

Abstract
In AD, an imbalance between Aβ production and removal drives elevated brain Aβ levels and eventual amyloid plaque deposition. APP undergoes nonamyloidogenic processing via α-cleavage at the plasma membrane, amyloidogenic β- and γ-cleavage within endosomes to generate Aβ, or lysosomal degradation in neurons. Considering multiple reports implicating impaired lysosome function as a driver of increased amyloidogenic processing of APP, we explored the efficacy of targeting transcription factor EB (TFEB), a master regulator of lysosomal pathways, to reduce Aβ levels. CMV promoter-driven TFEB, transduced via stereotactic hippocampal injections of adeno-associated virus particles in APP/PS1 mice, localized primarily to neuronal nuclei and upregulated lysosome biogenesis. This resulted in reduction of APP protein, the α and β C-terminal APP fragments (CTFs), and in the steady-state Aβ levels in the brain interstitial fluid. In aged mice, total Aβ levels and amyloid plaque load were selectively reduced in the TFEB-transduced hippocampi. TFEB transfection in N2a cells stably expressing APP695, stimulated lysosome biogenesis, reduced steady-state levels of APP and α- and β-CTFs, and attenuated Aβ generation by accelerating flux through the endosome-lysosome pathway. Cycloheximide chase assays revealed a shortening of APP half-life with exogenous TFEB expression, which was prevented by concomitant inhibition of lysosomal acidification. These data indicate that TFEB enhances flux through lysosomal degradative pathways to induce APP degradation and reduce Aβ generation. Activation of TFEB in neurons is an effective strategy to attenuate Aβ generation and attenuate amyloid plaque deposition in AD.
SIGNIFICANCE STATEMENT A key driver for AD pathogenesis is the net balance between production and clearance of Aβ, the major component of amyloid plaques. Here we demonstrate that lysosomal degradation of holo-APP influences Aβ production by limiting the availability of APP for amyloidogenic processing. Using viral gene transfer of transcription factor EB (TFEB), a master regulator of lysosome biogenesis in neurons of APP/PS1 mice, steady-state levels of APP were reduced, resulting in decreased interstitial fluid Aβ levels and attenuated amyloid deposits. These effects were caused by accelerated lysosomal degradation of endocytosed APP, reflected by reduced APP half-life and steady-state levels in TFEB-expressing cells, with resultant decrease in Aβ production and release. Additional studies are needed to explore the therapeutic potential of this approach.
Keywords: Alzheimer's disease, amyloid, amyloid precursor protein, lysosome, TFEB
Introduction
In AD, increased generation of the Aβ peptide in neurons synergizes with impaired Aβ removal from the interstitial fluid (ISF) in driving extracellular amyloid plaque formation (Haass et al., 2012) and the resultant neuropathogenesis (Bloom, 2014). After synthesis in the endoplasmic reticulum, APP is trafficked from the Golgi to the plasma membrane, where it is cleaved by α-secretases within the Aβ peptide fragment region, in a manner that precludes Aβ generation (i.e., nonamyloidogenic or α-cleavage; Rajendran and Annaert, 2012). Alternately, APP is endocytosed, and sequentially cleaved by a β-secretase (BACE1) and then the γ-secretase complex (comprised of presenilins, nicastrin, APH-1, and PEN2) within endosomes, to generate Aβ peptides (amyloidogenic cleavage; Rajendran and Annaert, 2012). Accumulating evidence suggests that dysregulation of endocytosis (Harold et al., 2009; Ginsberg et al., 2010) with accumulation of large endosomes within neurons, as observed in preclinical stages of AD pathology (Cataldo et al., 2000) and in neurons transdifferentiated from fibroblasts of patients with AD (Israel et al., 2012), increases the proclivity for amyloidogenic processing of APP.
Endocytosed APP can be sorted back to the Golgi (Morel et al., 2013) or trafficked to lysosomes along with other endosomal cargo (Théry et al., 2002). Indeed, a large fraction of full-length APP is degraded within lysosomes (Caporaso et al., 1992, 1994; Golde et al., 1992; Haass et al., 1992). Experimental inhibition of lysosome function markedly prolongs APP half-life (Caporaso et al., 1992) and impairs endocytotic flux with accumulation of enlarged vesicles in dystrophic neurites mimicking AD pathology (Lee et al., 2011), with increased BACE1 (Chia et al., 2013) and β-C-terminal fragment (Golde et al., 1992; Sannerud et al., 2011) resulting in markedly increased Aβ generation. Relevant to this discussion is that impaired lysosomal function is observed with aging (Kato et al., 1998; Cuervo and Dice, 2000; Wolfe et al., 2013) and with loss of presenilins (due to mutations causing familial AD; Lee et al., 2010; Neely et al., 2011; Coen et al., 2012), and is suspected with polymorphisms in genes coding for lysosomal proteins, which confer increased risk for sporadic AD, including phospholipase D3 (Cruchaga et al., 2014), cystatin C (Hua et al., 2012), and Cathepsin D (Schuur et al., 2011). Also, levels of APP, its β-CTF, and Aβ are increased in mouse models of lysosome storage disorders, namely, Neimann-Pick disease (Kodam et al., 2010), mucolipidoses (Keilani et al., 2012), and sphingolipidoses (Tamboli et al., 2011). These observations incriminate lysosomal dysfunction as the common cellular mechanism in provoking increased Aβ generation and impaired Aβ removal (Funk and Kuret, 2012; Peric and Annaert, 2015).
As a therapeutic strategy to address lysosomal insufficiency in astrocytes, we previously evaluated activation of transcription factor EB (TFEB), a master inducer of lysosomal degradative pathways (Settembre et al., 2013a), and demonstrated that astrocytic TFEB expression stimulates Aβ uptake and its lysosomal degradation to reduce ISF Aβ (Xiao et al., 2014). In this study, we evaluated the efficacy of neuronal TFEB activation in enhancing lysosomal degradation of APP to reduce Aβ levels and attenuate amyloid plaque pathogenesis.
Materials and Methods
Studies in animal models.
APP/PS1 transgenic mice (B6C3-Tg (APPswe/PS1ΔE9)85Dbo/Mmjax) of either sex were obtained from The Jackson Laboratory (Jankowsky et al., 2004) and injected with AAV8 viral particles (generated by the Hope Center Viral Core at Washington University) driving expression of TFEB (AAV8-CMV-FLAG-TFEB) or GFP as control (AAV8-CMV-GFP), each with 2 μl of 1.5 × 1012 viral genomes per milliliter into bilateral hippocampi at 6 months of age, as described previously (Xiao et al., 2012). Wild-type mice of either sex were also injected with AAV8-CMV-FLAG-TFEB or AAV8-CMV-GFP at 4 months of age to assess the effect of TFEB transduction on endogenous APP processing after 4 months. Before injection, AAV particles were subjected to two rounds of cesium chloride gradient purification and titers were quantified by assessment of viral genomes. For plaque load studies, TFEB or control-transduced APP/PS1 mice (six males and eight females per group) were killed 4 months later. One hemisphere was fixed and coronal sections (50 μm) were cut for histological analysis (X-34 staining and HJ3.4 immunostaining). The other hemisphere was snap frozen on dry ice and stored at −80°C for biochemical analysis (ELISA and immunoblotting). To assess the abundance of Aβ species in predepositing mice, the abovementioned AAV viral vectors were injected into the hippocampi of APP/PS1 mice of either sex at 2 months of age and the animals were killed 1 month later. All animal care and surgical procedures were approved by the Animal Studies Committee of Washington University School of Medicine in accordance with guidelines of the NIH.
In vivo microdialysis.
AAV particles driving expression of TFEB (AAV8-CMV-FLAG-TFEB) or GFP as control (AAV8-CMV-GFP) were transduced by stereotactically guided injection in the hippocampus of 2-month-old APP/PS1 transgenic mice of either sex followed by in vivo microdialysis, 1 month later, as previously described (Cirrito et al., 2003, 2011). All studies were initiated at the same time of day. Briefly, a guide cannula (BR-style; Bioanalytical Systems) was implanted and cemented with the tip at coordinates: bregma −3.1 mm, midline −2.5 mm, 1.2 mm below dura at a 12° angle. A 2 mm microdialysis probe was then inserted into the hippocampus that contained a 38 kDa MWCO semipermeable membrane (Bioanalytical Systems) allowing molecules smaller than this cutoff to diffuse into the probe. Aβ capable of entering the probe is dubbed “exchangeable Aβ” (eAβ; Cirrito et al., 2003). The probe was flushed with 0.15% bovine serum albumin (Sigma) in an artificial CSF perfusion buffer at a constant rate (1.0 μl/min). The effluate was collected into a refrigerated fraction collector and assayed by sandwich ELISA for Aβx-40 peptides at the end of each experiment. During microdialysis, animals were housed in specially designed cages to permit free movement and ad libitum food and water while ISF Aβ was sampled. Baseline levels of ISF Aβ were sampled every 90 min between hours 9 and 16 (after the microdialysis probe is inserted) and averaged to determine the “baseline ISF Aβ level” in each mouse. Absolute in vivo concentration of ISF eAβ for each mouse was determined by correcting for the 20.8% recovery (1.0 μl/min) as obtained by the interpolated zero flow method described previously (Menacherry et al., 1992; Cirrito et al., 2003). At hour 16 (t = 0), a γ-secretase inhibitor, Compound E (200 nm reverse microdialysis; synthesized by AsisChem), was administered directly to the hippocampus by adding the drug to the microdialysis perfusion buffer. ISF Aβ was then sampled every 60 min for an additional 8 h. This enabled measurement of the elimination half-life of endogenous ISF Aβ in vivo as described previously (Cirrito et al., 2003).
Immunohistochemistry.
Frozen brain sections were incubated in 0.3% H2O2 in TBS for 10 min and blocked with 3% dry milk in TBS-X (0.25% Triton X-100 in TBS) for 1 h, followed by incubation with HJ3.4 antibody (anti-Aβ-1-13; Roh et al., 2012) overnight. Thereafter solution A from Vectastain ABC kit (1:400) was applied for 1 h, followed by 0.025% DAB in 0.25% NiCl2 and 0.05% H2O2 for 10–15 min. The slices were placed on glass slides, dried overnight, dehydrated and mounted, and imaged with confocal microscopy (Zeiss LSM).
X-34 plaque staining.
Brain slices were mounted on SuperFrost Plus slides, permeabilized with 0.25% Triton X-100 for 30 min, and stained with X-34 (dissolved in 40% ethanol, 60% water, pH 10.0; a generous gift from Robert Mach, Washington University, St. Louis, MO) for 20 min. Thereafter the tissue was thoroughly rinsed in PBS and mounted with Fluoromount mounting media.
Amyloid plaque quantification.
Fifty micrometer thick brain sections were collected every 300 μms from the rostral anterior commissure to caudal hippocampus. For plaque imaging, sections were stained with X-34 or immunostained with HJ3.4 anti-Aβ antibodies. High-resolution digital images of the stained brain slices were obtained with the NanoZoomer Digital Scanner (Hamamatsu Photonics). The total area of plaque coverage was measured using NIH ImageJ in the region of the hippocampus or piriform cortex (devoid of viral transduction) and expressed as percentage total area for each slice. Results from n = 4 sections were averaged to represent each animal.
Immunofluorescence.
Paraffin-embedded brain sections were washed with PBS and permeabilized with 0.3% Triton X-100 in PBS for 20 min followed by blocking. For double labeling, a mixture of the following antibodies was used: mouse anti-TFEB (MyBioSource, 1:100), rabbit anti-NeuN (Abcam; 1:100), rabbit anti-GFAP (Sigma; 1:100), and rabbit anti-Iba1 (Wako Chemicals; 1:1000). A secondary antibody mixture of Alexa Fluor 488-conjugated goat anti-mouse IgG and Alexa Fluor 594-conjugated goat anti-rabbit IgG (Invitrogen) was applied. Sections were imaged by confocal microscopy (Zeiss LSM).
Neuronal counts.
Stereological analysis for neuronal counts was performed on sections stained with 0.05% cresyl violet solution, as previously described (Xiao et al., 2014). Briefly, cresyl violet-stained cells with nuclei imaged within the inclusive zone of each dissector frame were counted in four equally spaced sections (50 μm apart) in the dorsal hippocampus of strata pyramidale CA1 and CA3, using a computer-based stereology system (Stereo Investigator; MicroBrightField) with the optical fractionator (West et al., 1991).
In vitro studies.
N2a cells stably expressing the human APP695 construct (N2a-APP695) were grown in DMEM/Opti-MEM (50:50) supplemented with 5% FBS and 200 μg/ml G418 and transfected with FLAG-tagged murine TFEB or empty vector, as described previously (Xiao et al., 2014). We observed >90% efficiency for transfection of exogenous TFEB (or GFP as control) in these in vitro studies. For assessment of APP half-life by cycloheximide chase assay, N2a-APP695 cells transfected with TFEB (or control) were treated with 50 μg/ml cycloheximide followed by preparation of cellular extracts at various time intervals for immunoblotting for APP. To inhibit lysosome acidification and function, studies were performed in the presence of bafilomycin A1 (100 nm), as previously described (Xiao et al., 2014). Studies with cell-surface biotinylation to assess internalization of APP were performed as described previously (Xiao et al., 2012). Briefly, cells cultured in 6-well plates were washed with cold PBS and surface proteins were labeled with the nonmembrane-permeant, cleavable biotin derivative, sulfo-NHS-SS-biotin (1 mg/ml in PBS). Cells were kept at 4°C for 30 min in the dark and gently rocked during the incubation period. The biotin reagent was quenched by treating the cells with two 15 min washes of 0.1 m glycine in PBS. Cells were rinsed with PBS and lysed in RIPA buffer containing 50 mm Tris-HCl, pH 7.5; 150 mm NaCl; 1% (v/v) Nonidet P-40; 0.5% (w/v) deoxycholate; and 1× protease inhibitor mixture. Lysates were incubated with Dynabeads MyOne Streptavidin T1 (Invitrogen) for 2 h in a rotary mixer to isolate biotin-labeled proteins. A cohort of surface biotin-labeled cells was incubated for 0, 1, 2, 3, 5, 7.5, 10, 20, and 30 min at 37°C to allow internalization, followed by rapid cooling on ice to stop endocytosis. To cleave biotin exposed at the cell surface, these cells were incubated three times for 20 min at 4°C with 50 mm 2-mercaptoethanesulfonic acid (Sigma) in 50 mm Tris-HCl, pH 8.7; 100 mm NaCl; and 2.5 mm CaCl2. After thorough rinsing with PBS containing 20 mm HEPES, cells were lysed in RIPA buffer, and internalized biotinylated proteins were immunoprecipitated with streptavidin followed by immunoblotting for APP (CT695; Invitrogen). Uptake of biotin-labeled transferrin (T3915; Sigma) was assessed in parallel in TFEB and vector-transfected cells to determine the effect of TFEB on clathrin-mediated endocytosis. Immunofluorescence was performed on C17.2 cells cotransfected with Cer-APP-YFP and TFEB or control vector and incubated with LysoTracker Red (L7526; Invitrogen) to label the lysosome compartment, and live cells were examined by confocal microscope (Zeiss LSM).
Aβ ELISA.
Aβ species in cell lysates and media from transfected N2a-APP695 were detected by sandwich ELISA, as previously described (Xiao et al., 2014). To detect total Aβ in the hippocampus in 3-month-old mice, dissected tissue was sequentially homogenized in PBS followed by RIPA buffer to obtain detergent-soluble Aβ at an age when plaques are not observed (Yan et al., 2009), and samples were pooled for analysis. In aged mice (10 months old, when plaques are abundant), hippocampal tissues were sequentially homogenized in PBS followed by 5 m guanidine in TBS, pH 8.0 (to extract fibrillar and membrane-bound Aβ). For ELISA assays, Aβx-40 and Aβx-42 peptides were captured with mouse monoclonal-coating antibodies HJ2 (anti-Aβ35-40) and HJ7.4 (anti-Aβ37-42) or HJ5.1 (anti-Aβ13-28), respectively, as previously described (Kim et al., 2009; Xiao et al., 2014). HJ5.1 (anti-Aβ13-28), a biotinylated antibody targeting the central domain, or HJ3.5 (anti-Aβ1-13, provided by Dr. David Holtzman, Washington University School of Medicine, St. Louis, MO), which targets the N-terminal amino acids, was used as the detecting antibody, followed by streptavidin-poly-HRP-40 (Fitzgerald Industries). All ELISA assays were developed using Super Slow ELISA TMB (Sigma) and absorbance read on a Bio-Tek Epoch plate reader at 650 nm. Standard curves were generated from synthetic human Aβ1–40 or Aβ1–42 peptides (American Peptide).
Quantitative PCR.
Real-time PCR was performed as described previously (Ma et al., 2012). Briefly, total RNA was prepared from transfected N2a-APP695 cells or AAV-transduced hippocampus using an RNA-easy mini kit (Qiagen), and cDNA was synthesized with 1 μg of total RNA using the SuperScript III first-strand synthesis system (Invitrogen). One microliter of cDNA template was mixed with 12.5 μl of 2 × SYBR Green PCR Master Mix (Invitrogen) and subjected to quantitative PCR in triplicate under the following conditions: 50°C, 2 min; 95°C, 10 min; followed by 40 cycles of 95°C, 15 s; 60°C, 1 min in the ABI7500 Fast RealTime PCR system. The housekeeping gene GAPDH was also amplified in parallel as a reference for the quantification of transcripts. Primer sequences used have been previously described (Xiao et al., 2014).
Immunoblotting.
Protein samples were run on 4–12% Bis-Tris gels. Blots were probed with the following antibodies: FLAG (Sigma; 1:1000), TFEB (MyBioSource; 1:500), LAMP1 (Santa Cruz Biotechnology; 1:500), Cathepsin B (Abcam; 1:500), Cathepsin D (a generous gift from Stuart Kornfeld, Washington University, St. Louis, MO; 1:2000), APP CT695 (to detect both full-length APP and C-terminal fragments; Invitrogen; 1:500), 6E10 (to detect sAPPα; Covance; 1:1000), Adam10 (Santa Cruz Biotechnology; 1:100), Adam17 (Millipore; 1:1000), BACE1 (Cell Signaling Technology; 1:1000), Psen1 (Santa Cruz Biotechnology; 1:200), nicastrin (Thermo Scientific; 1:1000), IDE (Abcam; 1:2000), neprilysin (Millipore; 1:500), MMP2 (Abcam; 1:1000), MMP9 (Abcam; 1:1000), HRP-conjugated biotin (Cell Signaling Technology; 1:1000), and actin (Sigma; 1:2000). Normalized band intensity was quantified using ImageJ software.
Statistical analysis.
All data are shown as Mean ± SEM. Data were analyzed by two-tailed Student's t test for detecting significant differences between two groups. Differences among multiple groups were analyzed by one-way and two-way ANOVA for one and two independent variables, respectively, followed by post hoc Tukey's test. Statistical significance was pegged at p < 0.05 (*p < 0.05 and **p < 0.01).
Results
AAV-CMV-TFEB transduces TFEB specifically in neurons of APP/PS1 mice with upregulated lysosome abundance
Lysosomal dysfunction is speculated to play a central role in enhanced Aβ generation in neurons (Lee et al., 2010; Neely et al., 2011; Coen et al., 2012; Funk and Kuret, 2012; McBrayer and Nixon, 2013; Wolfe et al., 2013) or its impaired clearance (Wyss-Coray et al., 2003; Majumdar et al., 2011; Lee et al., 2012; Kanekiyo et al., 2013). In previous studies, we demonstrated that targeting expression of TFEB, a master inducer of lysosomal biogenesis (Sardiello et al., 2009; Settembre et al., 2011, 2013b), specifically to astrocytes, stimulated lysosome biogenesis and function resulting in accelerated Aβ uptake and degradation (Xiao et al., 2014). In the current study, we explored the hypothesis that enhancing lysosomal biogenesis in neurons with exogenous expression of TFEB will stimulate lysosomal degradation of holo-APP to attenuate Aβ production. Accordingly, we performed stereotactically guided injections of AAV8 particles encoding a CMV promoter-driven construct to transduce TFEB (AAV8-CMV-FLAG-TFEB; and AAV8-CMV-GFP as control) in the hippocampus of 6-month-old APP/PS1 mice, at a stage of early plaque deposition and growth (Yan et al., 2009). Stereotactic AAV injections consistently resulted in widespread transduction in the hippocampus (data not shown). As we observed previously (Xiao et al., 2012), the CMV promoter-driven construct specifically resulted in TFEB expression in neurons with a neuron-specific marker, NeuN, and not in astrocytes or microglia (Fig. 1A). Importantly, exogenous TFEB localized to the neuronal nuclei (Fig. 1A) and hippocampal extracts from AAV8-TFEB-transduced mice demonstrate robust overexpression with TFEB (11-fold increase over control, p < 0.01; N = 3 per group) with increased abundance of lysosomal proteins, LAMP1, and Cathepsin B and Cathepsin D (Fig. 1B,C), two lysosomal proteases that have been implicated in APP degradation (Higaki et al., 1996; Mueller-Steiner et al., 2006).
Figure 1.
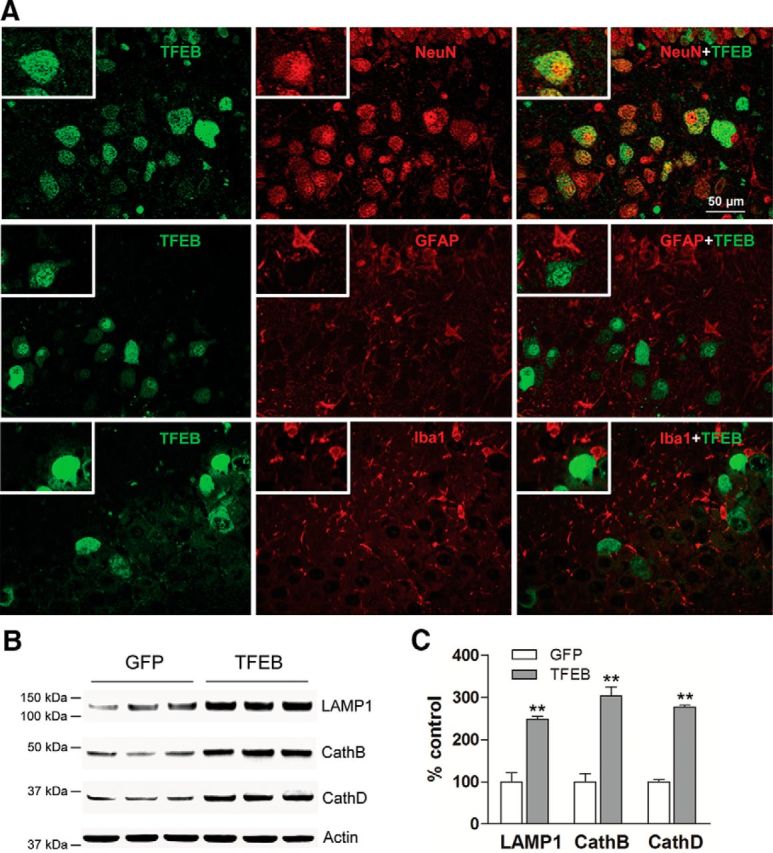
AAV8-CMV-TFEB drives lysosome biogenesis in neurons. A, Representative confocal images demonstrating expression of TFEB (green) in various CNS cell types marked by the expression of NeuN (neurons; red, top), GFAP (astrocytes; red, middle), and Iba-1 (microglia; red, bottom) in the hippocampus of APP/PS1 mice injected with AAV8-CMV-FLAG-TFEB particles. The boxed inserts (upper left corners) demonstrate magnified images of TFEB-expressing cells. B, C, Immunoblots (B) with quantification (C) of lysosomal proteins in extracts from hippocampi transduced with AAV8-CMV-FLAG-TFEB (TFEB) or AAV8-CMV-GFP (GFP). N = 3 per group; **p < 0.01.
Neuronal TFEB expression drives reduced APP abundance, in vivo
APP695 is synthesized and processed primarily in neurons, where its sequential cleavage via the β- and γ-secretases results in generation of amyloidogenic Aβ peptides (Hartmann et al., 1997; Kamenetz et al., 2003). Alternately, APP processing can proceed via α-secretases in a manner that precludes Aβ generation (Haass et al., 2012). Studies have demonstrated that a large fraction of full-length APP protein is also degraded within lysosomes, which may limit the pool of APP available for α-, β-, and γ-cleavage (Caporaso et al., 1992, 1994; Golde et al., 1992). Given that TFEB drives upregulation of the lysosomal machinery, we examined the effect of neuronal TFEB expression on abundance of APP and its various cleavage fragments. TFEB-transduced hippocampal extracts demonstrated a 28% reduction in APP protein abundance, accompanied by a matching 31 and 36% reduction in α-CTF and β-CTF (Fig. 2A,B). Given previous observations that amyloid pathogenesis is accelerated in female mice (Callahan et al., 2001), we examined the effects of TFEB transduction in male and female mice separately. We observed comparable reductions in APP in the two sexes (by 29% in males vs 30% in females; data not shown). These data show that the effect of neuronal TFEB expression on APP abundance is independent of gender.
Figure 2.

Neuronal TFEB transduction results in reduced APP abundance. A, B, Immunoblot (A) with quantitation (B) of APP and its α- and β-CTFs in APP/PS1 mouse hippocampi transduced with AAV8-CMV-FLAG-TFEB (TFEB) or AAV8-CMV-GFP (GFP). N = 6 per group (n = 3 males and n = 3 females); **p < 0.01. Representative immunoblot is from male mouse tissues. C, Expression of transcripts coding for various components of the APP-processing machinery in hippocampal tissues transduced with AAV8-CMV-FLAG-TFEB (TFEB) or AAV8-CMV-GFP (GFP). N = 5 per group. No statistically significant differences were noted for any of the transcripts. D, E, Immunoblot (D) and quantitation (E) of APP-processing machinery proteins in hippocampi transduced with TFEB (or GFP as control, as in A). N = 3 per group. F, Neuronal counts in the CA1 and CA3 layers of the hippocampi from AAV8-CMV-FLAG-TFEB (TFEB) and AAV8-CMV-GFP (GFP)-transduced APP/PS1 mice. N = 4 per group.
The reduction in APP abundance was not caused by a difference in the transcript levels for the transgenically expressed human APP695 protein (Fig. 2C) or in the expression of α-secretases and components of the β- and γ-secretase machinery between the TFEB and control-transduced hippocampi (Fig. 2C–E). Importantly, TFEB-transduced hippocampi did not demonstrate a difference in neuronal counts compared with control (Fig. 2F), ruling out neuronal toxicity as the underlying cause for these observations. Together with the proportionately comparable reductions in various CTFs, these data suggest that TFEB expression facilitates holo-APP degradation via pathways that do not involve APP processing by α-cleavage or sequential β- and γ-cleavage.
Neuronal TFEB expression reduces ISF Aβ levels, in vivo
Aβ peptides are released into the ISF and removed via cellular uptake and degradation, extracellular proteolysis, or transport across the blood–brain barrier. Consequently, the steady-state level of Aβ in the ISF is determined by the balance between its rate of production and degradation. Considering that exogenous TFEB expression results in a reduction in total APP levels with a parallel reduction in abundance of its cleaved fragments (Fig. 2A,B), we hypothesized that TFEB transduction will attenuate Aβ production resulting in reduced Aβ steady-state levels in the ISF. Accordingly, we transduced 2-month-old APP/PS1 mice with AAV8-CMV-FLAG-TFEB and control AAV8-CMV-GFP viral particles and performed in vivo microdialysis 1 month later to determine whether neuronal TFEB expression impacts ISF Aβ at a time point before deposition of plaques (Yan et al., 2009) to avoid a potential confounding influence from exchangeable plaque-bound Aβ pool. TFEB-transduced mice demonstrated a 57% reduction in steady-state ISF Aβ levels compared with controls (Fig. 3A). After steady-state measures of ISF Aβ were obtained, a potent γ-secretase inhibitor, Compound E, was administered to inhibit APP cleavage to Aβ (Cirrito et al., 2011), and ISF was sampled for an additional period of 8 h to determine the elimination rate (half-life) of pre-existing Aβ (Fig. 3A), which was observed to follow first-order kinetics in both control and TFEB-transduced mice (Fig. 3B). We observed a modest 24% reduction in half-life of ISF Aβ in CMV-TFEB-transduced hippocampi, indicating that neuronal TFEB transduction also accelerates Aβ removal from ISF (Fig. 3B). That steady-state ISF Aβ levels are reduced by 57%, but half-life is reduced by only 24%, strongly suggests that TFEB has only a minor role in enhancing ISF Aβ clearance with the primary effect instead on reducing Aβ production. Indeed, previous studies have demonstrated that neurons take up Aβ via endocytosis and macropinocytosis, and it is conceivable that TFEB transduction stimulates Aβ uptake in neurons via the latter similar to our studies with astrocyte-targeted TFEB expression (Xiao et al., 2014). Importantly, the effect of exogenous TFEB on reduction in ISF Aβ levels and Aβ half-life was comparable in both sexes (data not shown). We also explored the possibility that TFEB transduction could enhance extracellular degradation of Aβ, but did not observe any alteration in the transcript levels for known Aβ-metabolizing enzymes (MMP2, MMP9, IDE, or neprilysin) in TFEB-transduced hippocampi (Fig. 3C). Analogously, the protein abundance for these enzymes was unchanged (MMP2, MMP9, and IDE) or modestly reduced (neprilysin; Fig. 3D,E), suggesting that regulation of extracellular proteases is unlikely to contribute to a reduction in ISF Aβ levels with neuronal TFEB transduction.
Figure 3.

Neuronal TFEB transduction reduces ISF Aβ levels, in vivo. A, Assessment of Aβ levels by in vivo microdialysis in 3-month-old APP/PS1 mice transduced with AAV8-CMV-FLAG-TFEB (TFEB) and AAV8-CMV-GFP (GFP) with serial hourly measurements. N = 8 mice in TFEB group, n = 9 mice in GFP group. At t = 0, mice were continually administered Compound E directly to the hippocampus (200 nm, reverse microdialysis) followed by hourly sampling for Aβ. Inset shows mean absolute in vivo “exchangeable” Aβ (eAβx-40) concentration averaged over a 9 h period before drug administration; *p < 0.05. B, Semilog plot of decline in percentage basal ISF Aβ levels during administration of Compound E in animals in A. Inset shows Aβ half-life in the two groups; *p < 0.05. C–E, Transcript levels (C) and protein abundance [immunoblot (D) with quantitation (E)] for enzymes implicated in extracellular Aβ metabolism in hippocampal tissue from AAV8-CMV-FLAG-TFEB (TFEB) and AAV8-CMV-GFP (GFP)-transduced APP/PS1 mice. N = 5 per group; **p < 0.01.F, G, Aβ40 and Aβ42 levels in dissected hippocampal tissues from AAV8-CMV-FLAG-TFEB (TFEB) and AAV8-CMV-GFP (GFP)-transduced APP/PS1 mice at 3 months of age. Tissue was homogenized first in PBS (D) then in RIPA (E) quantified with ELISA assay. HJ2 and HJ7.4 antibodies were used for capture Aβ40 and Aβ42, respectively, and HJ5.1 antibody was used for detection. N = 6 mice per group; **p < 0.01.
Importantly, total immunodetectable Aβ levels were also reduced in the hippocampal extracts from TFEB-transduced mice at this early stage of amyloid pathogenesis, compared with controls (Fig. 3F,G), confirming that the reduction in steady-state levels of ISF Aβ was not due to intracellular Aβ accumulation. Together with the reduction in APP abundance and that of its various cleaved fragments (Fig. 2A,B), these data point to a predominant effect of neuronal TFEB expression on attenuating Aβ generation with a modest effect on accelerating Aβ removal as the primary driver of reduced steady-state ISF Aβ levels.
Neuronal TFEB expression reduces Aβ levels and amyloid plaques in APP/PS1 transgenic mice
ISF Aβ levels correlate with amyloid plaque pathology (Bero et al., 2011), supporting the hypothesis that Aβ aggregates into amyloid plaques in a concentration-dependent manner, in vivo. Indeed, studies from our laboratory as well as others have demonstrated that strategies to reduce ISF Aβ levels drive a reduction in amyloid plaque deposition (Cramer et al., 2012; Sheline et al., 2014; Xiao et al., 2014). Accordingly, we posited that neuronal TFEB transduction will drive reduction in fibrillar Aβ aggregates and attenuate deposition of amyloid plaques in aging APP/PS1 mice. To investigate this premise, we subjected TFEB-transduced (and control) hippocampal tissue to sequential homogenization in PBS followed by guanidine to quantify Aβ in PBS-soluble and PBS-insoluble fractions. In addition, we performed Aβ immunohistochemistry and X-34 staining to assess Aβ and amyloid load, respectively. As shown (Fig. 4A–C), TFEB transduction resulted in a 40 and 50% reduction in soluble Aβ40 and Aβ42, and 36 and 35% reduction in insoluble Aβ40 and Aβ42, respectively, compared with controls, by an ELISA assay to differentially capture the two species by specific C-terminal antibodies. To confirm these findings, we also used an alternate Aβ peptide capture and detection strategy using a combination of antibodies targeting distinct epitopes at the central domain (HJ5.1) and N terminus (HJ3.5), as previously described (Xiao et al., 2014), and observed a 29 and 31% reduction in the soluble and insoluble levels of all Aβ species in TFEB-transduced hippocampi (Fig. 4A–C). This suggests a uniform reduction in Aβ levels rather than a reduction in specific truncated fragments with exogenous TFEB expression.
Figure 4.
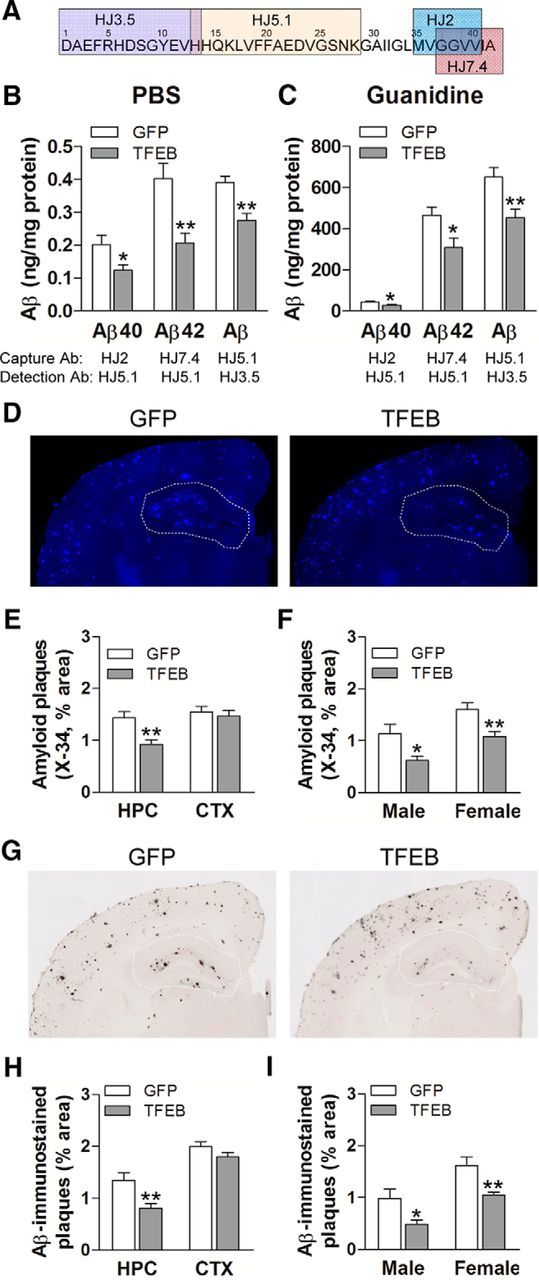
Neuronal TFEB transduction reduces amyloid plaque load in APP/PS1 mice. A, Schematic representation of specific antibodies used in ELISA. B, C, Aβ40 and Aβ42 levels in dissected hippocampal tissues from AAV8-CMV-FLAG-TFEB (TFEB) and AAV8-CMV-GFP (GFP)-transduced mice (10 months of age). Tissue was homogenized first in PBS (soluble levels, B), then in 5 mm guanidine (insoluble levels, C) quantified with ELISA assay. HJ2 and HJ7.4 antibodies were used for capture Aβ40 and Aβ42, respectively, and HJ5.1 antibody was used for detection. Total Aβ levels were also measured with a combination of HJ5.1 antibody for capture and HJ3.5 antibody for detection, as indicated in the schematic. N = 8 mice per group; *p < 0.05, **p < 0.01. D, Representative X-34-stained images from APP/PS1 mice treated as in A. The area of the hippocampus is outlined with a dotted line. E, F, Quantification of X-34-stained plaque burden in the hippocampus (HPC) in mice treated as in A (E) and plaque burden stratified by sex (F). N = 14 (6 male and 8 female) mice per group;*p < 0.05, **p < 0.01. G, Representative Aβ-immunostained images from mice treated as in B. H, I, Quantification of Aβ-stained plaque burden in the hippocampus in mice treated as in A (H) and plaque burden stratified by sex (I). N = 14 (6 male and 8 female) mice per group; *p < 0.05, **p < 0.01. HPC, Hippocampus; CTX, cortex.
This was accompanied by a 35% reduction in X-34-stained amyloid deposits (Fig. 4D,E) and a 40% reduction in Aβ-immunostained amyloid plaque load (Fig. 4G,H). Importantly, as we have observed previously with astrocytic expression of exogenous TFEB (Xiao et al., 2014), the effect of neuronal TFEB transduction on amyloid deposition was localized to the hippocampus (see area outlined with dotted line in Fig. 4D and G and quantitation in E and H), which was targeted for TFEB expression by stereotactic injection and not in the cortical areas that are distant from the site of AAV transduction. It is interesting to note that the degree of reduction in plaque load mirrored that of the reduction in Aβ levels (Fig. 4B,C) in the aged mice and that of the reduction in ISF steady-state Aβ (Fig. 3A,B) and hippocampal Aβ in the predepositing mice (Fig. 3F,G), attesting to a consistent effect of TFEB on Aβ generation throughout the various stages of amyloid pathogenesis. It is also noteworthy that TFEB-transduction had a comparable effect on amyloid plaque load reduction in either sex (Fig. 4F,I), as we have previously observed (Xiao et al., 2014).These data suggest that TFEB expression in neurons attenuates amyloid plaque deposition via reducing Aβ generation.
Neuronal TFEB transduction reduces APP and Aβ levels in wild-type mice
To exclude the possibility that TFEB primarily affects the processing of the mutant APP695 protein, which is co-overexpressed with a familial AD risk conferring ΔE9 mutant presenilin-1 protein in the APP/PS1 mice (Jankowsky et al., 2004), we examined the effect of neuronally transduced TFEB on the abundance of endogenous wild-type APP protein and its α- and β-CTFs in hippocampi of wild-type mice versus those in GFP-transduced hippocampi as controls. Remarkably, TFEB transduction led to a comparable degree of reduction in abundance of total endogenous APP and levels of the α- and β-CTFs (Fig. 5A,B) mimicking its effect in APP/PS1 transgenic mice (Fig. 2A,B). This translated into a 27 and 21% reduction in Aβ40 and Aβ42 species (Fig. 5C), respectively, indicating that TFEB also alters the metabolism of wild-type APP protein to reduce Aβ generation.
Figure 5.

Neuronal TFEB transduction reduces levels of APP, its CTFs, and Aβ species in wild-type mice. A, B, Immunoblot (A) with quantitation (B) of APP, and its α- and β-CTFs in wild-type mouse hippocampi transduced with AAV8-CMV-FLAG-TFEB (TFEB) or AAV8-CMV-GFP (GFP) at 8 months of age. N = 3 per group; *p < 0.05. C, Aβ40 and Aβ42 levels in dissected hippocampal tissues homogenized in RIPA buffer, from mice transduced as in A. HJ2 and HJ7.4 antibodies were used for capture Aβ40 and Aβ42, respectively, and HJ5.1 antibody was used for detection. N = 6 per group; *p < 0.05.
TFEB expression attenuates Aβ generation, in vitro
To confirm that TFEB expression results in reduced Aβ generation, we transfected N2a cells stably expressing APP695 with TFEB or vector control, and examined APP processing. TFEB-transfected cells demonstrated increased lysosome abundance, as indicated by upregulation of lysosomal proteins (Figure 6A,B) driven by eightfold overexpression of TFEB, as we observed previously in C17.2 cells (Xiao et al., 2014). TFEB-transfected cells demonstrated a reduction in APP abundance, as well as reduced levels of α- and β-CTFs and sAPPα in the medium (Fig. 6C,D). This was not associated with a change in APP transcript (TFEB/GAPDH) mRNA 1.04 ± 0.04-fold in TFEB transfected vs 1.0 ± 0.01-fold in control, p = 0.38, N = 3 per group), indicating that TFEB stimulates increased degradation of APP, which is consistent with the observations in TFEB-transduced hippocampi (Fig. 2). Confocal imaging of a dual fluorescent APP construct (Sannerud et al., 2011) also demonstrated a qualitative upregulation of lysosome abundance with reduction in full-length APP protein in TFEB-transfected cells, along with increased localization of APP in lysosomes (Fig. 6E). Colocalization of full-length APP (bearing both blue and green fluorescent tags) with LysoTracker Red-labeled lysosomes (see arrows) was qualitatively observed to be upregulated in TFEB-expressing cells (Fig. 6E, arrows). This was associated with reduced Aβ abundance, both intracellularly (Fig. 7A) and in the medium (Fig. 7B). Importantly, TFEB-transfected cells demonstrated a reduction in Aβ appearance in the medium (Fig. 7C,D), indicating that TFEB primarily attenuates Aβ generation, and reduced intracellular Aβ levels are not a result of increased Aβ secretion.
Figure 6.
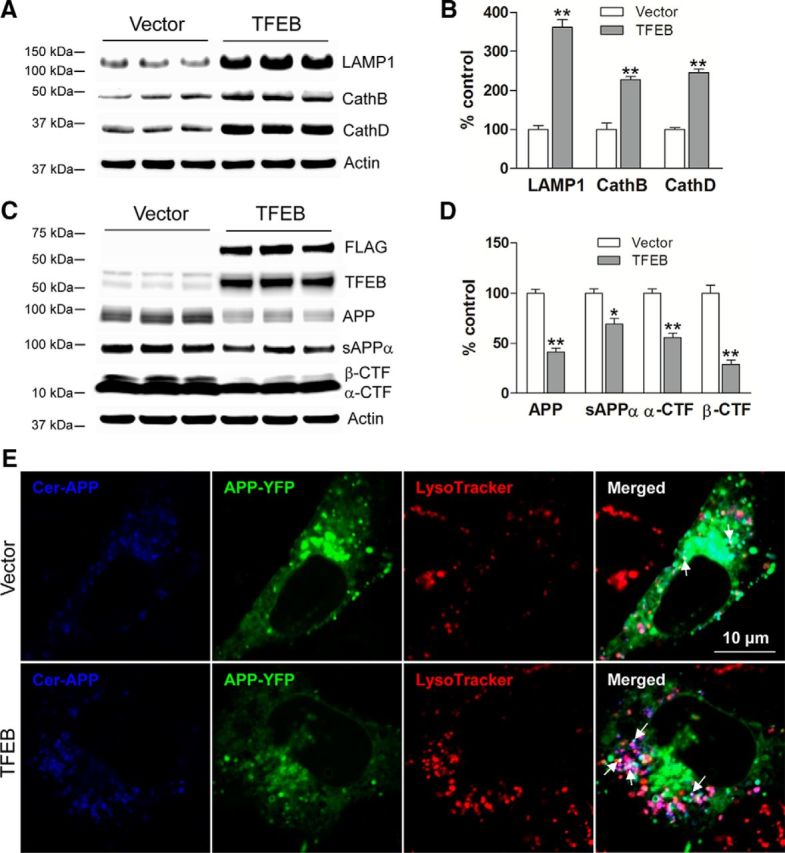
TFEB expression increases lysosomes and reduces levels of APP and its cleaved CTFs, in vitro. A, B, Immunoblots (A) with quantification (B) of lysosomal proteins in N2a-APP695 cells transfected with TFEB or vector control (for 48 h). C, D, Immunoblot (C) with quantitation (D) of APP and its α- and β-CTFs and of sAPPα fragment in the overlying medium, collected over a duration of 6 h in cells treated as in A. N = 3 per group; *p < 0.05, **p < 0.01. E, N2a cells were transfected with a dual fluorescent APP construct (Sannerud et al., 2011) with or without the TFEB construct and stained with LysoTracker Red. As shown in these confocal images, colocalization of full-length APP (both blue and green tags) with LysoTracker Red-labeled lysosomes (see arrows) was qualitatively increased in TFEB-expressing cells (see arrows) compared with empty vector-expressing cells.
Figure 7.

TFEB expression attenuates Aβ generation, in vitro. A, B, Abundance of Aβ40 and Aβ42 species in the cell lysates (A) and medium (B) in N2a-APP695 cells transfected with TFEB or vector control. C, D, Accumulation of Aβ40 (C) and Aβ42 (D) species in the overlying medium of cells treated as in A, over the indicated duration. N = 5 per group; *p < 0.05, **p < 0.01.
TFEB accelerates APP flux down the endosome-lysosome pathway
The intracellular itinerary of APP follows a pathway from the Golgi (trans-Golgi network), wherein it is post-translationally modified after synthesis in the ER, to the plasma membrane where it can be cleaved via a nonamyloidogenic α-cleavage. Alternatively, APP can be endocytosed, and multiple studies have demonstrated that endocytosis of APP is essential for its colocalization with β- and γ-secretases within endosomes and multivesicular bodies (formed by trafficking of endosomes into intraluminal vesicles via sequential activity of endosomal-sorting complexes required for transport, which are the primary sites for Aβ generation via amyloidogenic cleavage; Rajendran and Annaert, 2012). Studies have suggested that impairment in endosomal flux, likely secondary to lysosome dysfunction, results in increased transit time within this organelle, which increases the propensity for β- and γ-cleavage and, hence, Aβ generation (Cataldo et al., 2000; Rajendran and Annaert, 2012; Morel et al., 2013; Peric and Annaert, 2015). Indeed, after endocytosis, studies have demonstrated that a significant proportion of APP is targeted to the lysosomes (Benowitz et al., 1989; Haass et al., 1992; Caporaso et al., 1994) and APP levels rapidly build up in the cells in the presence of lysosomal acidification inhibitors (Golde et al., 1992), suggesting that lysosomal degradation drives APP proteolysis to preclude formation of Aβ peptides (Caporaso et al., 1992). Given previous reports that TFEB expression can stimulate endocytosis (Peña-Llopis et al., 2011), a potential adverse consequence of TFEB activation could be increased APP endocytosis resulting in enhanced β- and γ-cleavage and accelerated Aβ generation, as observed with other experimental manipulations to stimulate endocytosis (Grbovic et al., 2003; Schneider et al., 2008; Xiao et al., 2012). Alternatively, TFEB may stimulate flux through the endosome-lysosome pathway to prevent APP accumulation in endosomes and stimulate its proteolysis in lysosomes. To evaluate these possibilities, we examined the effect of TFEB on APP internalization. N2a-APP95 cells were transfected with TFEB or vector control, and cell-surface APP was labeled by biotinylation at 4°C (to prevent endocytosis), followed by rewarming to stimulate its internalization by endocytosis (Xiao et al., 2012). At various time points post rewarming, the cells were treated with 2-mercaptoethanesulfonic acid to remove cell-surface biotinylated APP molecules, permitting assessment of kinetics of appearance and subsequent degradation of total internalized biotin-tagged APP via streptavidin capture and immunoblotting. As shown in Figure 8, A and B, cells transfected with TFEB demonstrated reduced levels of both surface and internalized APP, such that the ratio of internalized to surface APP was not altered compared with control.
Figure 8.

TFEB expression reduces steady-state cell-surface and intracellular APP levels without altering its endocytosis. A, B, Immunoblot (A) and quantitation (B) of cell-surface and internalized APP in N2a-APP695 cells transfected with TFEB or vector control, after cell-surface APP was labeled by biotinylation at 4°C (to prevent endocytosis) followed by rewarming (to 37°C) to stimulate its internalization by endocytosis (for 10 min). Cell-surface APP was assessed in cells maintained at 4°C. To assess internalized APP, cells were treated with 2-mercaptoethanesulfonic acid to remove cell-surface biotinylated APP molecules, followed by streptavidin capture and immunoblotting (see Materials and Methods). Inset shows ratio of internalized to surface APP; *p < 0.05. C–E, Immunoblot (C) and quantitation (D) of intracellular APP as a fraction of cell-surface APP to demonstrate kinetics of intracellular Aβ in cells transfected as in A and kinetics of uptake of biotinylated transferrin [immunoblot (C) with quantitation (E)] in cells treated as in A. N = 3 per group. No statistically significant differences were observed by two-way ANOVA.
To determine whether the reduced steady-state levels of APP are due to accelerated endocytosis or enhanced flux into the lysosomes for degradation, we determined the kinetics of intracellular APP appearance in this assay (Fig. 8C,D). Interestingly, APP was rapidly endocytosed and the rate of increase of intracellular APP levels (from T = 1 to T = 7.5 min) was not different between TFEB-expressing cells and controls (Fig. 8C,D). These data are consistent with a lack of effect of TFEB on clathrin-mediated endocytosis, consistent with our previous observations (Xiao et al., 2014). To determine whether TFEB expression affects the kinetics of other transmembrane proteins taken up by clathrin-mediated endocytosis, we examined the uptake of exogenously applied biotin-labeled transferrin. Transferrin binds to the transferrin receptor and is endocytosed followed by recycling to the cell surface (Ciechanover et al., 1983). The kinetics of intracellular appearance of transferrin was not affected by TFEB expression (Fig. 8E), confirming that TFEB does not accelerate clathrin-mediated endocytosis.
In contrast, internalized APP levels at 30 min were 32% lower in TFEB-transfected cells versus 14% lower in control vector-transfected cells from their respective peak levels at 7.5 min (p = 0.017, n = 3 experimental trails per group), suggesting that TFEB accelerates degradation of APP, which is targeted for lysosomal degradation, in contrast to transferrin that gets recycled to the cell surface (Ciechanover et al., 1983). Together with the observed reduction in APP and its various cleaved fragments (Fig. 6C,D) and attenuated Aβ generation (Fig. 7A–D), these observations suggest that TFEB expression does not affect APP endocytosis, but stimulates increased flux of endocytosed APP down the endosome-lysosome pathway without buildup of APP protein in intracellular compartments, resulting in its lysosomal degradation and reduced abundance of APP at steady state.
TFEB enhances lysosomal degradation of APP
To assess the role of lysosomes in APP degradation, we treated TFEB-transfected (and control) cells with bafilomycin A1 to inhibit lysosome acidification and evaluated APP abundance. As shown in Figure 9A, bafilomycin treatment resulted in increased APP levels (by 42% compared with control) and largely prevented the TFEB-induced reduction in APP abundance (Fig. 9A,B). This was associated with increase in both α- and β-CTF levels (Fig. 9A,C,D) as well as increased generation of Aβ40 (Fig. 9E) and Aβ42 (Fig. 9F), as inhibiting lysosomal degradation of APP makes it increasingly available as a substrate for processing via the secretases. We next determined APP half-life by treating cells with cycloheximide to inhibit protein synthesis and follow APP abundance. As shown in Figure 9, G and H, TFEB transfection reduced APP half-life by 42% compared with control indicating enhanced APP degradation. Intriguingly, the levels of exogenous TFEB also declined rapidly, consistent with the observations that activated TFEB is degraded rapidly (Roczniak-Ferguson et al., 2012) possibly via the ubiquitin-proteasome pathway (Maia et al., 2015). It was also interesting to note that the abundance of APP in bafilomycin-treated TFEB-expressing cells was lower than the respective levels in vector controls (Fig. 9B), again pointing to the reduction in steady-state levels of APP with TFEB expression, before application of bafilomycin to inhibit lysosome acidification. Expectedly, this resulted in a relative decline in the CTFs and Aβ peptides in this setting (Fig. 9C–F).
Figure 9.

TFEB expression enhances lysosomal degradation of APP. A–F, Immunoblot (A) with quantitation of APP (B), α-CTF (C), β-CTF (D), Aβ40 (E), and Aβ42 (F) in N2a-APP695 cells transfected with TFEB or vector control and cultured in the presence of bafilomycin A1 (Baf) or diluent for 4 h. P values shown are by post hoc test after one-way ANOVA. G, N2a-APP695 cells were transfected with TFEB or vector control and treated with cycloheximide (CHX; 50 μg/ml) at t = 0. Cells were collected at the indicated times to evaluate APP abundance. H, APP abundance expressed as a percentage of baseline (t = 0 after addition of cycloheximide) in cells treated as in C. N = 3 per group. Inset shows half-life of APP; *p < 0.05. I, N2a-APP695 cells were transfected with TFEB or vector control, and were pretreated with bafilomycin A1 or diluent (100 nm for 30 min) and treated with cycloheximide (50 μg/ml) at t = 0. Cells were collected at the indicated times to evaluate APP abundance. J, K, Quantitation of APP abundance at various times in cells treated as in I, in the vector-transfected (J) or TFEB-transfected (K) groups. N = 4 per group. L, Half-life of APP in bafilomycin A1-treated cells expressed as a percentage of diluent-treated group. N = 4 per group; *p < 0.05.
As described previously (Caporaso et al., 1992), inhibition of lysosome acidification with bafilomycin A1 prolonged APP half-life, demonstrating that a significant fraction of APP is degraded via the lysosomes (Fig. 9I–K). Importantly, APP half-life was also markedly prolonged by 61% in the presence of bafilomycin A1 in TFEB-transfected cells (Fig. 9L). In summary, these studies demonstrate that TFEB expression accelerates endogenous pathways for holo-APP degradation via lysosomes, preventing its amyloidogenic processing into Aβ.
Discussion
Contemporary approaches to reducing Aβ generation in neurons have focused upon shifting APP processing to nonamyloidogenic pathways or sorting APP away from the endosomes where amyloidogenic cleavage takes place (Haass et al., 2012). Our study demonstrates the efficacy of accelerating holo-APP degradation in lysosomes as a strategy to reduce Aβ generation. Indeed, exogenous TFEB expression stimulates lysosome biogenesis in vitro, reduces full-length APP and its cleaved fragments, attenuates Aβ generation and release in the medium (Figs. 6, 7), and markedly shortens APP half-life (Fig. 9) in a lysosome-dependent manner. These effects are likely secondary to acceleration of lysosomal degradation of endocytosed APP, as suggested by reduction of steady-state APP at both the surface and within intracellular compartments (Fig. 8), and reduced APP half-life with TFEB expression (Fig. 9). Mirroring these in vitro findings, AAV-mediated neuronal TFEB transduction in the hippocampus of APP/PS1 mice also drives upregulation of lysosome abundance with reduction in APP and its cleaved fragments (Figs. 1, 2). This translates into reduced steady-state ISF Aβ levels (Fig. 3) and attenuated amyloid deposits specifically in the AAV-targeted hippocampus (Fig. 4). Remarkably, the magnitude of reduction in ISF Aβ and amyloid plaque load with neuronal TFEB transduction compares favorably with that observed with astrocytic TFEB transduction to accelerate Aβ degradation (Xiao et al., 2014), highlighting the potential for therapeutic synergy with these dual approaches.
Our data indicate that lysosomal degradation of APP is an important component of APP metabolism. Indeed, studies with pulse-chase radioactive labeling of APP revealed that full-length APP is degraded with a half-life of ∼90 min (consistent with our observations in Fig. 9H), and inhibition of lysosomal acidification with chloroquine prolongs its half-life (Caporaso et al., 1992) and accumulation within lysosomes (Caporaso et al., 1994). Remarkably, chloroquine treatment resulted in marked increase in the appearance of Aβ in the medium, providing the earliest clue on the essential role of lysosomes in facilitating breakdown of APP. Also, biotinylated cell-surface APP was recovered within lysosomes, along with its various CTFs, providing further evidence that APP and its CTFs are targeted to lysosomes (Haass et al., 1992). Indeed, BACE1, the rate-limiting step for amyloidogenic cleavage of APP, and its product, βCTF, the precursor for Aβ, are also degraded within lysosomes (Chen et al., 2000; Koh et al., 2005), indicating that flux into the lysosomes may be the critical determinant of whether APP metabolism leads to Aβ generation or its nonamyloidogenic degradation.
Endocytosis of APP via clathrin-mediated mechanisms is essential for Aβ generation, and endosomes are the primary sites for its amyloidogenic processing (Koo and Squazzo, 1994; Perez et al., 1999; Schneider et al., 2008). Given that endosomal cargo is typically sorted to other vesicles or targeted to lysosomes for degradation (Théry et al., 2002; Hurley, 2008), the mechanisms whereby APP is channeled toward Aβ generation rather than lysosomal degradation bear careful examination. Endocytosis is accelerated with upregulation of endocytotic GTPase effectors, rab5 and rab7, in vulnerable regions of the brain from individuals with mild cognitive impairment and AD (Ginsberg et al., 2010) and gain-of-function polymorphisms in endocytosis pathway genes confer increased risk for AD (Harold et al., 2009). Concomitantly, studies also point to impairment in flux through the endosome-lysosome pathway (for review, see Peric and Annaert, 2015) resulting in accumulation of endosomes (as observed in neurons in humans before development of AD pathology; Cataldo et al., 2000) and in transdifferentiated neurons from patients' skin fibroblasts (Israel et al., 2012). Emerging evidence indicates that lysosome dysfunction may underlie the observed impairment in endosomal flux (Funk and Kuret, 2012; Peric and Annaert, 2015). These events permit prolonged colocalization of APP with the amyloidogenic β- and γ-secretase machinery to permit Aβ generation (Rajendran and Annaert, 2012). Our findings indicate that activation of TFEB, a transcriptional activator of multiple lysosomal and trafficking genes (Palmieri et al., 2011; Settembre et al., 2013a), corrects the underlying lysosomal impairment to stimulate flux through the endosome-lysosome pathway to enhance APP degradation and reduce Aβ generation.
Our data do not exclude the possibility that TFEB enhances a pathway for trafficking of APP directly from Golgi to lysosomes, while bypassing the plasma membrane and endosomes, where amyloidogenic cleavage of APP takes place. Consistent with this hypothesis, APP has been demonstrated to interact with sortilin, a Vps10p family receptor, in a manner that increases its lysosomal targeting and reduces its abundance in lipid rafts, the primary sites for APP endocytosis (Kojro et al., 2001; Yang et al., 2013) and with a related protein, SORLA, which regulates APP localization in endosomes and Aβ generation in neurons (Burgert et al., 2013). It is intriguing to speculate that proteins may regulate physiologic trafficking of APP to the lysosomes in a manner mimicking their role in targeting of other lysosomal proteins (Coutinho et al., 2012; Burgert et al., 2013); further studies will be required to address this systematically.
TFEB also stimulates autophagy (Settembre et al., 2011), a lysosomal degradative pathway that is upregulated in advanced stages of AD pathology (Nixon and Yang, 2011). In neurons, both endosomes and autophagosomes traffic to the lysosomes (Lee et al., 2011), and accumulation of autophagosomes due to impaired fusion with lysosomes may also contribute to enhanced Aβ generation (Yu et al., 2005). A careful examination of neuronal autophagy with modulation of autophagic flux in rat cortical neurons revealed that while interventions to induce autophagosome formation did not affect APP metabolism, impairment in lysosome function increased APP-βCTF accumulation with increased Aβ generation, suggesting that accumulation of autophagosomes rather than upregulation of autophagosome formation accelerates amyloidogenic APP processing (Boland et al., 2010). Importantly, TFEB stimulates autophagosome-lysosome fusion and degradation of autophagosome contents (Settembre et al., 2011). Therefore, while we did not directly examine the effects of TFEB activation on neuronal autophagy, our data are also consistent with efficient trafficking of APP into the lysosomes down this pathway to attenuate Aβ generation.
Recently, AAV-mediated neuronal TFEB activation was not found to affect amyloid plaque deposition in the 5XFAD mouse model, whereas TFEB activation was sufficient to drive clearance of Tau aggregates to attenuate AD pathology (Polito et al., 2014). Conceivably, the lack of effect of TFEB activation on amyloid pathology is a function of the rapid development of amyloid pathology in this model (within a few weeks after birth), compared with the APP/PS1 mice where amyloid plaques first appear at 6 months of age (Yan et al., 2009) or the differences in the level of TFEB transduction and activation. Indeed, other studies have demonstrated that inhibition of GSK3α and β isoforms restores lysosomal acidification with attenuation of Aβ levels and plaque pathology in the 5XFAD model with increased APP degradation (Avrahami et al., 2013), likely driven by TFEB activation (Parr et al., 2012).
It is interesting to note that Aβ half-life was also reduced with neuronal TFEB expression (Fig. 3A,B). Indeed, neurons also take up and degrade Aβ, albeit their contribution to Aβ removal from the ISF is proportionately smaller compared with that of glial cell types (Kanekiyo and Bu, 2014). TFEB transduction in neurons may stimulate Aβ uptake by macropinocytosis (Holmes et al., 2013) as we have previously observed in TFEB-transduced astrocytes (Xiao et al., 2014).
Our study highlights the therapeutic potential of neuronal TFEB activation to reduce Aβ generation and amyloid plaque pathology. While evidence for endosome-lysosome pathway dysfunction in human AD is predominantly correlative, recent work demonstrates that Aβ levels in the CSF increase at the time of initial plaque deposition in AD animal models (Maia et al., 2015). This is followed by a subsequent decline in CSF Aβ, believed to be from Aβ sequestration by amyloid plaques. A similar decline in Aβ is also observed preceding the development of dementia in patients with dominantly inherited AD (Fagan et al., 2014). These findings lend further support to increased Aβ generation as an early driver of pathology and underscore the potential for targeting endosomal-lysosome dysfunction early in AD pathogenesis. Importantly, we did not observe adverse effects with neuronal TFEB transduction with the viral gene transfer approach, and no prior adverse consequences of TFEB expression in CNS cell types have been described (Tsunemi et al., 2012; Decressac et al., 2013; Polito et al., 2014; Xiao et al., 2014). Nevertheless, examination of the effects of global TFEB activation on neuronal pathology and cognitive parameters over long-term studies is warranted to further assess its therapeutic potential. A strategy of TFEB-mediated suppression of Aβ generation in neurons is likely to be synergistic with acceleration of Aβ uptake and degradation by astrocytes (Xiao et al., 2014) in lowering ISF Aβ levels. In this context, studies are also needed to determine the effect of exogenous TFEB on microglial phagocytosis of amyloid material, with its potential to accelerate removal of amyloid deposits, as a veritable arm of a multipronged approach to therapeutically target TFEB activation in multiple CNS cell types in AD.
Footnotes
This study was supported by grants from the Alzheimer's Association (NIRG 12-242588) to A.D., Brightfocus Foundation (A2012151) to J.R.C., and from the National Institutes of Health (R21 NS082529) to J.-M.L and (R01 AG042513 and R21 AG045691) to J.R.C. We thank Wim Annaert, VIB (Flanders Institute for Biotechnology), Leuven, Belgium, for providing the dual fluorescent APP construct and Stuart Kornfeld, Washington University, for scientific input.
The authors declare no competing financial interests.
References
- Avrahami L, Farfara D, Shaham-Kol M, Vassar R, Frenkel D, Eldar-Finkelman H. Inhibition of glycogen synthase kinase-3 ameliorates beta-amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the Alzheimer disease mouse model: in vivo and in vitro studies. J Biol Chem. 2013;288:1295–1306. doi: 10.1074/jbc.M112.409250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benowitz LI, Rodriguez W, Paskevich P, Mufson EJ, Schenk D, Neve RL. The amyloid precursor protein is concentrated in neuronal lysosomes in normal and Alzheimer disease subjects. Exp Neurol. 1989;106:237–250. doi: 10.1016/0014-4886(89)90156-8. [DOI] [PubMed] [Google Scholar]
- Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, Lee JM, Holtzman DM. Neuronal activity regulates the regional vulnerability to amyloid-beta deposition. Nat Neurosci. 2011;14:750–756. doi: 10.1038/nn.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom GS. Amyloid-beta and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014;71:505–508. doi: 10.1001/jamaneurol.2013.5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland B, Smith DA, Mooney D, Jung SS, Walsh DM, Platt FM. Macroautophagy is not directly involved in the metabolism of amyloid precursor protein. J Biol Chem. 2010;285:37415–37426. doi: 10.1074/jbc.M110.186411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgert T, Schmidt V, Caglayan S, Lin F, Füchtbauer A, Füchtbauer EM, Nykjaer A, Carlo AS, Willnow TE. SORLA-dependent and -independent functions for PACS1 in control of amyloidogenic processes. Mol Cell Biol. 2013;33:4308–4320. doi: 10.1128/MCB.00628-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan MJ, Lipinski WJ, Bian F, Durham RA, Pack A, Walker LC. Augmented senile plaque load in aged female beta-amyloid precursor protein-transgenic mice. Am J Pathol. 2001;158:1173–1177. doi: 10.1016/S0002-9440(10)64064-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso GL, Gandy SE, Buxbaum JD, Greengard P. Chloroquine inhibits intracellular degradation but not secretion of Alzheimer beta/A4 amyloid precursor protein. Proc Natl Acad Sci U S A. 1992;89:2252–2256. doi: 10.1073/pnas.89.6.2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso GL, Takei K, Gandy SE, Matteoli M, Mundigl O, Greengard P, De Camilli P. Morphologic and biochemical analysis of the intracellular trafficking of the Alzheimer beta/A4 amyloid precursor protein. J Neurosci. 1994;14:3122–3138. doi: 10.1523/JNEUROSCI.14-05-03122.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer's disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol. 2000;157:277–286. doi: 10.1016/S0002-9440(10)64538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, McPhie DL, Hirschberg J, Neve RL. The amyloid precursor protein-binding protein APP-BP1 drives the cell cycle through the S-M checkpoint and causes apoptosis in neurons. J Biol Chem. 2000;275:8929–8935. doi: 10.1074/jbc.275.12.8929. [DOI] [PubMed] [Google Scholar]
- Chia PZ, Toh WH, Sharples R, Gasnereau I, Hill AF, Gleeson PA. Intracellular itinerary of internalised beta-secretase, BACE1, and its potential impact on beta-amyloid peptide biogenesis. Traffic. 2013;14:997–1013. doi: 10.1111/tra.12088. [DOI] [PubMed] [Google Scholar]
- Ciechanover A, Schwartz AL, Dautry-Varsat A, Lodish HF. Kinetics of internalization and recycling of transferrin and the transferrin receptor in a human hepatoma cell line. Effect of lysosomotropic agents. J Biol Chem. 1983;258:9681–9689. [PubMed] [Google Scholar]
- Cirrito JR, May PC, O'Dell MA, Taylor JW, Parsadanian M, Cramer JW, Audia JE, Nissen JS, Bales KR, Paul SM, DeMattos RB, Holtzman DM. In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-beta metabolism and half-life. J Neurosci. 2003;23:8844–8853. doi: 10.1523/JNEUROSCI.23-26-08844.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, Disabato BM, Restivo JL, Verges DK, Goebel WD, Sathyan A, Hayreh D, D'Angelo G, Benzinger T, Yoon H, Kim J, Morris JC, Mintun MA, Sheline YI. Serotonin signaling is associated with lower amyloid-beta levels and plaques in transgenic mice and humans. Proc Natl Acad Sci U S A. 2011;108:14968–14973. doi: 10.1073/pnas.1107411108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coen K, Flannagan RS, Baron S, Carraro-Lacroix LR, Wang D, Vermeire W, Michiels C, Munck S, Baert V, Sugita S, Wuytack F, Hiesinger PR, Grinstein S, Annaert W. Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. J Cell Biol. 2012;198:23–35. doi: 10.1083/jcb.201201076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho MF, Prata MJ, Alves S. A shortcut to the lysosome: the mannose-6-phosphate-independent pathway. Mol Genet Metab. 2012;107:257–266. doi: 10.1016/j.ymgme.2012.07.012. [DOI] [PubMed] [Google Scholar]
- Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, Zinn AE, Casali BT, Restivo JL, Goebel WD, James MJ, Brunden KR, Wilson DA, Landreth GE. ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science. 2012;335:1503–1506. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruchaga C, Karch CM, Jin SC, Benitez BA, Cai Y, Guerreiro R, Harari O, Norton J, Budde J, Bertelsen S, Jeng AT, Cooper B, Skorupa T, Carrell D, Levitch D, Hsu S, Choi J, Ryten M, Ryten M, Hardy J, et al. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer's disease. Nature. 2014;505:550–554. doi: 10.1038/nature12825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM, Dice JF. When lysosomes get old. Exp Gerontol. 2000;35:119–131. doi: 10.1016/S0531-5565(00)00075-9. [DOI] [PubMed] [Google Scholar]
- Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, Björklund A. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc Natl Acad Sci U S A. 2013;110:E1817–1826. doi: 10.1073/pnas.1305623110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan AM, Xiong C, Jasielec MS, Bateman RJ, Goate AM, Benzinger TL, Ghetti B, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Salloway S, Schofield PR, Sperling RA, Marcus D, Cairns NJ, Buckles VD, Ladenson JH, Morris JC, et al. Longitudinal change in CSF biomarkers in autosomal-dominant Alzheimer's disease. Sci Transl Med. 2014;6:226ra30. doi: 10.1126/scitranslmed.3007901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk KE, Kuret J. Lysosomal fusion dysfunction as a unifying hypothesis for Alzheimer's disease pathology. Int J Alzheimers Dis. 2012;2012:752894. doi: 10.1155/2012/752894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD, Mufson EJ, Counts SE, Wuu J, Alldred MJ, Nixon RA, Che S. Regional selectivity of rab5 and rab7 protein upregulation in mild cognitive impairment and Alzheimer's disease. J Alzheimers Dis. 2010;22:631–639. doi: 10.3233/JAD-2010-101080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golde TE, Estus S, Younkin LH, Selkoe DJ, Younkin SG. Processing of the amyloid protein precursor to potentially amyloidogenic derivatives. Science. 1992;255:728–730. doi: 10.1126/science.1738847. [DOI] [PubMed] [Google Scholar]
- Grbovic OM, Mathews PM, Jiang Y, Schmidt SD, Dinakar R, Summers-Terio NB, Ceresa BP, Nixon RA, Cataldo AM. Rab5-stimulated up-regulation of the endocytic pathway increases intracellular beta-cleaved amyloid precursor protein carboxyl-terminal fragment levels and Abeta production. J Biol Chem. 2003;278:31261–31268. doi: 10.1074/jbc.M304122200. [DOI] [PubMed] [Google Scholar]
- Haass C, Koo EH, Mellon A, Hung AY, Selkoe DJ. Targeting of cell-surface beta-amyloid precursor protein to lysosomes: alternative processing into amyloid-bearing fragments. Nature. 1992;357:500–503. doi: 10.1038/357500a0. [DOI] [PubMed] [Google Scholar]
- Haass C, Kaether C, Thinakaran G, Sisodia S. Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med. 2012;2:a006270. doi: 10.1101/cshperspect.a006270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet. 2009;41:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann T, Bieger SC, Brühl B, Tienari PJ, Ida N, Allsop D, Roberts GW, Masters CL, Dotti CG, Unsicker K, Beyreuther K. Distinct sites of intracellular production for Alzheimer's disease A beta40/42 amyloid peptides. Nat Med. 1997;3:1016–1020. doi: 10.1038/nm0997-1016. [DOI] [PubMed] [Google Scholar]
- Higaki J, Catalano R, Guzzetta AW, Quon D, Navé JF, Tarnus C, D'Orchymont H, Cordell B. Processing of beta-amyloid precursor protein by cathepsin D. J Biol Chem. 1996;271:31885–31893. doi: 10.1074/jbc.271.50.31885. [DOI] [PubMed] [Google Scholar]
- Holmes BB, DeVos SL, Kfoury N, Li M, Jacks R, Yanamandra K, Ouidja MO, Brodsky FM, Marasa J, Bagchi DP, Kotzbauer PT, Miller TM, Papy-Garcia D, Diamond MI. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci U S A. 2013;110:E3138–3147. doi: 10.1073/pnas.1301440110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y, Zhao H, Lu X, Kong Y, Jin H. Meta-analysis of the cystatin C(CST3) gene G73A polymorphism and susceptibility to Alzheimer's disease. Int J Neurosci. 2012;122:431–438. doi: 10.3109/00207454.2012.672502. [DOI] [PubMed] [Google Scholar]
- Hurley JH. ESCRT complexes and the biogenesis of multivesicular bodies. Curr Opin Cell Biol. 2008;20:4–11. doi: 10.1016/j.ceb.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, Hefferan MP, Van Gorp S, Nazor KL, Boscolo FS, Carson CT, Laurent LC, Marsala M, Gage FH, Remes AM, Koo EH, Goldstein LS. Probing sporadic and familial Alzheimer's disease using induced pluripotent stem cells. Nature. 2012;482:216–220. doi: 10.1038/nature10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner SL, Younkin SG, Borchelt DR. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. 2004;13:159–170. doi: 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/S0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Kanekiyo T, Bu G. The low-density lipoprotein receptor-related protein 1 and amyloid-beta clearance in Alzheimer's disease. Front Aging Neurosci. 2014;6:93. doi: 10.3389/fnagi.2014.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekiyo T, Cirrito JR, Liu CC, Shinohara M, Li J, Schuler DR, Holtzman DM, Bu G. Neuronal clearance of amyloid-beta by endocytic receptor LRP1. J Neurosci. 2013;33:19276–19283. doi: 10.1523/JNEUROSCI.3487-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato Y, Maruyama W, Naoi M, Hashizume Y, Osawa T. Immunohistochemical detection of dityrosine in lipofuscin pigments in the aged human brain. FEBS Lett. 1998;439:231–234. doi: 10.1016/S0014-5793(98)01372-6. [DOI] [PubMed] [Google Scholar]
- Keilani S, Lun Y, Stevens AC, Williams HN, Sjoberg ER, Khanna R, Valenzano KJ, Checler F, Buxbaum JD, Yanagisawa K, Lockhart DJ, Wustman BA, Gandy S. Lysosomal dysfunction in a mouse model of Sandhoff disease leads to accumulation of ganglioside-bound amyloid-beta peptide. J Neurosci. 2012;32:5223–5236. doi: 10.1523/JNEUROSCI.4860-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Castellano JM, Jiang H, Basak JM, Parsadanian M, Pham V, Mason SM, Paul SM, Holtzman DM. Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular A beta clearance. Neuron. 2009;64:632–644. doi: 10.1016/j.neuron.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodam A, Maulik M, Peake K, Amritraj A, Vetrivel KS, Thinakaran G, Vance JE, Kar S. Altered levels and distribution of amyloid precursor protein and its processing enzymes in Niemann-Pick type C1-deficient mouse brains. Glia. 2010;58:1267–1281. doi: 10.1002/glia.21001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh YH, von Arnim CA, Hyman BT, Tanzi RE, Tesco G. BACE is degraded via the lysosomal pathway. J Biol Chem. 2005;280:32499–32504. doi: 10.1074/jbc.M506199200. [DOI] [PubMed] [Google Scholar]
- Kojro E, Gimpl G, Lammich S, Marz W, Fahrenholz F. Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha-secretase ADAM 10. Proc Natl Acad Sci U S A. 2001;98:5815–5820. doi: 10.1073/pnas.081612998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo EH, Squazzo SL. Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J Biol Chem. 1994;269:17386–17389. [PubMed] [Google Scholar]
- Lee CY, Tse W, Smith JD, Landreth GE. Apolipoprotein E promotes beta-amyloid trafficking and degradation by modulating microglial cholesterol levels. J Biol Chem. 2012;287:2032–2044. doi: 10.1074/jbc.M111.295451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, Uchiyama Y, Westaway D, Cuervo AM, Nixon RA. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146–1158. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Sato Y, Nixon RA. Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer's-like axonal dystrophy. J Neurosci. 2011;31:7817–7830. doi: 10.1523/JNEUROSCI.6412-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Liu H, Foyil SR, Godar RJ, Weinheimer CJ, Hill JA, Diwan A. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation. 2012;125:3170–3181. doi: 10.1161/CIRCULATIONAHA.111.041814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maia LF, Kaeser SA, Reichwald J, Lambert M, Obermüller U, Schelle J, Odenthal J, Martus P, Staufenbiel M, Jucker M. Increased CSF Abeta during the very early phase of cerebral Abeta deposition in mouse models. EMBO Mol Med. 2015;7:895–903. doi: 10.15252/emmm.201505026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar A, Capetillo-Zarate E, Cruz D, Gouras GK, Maxfield FR. Degradation of Alzheimer's amyloid fibrils by microglia requires delivery of ClC-7 to lysosomes. Mol Biol Cell. 2011;22:1664–1676. doi: 10.1091/mbc.E10-09-0745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBrayer M, Nixon RA. Lysosome and calcium dysregulation in Alzheimer's disease: partners in crime. Biochem Soc Trans. 2013;41:1495–1502. doi: 10.1042/BST20130201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menacherry S, Hubert W, Justice JB., Jr In vivo calibration of microdialysis probes for exogenous compounds. Anal Chem. 1992;64:577–583. doi: 10.1021/ac00030a003. [DOI] [PubMed] [Google Scholar]
- Morel E, Chamoun Z, Lasiecka ZM, Chan RB, Williamson RL, Vetanovetz C, Dall'Armi C, Simoes S, Point Du Jour KS, McCabe BD, Small SA, Di Paolo G. Phosphatidylinositol-3-phosphate regulates sorting and processing of amyloid precursor protein through the endosomal system. Nat Commun. 2013;4:2250. doi: 10.1038/ncomms3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller-Steiner S, Zhou Y, Arai H, Roberson ED, Sun B, Chen J, Wang X, Yu G, Esposito L, Mucke L, Gan L. Antiamyloidogenic and neuroprotective functions of cathepsin B: implications for Alzheimer's disease. Neuron. 2006;51:703–714. doi: 10.1016/j.neuron.2006.07.027. [DOI] [PubMed] [Google Scholar]
- Neely KM, Green KN, LaFerla FM. Presenilin is necessary for efficient proteolysis through the autophagy-lysosome system in a gamma-secretase-independent manner. J Neurosci. 2011;31:2781–2791. doi: 10.1523/JNEUROSCI.5156-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA, Yang DS. Autophagy failure in Alzheimer's disease—locating the primary defect. Neurobiol Dis. 2011;43:38–45. doi: 10.1016/j.nbd.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, Sardiello M, Ballabio A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet. 2011;20:3852–3866. doi: 10.1093/hmg/ddr306. [DOI] [PubMed] [Google Scholar]
- Parr C, Carzaniga R, Gentleman SM, Van Leuven F, Walter J, Sastre M. Glycogen synthase kinase 3 inhibition promotes lysosomal biogenesis and autophagic degradation of the amyloid-beta precursor protein. Mol Cell Biol. 2012;32:4410–4418. doi: 10.1128/MCB.00930-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña-Llopis S, Vega-Rubin-de-Celis S, Schwartz JC, Wolff NC, Tran TA, Zou L, Xie XJ, Corey DR, Brugarolas J. Regulation of TFEB and V-ATPases by mTORC1. EMBO J. 2011;30:3242–3258. doi: 10.1038/emboj.2011.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez RG, Soriano S, Hayes JD, Ostaszewski B, Xia W, Selkoe DJ, Chen X, Stokin GB, Koo EH. Mutagenesis identifies new signals for beta-amyloid precursor protein endocytosis, turnover, and the generation of secreted fragments, including Abeta42. J Biol Chem. 1999;274:18851–18856. doi: 10.1074/jbc.274.27.18851. [DOI] [PubMed] [Google Scholar]
- Peric A, Annaert W. Early etiology of Alzheimer's disease: tipping the balance toward autophagy or endosomal dysfunction? Acta Neuropathol. 2015;129:363–381. doi: 10.1007/s00401-014-1379-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polito VA, Li H, Martini-Stoica H, Wang B, Yang L, Xu Y, Swartzlander DB, Palmieri M, di Ronza A, Lee VM, Sardiello M, Ballabio A, Zheng H. Selective clearance of aberrant tau proteins and rescue of neurotoxicity by transcription factor EB. EMBO Mol Med. 2014;6:1142–1160. doi: 10.15252/emmm.201303671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran L, Annaert W. Membrane trafficking pathways in Alzheimer's disease. Traffic. 2012;13:759–770. doi: 10.1111/j.1600-0854.2012.01332.x. [DOI] [PubMed] [Google Scholar]
- Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, Walther TC, Ferguson SM. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 2012;5:ra42. doi: 10.1126/scisignal.2002790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh JH, Huang Y, Bero AW, Kasten T, Stewart FR, Bateman RJ, Holtzman DM. Disruption of the sleep-wake cycle and diurnal fluctuation of beta-amyloid in mice with Alzheimer's disease pathology. Sci Transl Med. 2012;4:150ra122. doi: 10.1126/scitranslmed.3004291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sannerud R, Declerck I, Peric A, Raemaekers T, Menendez G, Zhou L, Veerle B, Coen K, Munck S, De Strooper B, Schiavo G, Annaert W. ADP ribosylation factor 6 (ARF6) controls amyloid precursor protein (APP) processing by mediating the endosomal sorting of BACE1. Proc Natl Acad Sci U S A. 2011;108:E559–568. doi: 10.1073/pnas.1100745108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, Banfi S, Parenti G, Cattaneo E, Ballabio A. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–477. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- Schneider A, Rajendran L, Honsho M, Gralle M, Donnert G, Wouters F, Hell SW, Simons M. Flotillin-dependent clustering of the amyloid precursor protein regulates its endocytosis and amyloidogenic processing in neurons. J Neurosci. 2008;28:2874–2882. doi: 10.1523/JNEUROSCI.5345-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuur M, Ikram MA, van Swieten JC, Isaacs A, Vergeer-Drop JM, Hofman A, Oostra BA, Breteler MM, van Duijn CM. Cathepsin D gene and the risk of Alzheimer's disease: a population-based study and meta-analysis. Neurobiol Aging. 2011;32:1607–1614. doi: 10.1016/j.neurobiolaging.2009.10.011. [DOI] [PubMed] [Google Scholar]
- Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, Ballabio A. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol. 2013a;14:283–296. doi: 10.1038/nrm3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, Huynh T, Carissimo A, Palmer D, Klisch TJ, Wollenberg AC, Di Bernardo D, Chan L, Irazoqui JE, Ballabio A. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol. 2013b;15:647–658. doi: 10.1038/ncb2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheline YI, West T, Yarasheski K, Swarm R, Jasielec MS, Fisher JR, Ficker WD, Yan P, Xiong C, Frederiksen C, Grzelak MV, Chott R, Bateman RJ, Morris JC, Mintun MA, Lee JM, Cirrito JR. An antidepressant decreases CSF Abeta production in healthy individuals and in transgenic AD mice. Sci Transl Med. 2014;6:236re4. doi: 10.1126/scitranslmed.3008169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamboli IY, Hampel H, Tien NT, Tolksdorf K, Breiden B, Mathews PM, Saftig P, Sandhoff K, Walter J. Sphingolipid storage affects autophagic metabolism of the amyloid precursor protein and promotes Abeta generation. J Neurosci. 2011;31:1837–1849. doi: 10.1523/JNEUROSCI.2954-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Théry C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2:569–579. doi: 10.1038/nri855. [DOI] [PubMed] [Google Scholar]
- Tsunemi T, Ashe TD, Morrison BE, Soriano KR, Au J, Roque RA, Lazarowski ER, Damian VA, Masliah E, La Spada AR. PGC-1alpha rescues Huntington's disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci Transl Med. 2012;4:142ra97. doi: 10.1126/scitranslmed.3003799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- Wolfe DM, Lee JH, Kumar A, Lee S, Orenstein SJ, Nixon RA. Autophagy failure in Alzheimer's disease and the role of defective lysosomal acidification. Eur J Neurosci. 2013;37:1949–1961. doi: 10.1111/ejn.12169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, Silverstein SC, Husemann J. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med. 2003;9:453–457. doi: 10.1038/nm838. [DOI] [PubMed] [Google Scholar]
- Xiao Q, Gil SC, Yan P, Wang Y, Han S, Gonzales E, Perez R, Cirrito JR, Lee JM. Role of phosphatidylinositol clathrin assembly lymphoid-myeloid leukemia (PICALM) in intracellular amyloid precursor protein (APP) processing and amyloid plaque pathogenesis. J Biol Chem. 2012;287:21279–21289. doi: 10.1074/jbc.M111.338376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Q, Yan P, Ma X, Liu H, Perez R, Zhu A, Gonzales E, Burchett JM, Schuler DR, Cirrito JR, Diwan A, Lee JM. Enhancing astrocytic lysosome biogenesis facilitates Abeta clearance and attenuates amyloid plaque pathogenesis. J Neurosci. 2014;34:9607–9620. doi: 10.1523/JNEUROSCI.3788-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan P, Bero AW, Cirrito JR, Xiao Q, Hu X, Wang Y, Gonzales E, Holtzman DM, Lee JM. Characterizing the appearance and growth of amyloid plaques in APP/PS1 mice. J Neurosci. 2009;29:10706–10714. doi: 10.1523/JNEUROSCI.2637-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Virassamy B, Vijayaraj SL, Lim Y, Saadipour K, Wang YJ, Han YC, Zhong JH, Morales CR, Zhou XF. The intracellular domain of sortilin interacts with amyloid precursor protein and regulates its lysosomal and lipid raft trafficking. PLoS One. 2013;8:e63049. doi: 10.1371/journal.pone.0063049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee JH, Mohan PS, Mercken M, Farmery MR, Tjernberg LO, Jiang Y, Duff K, Uchiyama Y, Näslund J, Mathews PM, Cataldo AM, Nixon RA. Macroautophagy—a novel Beta-amyloid peptide-generating pathway activated in Alzheimer's disease. J Cell Biol. 2005;171:87–98. doi: 10.1083/jcb.200505082. [DOI] [PMC free article] [PubMed] [Google Scholar]