 2 patients (20%) required a dose reduction to 500 mg twice daily due to adverse effects (severe constipation in 1 patient and myalgia in another patient), with complete symptom resolution after dose reduction. Adverse effects included constipation (30%), nausea (20%), headache (20%), and myalgia (10%; Table 4). No significant arrhythmias were encountered during the study period, and QTc monitoring revealed no significant prolongation during drug administration. No major adverse events occurred during the 3-month study period.
Table 4.
Frequency of adverse effects associated with ranolazine therapy
| Adverse effect | No. (%) of patients |
|---|---|
| Constipation | 2 (20) |
| Nausea | 2 (20) |
| Headache | 2 (20) |
| Myalgia | 1 (10) |
Discussion
In a small, prospective, observational pilot study of patients with symptomatic WHO group I PAH, the addition of ranolazine was safe and well tolerated. Apart from a medication interaction effect (with subsequent discontinuation of ranolazine) in 1 study patient, there were no medication interactions or major adverse effects. Furthermore, ranolazine therapy was associated with an improvement in symptoms, with overall improvement in WHO functional class as well as positive RV remodeling, and with improvements in RV function at rest and during exercise.
In patients with significant PAH, the presence of RV ischemia is common as compensated PAH progresses to RV failure.18 Normally, the RV differs from the left ventricle (LV) in its pattern of coronary blood flow during the cardiac cycle. The left coronary artery blood flow occurs primarily in diastole due to the lack of a pressure gradient between the aorta and the LV in systole, whereas right coronary blood flow occurs throughout the cardiac cycle, because aortic pressure is much higher than RV pressure during systole and diastole, thereby driving coronary blood flow continuously. In patients with PAH, however, as RV systolic pressure increases and begins to match aortic systolic pressure, there is no longer a gradient between the aorta and RV. Therefore, the RV, which is normally perfused throughout the cardiac cycle, only receives coronary blood flow in diastole.5 Thus, in patients with advanced PAH, the RV becomes progressively ischemic as PA pressures (and RV pressures) increase, which results in further deterioration of an already vulnerable RV. Elevation of RV end-diastolic pressure also contributes to RV ischemia in these patients by impeding coronary blood flow in diastole, which is worsened with increased heart rate and reduced time in diastole during exercise.5,18 Although others have demonstrated that worse RV longitudinal strain at rest is associated with poor outcomes in PAH, we demonstrate, for the first time, improvement in RV longitudinal strain at peak exercise after 3 months of ranolazine therapy.19-21
Impaired RV function predicts clinical worsening in PAH more accurately than does the magnitude of elevated pulmonary vascular resistance.22,23 RV dysfunction is more prevalent in patients with SSc,24 which was the etiology of PAH in the majority of patients enrolled in our study (73%). It is important to note that the high proportion of patients with connective tissue disease in our cohort is a reflection of the PAH referral patterns at our center (in which the predominant etiology of WHO group I PAH is SSc) and differs from other PAH clinical trials, which have typically enrolled patients with idiopathic PAH. Furthermore, in SSc-associated PAH, mortality is higher and RV contractility may be lower secondary to RV microvascular dysfunction, rather than being attributable to a reduction in perfusion pressure due to PAH.25,26
Pulmonary vasodilators, such as prostacyclins, endothelin receptor antagonists, and phosphodiesterase inhibitors, improve exercise capacity in PAH and may indirectly improve RV function via afterload reduction,27 but they have not been convincingly shown to directly improve RV function. On the basis of small studies in PAH, digoxin may be beneficial as an RV inotrope in PAH,28 but it may increase myocardial oxygen demand. Currently, there are no treatments approved for PAH that directly improve RV function without increasing myocardial oxygen demand.
Ranolazine is an approved medication for the treatment of chronic stable angina in patients with coronary artery disease. Ranolazine reduces calcium overload of ischemic and failing myocytes through inhibition of the late sodium current (INa).9 These findings are based on data from a canine model that demonstrated ranolazine-induced improvement in abnormal repolarization and contraction in LV myocytes.29 Furthermore, in isolated myocardium from failing human hearts, ranolazine improved LV diastolic parameters.30 As stated above, we found no change in left heart parameters with ranolazine therapy in our study; however, this may be due to the fact that the late INa current becomes more accentuated in diseased, ischemic cardiomyocytes. In PAH, because the RV is the site of injury, the RV is likely where the late INa current is accentuated and where beneficial effects of ranolazine would be expected. Therefore, ranolazine could potentially ameliorate calcium overloaded RV myoctyes, which cause RV diastolic dysfunction. By reducing RV diastolic tension, ranolazine may improve myocardial blood flow during diastole in the ischemic RV, thereby relieving angina while simultaneously improving RV performance and contractility. This latter effect may increase stroke volume and cardiac output that could translate into benefits in exercise capacity.
In our pilot study, we found that administration of 3 months of ranolazine therapy, accompanied by stable background pulmonary vasodilator therapy, was associated with improvements in RV structure and systolic function both at rest and during exercise. Although there was a trend toward increased right atrial pressure by invasive hemodynamic testing at the conclusion of the study, these differences did not meet statistical significance; nevertheless, given the small sample size, evaluation of change in right atrial pressure will be important for future studies of ranolazine in PAH. In addition, ranolazine demonstrated a trend toward increase in invasively measured cardiac output, although this change did not meet statistical significance. These changes in RV structure and function after 3 months of ranolazine therapy were not associated with an improvement in KCCQ summary score, but they were associated with statistically significant improvements in WHO functional class and a trend toward increased 6-minute walk distance and exercise capacity, with an increase from 383 to 419 m (Δ = 36 m), which is similar to the magnitude of improvement in contemporary PAH clinical trials of phosphodiesterase inhibitors and endothelin receptor antagonists.31-34 The lack of consistent improvement across all measured functional parameters may be a result of our sample size and limited power to detect differences, or the inability of the KCCQ, which was developed for left heart failure, to demonstrate symptomatic differences in PAH. If verified in larger, randomized clinical trials, ranolazine and other late INa inhibitors and fatty acid oxidation inhibitors may prove to be beneficial as agents for the direct improvement of RV function for PAH and possibly other (secondary) forms of pulmonary hypertension as well.
In addition to its effects on the late INa current, ranolazine has additionally been shown to inhibit fatty acid oxidation and activate pyruvate dehydrogenase (PDH) in perfused, normoxic rat models.35,36 RV ischemia triggers metabolic remodeling in the RV, which drives aerobic glycolysis with resultant impairment of energetics and reduction of RV function and results in a vicious cycle promoting ongoing RV ischemia. Aerobic glycolysis is upregulated, similar to cancer physiology, in PAH, specifically in the lung vasculature and RV, resulting from pyruvate dehydrogenase kinase-mediated inhibition of PDH results in reduced contractility in the RV myocytes.37 A small study of primary pulmonary hypertension in humans demonstrated marked upregulation of 18Fluorodeoxyglucose accumulation in the RV free wall using gated positron emission tomography to support energy homeostasis limited by aerobic glycolysis, which was associated with the severity of RV pressure overload.7 In an animal model of RVH induced by PA banding in adult Sprague-Dawley rats, fatty acid oxidation was increased in RV myocytes.38 Administration of ranolazine for as little as 1 week enhanced glucose oxidation and resulted in reduced RV hypertrophy and improved RV function without causing QTc prolongation.38 The high rates of fatty acid oxidation inhibited PDH, which has been demonstrated as another mechanism for the glycolytic switch in RV metabolism in PAH. This is further supported by the complete resistance of development of PAH in response to acute hypoxia in an animal model with absolute deficiency of the metabolic enzyme, malonyl-coenzyme A decarboxylase, resulting in fatty acid oxidation inhibition.39
Our study should be interpreted in the context of several potential limitations. Most importantly, our study was an unblinded, single-arm pilot study with concomitant administration of standard-of-care PAH-specific therapy. Thus, we cannot exclude a placebo effect or a lagging benefit of vasodilator therapy, and we cannot definitively determine whether the improvements in functional class and RV structure and function were truly due to ranolazine. However, all patients were receiving stable background pulmonary vasodilator therapy with no changes in medication regimen during the 3-month study. Additional limitations include a small sample size, which likely decreased our power to detect statistically significant changes in exercise capacity and hemodynamic characteristics, and the inclusion of patients from a single academic center. In addition, patients with interstitial lung disease were included in our study; however, they were unlikely to have symptoms related to lung disease given only mild restrictive defects on pulmonary function testing with forced vital capacity greater than 70% and computed tomography with evidence of modest parenchymal disease before enrollment. Although ranolazine was generally well tolerated and without major adverse effects, it is important to note that the study drug was discontinued in 1 patient because of a medication interaction, and adverse effects—including constipation, nausea, headache, and myalgia (Table 4)—did occur, leading to a ranolazine dose reduction in 2 study patients. Finally, it is important to note that, in our cohort, despite the presence of RV dysfunction on echocardiographic examination as an inclusion criteria, mean baseline 6-minute walk distance was 383 m, mean right atrial pressure was 9 mmHg, and mean cardiac index was 2.54 L/min/m2, highlighting an overall low-risk group.33 Furthermore, the mean Registry to Evaluate Early and Long-term PAH Disease Management (REVEAL) registry-based risk score in the cohort was very low (2.5) with a predicted 95%–100% survival at 1 year.34 The low-risk nature of the study patients may have occurred in part because of the requirement that patients be able to perform exercise during both the 6-minute walk test and bicycle stress echocardiogram. It is possible that sicker patients with PAH, who have more advanced RV dysfunction, would demonstrate even greater benefit from ranolazine than demonstrated in our study; however, additional studies are needed to fully explore the role of ranolazine in high-risk patients with PAH. Our examination of ranolazine in PAH was a pilot study to assess feasibility, safety, and tolerability of ranolazine in patients with WHO group 1 PAH. Larger randomized studies of ranolazine in PAH must be completed before ranolazine can be recommended for use in PAH.10
In a small prospective, observational pilot study in WHO group I PAH, we have shown that treatment with ranolazine for 3 months demonstrated improvement in functional class and RV structure and function. Furthermore, ranolazine was safe overall and well tolerated. These findings are promising and support prospective randomized clinical trials of ranolazine (or related drugs) in PAH as an adjunctive therapy focused on ameliorating symptoms and directly improving RV function.
Acknowledgments
We thank Jo Anne and Stephen A. Schiller for their generous research funding in support of this work.
Source of Support: This study was funded by an investigator-initiated grant from Gilead Sciences to SJS.
Conflict of Interest: SJS reports receiving a research grant from Gilead Sciences. All other authors: none declared.
References
- 1.Shah SJ. Pulmonary hypertension. JAMA 2012;308(13):1366–1374. [DOI] [PubMed]
- 2.Voelkel NF, Quaife RA, Leinwand LA, et al. Right ventricular function and failure: report of a National Heart, Lung, and Blood Institute working group on cellular and molecular mechanisms of right heart failure. Circulation 2006;114(17):1883–1891. [DOI] [PubMed]
- 3.Kane GC, Maradit-Kremers H, Slusser JP, Scott CG, Frantz RP, McGoon MD. Integration of clinical and hemodynamic parameters in the prediction of long-term survival in patients with pulmonary arterial hypertension. Chest 2011;139(6):1285–1293. [DOI] [PubMed]
- 4.Kurzyna M, Zylkowska J, Fijalkowska A, et al. Characteristics and prognosis of patients with decompensated right ventricular failure during the course of pulmonary hypertension. Kardiol Pol 2008;66(10):1033–1039; discussion 1040–1031. [PubMed]
- 5.van Wolferen SA, Marcus JT, Westerhof N, et al. Right coronary artery flow impairment in patients with pulmonary hypertension. Eur Heart J 2008;29(1):120–127. [DOI] [PubMed]
- 6.Archer SL, Fang YH, Ryan JJ, Piao L. Metabolism and bioenergetics in the right ventricle and pulmonary vasculature in pulmonary hypertension. Pulm Circ 2013;3(1):144–152. [DOI] [PMC free article] [PubMed]
- 7.Oikawa M, Kagaya Y, Otani H, et al. Increased [18F]fluorodeoxyglucose accumulation in right ventricular free wall in patients with pulmonary hypertension and the effect of epoprostenol. J Am Coll Cardiol 2005;45(11):1849–1855. [DOI] [PubMed]
- 8.Haddad F, Doyle R, Murphy DJ, Hunt SA. Right ventricular function in cardiovascular disease, part II: pathophysiology, clinical importance, and management of right ventricular failure. Circulation 2008;117(13):1717–1731. [DOI] [PubMed]
- 9.Stone PH. Ranolazine: new paradigm for management of myocardial ischemia, myocardial dysfunction, and arrhythmias. Cardiol Clin 2008;26(4):603–614. [DOI] [PubMed]
- 10.Gomberg-Maitland M, Bull TM, Saggar R, et al. New trial designs and potential therapies for pulmonary artery hypertension. J Am Coll Cardiol 2013;62(25 suppl):D82–D91. [DOI] [PMC free article] [PubMed]
- 11.Liles JT, Oliver J, Forrer S, Chisholm JW, Belardinelli L, Dhalla AK. Beneficial effects of ranolazine in a model of pulmonary hypertension and right-sided heart failure. Am J Respir Crit Care Med 2011;183:A4977.
- 12.Rudski LG, Lai WW, Afilalo J, et al. Guidelines for the echocardiographic assessment of the right heart in adults. J Am Soc Echocardiogr 2010;23(7):685–713; quiz 786–688. [DOI] [PubMed]
- 13.Green CP, Porter CB, Bresnahan DR, Spertus JA. Development and evaluation of the Kansas City Cardiomyopathy Questionnaire: a new health status measure for heart failure. J Am Coll Cardiol 2000;35(5):1245–1255. [DOI] [PubMed]
- 14.Lang RM, Bierig M, Devereux RB, et al. Recommendations for chamber quantification. J Am Soc Echocardiogr 2005;18(12):1440–1463. [DOI] [PubMed]
- 15.Nagueh SF, Appleton CP, Gillebert TC, et al. Recommendations for the evaluation of left ventricular diastolic function by echocardiography. J Am Soc Echocardiogr 2009;22(2):107–133. [DOI] [PubMed]
- 16.Meris A, Faletra F, Conca C, et al. Timing and magnitude of regional right ventricular function: a speckle tracking-derived strain study of normal subjects and patients with right ventricular dysfunction. J Am Soc Echocardiogr 2010;23(8):823–831. [DOI] [PubMed]
- 17.Hoeper MM, Maier R, Tongers J, et al. Determination of cardiac output by the Fick method, thermodilution, and acetylene rebreathing in pulmonary hypertension. Am J Respir Crit Care Med 1999;160(2):535–541. [DOI] [PubMed]
- 18.Barst RJ. Evaluation and treatment for angina in pulmonary arterial hypertension. Am J Med 2004;116(6):427–428. [DOI] [PubMed]
- 19.Borges AC, Knebel F, Eddicks S, et al. Right ventricular function assessed by two-dimensional strain and tissue Doppler echocardiography in patients with pulmonary arterial hypertension and effect of vasodilator therapy. Am J Cardiol 2006;98(4):530–534. [DOI] [PubMed]
- 20.Sachdev A, Villarraga HR, Frantz RP, et al. Right ventricular strain for prediction of survival in patients with pulmonary arterial hypertension. Chest 2011;139(6):1299–1309. [DOI] [PubMed]
- 21.Fine NM, Chen L, Bastiansen PM, et al. Outcome prediction by quantitative right ventricular function assessment in 575 subjects evaluated for pulmonary hypertension. Circ Cardiovasc Imaging 2013;6(5):711–721. [DOI] [PubMed]
- 22.van de Veerdonk MC, Kind T, Marcus JT, et al. Progressive right ventricular dysfunction in patients with pulmonary arterial hypertension responding to therapy. J Am Coll Cardiol 2011;58(24):2511–2519. [DOI] [PubMed]
- 23.Forfia PR, Fisher MR, Mathai SC, et al. Tricuspid annular displacement predicts survival in pulmonary hypertension. Am J Respir Crit Care Med 2006;174(9):1034–1041. [DOI] [PubMed]
- 24.Kawut SM, Taichman DB, Archer-Chicko CL, Palevsky HI, Kimmel SE. Hemodynamics and survival in patients with pulmonary arterial hypertension related to systemic sclerosis. Chest 2003;123(2):344–350. [DOI] [PubMed]
- 25.Bogaard HJ, Natarajan R, Henderson SC, et al. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation 2009;120(20):1951–1960. [DOI] [PubMed]
- 26.Overbeek MJ, Lankhaar JW, Westerhof N, et al. Right ventricular contractility in systemic sclerosis-associated and idiopathic pulmonary arterial hypertension. Eur Resp J 2008;31(6):1160–1166. [DOI] [PubMed]
- 27.Galiè N, Hoeper MM, Humbert M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 2009;30(20):2493–2537. [DOI] [PubMed]
- 28.Rich S, Seidlitz M, Dodin E, et al. The short-term effects of digoxin in patients with right ventricular dysfunction from pulmonary hypertension. Chest 1998;114(3):787–792. [DOI] [PubMed]
- 29.Undrovinas AI, Belardinelli L, Undrovinas NA, Sabbah HN. Ranolazine improves abnormal repolarization and contraction in left ventricular myocytes of dogs with heart failure by inhibiting late sodium current. J Cardiovasc Electrophysiol 2006;17(suppl 1):S169–S177. [DOI] [PMC free article] [PubMed]
- 30.Sossalla S, Wagner S, Rasenack EC, et al. Ranolazine improves diastolic dysfunction in isolated myocardium from failing human hearts–role of late sodium current and intracellular ion accumulation. J Mol Cell Cardiol 2008;45(1):32–43. [DOI] [PubMed]
- 31.Galiè N, Olschewski H, Oudiz RJ, et al. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 2008;117(23):3010–3019. [DOI] [PubMed]
- 32.Galiè N, Ghofrani HA, Torbicki A, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med 2005;353(20):2148–2157. [DOI] [PubMed]
- 33.McLaughlin VV, Gaine SP, Howard LS, et al. Treatment goals of pulmonary hypertension. J Am Coll Cardiol 2013;62(25 suppl):D73–D81. [DOI] [PubMed]
- 34.Benza RL, Gomberg-Maitland M, Miller DP, et al. The REVEAL Registry risk score calculator in patients newly diagnosed with pulmonary arterial hypertension. Chest 2012;141(2):354–362. [DOI] [PubMed]
- 35.Clarke B, Wyatt KM, McCormack JG. Ranolazine increases active pyruvate dehydrogenase in perfused normoxic rat hearts: evidence for an indirect mechanism. J Mol Cell Cardiol 1996;28(2):341–350. [DOI] [PubMed]
- 36.McCormack JG, Barr RL, Wolff AA, Lopaschuk GD. Ranolazine stimulates glucose oxidation in normoxic, ischemic, and reperfused ischemic rat hearts. Circulation 1996;93(1):135–142. [DOI] [PubMed]
- 37.Ryan JJ, Archer SL. The right ventricle in pulmonary arterial hypertension: disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ Res 2014;115(1):176–188. [DOI] [PMC free article] [PubMed]
- 38.Fang YH, Piao L, Hong Z, et al. Therapeutic inhibition of fatty acid oxidation in right ventricular hypertrophy: exploiting Randle’s cycle. J Mol Med (Berl) 2012;90(1):31–43. [DOI] [PMC free article] [PubMed]
- 39.Sutendra G, Bonnet S, Rochefort G, et al. Fatty acid oxidation and malonyl-CoA decarboxylase in the vascular remodeling of pulmonary hypertension. Sci Transl Med 2010;2(44):44ra58. [DOI] [PubMed]