

Abstract
Transforming growth factor-β (TGF-β) strongly promotes renal tubulointerstitial fibrosis, but the cellular target that mediates its profibrotic actions has not been clearly identified. While in vitro data suggest that TGF-β-induced matrix production is mediated by renal fibroblasts, the role of these cells in TGF-β-dependent tubulointerstitial fibrosis following renal injury is not well defined. To address this, we deleted the TGF-β type II receptor in matrix-producing interstitial cells using two different inducible Cre models: COL1A2-Cre with a mesenchymal enhancer element and tenascin-Cre which targets medullary interstitial cells and either the mouse unilateral ureteral obstruction or aristolochic acid renal injury model. Renal interstitial cells lacking the TGF-β receptor had significantly impaired collagen I production, but unexpectedly, overall tissue fibrosis was unchanged in the conditional knockouts after renal injury. Thus, abrogating TGF-β signaling in matrix-producing interstitial cells is not sufficient to reduce fibrosis after renal injury.
Keywords: obstruction, chronic kidney disease, growth factors, renal injury
Introduction
Progressive tubulointerstitial fibrosis is the common mechanism whereby renal injuries of diverse etiologies lead to end-stage renal disease. The accumulation of extracellular matrix (ECM) proteins such as collagen I is the hallmark of fibrosis, and transforming growth factor-β (TGF-β) plays a central role in ECM production in the kidney. There are three TGF-β isoforms (−β1, −β2, −β3), all of which bind to the TGF-β type II receptor (TβRII), which phosphorylates the type I receptor. This activated receptor complex mediates diverse biological effects through both Smad-dependent and independent signaling pathways.(1–3) Mice overexpressing TGF-β1 in the renal tubular epithelium spontaneously develop significant tubulointerstitial fibrosis.(4) Conversely, a blocking antibody to TGF-β isoforms attenuates fibrosis after renal injury, supporting a strong pro-fibrotic role for this growth factor following renal injury.(5, 6)
TGF-β’s pro-fibrotic effects are thought to be primarily mediated by myofibroblasts, mesenchymal cells with contractile properties and potent producers of ECM. A number of different interstitial cells (fibroblasts, pericytes, infiltrating bone marrow-derived cells) may transform into myofibroblasts.(7) TGF-β1 has been implicated in the transformation of these matrix-producing interstitial cells (MPIC) into myofibroblasts and their synthesis of collagen I.(8–10) In vitro data shows that TGF-β increases collagen I and fibronectin synthesis by MPIC,(11, 12) but how TGF-β signaling in MPIC in vivo modulates the response to renal injury has not been well studied. Efforts to define the role of TGF-β signaling in MPIC in vivo have been hindered by the lack of specific markers for this population. S100A4/FSP-1 has been used as a fibroblast marker, but recent studies have shown this protein marks leukocytes as well.(13, 14) Ecto-5′-nucleotidase (CD73), platelet-derived growth factor receptor-β (PDGFRβ), and CD90 are other commonly used markers that lack specificity as they are also expressed by proximal tubules (CD73), certain T cells (CD73 and CD90), vascular smooth muscle cells (PDGFRβ), and mesangial cells (CD73, PDGFRβ, and CD90).(15–17) Recently, the TGF-β type II receptor (TβRII), necessary for downstream signaling, was deleted in mice using Cre driven by the promoter of α-smooth muscle actin (α-SMA), a commonly used marker of myofibroblasts.(9) However, the α-SMA-Cre was not inducible, and α-SMA is expressed early in embryogenesis in cells not typically considered myofibroblasts (e.g. cardiomyocytes).(18) Additionally, α-SMA may not be the best marker for MPIC as α-SMA expression was observed in some renal tubular epithelial cells and vascular cells after injury,(9) and there are mixed reports regarding its correlation with collagen I production.(18–20)
In this study, we defined how TGF-β signaling in MPIC alters fibrosis by deleting TβRII using mice containing Cre driven by the promoters of ECM components. We chose the COL1A2-Cre/ERT (abbreviated COL-Cre) in which the COL1A2 promoter is driven by a mesenchymal upstream enhancer(21, 22) as well as Tenascin C-Cre/ERT (TNC-Cre), a newly described mouse that targets medullary MPIC, a small population in the healthy adult kidney that greatly expands in areas of fibrosis.(23–25) As medullary and cortical interstitial cells have distinct morphologic and functional roles, TNC-Cre allows delineation of medullary MPIC’s role in renal injury. The COL-Cre and TNC-Cre mouse models are ideally suited for targeting MPIC because their promoters are functionally associated with matrix production and they are tamoxifen-inducible, which is important because many mesenchymal markers are expressed early in development.
Contrary to expectations, deleting TβRII using COL-Cre or TNC-Cre did not affect fibrosis after either unilateral ureteral obstruction (UUO) or aristolochic acid-induced nephropathy, models which both upregulate TGF-β signaling.(26) This was despite the fact that TβRII-null MPIC had decreased collagen I transcripts in vivo and reduced collagen I production in vitro. These data suggest that inhibiting TGF-β signaling in MPIC is not sufficient to halt the progression of renal fibrosis following injury.
Results
Renal MPIC population increases after injury
We verified that COL-Cre and TNC-Cre activity was present in MPIC by crossing these mice with the mT/mG reporter which has ubiquitous membrane-bound red fluorescence that is converted to green (GFP) by Cre activity.(27) As recombination efficiency is a concern with an inducible system, we used a high dose tamoxifen regimen (see Methods) previously proven to induce efficient recombination in the adult mouse.(28) In the uninjured COL-Cre and TNC-Cre mice, Cre activity localized to peritubular cells primarily in the medullary interstitium (Figure 1A). These GFP+ cells were few in number, consistent with the paucity of collagen I-producing interstitial cells in the healthy kidney. COL-Cre mice had glomerular GFP staining as previously described,(21) but no Cre activity was detectable in the cortex of TNC-Cre mice (Figure 1A).
Figure 1.
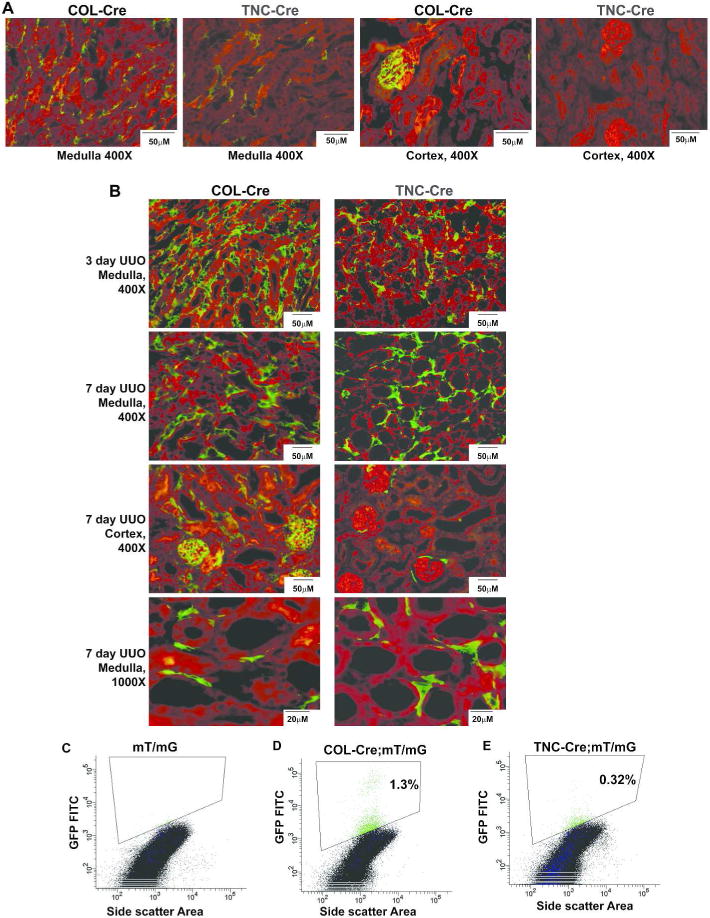
Expression and localization of COL1A2-Cre/ERT+ (abbreviated COL-Cre) and TenascinC-Cre/ERT+ (abbreviated TNC-Cre) cells. The mT/mG mouse was crossed with COL-Cre and TNC-Cre-containing mice, and Cre activity converts ubiquitous membrane-bound red fluorescence to green (A, B). Distribution of Cre activity for COL-Cre and TNC-Cre is shown in frozen sections of uninjured kidneys (A) as well as in obstructed kidneys (B). Kidneys obstructed for 3 days were converted to single cell suspensions and GFP+ cells were measured. The gating strategy, exclusion of dead cells and doublets, and forward and side scatter characteristics of GFP+ cells are shown in Supplemental Figure 1. An obstructed kidney from mT/mG-containing mouse without Cre is used as negative control (C). The percentage of GFP+ cells as a percentage of viable, single cells is listed for COL-Cre (D) and TNC-Cre (E).
Three days after UUO-induced injury, the number of medullary COL-Cre and TNC-Cre cells expanded significantly, but the COL-Cre mice had more GFP+ medullary interstitial cells than did the TNC-Cre mice (Figure 1B). Far fewer GFP+ interstitial cells were in the cortex of TNC-Cre compared to COL-Cre mice after UUO (Figure 1B). To quantify the number of GFP+ cells in COL-Cre and TNC-Cre mice after UUO, flow cytometry was performed on kidneys at 3 days after UUO (Figure 1C–E), a time point consistent with the dramatic increase in GFP+ cells (Figure 1B). Gating on viable cells was achieved using DAPI and doublets were excluded by pulse width characteristics (Supplemental Figure 1). To gate the GFP+ population (i.e. COL-Cre+ and TNC-Cre+ cells), obstructed and tamoxifen-injected mice with mT/mG but lacking Cre were used as a negative control (Figure 1C). Consistent with the GFP staining in Figure 1B, the COL-Cre genetic model targets more cells than TNC-Cre. Thus, GFP+ cells localized to the renal interstitium in both genetic models, significantly increased in number following UUO, and were more numerous in the COL-Cre compared to the TNC-Cre mouse.
Number of COL-Cre+ cells similar to α-SMA+ and PDGFRβ+ cells
To determine how efficiently our genetic models marked MPIC, we should ideally compare the expression of GFP+ with a marker specific for MPIC. Though no perfect marker exists, we chose α-SMA and PDGFRβ because both are associated with collagen I-producing interstitial cells in the injured kidney. Expression of these markers was compared with GFP+ cells in the COL-Cre mouse as the TNC-Cre is limited to the medulla. At 7 days after UUO, the number of α-SMA+ and GFP+ cells was greatest in the cortico-medullary region (Figure 2A). Quantification revealed a similar amount of α-SMA+ and GFP+ area though only about 40% of the cells expressed both GFP and α-SMA (Figure 2B). Interestingly, there was also a spatial difference in distribution of α-SMA+ and GFP+ cells with a predominance of α-SMA+ cells in the cortex and GFP+ cells in the medulla (Figure 2A). The number of GFP+ and PDGFRβ+ cells, as assessed by flow cytometry (Figure 2C–H), increased slightly from 3 to 7 days after injury with roughly 30% more GFP+ cells at each time point (Figure 2H). Thus, the number of COL-Cre+ (i.e. GFP+) cells is equal to or greater than the number of α-SMA+ and PDGFRβ+ cells, suggesting robust recombination at the mT/mG locus.
Figure 2.

COL-Cre+ cells similar in number compared to α-SMA+ and PDGFRβ+ cells. Frozen sections from COL-Cre;mT/mG mice at 7 days after UUO were stained with α-SMA (A). The fluorescence as a % of total area was quantified for the corticomedullary area, the region with the most GFP+ and α-SMA+ cells (B). Means ± standard error (SE) are shown based on at least 5 pictures per kidney and a total of 3 mice (B). GFP+ and PDGFRβ+ cells were measured by flow cytometry in COL-Cre kidneys (C–H) obstructed for either 3 (C, D) or 7 (F, G) days with appropriate isotype-matched control for gating (E). The number of GFP+ and PDGFRβ+ cells, as a percentage of viable, single cells is shown at both 3 and 7 days with at least 3 animals per group (H).
High recombination efficiency among GFP+ cells in COL-Cre;Tgfbr2fl/fl and TNC-Cre;Tgfbr2fl/fl mice
To confirm that the recombination efficiency at the Tgfbr2 locus was similar to that of the mT/mG locus, we determined recombination among the GFP+ cells after crossing both COL-Cre and TNC-Cre mice with the Tgfbr2fl/fl mice. Recombination in the COL-Cre;Tgfbr2fl/fl and TNC-Cre;Tgfbr2fl/fl mice was verified by polymerase chain reaction (PCR) with primers that amplify a sequence present only after recombination occurs (Figure 3A, 3B). To define recombination among GFP+ cells, we sorted these cells from kidneys of obstructed COL-Cre;Tgfbr2fl/fl, COL-Cre;WT, TNC-Cre;Tgfbr2fl/fl and TNC-Cre;WT mice and passaged them up to 4 times. Cells derived from TNC-Cre;Tgfbr2fl/fl or COL-Cre;Tgfbr2fl/fl mice had virtually no TβRII expression (Figure 3C, 3E) and had minimal responses to TGF-β1 as measured by Smad2 phosphorylation (Figure 3D, 3F). Thus, the GFP+ cells have no appreciable TβRII expression and no detectable response to TGF-β1.
Figure 3.

High recombination efficiency among GFP+ cells in COL-Cre and TNC-Cre crossed with Tgfbr2fl/fl mice. DNA was extracted from kidneys of COL-Cre;Tgfbr2fl/fl (A), TNC-Cre;Tgfbr2fl/fl (B), and Tgfbr2fl/fl littermate mice (A,B) and PCR performed to detect recombination. (A, B) DNA from the γGT-Cre;Tgfbr2fl/fl murine renal cortex was used as a positive control. (B) White lines represent where lanes within the same gel were moved. (C–F) GFP+ cells from COL-Cre;WT, COL-Cre;Tgfbr2fl/fl, TNC-Cre;WT, and TNC-Cre;Tgfbr2fl/fl mice were isolated by FACS three days after UUO and grown as primary cells in culture. (C, E) Immunoblots of TβRII expression with immortalized fibroblasts isolated from Tgfbr2fl/fl uninjured mice used as positive control and the same cells treated with adeno-Cre used as negative control. (D,F) Immunoblots measured Smad2 phosphorylation by these primary cells in response to TGF-β1 stimulation for 48hrs with focal adhesion kinase (FAK) used as loading control. (F) White lines represent where lanes within the same gel were moved.
Deleting TβRII reduces collagen I synthesis by MPIC in vitro and in Vivo
We investigated whether or not TGF-β1’s stimulatory effect on collagen I production reported in vitro(11, 12) could be recapitulated in FACS-sorted primary MPIC cells described above. Cells from COL-Cre;WT and TNC-Cre;WT mice expressed markers consistent with MPIC and had significantly more collagen I expression at baseline compared to the cells lacking TβRII (Figure 4A–4C). Moreover, both COL-Cre;WT and TNC-Cre;WT cells had increased collagen I expression after incubation with TGF-β1 for 48hrs; however, this response was absent in the cells sorted from COL-Cre;Tgfbr2fl/fl and TNC-Cre;Tgfbr2fl/fl mice (Figure 4B and 4C). We immortalized these cells and assessed their response to other growth factors (e.g. connective tissue growth factor (CCN2), PDGF). Although cells from both COL-Cre;WT and COL-Cre;Tgfbr2fl/fl mice had morphologic changes in response to growth factors other than TGF-β, there was not a robust expression of collagen I by either set of cells (Supplemental Figure 2). Thus, collagen I synthesis in vitro was heavily dependent upon TβRII-dependent TGF-β signaling but not other growth factors in vitro.
Figure 4.

TβRII null cells isolated from obstructed kidneys have reduced collagen I production in vitro. (A) GFP+ cells sorted from obstructed COL-Cre;WT and TNC-Cre;WT mice and grown as primary cell cultures expressed PDGFRβ, vimentin, and α-SMA, consistent with renal fibroblasts, but not zona occludens-1 (ZO-1), an epithelial marker, with inner medullary collecting duct (IMCD) and proximal tubule epithelial cells (PTEC) as controls. White lines denote lanes moved within a blot. (B, C) Primary cells sorted from COL-Cre;WT, COL-Cre;Tgfbr2fl/fl, TNC-Cre;WT, and TNC-Cre;Tgfbr2fl/fl mice were stimulated with TGF-β1 for 48 hours and immunoblotted for collagen I. (D, E) GFP+ cells were sorted from COL-Cre;WT (n=5) and COL-Cre;Tgfbr2fl/fl (n=5) mice 3 days after UUO, and RNA was isolated and reverse transcribed to cDNA. RT-PCR was performed using primers for COL1A1, COL1A2 (C), α-SMA, CCN2, and PAI-1 (D) and normalized to GAPDH, validated by comparison with a panel of housekeeping genes, with relative expression levels calculated using the delta-delta Ct formula.
As cell culture conditions differ from the injured kidney microenvironment, we sorted GFP+ cells from both obstructed COL-Cre;WT and COL-Cre;Tgfbr2fl/fl kidneys and immediately isolated RNA to measure collagen I transcripts. TβRII+ MPIC had twice the amount of COL1A1 and COL1A2 transcripts by real-time PCR as did MPIC lacking TβRII (Figure 4D). GFP+ cells from COL-Cre;Tgfbr2fl/fl mice had 40% reduced expression of α-SMA, a marker of activated fibroblasts shown to be upregulated by TGF-β in vitro (Figure 4E). Similarly, cells from COL-Cre;WT mice had almost threefold the cDNA levels of CCN2 and PAI-1 (plasminogen activator inhibitor-1), transcriptional targets of TGF-β signaling, compared to COL-Cre;Tgfbr2fl/fl mice (Figure 4E). Thus, these data demonstrate that COL-Cre+ cells lacking TβRII have reduced transcription of both TGF-β target genes and collagen I and cultured TβRII null primary MPIC have reduced collagen I protein expression.
Inhibiting TGF-β signaling in MPIC does not reduce fibrosis following UUO
To determine if TGF-β signaling in MPIC increases fibrosis in disease models, UUO was performed on TNC-Cre;Tgfbr2fl/fl, COL-Cre;Tgfbr2fl/fl and Tgfbr2fl/fl littermate mice. Surprisingly, there were no major differences between the genotypes in tubular dilation, epithelial flattening, or extracellular matrix accumulation at days 3 (data not shown) and 7 after UUO injury (Figure 5A). There were no differences in collagen I, fibronectin, and collagen IV expression by immunohistochemistry at 7 days after injury (Figure 5A). Similarly, no quantitative difference in collagen I expression was detected by immunoblots at day 7 and 14 after UUO between COL-Cre;Tgfbr2fl/fl and Tgfbr2fl/fl mice (Figure 5B–E) or TNC-Cre;Tgfbr2fl/fl and Tgfbr2fl/fl mice (Supplemental figure 3).
Figure 5.
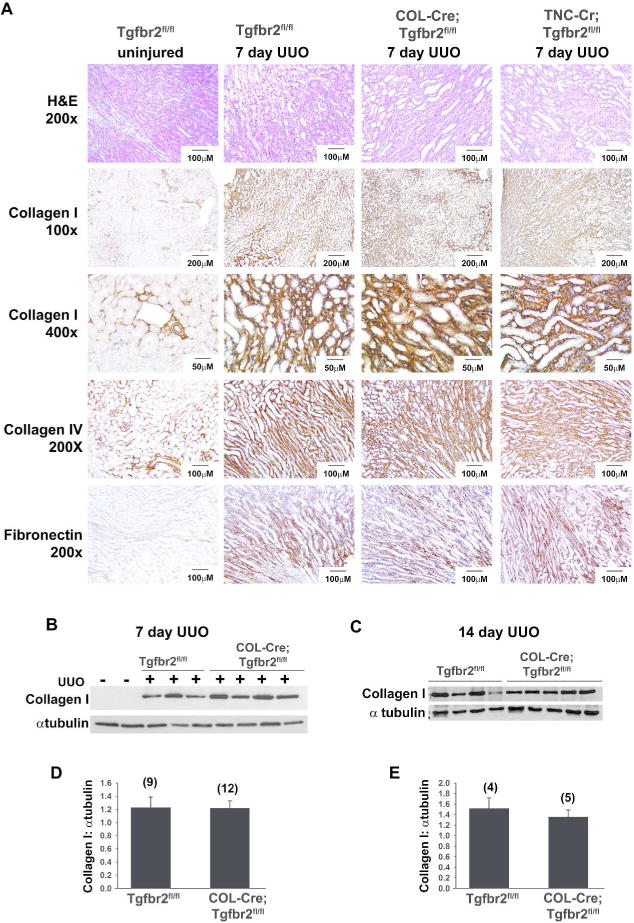
Deleting TβRII using COL-Cre or TNC-Cre does not protect against fibrosis after UUO. (A) Collagen I, collagen IV, and fibronectin staining were performed on 7 day UUO kidneys. Although separate Tgfbr2fl/fl mice (littermates) were used as controls for the COL-Cre;Tgfbr2fl/fl and TNC-Cre;Tgfbr2fl/fl mice, just one representative set is shown (A). A representative immunoblot of tissue lysates from unobstructed kidneys and 7 day post-UUO kidneys shows an upregulation of collagen I after injury, but no significant difference between genotypes (B). Densitometry was performed, and collagen I expressed as a ratio with α tubulin for loading control (D). Similarly, kidneys obstructed for 14 days were also immunoblotted with collagen I (C), and quantified (E). The number of kidneys quantified in each group is listed in parenthesis (D, E), and bars represent standard errors.
TGF-β signaling can alter cellular proliferation and exert both pro- and anti-inflammatory effects depending upon the cellular milieu, so we investigated whether deleting TβRII in COL-Cre cells alters the number of MPIC or inflammation after UUO. There were no major changes in GFP+ or PDGFRβ+ expression between COL-Cre and COL-Cre;Tgfbr2fl/fl mice at 3 days after UUO (Figure 6A, 6B, 6D). Staining of F4/80, a macrophage marker, revealed a small but statistically significant increase in F4/80+ cells (14.8% to 19%) when TβRII was deleted using COL-Cre (Figure 6C, 6E). Thus, deleting TβRII using COL-Cre does not ameliorate fibrosis or alter the number of MPIC, but does increase F4/80 infiltration.
Figure 6.

Deleting TβRII in fibroblasts does not change GFP or PDGFRβ+ expression but does alter inflammation. (A–C) Flow cytometry was performed on kidneys 3 days after UUO from COL-Cre;WT and COL-Cre;Tgfbr2fl/fl mice to measure GFP+, PDGFRβ+, and F4/80+ with the isotype control (IgG2a) in Figure 2G used for both PDGFRβ and F4/80. The percentage of GFP+ (D), PDGFRβ+ (D), and F4/80+ (E) cells among viable single cells was measured among 5 COL-Cre;WT and 3 COL-Cre;Tgfbr2fl/fl mice (* for p<0.05).
COL-Cre;Tgfbr2fl/fl mice are not protected from aristolochic acid-induced injury
Medullary and cortical fibroblasts are distinct populations,(16, 29) and UUO targets the collecting system with a predominance of medullary interstitial cells (Figure 1B, 2A). We then assessed the role of TGF-β signaling in MPIC after a cortical injury as heterogeneous responses to TGF-β1 have been noted by subpopulations of renal interstitial cells.(30) We injured COL-Cre;Tgfbr2fl/fl mice with aristolochic acid which targets the proximal tubule acutely, produces a delayed TGF-β-dependent fibrotic response, and causes end-stage renal disease in humans.(31, 32) Five weeks after aristolochic acid injections, there was an increased number of COL-Cre cells which was particularly robust in the cortico-medullary and medullary regions (Figure 7A). Both the COL-Cre;Tgfbr2fl/fl and Tgfbr2fl/fl mice had dilated cortical tubules with casts, an inflammatory infiltrate, and expansion of interstitial matrix (Figure 7B). Collagen I expression by immunoblots and macrophage infiltration by immunohistochemistry were similarly increased in injured COL-Cre;Tgfbr2fl/fl and Tgfbr2fl/fl mice (Figure 7C–F). Therefore, abrogating TGF-β signaling in COL-Cre+ cells did not alter the fibrotic response to either a medullary or cortical renal injury.
Figure 7.

COL-Cre;Tgfbr2fl/fl mice are not protected from fibrosis 5 weeks after aristolochic acid. (A) Frozen sections of COL-Cre+ cells (green fluorescence) in COL-Cre;Tgfbr2fl/fl mice with mT/mG reporter. (B) H&E staining and (C) collagen I and F4/80 staining were performed on COL-Cre;Tgfbr2fl/fl and Tgfbr2fl/fl kidneys after aristolochic acid injury. (D) Immunoblots of collagen I and FAK (loading control) of renal cortical lysates after aristolochic acid administration and quantified by densitometry (E). F4/80+ area was quantified using Image J on 10 high powered fields per mouse, n=4 per genotype (F).
Discussion
TGF-β is a major pro-fibrotic growth factor involved in renal tubulointerstitial fibrosis. Controversy surrounds the origin of MPIC that transform into myofibroblasts, but these collagen I producing interstitial cells are thought to be the mediators of TGF-β-dependent fibrosis. When we selectively deleted TβRII in mice using Cre driven by the COL1A2 and tenascin C promoters, there was surprisingly no difference in fibrosis after either UUO or aristolochic acid-induced injury although renal MPIC lacking TβRII had a significant reduction in collagen I production in vitro and transcription in vivo. Our results suggest that TGF-β signaling in MPIC is not required for the development of fibrosis.
Surprisingly, no reduction in fibrosis was noted when we deleted TβRII in MPIC using two different genetic models and two renal injuries. This unexpected finding raises questions about the adequacy of recombination. Establishing the recombination efficiency of MPIC is challenging given the absence of specific markers for this population. However, the number of COL1A2+ (i.e. GFP+) cells in the injured kidney equaled or exceeded the number of α-SMA+ cells and PDGFRβ+ cells, both commonly used markers for MPIC. It is possible that Cre has greater efficiency targeting mT/mG compared to the Tgfbr2 locus. Therefore, we isolated the GFP+ cells and verified that TβRII protein expression was virtually nonexistent, canonical TGF-β signaling abrogated, and collagen I production drastically reduced. Thus, it is unlikely that our results were due to low recombination rates.
There are several other possible explanations for our findings. First, renal interstitial cells also perform beneficial biological effects such as providing structural support and reparative growth factors to injured epithelium as well as having immunomodulatory properties.(33, 34) Deleting TβRII may compromise these functions, leading to an increase in production of pro-fibrotic factors such as (e.g. PDGF) by epithelial or inflammatory cells which may induce paracrine signaling on neighboring interstitial cells. Second, the deletion of TβRII in MPIC may result in compensatory increased autocrine signaling of other growth factors (e.g. PDGF, CCN2) proven to induce proliferation and collagen synthesis in fibroblasts.(8, 35) Many growth factors have pro-fibrotic effects on fibroblasts, and abrogating just TGF-β signaling may not be sufficient. Although TβRII−/− MPIC did not have an increased production of collagen I in response to some of these factors in vitro, others have described a similar lack of collagen I upregulation in vitro despite pro-fibrotic effects in vivo.(36–38) Immortalizing the MPIC may have altered their response to these growth factors though the response to TGF-β1 was similar to primary cells. Finally, cells other than fibroblasts, such as endothelial and epithelial cells, may play an important role in TGF-β-dependent fibrosis progression after kidney injury.(39, 40) In the aristolochic acid model, tubular epithelial cells were identified as the main source of collagen I expression.(41) Although healthy tubular epithelial cells do not produce collagen I, chronic tubular injury can lead to de-differentiation and ECM production, effects both associated with excess TGF-β signaling.(42) Previously, we reported that deleting TβRII in collecting duct epithelial cells worsened the response to UUO,(43) but this does not preclude the possibility that the tubular epithelium may be an important mediator of TGF-β-dependent injury. There is an important dose-dependent response to TGF-β,(37, 44) and just as excessive signaling is deleterious,(5, 45) the absence of any TβRII-dependent signaling may also have adverse consequences.
Our data suggest that MPIC comprise a heterogeneous population of cells, consistent with previous reports in the literature.(33, 46) We have refrained from defining any of these cells as fibroblasts, pericytes, or myofibroblasts because ultrastructural analysis would be necessary for proper classification, but these cells should be targeted for recombination if the collagen I promoter is active.(47) The term MPIC more accurately describes our diverse target population, composed of different cells, which produces ECM and is sensitive to TGF-β’s pro-fibrotic effects. Immunofluorescence has identified at least three distinct subsets of MPIC: COL+,α-SMA+; COL+,α-SMA−; and COL−,α-SMA+. Though α-SMA expression has been considered synonymous with collagen I production, our finding of a distinct COL+,α-SMA− population challenges this assumption. This population has been described in other fibrotic organs(48) and has been previously termed as proto-myofibroblasts, or cells that produce collagen I without α-SMA expression.(49) Though α-SMA is considered the best available marker for myofibroblasts, abundant literature suggests that this marker does not always correlate with ECM production.(18, 19, 50) Whether or not there are functional differences between the subgroups of MPIC and whether the diversity of MPIC is due to heterogeneous origins or functional plasticity are important questions for future research.
Recently, the α-SMA promoter was used to selectively delete TβRII, and a modest reduction in fibrosis after UUO was observed.(9) The difference between our results and those with the α-SMA promoter may have been mediated by the COL−,α-SMA+ subpopulation. The authors attributed the TGF-β-dependent effects in injury to non-proliferating α-SMA+ cells (predominantly from the bone marrow) rather than proliferating resident fibroblasts.(9) Assuming these are COL−,α-SMA+ cells, it is possible that these cells may have modulated the fibrotic response to injury through effects independent of direct collagen I production (e.g. immunomodulation).
The TNC-Cre mouse model was used to determine whether medullary interstitial cells play a different role in injury from those MPIC more broadly targeted by COL-Cre. This population has not been well studied in renal injury and, to the best of our knowledge, only one other study has genetically targeted this population.(51) Though cortical and medullary renal interstitial cells have distinct roles in the uninjured kidney (e.g. erythropoietin production), the similar phenotype in COL1A2-Cre and TNC-Cre implies that these cells may perform similar functions after injury.
Although TGF-β strongly promotes progression of tubulointerstitial fibrosis, our studies show that deleting TβRII in MPIC, the cells assumed to be responsible for TGF-β-dependent ECM production, does not ameliorate the fibrotic response to either UUO or aristolochic acid. More research is needed to define how TGF-β promotes renal fibrosis and the different functional characteristics of the heterogeneous collagen-producing interstitial cells. TGF-β is an attractive therapeutic target but clinical trials have been disappointing implying that a better understanding of how this pleiotropic growth factor mediates response to injury may provide insights into future anti-fibrotic strategies.
Methods
Animal Models
The Institutional Animal Care and Use Committee of Vanderbilt University approved all procedures which were consistent with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Tgfbr2floxed(52) and wild type mice were crossed with mice containing Cre under the control of the proα2(I) collagen promoter, COL1A2-Cre/ERT, as previously described(21) (generous gift from Neil Bhowmick), or mice with Cre driven by the Tenascin C promoter, TNC-Cre/ERT.(25) Both conditional knockout models are tamoxifen-inducible (Cre/ERT) and, along with their littermate controls, were injected with 4-OH tamoxifen (4mg/25gm mouse) intraperitoneally (IP) at age 5–7 weeks, every other day × 4, with a fifth injection after injury. Mice were further crossed with the mT/mG reporter mouse (Jackson laboratory).
Unilateral Ureteral Obstruction
UUO was performed on mice aged 8–10 weeks, backcrossed onto C57BL/6 × 10 generations with the exception of the mT/mG (mixed) crossed with the TNC-Cre(ERT). The right kidney was exposed dorsally via a flank incision, and two sutures were placed on the ureter just distal to the renal pelvis. A fifth injection of tamoxifen was administered IP the day after surgery, and mice were euthanized at 3, 7, or 14 days after surgery.
Aristolochic Acid Injury
Aristolochic acid (Sigma) was administered to mice IP 5mg/kg × 5 days with the fifth injection of tamoxifen 3 days after the last dose of aristolochic acid and sacrificed five weeks after the last dose of aristolochic acid.
Fluorescence-activated cell sorting (FACS) on tissue
For tissue preparation, the obstructed kidney was minced and incubated with 2mg/mL collagenase II (Sigma), 1mL DMEM, and DNAse (BioRad 10ul/mL) in a shaker at 37C for 1 hour. The tissue was filtered (70 μm), centrifuged, and incubated in red blood cell lysis buffer (15.5mM NH4Cl, 1mM KHCO3, 10 μM EDTA) at 37C for 5 minutes, neutralized with PBS, centrifuged, and resuspended in PBS with 1%FBS. The tissue was passed through a 50 μM strainer, and tissue from COL-Cre mice (and controls) was passed through a 24 μM filter to remove the glomeruli. The FACSAria II from BD Biosciences in the Research Flow Cytometry Core Laboratory at the Nashville VA Medical Center was used for sorting of GFP+ cells.
For flow cytometry, samples were incubated with Fc blocking antibody (Biolegend) on ice, then incubated with APC-conjugated anti-F4/80, anti-CD140b (PDGFRβ), or the appropriate APC-conjugated isotype controls (Biolegend) for 30 minutes at room temperature. Cells were analyzed on a FACS Canto II cytometer with FACSDiVa v6.1 software for data acquisition and analysis at the Nashville VA Medical Center’s Flow Cytometry Laboratory.
Immunohistochemistry, Frozen Sections, and Immunoblots
Mouse kidneys were fixed in 4% paraformaldehyde for 12–18hrs, paraffin embedded, and incubated with the following primary antibodies: collagen IV (Biodesign), collagen I, F4/80 (Abcam), and fibronectin (Rockland). To prepare frozen sections for mT/mG mice, tissue was fixed for 4–6hours in 4% paraformaldehyde, dehydrated in 30% sucrose overnight in 4C, mounted in O.C.T, and sectioned by cryostat. Slides were fixed for 20 minutes with 4% paraformaldehyde. Frozen sections were stained with α-SMA (DAKO), biotinylated, and incubated with streptavidin-conjugated Alexa-fluor350. To quantify GFP+, α-SMA+, and F4/80+ staining, images were taken with Olympus BX51 microscope and Image J with the Color Deconvolution function was used.
Obstructed kidneys were minced in lysis buffer (50mM Tris HCl, 150mM NaCl, 1mM EDTA, 2%SDS, 1%Triton, phosphatase and protease inhibitor cocktails (Sigma)), homogenized by sonication, and clarified by centrifugation. Protein quantification was done using Thermo BCA kit, and electrophoresis performed with SDS/PAGE, and incubated with the following primary antibodies: collagen I (MD Biosciences), TβRII and PDGFRβ (Santa Cruz), pSmad2 and total Smad 2 (Cell Signaling), vimentin (ThermoScientific), α-SMA (Sigma), ZO-1 (Invitrogen).
Cell culture
GFP+ cells were sorted by FACS from COL-Cre;WT, COL-Cre;Tgfbr2fl/fl, TNC-Cre;WT, and TNC-Cre;Tgfbr2fl/fl mice at 3 days post UUO. The primary cells were grown in DMEM/F12 media supplemented with 10% FBS and penicillin/streptomycin antibiotics. At passage 3–4, cells were put in 1%FBS ± various concentrations of TGF-β1 (R&D) for 48 hours, and lysates were made using RIPA buffer plus protease and phosphatase inhibitors (Sigma cocktail). Cells were immortalized by sv40-adenovirus, stimulated with PDGF-BB (R&D Systems), CTGF (gift from Pampee Young), or EGF (Millipore), and imaged with a Nikon Eclipse TE300 inverted scope.
Polymerase Chain Reaction
RNA was extracted from cells using the Qiagen RNeasy kit, and Bio-Rad’s iScript cDNA Synthesis kit generated cDNA. Quantitative real-time PCR was performed using the Bio-Rad CFX96 thermal cycler and primers are listed in Supplemental Table 1. The use of GAPDH as a housekeeping transcript was validated by comparison to a panel of commonly used reference cDNAs.
Statistical Analyses
The student’s t-test with unequal variance was used to compare two sets of data with p<0.05 considered statistically significant. Data are shown as means ± standard error. For real time PCR, the delta-delta Ct equation was used to determine relative expression.
Supplementary Material
Acknowledgments
This work was supported by a Career Development Award from the Department of Veteran’s Affairs, Veteran’s Health Administration, Office of Research and Development (LG), Pediatric Nephrology Center of Excellence (LG), VA Merit Awards, 2I01BX000320 (RCH) and 1I01BX002196 (RZ); the National Institutes of Health Grants RO1-DK51265 (RCH), RO1-DK95785 (RCH), RO1-DK083187 (RZ), RO1-DK075594 (RZ), RO1-DK069221 (RZ). This material is based upon work supported in part by the Department of Veterans Affairs, Veterans Health Administration, Biomedical Laboratory Research and Development. This material is the result of work supported with resources and the use of facilities at the VA Tennessee Valley Healthcare System. An abstract based on this work was presented at the American Society of Nephrology (November 2013) and at the Keystone Fibrosis Conference (March 2014).
Financial Disclosures: Agnes Fogo was a pathology consultant for the Genzyme FSGS clinical trial pilot phase for anti-TGF-beta therapy, an agent not used in this study.
Footnotes
Supplementary information is available at Kidney International’s website.
References
- 1.Wrana JL, Attisano L, Carcamo J, et al. TGF beta signals through a heteromeric protein kinase receptor complex. Cell. 1992;71(6):1003–14. doi: 10.1016/0092-8674(92)90395-s. [DOI] [PubMed] [Google Scholar]
- 2.Yamashita H, ten Dijke P, Franzen P, et al. Formation of hetero-oligomeric complexes of type I and type II receptors for transforming growth factor-beta. J Biol Chem. 1994;269(31):20172–8. [PubMed] [Google Scholar]
- 3.Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009;19(1):128–39. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koesters R, Kaissling B, Lehir M, et al. Tubular overexpression of transforming growth factor-beta1 induces autophagy and fibrosis but not mesenchymal transition of renal epithelial cells. Am J Pathol. 177(2):632–43. doi: 10.2353/ajpath.2010.091012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miyajima A, Chen J, Lawrence C, et al. Antibody to transforming growth factor-beta ameliorates tubular apoptosis in unilateral ureteral obstruction. Kidney Int. 2000;58(6):2301–13. doi: 10.1046/j.1523-1755.2000.00414.x. [DOI] [PubMed] [Google Scholar]
- 6.Ziyadeh FN, Hoffman BB, Han DC, et al. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proc Natl Acad Sci U S A. 2000;97(14):8015–20. doi: 10.1073/pnas.120055097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boor P, Ostendorf T, Floege J. Renal fibrosis: novel insights into mechanisms and therapeutic targets. Nature reviews Nephrology. 2010;6(11):643–56. doi: 10.1038/nrneph.2010.120. [DOI] [PubMed] [Google Scholar]
- 8.Wu CF, Chiang WC, Lai CF, et al. Transforming growth factor beta-1 stimulates profibrotic epithelial signaling to activate pericyte-myofibroblast transition in obstructive kidney fibrosis. Am J Pathol. 2013;182(1):118–31. doi: 10.1016/j.ajpath.2012.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lebleu VS, Taduri G, O’Connell J, et al. Origin and function of myofibroblasts in kidney fibrosis. Nature medicine. 2013;19(8):1047–53. doi: 10.1038/nm.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Desmouliere A, Geinoz A, Gabbiani F, et al. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol. 1993;122(1):103–11. doi: 10.1083/jcb.122.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ignotz RA, Endo T, Massague J. Regulation of fibronectin and type I collagen mRNA levels by transforming growth factor-beta. J Biol Chem. 1987;262(14):6443–6. [PubMed] [Google Scholar]
- 12.Roberts AB, Sporn MB, Assoian RK, et al. Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci U S A. 1986;83(12):4167–71. doi: 10.1073/pnas.83.12.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee K, Boyd KL, Parekh DV, et al. Cdc42 promotes host defenses against fatal infection. Infection and immunity. 2013;81(8):2714–23. doi: 10.1128/IAI.01114-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Inoue T, Plieth D, Venkov CD, et al. Antibodies against macrophages that overlap in specificity with fibroblasts. Kidney Int. 2005;67(6):2488–93. doi: 10.1111/j.1523-1755.2005.00358.x. [DOI] [PubMed] [Google Scholar]
- 15.Le Hir M, Kaissling B. Distribution and regulation of renal ecto-5′-nucleotidase: implications for physiological functions of adenosine. Am J Physiol. 1993;264(3 Pt 2):F377–87. doi: 10.1152/ajprenal.1993.264.3.F377. [DOI] [PubMed] [Google Scholar]
- 16.Kaissling B, Hegyi I, Loffing J, et al. Morphology of interstitial cells in the healthy kidney. Anatomy and embryology. 1996;193(4):303–18. doi: 10.1007/BF00186688. [DOI] [PubMed] [Google Scholar]
- 17.Taneda S, Hudkins KL, Topouzis S, et al. Obstructive uropathy in mice and humans: potential role for PDGF-D in the progression of tubulointerstitial injury. J Am Soc Nephrol. 2003;14(10):2544–55. doi: 10.1097/01.asn.0000089828.73014.c8. [DOI] [PubMed] [Google Scholar]
- 18.Hinz B. The myofibroblast: paradigm for a mechanically active cell. Journal of biomechanics. 2010;43(1):146–55. doi: 10.1016/j.jbiomech.2009.09.020. [DOI] [PubMed] [Google Scholar]
- 19.Roufosse C, Bou-Gharios G, Prodromidi E, et al. Bone marrow-derived cells do not contribute significantly to collagen I synthesis in a murine model of renal fibrosis. J Am Soc Nephrol. 2006;17(3):775–82. doi: 10.1681/ASN.2005080795. [DOI] [PubMed] [Google Scholar]
- 20.Lin SL, Kisseleva T, Brenner DA, et al. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol. 2008;173(6):1617–27. doi: 10.2353/ajpath.2008.080433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng B, Zhang Z, Black CM, et al. Ligand-dependent genetic recombination in fibroblasts: a potentially powerful technique for investigating gene function in fibrosis. Am J Pathol. 2002;160(5):1609–17. doi: 10.1016/S0002-9440(10)61108-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bou-Gharios G, Garrett LA, Rossert J, et al. A potent far-upstream enhancer in the mouse pro alpha 2(I) collagen gene regulates expression of reporter genes in transgenic mice. J Cell Biol. 1996;134(5):1333–44. doi: 10.1083/jcb.134.5.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Truong LD, Foster SV, Barrios R, et al. Tenascin is an ubiquitous extracellular matrix protein of human renal interstitium in normal and pathologic conditions. Nephron. 1996;72(4):579–86. doi: 10.1159/000188943. [DOI] [PubMed] [Google Scholar]
- 24.Ekblom P, Aufderheide E. Stimulation of tenascin expression in mesenchyme by epithelial-mesenchymal interactions. The International journal of developmental biology. 1989;33(1):71–9. [PubMed] [Google Scholar]
- 25.He W, Xie Q, Wang Y, et al. Generation of a Tenascin-C-CreER2 Knockin Mouse Line for Conditional DNA Recombination in Renal Medullary Interstitial Cells. PloS one. 2013;8(11):e79839. doi: 10.1371/journal.pone.0079839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou L, Fu P, Huang XR, et al. Mechanism of chronic aristolochic acid nephropathy: role of Smad3. Am J Physiol Renal Physiol. 2010;298(4):F1006–17. doi: 10.1152/ajprenal.00675.2009. [DOI] [PubMed] [Google Scholar]
- 27.Muzumdar MD, Tasic B, Miyamichi K, et al. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45(9):593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- 28.Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Developmental biology. 2002;244(2):305–18. doi: 10.1006/dbio.2002.0597. [DOI] [PubMed] [Google Scholar]
- 29.Grupp C, Troche I, Klass C, et al. A novel model to study renal myofibroblast formation in vitro. Kidney Int. 2001;59(2):543–53. doi: 10.1046/j.1523-1755.2001.059002543.x. [DOI] [PubMed] [Google Scholar]
- 30.Tang N, Cunningham K, Enger MD. TGF beta elicits opposite responses in clonal subpopulations of NRK-49F cells. Exp Cell Res. 1991;196(1):13–9. doi: 10.1016/0014-4827(91)90450-9. [DOI] [PubMed] [Google Scholar]
- 31.Huang L, Scarpellini A, Funck M, et al. Development of a chronic kidney disease model in C57BL/6 mice with relevance to human pathology. Nephron extra. 2013;3(1):12–29. doi: 10.1159/000346180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rui HL, Wang YY, Cheng H, et al. JNK-dependent AP-1 activation is required for aristolochic acid-induced TGF-beta1 synthesis in human renal proximal epithelial cells. Am J Physiol Renal Physiol. 2012;302(12):F1569–75. doi: 10.1152/ajprenal.00560.2011. [DOI] [PubMed] [Google Scholar]
- 33.Boor P, Floege J. The renal (myo-)fibroblast: a heterogeneous group of cells. Nephrol Dial Transplant. 2012;27(8):3027–36. doi: 10.1093/ndt/gfs296. [DOI] [PubMed] [Google Scholar]
- 34.Cappellesso-Fleury S, Puissant-Lubrano B, Apoil PA, et al. Human fibroblasts share immunosuppressive properties with bone marrow mesenchymal stem cells. Journal of clinical immunology. 2010;30(4):607–19. doi: 10.1007/s10875-010-9415-4. [DOI] [PubMed] [Google Scholar]
- 35.Gao X, Li J, Huang H, et al. Connective tissue growth factor stimulates renal cortical myofibroblast-like cell proliferation and matrix protein production. Wound repair and regeneration: official publication of the Wound Healing Society [and] the European Tissue Repair Society. 2008;16(3):408–15. doi: 10.1111/j.1524-475X.2008.00380.x. [DOI] [PubMed] [Google Scholar]
- 36.Chen YT, Chang FC, Wu CF, et al. Platelet-derived growth factor receptor signaling activates pericyte-myofibroblast transition in obstructive and post-ischemic kidney fibrosis. Kidney Int. 2011;80(11):1170–81. doi: 10.1038/ki.2011.208. [DOI] [PubMed] [Google Scholar]
- 37.Alvarez RJ, Sun MJ, Haverty TP, et al. Biosynthetic and proliferative characteristics of tubulointerstitial fibroblasts probed with paracrine cytokines. Kidney Int. 1992;41(1):14–23. doi: 10.1038/ki.1992.3. [DOI] [PubMed] [Google Scholar]
- 38.Kordes C, Brookmann S, Haussinger D, et al. Differential and synergistic effects of platelet-derived growth factor-BB and transforming growth factor-beta1 on activated pancreatic stellate cells. Pancreas. 2005;31(2):156–67. doi: 10.1097/01.mpa.0000168222.05591.a0. [DOI] [PubMed] [Google Scholar]
- 39.Yang J, Lin SC, Chen G, et al. Adiponectin Promotes Monocyte-to-Fibroblast Transition in Renal Fibrosis. J Am Soc Nephrol. 2013;24(10):1644–59. doi: 10.1681/ASN.2013030217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rastaldi MP, Ferrario F, Giardino L, et al. Epithelial-mesenchymal transition of tubular epithelial cells in human renal biopsies. Kidney Int. 2002;62(1):137–46. doi: 10.1046/j.1523-1755.2002.00430.x. [DOI] [PubMed] [Google Scholar]
- 41.Fragiadaki M, Witherden AS, Kaneko T, et al. Interstitial fibrosis is associated with increased COL1A2 transcription in AA-injured renal tubular epithelial cells in vivo. Matrix biology: journal of the International Society for Matrix Biology. 2011;30(7–8):396–403. doi: 10.1016/j.matbio.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 42.Terashima H, Kato M, Yasumo H, et al. A sensitive short-term evaluation of antifibrotic effects using newly established type I collagen reporter transgenic rats. Am J Physiol Renal Physiol. 2010;299(4):F792–801. doi: 10.1152/ajprenal.00141.2009. [DOI] [PubMed] [Google Scholar]
- 43.Gewin L, Bulus N, Mernaugh G, et al. TGF-beta receptor deletion in the renal collecting system exacerbates fibrosis. J Am Soc Nephrol. 21(8):1334–43. doi: 10.1681/ASN.2010020147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma LJ, Jha S, Ling H, et al. Divergent effects of low versus high dose anti-TGF-beta antibody in puromycin aminonucleoside nephropathy in rats. Kidney Int. 2004;65(1):106–15. doi: 10.1111/j.1523-1755.2004.00381.x. [DOI] [PubMed] [Google Scholar]
- 45.Galarreta CI, Thornhill BA, Forbes MS, et al. Transforming growth factor-beta1 receptor inhibition preserves glomerulotubular integrity during ureteral obstruction in adults but worsens injury in neonatal mice. Am J Physiol Renal Physiol. 2013;304(5):F481–90. doi: 10.1152/ajprenal.00496.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chang HY, Chi JT, Dudoit S, et al. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc Natl Acad Sci U S A. 2002;99(20):12877–82. doi: 10.1073/pnas.162488599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaissling B, Le Hir M. The renal cortical interstitium: morphological and functional aspects. Histochemistry and cell biology. 2008;130(2):247–62. doi: 10.1007/s00418-008-0452-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Magness ST, Bataller R, Yang L, et al. A dual reporter gene transgenic mouse demonstrates heterogeneity in hepatic fibrogenic cell populations. Hepatology. 2004;40(5):1151–9. doi: 10.1002/hep.20427. [DOI] [PubMed] [Google Scholar]
- 49.Tomasek JJ, Gabbiani G, Hinz B, et al. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nature reviews Molecular cell biology. 2002;3(5):349–63. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 50.Okada H, Ban S, Nagao S, et al. Progressive renal fibrosis in murine polycystic kidney disease: an immunohistochemical observation. Kidney Int. 2000;58(2):587–97. doi: 10.1046/j.1523-1755.2000.00205.x. [DOI] [PubMed] [Google Scholar]
- 51.DiRocco DP, Kobayashi A, Taketo MM, et al. Wnt4/beta-catenin signaling in medullary kidney myofibroblasts. J Am Soc Nephrol. 2013;24(9):1399–412. doi: 10.1681/ASN.2012050512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chytil A, Magnuson MA, Wright CV, et al. Conditional inactivation of the TGF-beta type II receptor using Cre:Lox. Genesis. 2002;32(2):73–5. doi: 10.1002/gene.10046. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.