

Abstract Abstract
Pulmonary capillary hemangiomatosis (PCH) is a rare form of pulmonary arterial hypertension (PAH) characterized by pulmonary capillary proliferation and pseudoinvasion of collagenous septal structures. PCH is often accompanied by veno-occlusive changes and pulmonary hypertensive arterial remodeling. The clinical and pathological diagnosis of PCH can be subtle and easily missed. Most reported cases of PCH have been associated with resting PAH. We report the cases of 3 patients who initially presented with exertional dyspnea with normal to mildly elevated resting pulmonary arterial pressures and marked intrapulmonary shunting. In all 3 patients, invasive cardiopulmonary exercise testing was suggestive of pulmonary vascular disease. Owing to abnormalities on invasive exercise testing, lung biopsies were performed; these were diagnostic of PCH, and the patients were referred for lung transplantation. We describe unique features of these 3 cases—including novel pathological findings and the presence of intrapulmonary shunting in all 3 patients—and we discuss the role of cardiopulmonary exercise testing in the evaluation of PCH.
Keywords: pulmonary capillary hemangiomatosis, pulmonary arterial hypertension, cardiopulmonary exercise test, intrapulmonary shunt, pulmonary arteriovenous malformation
Pulmonary capillary hemangiomatosis (PCH) is a form of pulmonary arterial hypertension (PAH) characterized by pulmonary capillary duplication and proliferation and a rapidly progressive, often fatal clinical course. Alveolar capillaries can invade the pulmonary interstitium, pulmonary veins and arteries, alveolar walls and septae, and other intrathoracic structures in a neoplastic manner, often with accompanying pulmonary veno-occlusive changes. This leads to hypoxemia and dyspnea.1-4 Despite pathological abnormalities in the capillaries and veins, both PCH and pulmonary veno-occlusive disease (PVOD) are categorized as World Health Organization group 1′ PAH and are typically associated with elevated pulmonary arterial pressures.1-5
In many published case reports of PCH, the diagnosis was not established prior to lung transplantation or death,2,5-7 suggesting that the disease is likely underdiagnosed. Potential reasons for this include nonspecific clinical and radiographic features,8 reluctance to biopsy the lung in patients with moderate or severe PAH, and the subtle pathological features that establish the diagnosis.
Invasive cardiopulmonary exercise testing with a pulmonary arterial catheter (level 3 CPET) has been shown to be helpful in the evaluation of unexplained dyspnea and in the diagnosis of pulmonary vascular disease.9-12 Reduced maximal oxygen consumption, elevated mean pulmonary arterial pressure–cardiac output slope, elevated minute ventilation/carbon dioxide (V̇E/V̇co2) slope, and low end-tidal partial pressure of carbon dioxide (PETco2) are abnormal findings on level 3 CPET suggestive of pulmonary vascular disease. Additionally, exercise capacity can be an important prognostic indicator.9-12 The CPET characteristics of PCH, however, have been described previously in only one patient, whose disease was diagnosed postmortem.13
Here we present 3 cases of patients who presented with exertional dyspnea and desaturation, with severely reduced diffusion capacity of the lungs for carbon monoxide, corrected for hemoglobin (DLco[Hb]) accompanied by normal spirometry. The patients underwent level 3 CPET to further characterize the etiology of their dyspnea, and testing was suggestive of pulmonary vascular disease. Given these exercise test results, lung biopsies were performed despite minimal imaging abnormalities, and these were diagnostic of PCH.
Case descriptions
Cases were identified from the Massachusetts General Hospital pulmonary hypertension clinic. Lung biopsy specimens were fixed in 5% buffered formalin, embedded in paraffin, and sectioned at 5 uM. Explanted lungs were inflated with 5% buffered formalin to a pressure of 25 cm prior to sectioning and standard processing. Lung tissues were stained with hematoxylin and eosin, and selected sections were stained with Weigert elastic stain. An indirect immunoperoxidase technique was applied to selected sections, with primary monoclonal antibodies directed against CD3, CD31, and tryptase antigens at predetermined optimal dilutions and stained with horseradish peroxidase.
Case 1
A 37-year-old man who never smoked, with no significant medical history, prior exposures, or family history of pulmonary disease, presented with exertional dyspnea of several years’ duration. Physical examination was notable for a resting oxygen saturation of 91% with exertional desaturation to 84% while he was breathing room air. Cardiac and pulmonary examination findings were normal.
Pulmonary function test results were normal, with the exception of a DLco[Hb] of 12.29 mL/min/mmHg (34% predicted). Transthoracic echocardiogram revealed a normal left ventricular ejection fraction and an estimated right ventricular systolic pressure (RVSP) of 28 mmHg plus central venous pressure (CVP) with normal right heart size and function. A contrast-enhanced echocardiogram was positive for right-to-left intrapulmonary shunting with appearance of saline contrast within the left atrium within 4 cardiac cycles. A chest computed tomography (CT) scan demonstrated enlarged pulmonary arteries and diffuse centrilobular ground-glass opacities. A ventilation-perfusion (V̇/Q̇) scan revealed normal ventilation with heterogeneous distribution of tracer in the perfusion phase without segmental or subsegmental V̇/Q̇ mismatch to suggest pulmonary embolism. There was tracer uptake in the brain consistent with right-to-left shunting. The patient was referred for level 3 CPET to characterize his exertional hypoxemia and to determine the etiology of his dyspnea.
The results of level 3 CPET with upright cycle ergometry demonstrated mild resting PAH (mPAP: 30 mmHg) with a markedly abnormal pulmonary vascular response to exercise (Table 1) and exercise-induced oxygen desaturation. CPET was repeated while the patient was breathing 100% oxygen, and there was no decrease in arterial partial pressure of oxygen during exercise, effectively excluding the presence of an intracardiac shunt. The patient underwent thoracoscopic lung biopsy for further evaluation.
Table 1.
Cardiopulmonary exercise test data
| Patient 1 | Patient 2 | Patient 3 | ||||
|---|---|---|---|---|---|---|
| Parametersa | Rest | Exercise | Rest | Exercise | Rest | Exercise |
| Exercise capacity | ||||||
| RER (>1) | NA | 1.13 | NA | 1.16 | NA | 1.09 |
| V̇o2max (>80% predicted), mL/kg/min (% predicted) | NA | 20.0 (69) | NA | 16.1 (49) | NA | 20.5 (65) |
| AT (>40% predicted peak V̇o2), % predicted | NA | 34 | NA | 24 | NA | 35 |
| Hemodynamics | ||||||
| mPAP (<25), mmHg | 28 | 72 | 15 | 34 | 22 | 49 |
| PVR (<240 at rest, <200 with exercise), dyn | 426 | 215 | 140 | 89 | 252 | 186 |
| ΔmPAP/ΔCO (<2) | NA | 3.3 | NA | 4.5 | NA | 3.2 |
| Ventilatory parameters | ||||||
| Pao2 (87–99), mmHg | 81 | 41 | 62 | 52 | 72 | 38 |
| A-a gradient (5–15), mmHg | 37 | 87 | 56 | 75 | 39 | 86 |
| V̇E/V̇co2 slope (<33) | NA | 54 | NA | 81 | NA | 40 |
| PETco2 (>36 at rest, >39 at AT), mmHg | 28 | 26 | 20 | 19 | 28 | 30 |
RER (>1) indicates that all studies were maximal effort. All patients had a reduced V̇o2max and an early anaerobic threshold. Two out of three patients had normal resting pulmonary arterial pressures. All three patients had an elevated ΔmPAP/ΔCO, elevated V̇E/V̇co2 slope, and significant exertional hypoxemia with an elevated A-a gradient and a low PETco2. RER: respiratory exchange ratio; NA: not applicable; V̇o2max: maximal oxygen consumption; AT: anaerobic threshold; mPAP: mean pulmonary arterial pressure; PVR: pulmonary vascular resistance; CO: cardiac output; Pao2: partial pressure of oxygen; A-a: alveolar-arterial; V̇E: minute ventilation; V̇co2: carbon dioxide production; PETco2: partial pressure of end-tidal carbon dioxide.
Values in parentheses are reference values for our exercise laboratory.
The lung biopsy specimen revealed patchy alveolar capillary duplication and rare obliterated small pulmonary venules consistent with the diagnosis of PCH (Fig. 1). Mild interstitial T cell alveolitis and a marked increase in interstitial mast cells were also present.
Figure 1.

A, Lung shows focal proliferation and duplication of alveolar capillaries with thickened walls and dilated lumens that are characteristic of pulmonary capillary hemangiomatosis. B, CD31 immunostain highlights the proliferating endothelial cells. C, An elastic stain shows normal-caliber pulmonary artery adjacent to a bronchiole. D, CD3 immunostain highlights the T cell alveolitis. E, An immunostain for tryptase highlights the expanded interstitial mast cell population.
Because of mild PAH, the patient was prescribed sildenafil. He also received doxycycline and prednisone, with no improvement in symptoms and a progression in the severity of his hypoxemia. Repeat right heart catheterization 9 months later, while he was taking sildenafil, demonstrated worsening pulmonary hypertension (PH), with a mean resting pulmonary arterial pressure of 39 mmHg and a pulmonary vascular resistance of 393 dyn. On a 6-minute walk test (16 months after CPET), the patient experienced oxygen desaturation to 67% while receiving 8 L/min supplemental oxygen. He subsequently underwent bilateral lung transplantation. The examination of the pathology of the explant confirmed the diagnosis of PCH.
Case 2
A 52-year-old woman who never smoked and who had a history of breast cancer in remission presented with a 1-year history of rapidly progressive dyspnea on exertion. Physical examination was notable for oxygen saturations of 87%–91% while breathing room air and mild digital clubbing. Cardiac and pulmonary examination results were normal.
Pulmonary function test results were normal, with the exception of a DLco[Hb] of 10.61 mL/min/mmHg (47% predicted). Chest CT scan demonstrated arteriovenous malformations (AVMs) in the lingula and right lower lobe with multiple ground-glass nodules and superimposed mosaic attenuation. Transthoracic echocardiogram revealed normal left ventricular function, normal right heart size and function with an estimated RVSP of 29 mmHg plus CVP, and a large amount of right-to-left shunting within 4 cardiac cycles following the injection of agitated saline. A V̇/Q̇ scan showed an unusual pattern of diffuse perfusion heterogeneity (Fig. 2). There was also tracer uptake in the kidneys and thyroid consistent with right-to-left shunting. Additional workup for primary liver disease, which included aminotransferase and bilirubin levels, international normalized ratio, and abdominal CT scan, yielded negative results.
Figure 2.

Perfusion images from ventilation perfusion scan in patient 2 demonstrating diffuse heterogeneous perfusion abnormalities.
The patient underwent level 3 CPET with upright cycle ergometry. Results were consistent with a markedly abnormal pulmonary vascular response to exercise despite normal resting and exercise hemodynamics (Table 1).
Lung biopsy revealed a complex pattern of pathology with patchy respiratory bronchiolitis and mild emphysema. There was grade 3/6 hypertensive pulmonary arteriopathy and evidence of multifocal acute and old hemorrhages with hemosiderin-laden macrophages. Several alveolar septa showed duplicated capillaries consistent with PCH. The pulmonary veins and venules showed patchy obliterative changes, and the visceral pleura was fibrotic. As in the first case, both mild, nonspecific T cell interstitial inflammation (Fig. 3) and an increased number of mast cells (not shown) were present.
Figure 3.
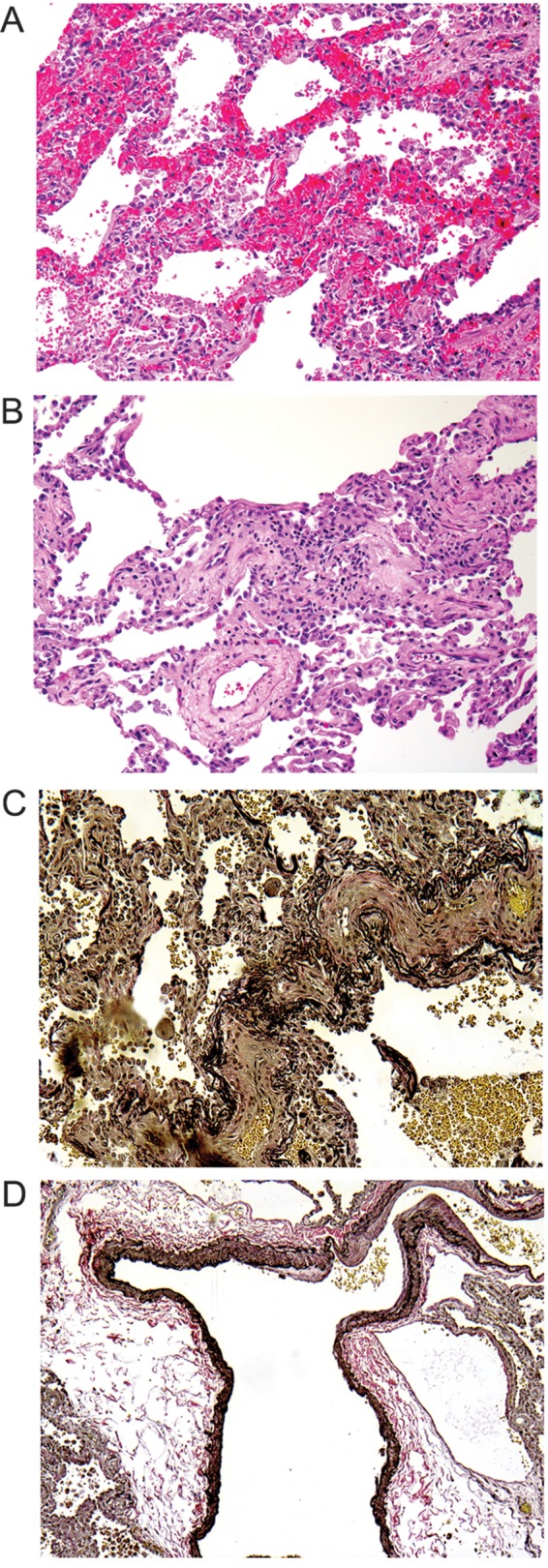
A, Lung shows the features of pulmonary capillary hemangiomatosis. B, Interstitial lymphocytic alveolitis is seen in a hematoxylin and eosin–stained section. C, Veno-occlusive changes of small venules are demonstrated by elastic stain. D, An arteriovenous malformation is highlighted by an elastic stain.
The result of genetic testing for mutations in eukaryotic translation initiation factor 2 α-kinase 4 (EIF2AK4) was negative. The patient underwent coiling of the lingular AVM with no improvement in symptoms or hypoxemia. She was prescribed doxycycline and prednisone, with no improvement in symptoms. Over the following weeks, her hypoxemia worsened, and 8 L/min supplemental oxygen was required to maintain arterial saturations of >90%. The patient successfully underwent urgent lung transplantation. Pathological examination of the explanted lung revealed duplications of alveolar capillaries as well as intimal fibrosis and arterialization of small and medium-sized pulmonary veins and intimal fibrosis of pulmonary arteries, in a background of patchy alveolar hemosiderosis, and a few scattered AVMs. The findings were judged to represent overlapping features of PCH and PVOD.
Case 3
A 46-year-old man with no significant medical history presented to the pulmonary clinic with dyspnea on exertion for several years. Physical examination was notable for an oxygen saturation of 94% while breathing room air, clubbing, and normal cardiovascular and pulmonary examinations.
Pulmonary function test results were normal, with the exception of a DLco[Hb] of 9.72 mL/min/mmHg (31% predicted). The chest CT scan was normal. Transthoracic echocardiogram revealed an estimated RVSP of 27 mmHg plus CVP with normal right heart size and function; the bubble study was positive, with right-to-left shunting after 4 cardiac cycles, suggestive of intrapulmonary shunting.
Level 3 CPET revealed borderline-normal resting hemodynamics and a markedly abnormal pulmonary vascular response to exercise with exertional desaturation (Table 1). A 100% oxygen shunt study was negative for exercise-induced intracardiac shunting.
The patient underwent thoracoscopic lung biopsy. Pathological study revealed capillary proliferation, mild interstitial fibrosis, and hemosiderin-laden macrophages consistent with PCH. His lung also showed T cell alveolitis and increased mast cells (not shown).
The patient was given tadalafil, doxycycline, and prednisone, but he has had progression of symptoms and hypoxemia and has been listed for lung transplantation.
Discussion
These cases highlight the utility of invasive CPET in the workup of occult pulmonary vascular disease and the diagnosis of PCH, particularly in the absence of significantly elevated resting pulmonary arterial pressures. Specific features that were characteristic of our patients’ presentations included severely reduced DLco[Hb], significant exertional desaturation, a steep mPAP-flow response to exercise, an elevated V̇E/V̇co2 slope, reduced PETco2, and intrapulmonary shunting on contrast-enhanced echocardiogram.
Two of the 3 patients presented with normal or borderline pulmonary arterial pressures at rest (mPAP: ≤25 mmHg) and therefore would not meet current diagnostic criteria for PAH based on the 5th World Symposium Updated Clinical Classification of Pulmonary Hypertension.5 PCH in the absence of significant PH has been previously described in only 2 cases, although neither of those patients had an invasive hemodynamic evaluation.6,14 Our cases clearly demonstrate that pathological findings of PCH can be present in the absence of significant resting PH. Exercise testing may be helpful in identification of these patients, and it informs the need for early intervention, including prompt referral for lung transplantation.
Another interesting and unexpected finding in these cases that, to our knowledge, has not previously been described was the presence of late right-to-left shunting on contrast-enhanced transthoracic echocardiography, suggesting intrapulmonary shunting. Patient 2 also had discrete pulmonary AVMs that were coiled without symptomatic or physiological benefit. The concomitant presence of both PCH and pulmonary AVMs was previously described by one of us (RLK) in a patient with hereditary hemorrhagic telangiectasia (HHT).15 However, none of the patients in our case series met the Curaçao clinical diagnostic criteria for HHT.16 Patient 2 was referred to an HHT specialist, but genetic testing for HHT was not performed, given the absence of other features. Additionally, none of our patients had evidence of primary liver disease, which can also be associated with intrapulmonary shunting due to pulmonary microvascular dilation.17
Mechanistically, intrapulmonary shunting may be due to capillary proliferation with vasodilation or the development of microscopic pulmonary AVMs and may be a newly described feature of PCH. Because our case series is small and few prior case reports have commented on the presence or absence of shunting, the prevalence of pulmonary AVMs in patients with PCH/PVOD is not known. Physiologically, it is possible that the presence of intrapulmonary AVMs, which are low-resistance vascular units, mitigated the PH and explains the normal resting hemodynamics despite advanced pulmonary vascular remodeling that was seen in our patients.
Prior case reports have described various radiographic abnormalities in patients with PCH/PVOD, including pulmonary artery enlargement, widespread centrilobular nodules, ground-glass opacities, septal thickening, and perfusion abnormalities on ventilation-perfusion scintigraphy.8,18-20 Our cases had a spectrum of radiographic findings ranging from normal (patient 3) to more typical findings of PCH, such as diffuse centrilobular nodules (patient 1), and to pulmonary AVMs (patient 2) with abnormal perfusion on V̇/Q̇ scan. The V̇/Q̇ scan in Figure 2 is a striking example of the marked perfusion abnormalities that can develop due to abnormalities in the pulmonary capillaries and venules.
These cases also have unique pathological features. In addition to the previously reported elements of PCH and veno-occlusive changes, all of the patients showed mild T cell alveolitis and a marked expansion of interstitial mast cells. Whether this is a primary pathogenetic feature of this disease or a reaction to the primary vascular changes is not known.
All 3 cases showed elements of PVOD consistent with the current theory that PVOD and PCH are often part of the same process rather than distinct entities. This concept is supported by the recent discovery that mutations in EIF2AK4, an amino acid availability sensor and regulator of angiogenesis, were present in familial and sporadic cases of both PCH and PVOD.21,22 Given characteristic pathological abnormalities consistent with PCH and a relatively low (20%) prevalence of mutations in patients with sporadic PCH,21 genetic testing for mutations in EIF2AK4 was performed in only 1 of the 3 patients (patient 2) since it was not felt that testing would change clinical management.
There are currently no proven medical treatments for PCH, and thus patients should be referred promptly for lung transplantation. While case reports have described improvement with doxycycline, interferon alfa, and imatinib in some patients, consistent benefit has not been demonstrated.23-26 Additionally, treatment of PAH in these patients is challenging since initiation of prostacyclin therapy can result in pulmonary edema.27 Two of our 3 patients tolerated phosphodiesterase inhibitors without the development of pulmonary edema and experienced mild symptomatic improvement. Unfortunately, all 3 patients experienced disease progression despite treatment with doxycycline and steroids. Two patients underwent successful lung transplantation, while the other patient is currently listed for transplant.
Conclusions
In summary, the diagnosis of PCH should be considered in patients with severely reduced DLco[Hb] and normal spirometry with unexplained exertional dyspnea and hypoxemia, even in the absence of significant resting PAH. CPET with measurement of exercise hemodynamics may be helpful in the early recognition of PCH. However, an accurate diagnosis of PCH requires biopsy confirmation, and pathologists must recognize that the disease can have subtle histological features. Additionally, intrapulmonary shunting may be a newly described feature of this disease, and an evaluation of intrapulmonary shunting should be included in the diagnostic workup of patients with PCH to determine its prevalence in this poorly understood and devastating disease.
Acknowledgments
We thank Dr. D. Hunter Best from the University of Utah School of Medicine for EIF2AK4 mutation testing.
Source of Support: Nil.
Conflict of Interest: None declared.
References
- 1.Wagenvoort CA, Beetstra A, Spijker J. Capillary haemangiomatosis of the lungs. Histopathology 1978;2(6):401–406. [DOI] [PubMed]
- 2.Tron V, Magee F, Wright JL, Colby T, Churg A. Pulmonary capillary hemangiomatosis. Hum Pathol 1986;17:1144–1150. [DOI] [PubMed]
- 3.Lantuejoul S, Sheppard MN, Corrin B, Burke MM, Nicholson AG. Pulmonary veno-occlusive disease and pulmonary capillary hemangiomatosis: a clinicopathologic study of 35 cases. Am J Surg Pathol 2006;30:850–857. [DOI] [PubMed]
- 4.Eltorky MA, Headley AS, Winer-Muram H, Garrett HE Jr., Griffin JP. Pulmonary capillary hemangiomatosis: a clinicopathologic review. Ann Thorac Surg 1994;57(3):772–776. [DOI] [PubMed]
- 5.Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, Gomez Sanchez MA, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013;62(25):D34–D41. [DOI] [PubMed]
- 6.Umezu H, Naito M, Yagisawa K, Hattori A, Aizawa Y. An autopsy case of pulmonary capillary hemangiomatosis without evidence of pulmonary hypertension. Virchows Arch 2001;439:586–592. [DOI] [PubMed]
- 7.Havlik DM, Massie LW, Williams WL, Crooks LA. Pulmonary capillary hemangiomatosis-like foci: an autopsy study of 8 cases. Am J Clin Pathol 2000;113(5):655–662. [DOI] [PubMed]
- 8.Frazier AA, Franks TJ, Mohammed TH, Ozbudak IH, Galvin JR. From the archives of the AFIP: pulmonary veno-occlusive disease and pulmonary capillary hemangiomatosis. RadioGraphics 2007;27:867–882. [DOI] [PubMed]
- 9.Lewis GD. Pulmonary vascular response patterns to exercise. Adv Pulm Hypertens 2010;9:92–101. [DOI] [PMC free article] [PubMed]
- 10.Lewis, GD, Bossone E, Naeije R, Grünig E, Saggar R, Lancellotti P, Ghio S, et al. Pulmonary vascular hemodynamic response to exercise in cardiopulmonary diseases. Circulation 2013;128:1470–1479. [DOI] [PubMed]
- 11.D’Alonzo GE, Gianotti LA, Pohil RL, Reagle RR, DuRee SL, Fuentes F, Dantzker DR. Comparison of progressive exercise performance of normal subjects and patients with primary pulmonary hypertension. Chest 1987;92:57–62. [DOI] [PubMed]
- 12.Yasunobu Y, Oudiz RJ, Sun XG, Hansen JE, Wasserman K. End-tidal Pco2 abnormality and exercise limitation in patients with primary pulmonary hypertension. Chest 2005;127(5):1637–1646. [DOI] [PubMed]
- 13.Aston K, Riddell GJ, Sheppard MN, Wells AU, Riley MS. Pulmonary capillary haemangiomatosis—an unusual cause of hypoxia. Respir Med Case Rep 2012;7:12–14. [DOI] [PMC free article] [PubMed]
- 14.Takiguchi Y, Uruma T, Hiroshima K, Motoori K, Watanabe R, Hamaoka T, Okada O, Kimura H, Kuriyama T. Stable pulmonary capillary haemangiomatosis without symptomatic pulmonary hypertension. Thorax 2001;56:815–817. [DOI] [PMC free article] [PubMed]
- 15.Varnholt H, Kradin R. Pulmonary capillary hemangiomatosis arising in hereditary hemorrhagic telangiectasia. Hum Pathol 2004;35(2):266–268. [DOI] [PubMed]
- 16.Shovlin CL, Guttmacher AE, Buscarini E, Faughnan ME, Hyland RH, Westermann CL, Kjeldsen AD, Plauchu H. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet 2000;91(1):66–67. [DOI] [PubMed]
- 17.Rodriguez-Roisin R, Krowka MJ. Hepatopulmonary syndrome: a liver-induced lung vascular disorder. N Engl J Med 2008;358:2378–2387. [DOI] [PubMed]
- 18.Dufour B, Maitre S, Humbert M, Capron F, Simonneau G, Musset D. High-resolution CT of the chest in four patients with pulmonary capillary hemangiomatosis or pulmonary venoocclusive disease. Am J Roentgenol 1998;171(5):1321–1324. [DOI] [PubMed]
- 19.Rush C, Langleben D, Schlesinger RD, Stern J, Wang NS, Lamoureux E. Lung scintigraphy in pulmonary capillary hemangiomatosis: a rare disorder causing primary pulmonary hypertension. Clin Nucl Med 1991;16(12):913–917. [DOI] [PubMed]
- 20.Bailey CL, Channick RN, Auger WR, Fedullo PF, Kerr KM, Yung GL, Rubin LJ. “High probability” perfusion lung scans in pulmonary venoocclusive disease. Am J Respir Crit Care Med 2000;162(5):1974–1978. [DOI] [PubMed]
- 21.Best DH, Sumner KL, Austin ED, Chung WK, Brown LM, Borczuk AC, Rosenzweig EB, et al. EIF2AK4 mutations in pulmonary capillary hemangiomatosis. Chest 2014;145(2):231–236. [DOI] [PMC free article] [PubMed]
- 22.Eyries M, Montani D, Girerd B, Perret C, Leroy A, Lonjou C, Chelghoum N, et al. EIF2AK4 mutations cause pulmonary veno-occlusive disease, a recessive form of pulmonary hypertension. Nat Genet 2014;46(1):65–69. [DOI] [PubMed]
- 23.Ginns LC, Robert DH, Mark EJ, Brusch JL, Marler JJ. Pulmonary capillary hemangiomatosis with atypical endotheliomatosis: successful antiangiogenic therapy with doxycycline. Chest 2003;124:2017–2022. [DOI] [PubMed]
- 24.White CW, Sondheimer HM, Crouch EC, Wilson H, Fan LL. Treatment of pulmonary hemangiomatosis with recombinant interferon alfa-2a. N Engl J Med 1989;320:1197–1200. [DOI] [PubMed]
- 25.Nayyar D, Muthiah K, Kumarasinghe G, Hettiarachchi R, Celermajer D, Kotlyar E, Keogh A. Imatinib for the treatment of pulmonary arterial hypertension and pulmonary capillary hemangiomatosis. Pulm Circ 2014;4(2):342–345. [DOI] [PMC free article] [PubMed]
- 26.Adachi S, Hirashiki A, Kondo T, Nakaguro M, Ogawa A, Miyaji K, Matsubara H, Yokoi T, Murohara T. Imatinib is partially effective for the treatment of pulmonary capillary hemangiomatosis. Intern Med 2014;53(6):603–607. [DOI] [PubMed]
- 27.Humbert M, Maitre S, Capron F, Rain B, Musset D, Simonneau G. Pulmonary edema complicating continuous intravenous prostacyclin in pulmonary capillary hemangiomatosis. Am J Respir Crit Care Med 1998;157:1681–1685. [DOI] [PubMed]