

Abstract
Objectives
To test the hypothesis that glucocorticoid receptor (GR) activation increases resistance to chemotherapy in high-grade serous ovarian cancer (HGS-OvCa) and that treatment with a GR antagonist will improve sensitivity to chemotherapy.
Methods
GR expression was assessed in OvCa cell lines by qRT-PCR and Western blot analysis and in xenografts and primary human tumors using immunohistochemistry (IHC). We also examined the effect of GR activation versus inhibition on chemotherapy-induced cytotoxicity in OvCa cell lines and in a xenograft model.
Results
With the exception of IGROV-1 cells, all OvCa cell lines tested had detectable GR expression by Western blot and qRT-PCR analysis. Twenty-five out of the 27 human primary HGS-OvCas examined expressed GR by IHC. No cell line expressed detectable progesterone receptor (PR) or androgen receptor (AR) by Western blot analysis. In vitro assays showed that in GR-positive HeyA8 and SKOV3 cells, dexamethasone (100 nM) treatment upregulated the pro-survival genes SGK1 and MKP1/DUSP1 and inhibited carboplatin/gemcitabine-induced cell death. Concurrent treatment with two GR antagonists, either mifepristone (100 nM) or CORT125134 (100 nM), partially reversed these effects. There was no anti-apoptotic effect of dexamethasone on chemotherapy-induced cell death in IGROV-1 cells, which did not have detectable GR protein. Mifepristone treatment alone was not cytotoxic in any cell line. HeyA8 OvCa xenograft studies demonstrated that adding mifepristone to carboplatin/gemcitabine increased tumor shrinkage by 48% compared to carboplatin/gemcitabine treatment alone (P=0.0004).
Conclusions
These results suggest that GR antagonism sensitizes GR+ OvCa to chemotherapy-induced cell death through inhibition of GR-mediated cell survival pathways.
Keywords: ovarian cancer, mifepristone, GR antagonist, chemotherapy
Introduction
Ovarian cancer is usually a fatal disease with an estimated 21,290 new cases and 14,180 deaths in the United States in 2015 (1). High-grade serous ovarian carcinoma (HGS-OvCa) is the most common subtype and accounts for 70% of ovarian cancer deaths (2). Despite advances in treatment, including surgical tumor debulking followed by platinum/taxane-based therapy, the majority of patients diagnosed with Stage III and IV disease eventually relapse and develop chemotherapy-resistant, incurable disease with a low 5-year survival rate of 18-27% (1). Therefore, it is important to develop approaches that will improve chemotherapy sensitivity of HGS-OvCa.
Glucocorticoid receptors (GRs) are expressed in a subset of epithelial tumors including OvCa and triple-negative breast cancer (TNBC) (3, 4). Our group and others have demonstrated that GR activation by dexamethasone (dex), a synthetic glucocorticoid, inhibits taxane-induced tumor cell death in breast, cervical, ovarian, and lung cancer cell lines and in xenograft models (5-8). GR activation directly activates the transcription of several genes including serum and glucocorticoid-regulated protein kinase (SGK1) and mitogen-activated protein kinase phosphatase-1 (MKP1)/dual-specificity phosphatase 1 (DUSP1), both of which encode proteins that promote epithelial cell survival during apoptosis (e.g. exposure to serum deprivation, radiation, or chemotherapy treatment) (9). We have demonstrated that treatment with dex induces expression of the SGK1 and MKP1/DUSP1 and decreases chemotherapy-induced tumor cell death in TNBC cell lines (3, 10), in OvCa (4), and in anti-androgen-induced cell death in castrate-resistant prostate cancer (11). Recently, data published from The Cancer Genome Atlas (TCGA) revealed that HGS-OvCa has a gene expression and somatic mutation profile similar to that of TNBC (12). Platinum-based therapies are actively used in the treatment of both ovarian cancer and TNBC (13). Therefore, we hypothesized that GR activation would inhibit chemotherapy-induced cell death in GR+ OvCa cell lines and that this effect might be reversed by GR antagonism. We also tested this hypothesis in an OvCa xenograft model and examined GR expression in primary HGS-OvCa samples.
Materials and Methods
Drugs
Water-soluble dex (D4902) was purchased from Sigma-Aldrich. Corcept Therapeutics (Menlo Park, CA) provided pharmaceutical-grade mifepristone (mif) and the nonsteroidal selective GR antagonist CORT125134. Pharmaceutical-grade gemcitabine (APP Pharmaceuticals) and carboplatin (APP Pharmaceuticals) were used.
Cells and Cell Culture
The human OvCa cell line SKOV3, and MDA-MB-231 and MCF-7 breast cancer cells were purchased from American Type Culture Collection (ATCC). The human OvCa cell lines Monty-1, HeyA8, CAOV-3, and IGROV-1 were a generous gift from Dr. Ernst Lengyel (The University of Chicago). The T47D breast cancer cell line was a generous gift from Dr. Olufunmilayo Olopade (The University of Chicago). All cells were maintained in Dulbecco's modified Eagle Medium (DMEM; Lonza) and supplemented with 10% fetal calf serum (FCS; Gemini Bio-Products) and antibiotics (1% penicillin-streptomycin, Lonza). All cell lines were cultured at 37°C in a humidified atmosphere in the presence of 5% CO2. Before treatment with glucocorticoid, mifepristone, CORT125134, and/or chemotherapy, cells were grown for 48 hours in DMEM supplemented with 2.5% charcoal-stripped FCS (CS-FCS) and 1% penicillin-streptomycin. All cell lines tested negative for mycoplasma with the ATCC Universal Mycoplasma Detection kit.
Cell Death Assay
OvCa cell lines (HeyA8 at 4 × 103 cells/well, SKOV3 at 4 × 103 cells/well, and IGROV-1 at 6 × 103 cells/well) were seeded in 96-well plates in 2.5% CS-FCS for 48 hours. Cells were then treated with vehicle (EtOH 0.1% v/v), dex (100 nmol/L) or mif (1 μmol/L) alone or dex/mif (dex 100 nmol/L and mif 1 μmol/L) starting 1 hour before treatment with carboplatin (120 nmol/L) and gemcitabine (250 nmol/L) for 72 hours. A cyanine dimer nucleic acid dye, YOYO-1 (Life Technologies, Y3601) that stains cellular nuclei if the cellular membrane is compromised was used to detect dead cells. Two images (1.90 × 1.52 mm) in separate regions of each well were captured with a 10× objective at 4-hour intervals using the ZOOM IncuCyte FLR HD real-time in vitro micro-imaging system (Essen Instruments). Dead cells (YOYO-1-positive) and total cell counts (detected using phase contrast) were enumerated using ImageJ Software (Version 1.48v) as reported previously (14). The “cytotoxic index” was calculated and represents the number of dead cells/total (live and dead) cells for each condition. Images collected between 0 and 72 hours post-treatment were used in the analysis. The cytotoxic index was log-transformed to satisfy the normality assumption. A two tailed t-test was used at the 72 hour time point to compare cell death between two treatment conditions – either dex/gem/carbo vs dex/mif/gem/carbo or dex/gem/carbo vs dex/CORT125134/gem/carbo. Each experiment was performed at least twice and each treatment had five replicate wells per experiment. One representative experiment is presented with standard error of the mean (±SEM) for the five wells. Similar findings were obtained in each independent experiment.
Quantitative Real-Time PCR (qRT-PCR)
CAOV3, HeyA8, Monty-1, SKOV3, and IGROV-1 cells were seeded at approximately 50% confluence and allowed to adhere overnight in DMEM with 10% FCS, then cultured in 2.5% CS-FCS for an additional 48 hours. Medium was removed and equal volumes of either vehicle (ethanol), dex (100 nmol/L) or dex/mif (100 nmol/L) diluted in DMEM supplemented with 2.5% CS-FCS was then added. After 4 hours of treatment, 500 μL of lysis buffer (Ambion by Life Technologies Pure Link™ RNA Mini Kit) supplemented with 2% 2-mercaptoethanol was added to each well to harvest RNA. Total RNA was extracted using the Qiagen All-Prep DNA/RNA Mini Kit. cDNA was then reverse transcribed from 0.5 μg of total RNA with Quanta reverse transcription reagents (Quanta Biosciences) using the GeneAmp PCR System 9700 (Applied BioSystems) per manufacturer's instruction. The cDNA was diluted in PerfeCTa SYBR Green FastMix (Quanta Biosciences), and quantitative real-time PCR (qRT-PCR) was carried out in a BioRad PCR System MyIQ (BioRad Life Sciences). The following primers were used: SGK1, 5′-AGGCCCATCCTTCTCTGTTT-3′ (forward) and 5′-TTCACTGCTCCCCTCAGTCT-3′ (reverse); MKP1/DUSP1, 5′-CCTGACAGCGCGGAATCT-3′ (forward) and 5′-GATTTCCACCGGGCCAC-3′ (reverse); NR3C1/GR, 5′-TCTGAACTTCCCTGGTCGAA-3′ (forward) and 5′-GTGGTCCTGTTGTTGCTGTT-3′ (reverse); Actin-B 5′-CAGCGGAACCGCTCATTGCCAATGG-3′ (forward) and 5′-TCACCCCCTGTGCCCATCTACGA-3′ (reverse); PR-AB, 5′-ACAGAATTCATGAGCCGGTCCGGGTG-3′ (forward) and 5′-ACAAGATCTCCACCCAGAGCCCGAGG-3′ (reverse); ER-alpha, 5′CCTGATCATGGAGGGTCAAA-5′ (forward) and 5′TGGGCTTACTGACCAACCTG-3′ (reverse); AR, ‘ATCCCAGTCCCACTTGTGTC’ (forward) and ‘GGTCTTCTGGGTGGAAAGT’ (reverse). Relative quantification of steady-state mRNA transcript expression was calculated according to the standard curve method, as described by Applied Biosystems User Bulletin 2, October 2001, based on the ΔΔCt approach (15). Transcript levels were normalized to Actin-B expression. Each experiment was performed three times and each treatment was performed in three technical replicate wells per individual experiment. One representative experiment is shown in figures with error bars representing SEM of the triplicate wells.
Western Blot Analysis
Cells were allowed to adhere overnight in media containing 10% FCS. The following day, medium was changed to 2.5% CS-FCS and cells were cultured for an additional 48 hours, followed by lysis in buffer containing PhosSTOP phosphatase inhibitor (Roche Life Science) and Complete EDTA protease inhibitor (Roche Life Science). Protein concentrations were measured using the BCA Assay Kit (Thermo Scientific) and 2× Laemmli buffer supplemented with 5% 2-mercaptoethanol was added to an equivalent volume of protein lysate. Proteins (60 μg per lane) were resolved by SDS-PAGE and then transferred to polyvinylidene difluoride membranes (Bio-Rad). The membranes were washed three times in 0.1% Tween 20 in TBS (TBS/T), and then incubated with blocking solution (5% bovine serum albumin (BSA; Fisher Scientific) in TBS/T) for 1 hour at room temperature. Primary antibodies were diluted in blocking solution and incubated with the membrane for 1 hour or overnight incubation at 4°C. Antibodies and concentrations were as follows: monoclonal rabbit anti-glucocorticoid receptor (GR) XP antibody (D8H2, 1:500, GR-XP Cell Signaling), rabbit monoclonal anti-androgen receptor (AR) antibody (N20, 1:2,000, Santa Cruz), mouse anti-estrogen receptor (ER) antibody (16460, 1:500, Abcam), rabbit anti-progesterone receptor (PR) antibody (D8Q2J, 1:1,000, Abcam), mouse monoclonal anti-beta actin (β-Actin) antibody (8226, 1:10,000, Sigma-Aldrich), or anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (G9295, 1:10,0000 Sigma Aldrich). After additional washing, membranes were incubated for one hour at room temperature with either Alexa Fluor 680 goat anti-rabbit (Invitrogen) or 800 goat anti-mouse (LI-COR) secondary antibody, rinsed and scanned using the Odyssey infrared imaging system (LI-COR) at a wavelength of 700 or 800 nm, respectively.
Female SCID Mouse Xenograft Model of Ovarian Cancer
All mouse experiments were carried out in accordance with the U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals and approved by the University of Chicago Institutional Animals Care and Use Committee. Suspensions of HeyA8 (1 × 106 in 500 μL of PBS) were injected intraperitoneally (i.p.) into 6-week-old female SCID mice (Taconic). On day 5 after injection, by which time the cells had metastasized as previously described (16), mice were randomized into three cohorts. Chemotherapy was administered on days 1 and 8 of a 21-day cycle for a total of two cycles. Placebo or mifepristone were administered alone the day before chemotherapy (day 0 and day 7) and then again one hour before chemotherapy. One group was treated with mifepristone (15 mg/kg) followed by i.p. injection of carboplatin (15 mg/kg) and gemcitabine (80 mg/kg) suspended in ethanol and sesame seed oil (1:10 v/v); the second group was treated with vehicle (ethanol and sesame seed oil 1:10 v/v) followed by chemotherapy; and a third group was treated with vehicle only. Chemotherapy-treated mice were sacrificed at days 38-39 while non-chemotherapy-treated controls were sacrificed sooner on days 21-25 due to tumor burden. Two separate experiments were performed for a total of twenty-six mice. Following sacrifice, tumors were dissected and cut lengthwise into mirror image sections. One section was either minced in lysis buffer (for protein analysis) or minced in RNase + beta-mercaptoethanol (BME) (qRT-PCR analysis) and frozen, and the other was fixed in 10% neutral-buffered formalin for immunohistochemistry (IHC). Following dissection of all visible tumors, individual mouse tumor burden weights were determined and a comparison performed between gem/carbo and mif/gem/carbo treatment groups using a t-test. Because three mice in the gem/carbo group and one mouse in the mif/gem/carbo group had to be sacrificed earlier than day 38-39 due to high tumor burden and were likely to have extremely large tumors on days 38-39, tumor weights for these animals were imputed with a weight exceeding all observed weights to ensure the highest rank (worst outcome) when treatment groups were further compared using Wilcoxon's rank-sum test as a sensitivity analysis
Histopathological Examination
Tumor xenograft samples were fixed in 10% buffered formalin for 24 hours immediately after necropsy and then embedded in paraffin. IHC studies were performed using 5 μm thick formalin-fixed deparaffinized sections that were rehydrated with xylene and serial dilutions of ethanol and distilled water. Samples were incubated in antigen retrieval buffer (S1699, DAKO) and heated in a steamer oven at 97°C for 20 minutes. The antigen-antibody binding was detected by the Bond Polymer Refine Detection system (DS9800, Leica Microsystem). Xenograft tumor tissue was evaluated by IHC for GR expression using an anti-GR-XP antibody (D8H2, 1:500, Cell Signaling) and the percentage of positively-staining xenografted tumor cells estimated.
Tissue Microarray and Immunohistochemical Staining
Human OvCa tissue microarrays were constructed by the Human Tissue Research Center (HTRC) of the University of Chicago with Institutional Review Board Approval as previously described (16, 17). All samples underwent pathology review to establish ovarian tumor subtype. H&E staining to confirm tumor as well as percentage and intensity of nuclear GR, ER, PR, and AR staining was performed on the formalin-fixed paraffin-embedded specimens as above. The following antibodies were used anti-GR-XP antibody (D8H2, 1:500, Cell Signaling), anti-ER-alpha antibody (6F11, 1:40, DAKO) anti-PR antibody (16, 1:200, DAKO) and anti-AR antibody (AR441, 1:300, DAKO). A modification of the H- (Histological) score as previously published (18, 19) was calculated to quantify GR, ER, PR, and AR expression in the primary human HGS-OvCa tissue samples. The H-score was calculated by multiplying the intensity of nuclear staining (0: no staining, 1: weak, 2: moderate and 3: strong staining) by the percentage of the tumor cells staining. The minimum H-score was 0 and the maximum was 300.
Results
GR, ER, PR, and AR expression in OvCa cell lines
We examined five OvCa cell lines for GR-alpha expression by Western analysis (Fig. 1A). The breast cancer cell line MDA-MB-231 was the positive control for GR expression. CAOV-3, HeyA8, Monty-1, and SKOV3 OvCa cell lines all demonstrated detectable GR-alpha immunoreactivity (20) while IGROV-1 cells did not. GR (NR3C1) steady-state mRNA expression was also evaluated using q-RT-PCR. CAOV-3, HeyA8, Monty-1, and SKOV3 OvCa cell lines all demonstrated detectable GR-alpha mRNA transcript while IGROV-1 had extremely low levels of GR mRNA transcripts (Fig. 1B).
Figure 1. GR expression by Western blot and qRT-PCR in ovarian cancer cell lines.
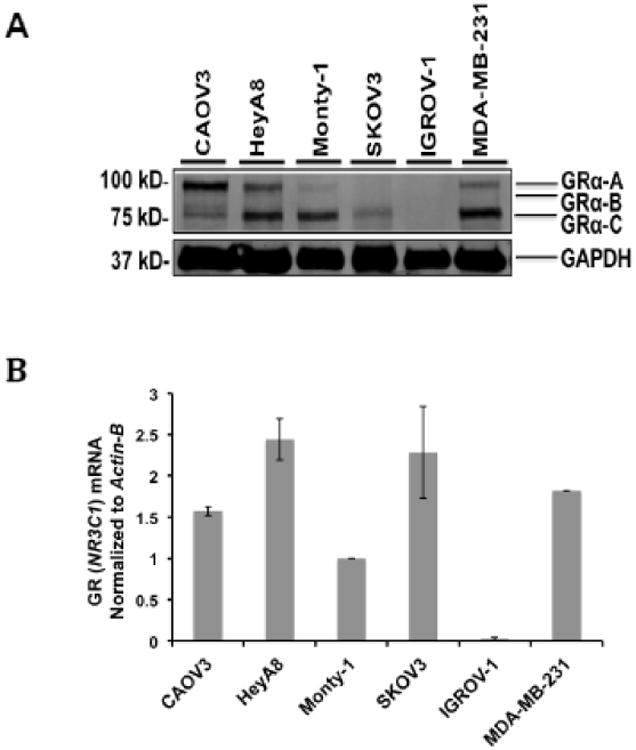
A) GR protein expression in OvCa cell lines by Western blot (B) GR mRNA transcript levels in OvCa cell lines. Transcript levels were first normalized to Actin-B mRNA expression and are shown as a ratio compared to Monty-1 cell line normalized GR (NR3C1) transcript expression. MDA-MB-231 GR (NR3C1) mRNA expression was used as a control. The error bars represent ± standard error of the mean (SEM).
We also assessed the same cell lines for estrogen receptor (ER), progesterone receptor (PR), and androgen receptor (AR) protein expression by Western blot (Fig. S1A-C). HeyA8, SKOV3, and Monty-1 had detectable ER expression (Fig. S1A) while no cell line examined demonstrated any PR-A or PR-B immunoreactivity (Fig. S1B). AR was also not detected by Western analysis (Fig. S1C). Steady-state mRNA levels of genes encoding ER-alpha and PR were also determined and correlated with the relative amount (high or low) of protein expression (Fig. S1A-B). Of note, the breast cancer cell line MCF-7 was used as positive control for ER-alpha and showed 60-fold more ER-alpha mRNA compared to the SKOV3 cell line (data not shown). Similarly, T47D cells were used as a positive control for PR and demonstrated 300-fold more PR mRNA relative to the Monty-1 cell line (data not shown). Thus ER-alpha and PR mRNA transcript expression in OvCa cell lines was relatively low compared to the commonly studied breast cancer cell lines.
GR-mediated anti-apoptotic gene expression
To determine whether the GR modulators mifepristone or CORT125134 could antagonize dex-induced/GR-mediated gene expression in OvCa cell lines, OvCa cell lines HeyA8, Monty-1, and SKOV3 were treated with vehicle (ethanol), dex (100 nM) +/-mif (100 nM) or dex +/- CORT125134 (100 nM) for 4 hours. Treatment with dex alone upregulated SGK1 steady-state mRNA levels by 3 to 18 fold when compared to dex/mif or dex/CORT125134 (P= <0.05 and P= <0.01, respectively, Fig. 2A-C). Parallel experiments were completed to examine dex (100 nM) treatment of MKP1/DUSP1 in HeyA8, Monty-1, and SKOV3 OvCa cell lines. In the HeyA8, Monty-1, and SKOV3 OvCa cell lines, MKP1/DUSP1 transcripts were increased by 4.5 to 13.8 fold after 4 hours of treatment with dex when compared to dex/mif or dex/CORT125134 (P <0.05 and P<0.01, respectively (Fig. 2A-C). Thus, treatment with the steroidal GR/PR antagonist mif or the nonsteroidal GR-specific antagonist CORT125134, in combination prevented dex-mediated upregulation of both GR target genes SGK1 and MKP1.
Figure 2. Evaluation of GR target genes SGK1 and MKP1 mRNA expression following dex, dex/mif, or dex/CORT125134 treatment.
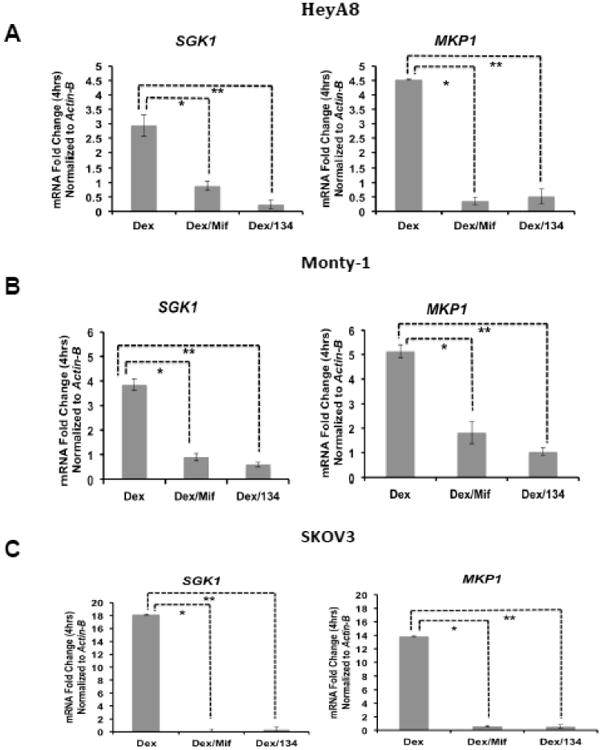
OvCa cell lines HeyA8 (A), Monty-1 (B), and SKOV3 (C) were treated for four hours with either vehicle (ethanol), dex (100 nM), dex (100 nM) with mif (100 nM) or dex with CORT125134 (100 nM). SGK1 and MKP1/DUSP1 mRNA expression was first normalized to Actin-B mRNA levels and is shown as a ratio relative to normalized transcript expression from vehicle-treated cells.
* P<0.05, ** P<0.01. NS, not significant. CORT125134 is abbreviated as 134.
GR activation inhibits chemotherapy-induced cell death and is reversed by GR antagonism
To determine if dex-mediated GR activation inhibits chemotherapy-induced cell death in HGS-OvCa cell lines, we measured total cell count and cell death continuously over 72 hours using the Incucyte™ Live Imaging system as previously described (3). GR activation following dex treatment significantly inhibited carboplatin/gemcitabine-induced cell death in GR-positive HeyA8 and SKOV3 cell lines (Fig. 3A-B). Of note, dex treatment alone did not increase cell death. There was no effect on chemotherapy-induced cell death in the GR-negative IGROV-1 with the addition of dex (Fig. 3C), suggesting that GR activation was responsible for the dex effect on cell survival in the GR+ cell lines. To test whether GR antagonism could reverse the dex effect, we co-treated cells with mifepristone or CORT125134 for one hour prior to adding chemotherapy. Both mif and CORT125134 partially or fully reversed the effect of dex-mediated GR activation in the GR+ HeyA8 and SKOV3 cells, but had no significant effect on the GR-negative IGROV-1 cells (Fig. 3A-C).
Figure 3. Continuous microscopic imaging analysis (Incucyte) of cell death following treatment with chemotherapy +/- mifepristone or +/- CORT125134.
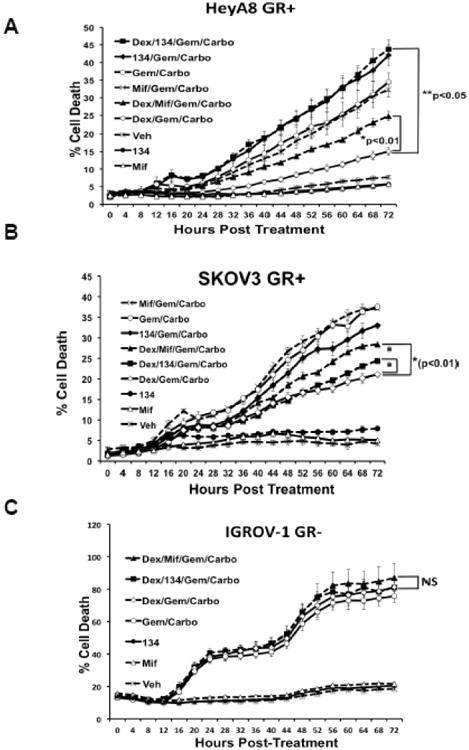
OvCa cell lines HeyA8 (A), SKOV3 (B), and IGROV-1 (C) were treated with dex (100nM) +/- mif (1 μM) or +/- CORT125134 (1 μM). This treatment was followed one hour later by gemcitabine (250 nM)/carboplatin (120 nM) treatment. Error bars represent ± SEM of triplicate wells.
GR antagonism increases chemotherapy sensitivity in an OvCa xenograft model
We used a previously published in vivo murine model of peritoneal HGS-OvCa tumor growth (16) to test the hypothesis that GR antagonism would enhance chemotherapy sensitivity in vivo. The experiment was repeated twice, with similar results, using a total of 26 mice treated with vehicle (n=6), gem/carbo (n=10) or mif/gem/carbo (n=10). Vehicle-treated (control) animals were sacrificed on days 21-25 due to tumor burden-related slow mobility and/or increased abdominal girth. Most mice receiving chemotherapy were sacrificed on days 38-39, with the exception of three mice sacrificed earlier in the gem/carbo alone group due to tumor-related morbidity (days 25, 34 and 35) and one mouse in the mif/gem/carbo group that died on day 38 prior to intended sacrifice. Among mice with measurable tumors on days 38-39, the mif/gem/carbo group had significantly smaller average total tumor burden (mean of 0.17g, range 0.10g-0.26g) compared to the gem/carbo alone group (mean of 0.36g, range 0.27g-0.58g; p=0.0004) by two-sample t-test (Fig. 4). As a sensitivity analysis, a large tumor weight (0.3g) was imputed for the four mice that were sacrificed and/or died early, and the Wilcoxon rank-sum test similarly showed that mif/gem/carbo treated mice had smaller total tumor burden compared to gem/carbo-treated mice (p=0.0016). Resected HeyA8 xenograft tumors were also evaluated by anti-GR IHC staining. All xenografted tumors had >95% moderately- to strongly-positive GR expression, regardless of treatment group (data not shown).
Figure 4. Carboplatin/gemcitabine +/- mifepristone treatment of mice bearing HeyA8 tumor xenografts.
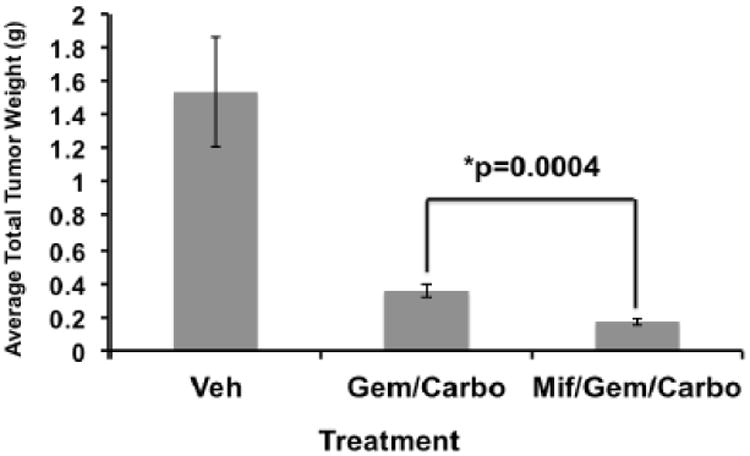
Combined data from two independent in vivo experiments. Total tumor burden is indicated at days 38-39 after initial treatment, P=0.0004 between gem/carbo (0.36g, range 0.27g-0.58g) and mif/gem/carbo (0.17g, range 0.10g-0.26g) groups. Error bars represent ± SEM of tumor weights in grams, g.
GR, ER, PR, and AR expression in a HGS-OvCa human tissue microarray
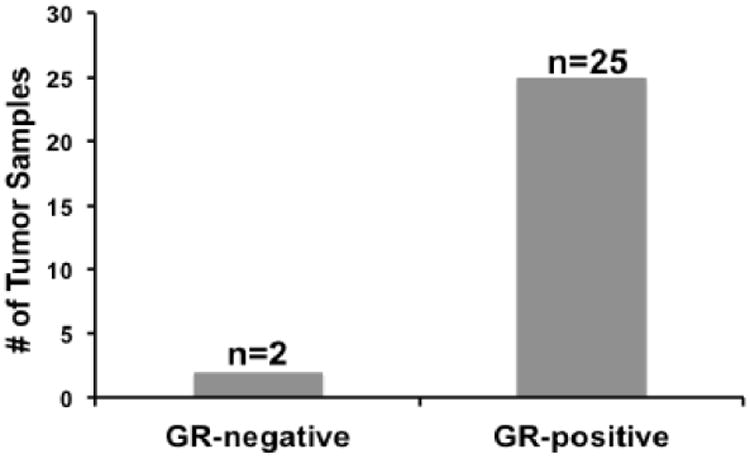
Human tissue microarrays from the University of Chicago tumor bank of twenty-seven patients confirmed to have HGS-OvCa were evaluated for GR expression by IHC. For this study, tissue microarrays were assessed using a modified H-score method to quantitate tumor GR, ER, PR, and AR expression (applying the previously described H-score method) (18, 19). Twenty-five samples (93%) had a GR H score >1 (Fig. 5). (See Fig. S2 for GR positive control and negative control i.e. secondary antibody alone). As observed in the OvCa cell lines, we found variable expression of GR (Fig. S3A), ER-alpha (Fig. S3B), and negative to low expression of PR and AR proteins by IHC (Fig. S3C-D).
Figure 5. Primary human HGS-OvCa GR expression by IHC.
Discussion
Here we demonstrate that, as previously reported for TNBC (3), treatment of GR+ OvCa cell lines with glucocorticoids results in attenuation of chemotherapy-induced cell death. Conversely, GR antagonism with the GR/PR antagonist, mifepristone, or the GR-selective nonsteroidal antagonist, CORT125134, prior to chemotherapy administration inhibits the tumor cell survival effect of GR activation. Moreover, mifepristone increased the efficacy of gemcitabine/carboplatin chemotherapy in a xenograft model, presumably by antagonizing the tumor cell survival effect of endogenous corticosterone-mediated tumor GR activation. When we initially selected our OvCa cell lines, we chose those we believed to originate from HGS-OvCas, because our hypothesis was that the findings in TNBC could be extended to OvCa. This hypothesis was spurred by the TCGA report regarding the genomic similarity of TNBC and HGS-OvCa. It has subsequently been reported that the OvCa cell lines we used may not all be of HGS origin (21, 22); however, the GR status of these cell lines still appears to correlate with the ability of glucocorticoid treatment to promote cell survival and conversely, with the ability of GR antagonism to increase chemotherapy sensitivity.
Mifepristone is a steroidal PR and GR mixed agonist/antagonist as well as a weak AR antagonist that is currently FDA-approved for emergency contraception and the treatment of Cushing's syndrome with glucose intolerance (23, 24). The cell lines used in our study appear to be PR-negative using a highly sensitive anti-PR antibody as well as by quantitative RT-PCR. Thus, the effects of mifepristone on tumor cell lines are not likely to be related to PR antagonism. In a previous healthy human pharmacokinetic analysis, a single oral dose of mifepristone 100-800 mg/day resulted in serum concentration levels of approximately 2.5 μM at 24 hours (25). A study examining mifepristone tissue concentrations in human endometrial villi following a single dose of mifepristone 200 mg yielded intracellular levels of 238 nM ± 113 (26). Telleria et al. previously reported that mifepristone treatment of OvCa cell lines at the much higher concentrations of 10-20 μM (versus 1 μM used in our studies) causes cell cycle arrest (27) and cytostasis (28). These authors also noted that the growth inhibitory effect of mifepristone did not require PR expression. At the lower concentration we used, mifepristone alone had no growth inhibitory effect in the HGS-OvCa lines we tested. Unfortunately, clinical trials in ovarian cancer with single agent mifepristone have not yielded positive results, suggesting that 10 μM levels may not be easily achieved within tumor tissue (29, 30).
We also examined human HGS-OvCa samples and found that most expressed significant GR. Woenckhaus et al. also evaluated 85 invasive ovarian cancers with various histopathological characteristics and found that there was variable GR expression (negative to strong) among OvCa tissues (31).
We believe our results warrant clinical investigation. A recently completed phase I randomized clinical trial was performed at the University of Chicago to evaluate the safety and tolerability of nab-paclitaxel (which does not required steroid premedication) +/- mifepristone in advanced breast cancer. However, a dose-limiting toxicity of neutropenia was noted in some patients receiving concomitant mifepristone and nab-paclitaxel. While this side effect was manageable, the nab-paclitaxel levels were quite variable in the combination arm, and may be related to unpredictable pharmacokinetic (PK) interaction between the two drugs and/or pharmacodynamic (PD) effects (32). Based on our laboratory results reported here with OvCa cell lines, we have initiated a Phase 1 clinical trial combining mifepristone with carboplatin/gemcitabine in women with advanced breast or OvCa.
Supplementary Material
Supplementary Figure 1: Western blot and mRNA steady-state levels in ER, PR, and AR in ovarian cancer cell lines. (A) ER expression by Western blot (left panel). An ER-FLAG-HEK cell line lysate was used as the ER+ control. ER-alpha transcripts measured by qRT-PCR are shown (right panel). MCF-7 mRNA was used as a positive control for ER-alpha expression (data not shown) (B) PR-A and PR-B protein expression by Western blot (left panel) and steady-state PR mRNA by qRT-PCR (right panel). T47D lysate or mRNA was used as a PR positive control (data not shown). (C) AR expression by Western blot. LNCaP prostate cancer cell lysate was used an AR positive control. Error bars represent ± SEM of three replicate qRT-PCR reactions.
Supplementary Figure 2: IHC staining for GR in primary HGS-OvCa. (A) Tumor cells staining positive for GR. (B) Negative control (secondary antibody alone).
Supplementary Figure 3: H-scores of GR, ER, PR, and AR in primary human HGS-OvCas. (A) GR H-score. (B) ER H-score. (C) PR H-score. (D) AR H-score.
Highlights.
GR activation promotes cell survival in GR-positive high-grade serous ovarian carcinoma (HGS-OvCa) cell lines treated with chemotherapy.
GR antagonism increases chemotherapy-induced cell death in a GR-positive HGS-OvCa xenograft model.
GR is expressed in the majority of primary HGS-OvCas examined.
Acknowledgments
We thank Drs. Robert Roe and Hazel Hunt (Corcept Therapeutics) for supplying pharmaceutical-grade mifepristone and CORT125134 and sharing their extensive expertise. We thank members of the Ernst Lengyel laboratory, especially Drs. Arniban Mitra and Hillary Kenny for assistance with OvCa xenograft modeling, and the Suzanne Conzen and Matthew Brady laboratories for valuable feedback and expertise. Terri Li and the University of Chicago Medicine Comprehensive Cancer Center's Human Tissue Resource Center (HTRC) provided essential expertise for the xenograft and human tissue microarray IHC staining. We also thank the Geoffrey Greene laboratory for the use of the Incucyte™. Finally, we thank the University of Chicago Cancer Research Foundation's (UCCRF) Women's Board for supporting this project.
Financial Support: Conquer Cancer Foundation ASCO
NIH Basic Research Training in Medical Oncology Grant T32-CA009566
NIH Clinical Therapeutics Training Grant T32-GM007019
The University of Chicago Medicine Comprehensive Cancer Center's Human Tissue Resource Center (HTRC), supported in part by the Cancer Center Support Grant CA-014599
The University of Chicago Cancer Research Foundation's (UCCRF) Women's Board
E. Stringer-Reasor received support from the Conquer Cancer Foundation of ASCO YIA award, the NIH Basic Research Training in Medical Oncology Grant (T32-CA009566), and the NIH Clinical Therapeutics Training Grant (T32-GM007019). G. Fleming and S. Conzen received research support from The University of Chicago Cancer Foundation Women's Board. S. Conzen and M. Kocherginsky are co-inventors on a patent entitled “Methods and Compositions Related to Glucocorticoid Receptor Antagonist in Breast Cancer” awarded to The University of Chicago and licensed by Corcept Therapeutics.
Grant Support: The study was supported by the University of Chicago Comprehensive Cancer Center NIH support grant CA-014599 (core facilities for human tissue) and The University of Chicago Cancer Research Foundation's (UCCRF) Women's Board. E. Stringer-Reasor was also supported by the Conquer Cancer Foundation of ASCO YIA award, the NIH Basic Research Training in Medical Oncology Grant (T32-CA009566), and the NIH Clinical Therapeutics Training Grant (T32-GM007019).
Footnotes
Disclosure of Potential Conflicts of Interest Statement: All other authors report no conflicts of interest. Study sources that provided financial support had no involvement in study design, collection of data, analysis, or interpretation.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA: A Cancer Journal for Clinicians. 2015;65(1):5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Bowtell DD. The genesis and evolution of high-grade serous ovarian cancer. Nature Reviews Cancer. 2010;10(11):803–8. doi: 10.1038/nrc2946. [DOI] [PubMed] [Google Scholar]
- 3.Skor MN, Wonder EL, Kocherginsky M, Goyal A, Hall BA, Cai Y, et al. Glucocorticoid receptor antagonism as a novel therapy for triple-negative breast cancer. Clinical Cancer Research. 2013;19(22):6163–72. doi: 10.1158/1078-0432.CCR-12-3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Melhem A, Yamada SD, Fleming GF, Delgado B, Brickley DR, Wu W, et al. Administration of glucocorticoids to ovarian cancer patients is associated with expression of the anti-apoptotic genes SGK1 and MKP1/DUSP1 in ovarian tissues. Clinical Cancer Research. 2009;15(9):3196–204. doi: 10.1158/1078-0432.CCR-08-2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herr I, Ucur E, Herzer K, Okouoyo S, Ridder R, Krammer PH, et al. Glucocorticoid cotreatment induces apoptosis resistance toward cancer therapy in carcinomas. Cancer Research. 2003;63(12):3112–20. [PubMed] [Google Scholar]
- 6.Wu W, Chaudhuri S, Brickley DR, Pang D, Karrison T, Conzen SD. Microarray analysis reveals glucocorticoid-regulated survival genes that are associated with inhibition of apoptosis in breast epithelial cells. Cancer Research. 2004;64(5):1757–64. doi: 10.1158/0008-5472.can-03-2546. [DOI] [PubMed] [Google Scholar]
- 7.Zhang C, Wenger T, Mattern J, Ilea S, Frey C, Gutwein P, et al. Clinical and mechanistic aspects of glucocorticoid-induced chemotherapy resistance in the majority of solid tumors. Cancer Biology & Therapy. 2007;6(2):278–87. doi: 10.4161/cbt.6.2.3652. [DOI] [PubMed] [Google Scholar]
- 8.Huang Y, Johnson KR, Norris JS, Fan W. Nuclear factor-kappaB/IkappaB signaling pathway may contribute to the mediation of paclitaxel-induced apoptosis in solid tumor cells. Cancer Research. 2000;60(16):4426–32. [PubMed] [Google Scholar]
- 9.Mikosz CA, Brickley DR, Sharkey MS, Moran TW, Conzen SD. Glucocorticoid receptor-mediated protection from apoptosis is associated with induction of the serine/threonine survival kinase gene, sgk-1. The Journal of Biological Chemistry. 2001;276(20):16649–54. doi: 10.1074/jbc.M010842200. [DOI] [PubMed] [Google Scholar]
- 10.Pang D, Kocherginsky M, Krausz T, Kim SY, Conzen SD. Dexamethasone decreases xenograft response to Paclitaxel through inhibition of tumor cell apoptosis. Cancer Biology & Therapy. 2006;5(8):933–40. doi: 10.4161/cbt.5.8.2875. [DOI] [PubMed] [Google Scholar]
- 11.Isikbay M, Otto K, Kregel S, Kach J, Cai Y, Vander Griend DJ, et al. Glucocorticoid receptor activity contributes to resistance to androgen-targeted therapy in prostate cancer. Hormones & Cancer. 2014;5(2):72–89. doi: 10.1007/s12672-014-0173-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang ZC, Birkbak NJ, Culhane AC, Drapkin R, Fatima A, Tian R, et al. Profiles of genomic instability in high-grade serous ovarian cancer predict treatment outcome. Clinical Cancer Research. 2012;18(20):5806–15. doi: 10.1158/1078-0432.CCR-12-0857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thon JN, Devine MT, Jurak Begonja A, Tibbitts J, Italiano JE., Jr High-content live-cell imaging assay used to establish mechanism of trastuzumab emtansine (T-DM1)--mediated inhibition of platelet production. Blood. 2012;120(10):1975–84. doi: 10.1182/blood-2012-04-420968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Williams JB, Pang D, Delgado B, Kocherginsky M, Tretiakova M, Krausz T, et al. A model of gene-environment interaction reveals altered mammary gland gene expression and increased tumor growth following social isolation. Cancer Prevention Research. 2009;2(10):850–61. doi: 10.1158/1940-6207.CAPR-08-0238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaur S, Kenny HA, Jagadeeswaran S, Zillhardt MR, Montag AG, Kistner E, et al. {beta}3-integrin expression on tumor cells inhibits tumor progression, reduces metastasis, and is associated with a favorable prognosis in patients with ovarian cancer. The American Journal of Pathology. 2009;175(5):2184–96. doi: 10.2353/ajpath.2009.090028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sawada K, Radjabi AR, Shinomiya N, Kistner E, Kenny H, Becker AR, et al. c-Met overexpression is a prognostic factor in ovarian cancer and an effective target for inhibition of peritoneal dissemination and invasion. Cancer Research. 2007;67(4):1670–9. doi: 10.1158/0008-5472.CAN-06-1147. [DOI] [PubMed] [Google Scholar]
- 18.McCarty KS, Jr, Szabo E, Flowers JL, Cox EB, Leight GS, Miller L, et al. Use of a monoclonal anti-estrogen receptor antibody in the immunohistochemical evaluation of human tumors. Cancer Research. 1986;46(8 Suppl):4244s–8s. [PubMed] [Google Scholar]
- 19.Abduljabbar R, Negm OH, Lai CF, Jerjees DA, Al-Kaabi M, Hamed MR, et al. Clinical and biological significance of glucocorticoid receptor (GR) expression in breast cancer. Breast cancer Research and Treatment. 2015;150(2):335–46. doi: 10.1007/s10549-015-3335-1. [DOI] [PubMed] [Google Scholar]
- 20.Lu NZ, Cidlowski JA. Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Molecular Cell. 2005;18(3):331–42. doi: 10.1016/j.molcel.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 21.Domcke S, Sinha R, Levine DA, Sander C, Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nature Communications. 2013;4:2126. doi: 10.1038/ncomms3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jacob F, Nixdorf S, Hacker NF, Heinzelmann-Schwarz VA. Reliable in vitro studies require appropriate ovarian cancer cell lines. Journal of Ovarian Research. 2014;7:60. doi: 10.1186/1757-2215-7-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spitz IM, Bardin CW. Mifepristone (RU 486)--a modulator of progestin and glucocorticoid action. The New England Journal of Medicine. 1993;329(6):404–12. doi: 10.1056/NEJM199308053290607. [DOI] [PubMed] [Google Scholar]
- 24.Fleseriu M, Molitch ME, Gross C, Schteingart DE, Vaughan TB, 3rd, Biller BM. A new therapeutic approach in the medical treatment of Cushing's syndrome: glucocorticoid receptor blockade with mifepristone. Endocrine Practice. 2013;19(2):313–26. doi: 10.4158/EP12149.RA. [DOI] [PubMed] [Google Scholar]
- 25.Heikinheimo O, Kekkonen R, Lahteenmaki P. The pharmacokinetics of mifepristone in humans reveal insights into differential mechanisms of antiprogestin action. Contraception. 2003;68(6):421–6. doi: 10.1016/s0010-7824(03)00077-5. [DOI] [PubMed] [Google Scholar]
- 26.Wang JD, Shi WL, Zhang GQ, Bai XM. Tissue and serum levels of steroid hormones and RU 486 after administration of mifepristone. Contraception. 1994;49(3):245–53. doi: 10.1016/0010-7824(94)90042-6. [DOI] [PubMed] [Google Scholar]
- 27.Goyeneche AA, Caron RW, Telleria CM. Mifepristone inhibits ovarian cancer cell growth in vitro and in vivo. Clinical Cancer Research. 2007;13(11):3370–9. doi: 10.1158/1078-0432.CCR-07-0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gamarra-Luques CD, Goyeneche AA, Hapon MB, Telleria CM. Mifepristone prevents repopulation of ovarian cancer cells escaping cisplatin-paclitaxel therapy. BMC Cancer. 2012;12:200. doi: 10.1186/1471-2407-12-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rocereto TF, Saul HM, Aikins JA, Jr, Paulson J. Phase II study of mifepristone (RU486) in refractory ovarian cancer. Gynecologic Oncology. 2000;77(3):429–32. doi: 10.1006/gyno.2000.5789. [DOI] [PubMed] [Google Scholar]
- 30.Rocereto TF, Brady WE, Shahin MS, Hoffman JS, Small L, Rotmensch J, et al. A phase II evaluation of mifepristone in the treatment of recurrent or persistent epithelial ovarian, fallopian or primary peritoneal cancer: a gynecologic oncology group study. Gynecologic Oncology. 2010;116(3):332–4. doi: 10.1016/j.ygyno.2009.10.071. [DOI] [PubMed] [Google Scholar]
- 31.Woenckhaus J, Franke FE, Hackethal A, Von Georgi R, Munstedt K. Glucocorticosteroid receptors in ovarian carcinomas. Oncology Reports. 2006;15(5):1137–40. [PubMed] [Google Scholar]
- 32.Nanda R, Chennameni P, Stringer E, Gibson J, Koetter K, Libao B, Skor M, Maranville J, Hoffman P, Obeid E, DiRienzo A, Fleming G, Conzen S. San Antonio Breast Cancer Symposium. San Antonio, Texas: Dec 11, 2013. A randomized phase I trial of nanoparticle-albumin bound paclitaxel (nab-paclitaxel, Abraxane) with or without mifepristone for advanced breast cancer. abstract P2-16-21. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Western blot and mRNA steady-state levels in ER, PR, and AR in ovarian cancer cell lines. (A) ER expression by Western blot (left panel). An ER-FLAG-HEK cell line lysate was used as the ER+ control. ER-alpha transcripts measured by qRT-PCR are shown (right panel). MCF-7 mRNA was used as a positive control for ER-alpha expression (data not shown) (B) PR-A and PR-B protein expression by Western blot (left panel) and steady-state PR mRNA by qRT-PCR (right panel). T47D lysate or mRNA was used as a PR positive control (data not shown). (C) AR expression by Western blot. LNCaP prostate cancer cell lysate was used an AR positive control. Error bars represent ± SEM of three replicate qRT-PCR reactions.
Supplementary Figure 2: IHC staining for GR in primary HGS-OvCa. (A) Tumor cells staining positive for GR. (B) Negative control (secondary antibody alone).
Supplementary Figure 3: H-scores of GR, ER, PR, and AR in primary human HGS-OvCas. (A) GR H-score. (B) ER H-score. (C) PR H-score. (D) AR H-score.