

ABSTRACT
Chronic infections are a serious health care problem, and bacterial persisters have been implicated in infection reoccurrence. Progress toward finding antipersister therapies has been slow, in part because of knowledge gaps regarding the physiology of these rare phenotypic variants. Evidence shows that growth status is important for survival, as nongrowing cultures can have 100-fold more persisters than growing populations. However, additional factors are clearly important, as persisters remain rare even in nongrowing populations. What features, beyond growth inhibition, allow persisters to survive antibiotic stress while the majority of their kin succumb to it remains an open question. To investigate this, we used stationary phase as a model nongrowing environment to study Escherichia coli persistence to ofloxacin. Given that the prevailing model of persistence attributes survival to transient dormancy and antibiotic target inactivity, we anticipated that persisters would suffer less damage than their dying kin. However, using genetic mutants, flow cytometry, fluorescence-activated cell sorting, and persistence assays, we discovered that nongrowing ofloxacin persisters experience antibiotic-induced damage that is indistinguishable from that of nonpersisters. Consistent with this, we found that these persisters required DNA repair for survival and that repair machinery was unnecessary until the posttreatment recovery period (after ofloxacin removal). These findings suggest that persistence to ofloxacin is not engendered solely by reduced antibiotic target corruption, demonstrate that what happens following antibiotic stress can be critical to the persistence phenotype, and support the notion that inhibition of DNA damage repair systems could be an effective strategy to eliminate fluoroquinolone persisters.
IMPORTANCE
In the absence of resistant mutants, infection reoccurrences can still occur because of persisters, rare bacterial cells that survive antibiotic treatments to repopulate infection sites. Persister survival is attributed to a transient state of dormancy in which a cell’s growth and metabolism are significantly reduced and many essential processes are thought to be inactive. Thus, dormancy is believed to protect persisters from antibiotic-induced damage and death. In this work, we show that in nongrowing populations, persisters to ofloxacin experience the same level of antibiotic-induced damage as cells that succumb to the treatment and that their survival critically depends on repair of this damage after the conclusion of treatment. These findings reveal that persistence to ofloxacin is not engendered solely by reduced antibiotic target corruption and highlight that processes following antibiotic stress are important to survival. We hypothesize that effective antipersister therapies may be developed on the basis of this knowledge.
INTRODUCTION
Health care-associated infections (HAIs) in the United States are a significant public health problem, taking almost 100,000 lives and costing 35 to 45 billion dollars annually (1, 2). Many HAIs involve bacterial biofilms, which have a tendency to relapse upon the conclusion of antibiotic treatment (3). The bacteria that repopulate these infections are often as sensitive as the original population to the antibiotic used, suggesting that bacterial persisters, rather than resistant mutants, are to blame for the recurring infections (4–9).
Bacterial persisters are rare phenotypic variants that can transiently tolerate supralethal antibiotic concentrations (10, 11). Persisters are present in most bacterial cultures (3, 5–8, 11–15), and they are detected by using antibiotic kill curves (5, 6, 11, 13, 16, 17). Initially, a rapid killing rate is observed, which depicts the death of normal cells. It then slows to a second, much lower rate of killing, demonstrating the presence of persisters.
Persisters’ impressive antibiotic tolerances have long been attributed to transient dormancy, in which temporary cessation of essential cell functions renders antibiotic primary targets inactive, thereby protecting bacteria from antibiotic-induced death (8, 11). Balaban and colleagues provided strong evidence supporting this theory, demonstrating that ampicillin persisters can originate from growth-inhibited cells that resume normal replication upon antibiotic removal (10). Recent work by Maisonneuve and colleagues also supports the association between slow growth and ampicillin tolerance (18). Small subpopulations of exponential-phase cultures displayed a highly activated yoeB/yefM operon or high RpoS levels, nearly complete growth cessation, tolerance to ampicillin treatment, and the ability to resume growth upon antibiotic removal. However, only 1 of the 15 or 16 highly activated and nonlysing cells was shown to resume growth to form microcolonies (18), suggesting that the remaining >90% of the nongrowing cells may have lacked the additional properties required for persistence. Shah and colleagues studied persistence as a function of rRNA expression in exponential-phase cultures, isolating cells with low green fluorescent protein (GFP) expression from the bulk, and observed an ~20-fold higher survival rate in this “dim” subpopulation. Although enriched, most of this subpopulation still succumbed to ofloxacin (19), indicating that the majority of the cells with low protein expression still lacked the characteristics necessary for persistence. Recently, we found that although low metabolic activity and a lack of cell division prior to antibiotic stress greatly increase the odds that a cell will be a persister, >99% of the dormant cells in an exponentially growing culture were still not persisters and bacteria that were growing rapidly prior to antibiotic treatment could also give rise to persisters (20). While these studies support a strong and important association between slow growth and persistence, they also highlight the fact that persistence is more complex than dormancy. The additional features, beyond growth inhibition, that allow persisters to weather antibiotic stress while the majority of their kin succumb to it remain ill defined.
Stationary-phase cultures have served as a valuable model of nongrowing populations, and useful knowledge about persistence has been obtained by studying these populations under antibiotic stress (21–23). Hansen and colleagues identified mutations, including several global regulators (e.g., fis, hns, and hnr), that altered tolerance to ofloxacin in stationary phase, indicating the complexity of persister formation (21). By screening a transposon library, Li and Zhang showed that inactivation of phoU, a regulator of phosphate metabolism, led to a general increase in susceptibility to numerous antibiotic and environmental stresses, particularly in stationary phase (22). Luidalepp and colleagues added to this knowledge base by demonstrating altered persister levels upon the modulation of central metabolism with ΔicdA, Δmdh, and ΔacnB and discovering that the effect of mutations on persistence could be highly dependent on the length of time in stationary phase (23). Although these studies contribute to the knowledge of persistence in stationary phase, the phenotypic qualities that allow survival remain unclear. Identifying the features that separate nongrowing persisters from nongrowing cells that die is important to understanding persister physiology and improving therapies that combat this detrimental phenotype.
In this study, we examined persistence to ofloxacin, a fluoroquinolone (FQ) antibiotic, in stationary-phase cultures. FQs are effective against nongrowing bacteria and possess broad-spectrum activity against topoisomerases of both Gram-positive and Gram-negative bacteria. Because of this ability, FQs are often used to treat serious infections (including HAIs) (24). The source of toxicity for this antibiotic class is FQ binding to topoisomerases, including DNA gyrase, which leads to DNA damage (25, 26). Specifically, ofloxacin binds DNA-bound DNA gyrase A and allows its endonuclease activity to proceed but inhibits its religation activity, thereby producing DNA breaks (27). Given the prevailing model of persistence, in which tolerance is achieved by transient inactivity of antibiotic primary targets, ofloxacin persisters should possess reduced DNA gyrase activity, limiting drug-induced corruption, DNA damage, and cell death. Evidence in support of this model for FQs has been obtained from exponentially growing cultures (28, 29). Dorr and colleagues discovered that specific DNA damage repair enzymes are required for persistence to ciprofloxacin in exponential-phase populations. However, they also determined that most persisters experienced modest ciprofloxacin-induced damage compared to that experienced by cells that died, which was evidenced by the use of a λ-derived DNA damage reporter (28). In a follow-up study, Dorr and colleagues identified the TisB toxin as the DNA damage-inducible element responsible for the majority of ciprofloxacin persistence (29). Collectively, these studies suggested that small amounts of ciprofloxacin-induced DNA damage in persisters stimulated the expression of TisB, which went on to disrupt the inner membrane, depress proton motive force and ATP synthesis, and reduce topoisomerase activity. Interestingly, Dorr and colleagues did not find TisB to be important for ciprofloxacin persistence in stationary phase (29), which suggested that the connection between FQ-induced DNA damage and persistence in exponential-phase populations may not apply to nongrowing populations.
To determine whether the extent of antibiotic-induced damage is defining for persistence to FQs in nongrowing populations, we analyzed the SOS response in ofloxacin-treated stationary-phase cultures of Escherichia coli at single-cell resolution. SOS induction occurs in response to DNA damage, and reporters of the SOS response have previously been used as indicators of DNA damage (28–30). Here, using four distinct transcriptional reporters, heterogeneous SOS induction was observed upon ofloxacin treatment and this heterogeneous activation was not found to result from heterogeneous protein synthesis between SOS-responding (R) and nonresponding (nR) subpopulations. Interestingly, using fluorescence-activated cell sorting (FACS), we found persisters to arise with comparable frequencies in the R and nR subpopulations. Further, upon removal of ofloxacin and introduction into fresh medium for 2 h, which stimulated an impressive induction of the SOS response in >90% of the population, persisters were again found to be equally distributed throughout the spectrum of SOS induction. These data surprisingly suggested that the extent of FQ-induced DNA damage in E. coli within nongrowing populations is not a feature that distinguishes persisters from cells that succumb to antibiotic treatment. Upon further dissection of this phenomenon, we discovered that neither the level of SOS machinery before nor SOS induction during ofloxacin treatment impacts persister levels. Rather, we showed that the abundance of DNA repair machinery and the cell’s ability to repair ofloxacin-induced DNA damage during recovery, which is the time period following the conclusion of antibiotic treatment, are critical to persistence. Together, this evidence implies that persisters to ofloxacin in stationary-phase populations sustain DNA damage that is equivalent to the damage in cells that die or lose their culturability as a result of antibiotic treatment and that the events after treatment are equally, if not more, important to cell survival as what occurs before or during treatment.
RESULTS
In this study, we investigated the response of persisters in stationary-phase populations to ofloxacin. Use of nongrowing cultures allowed us to dissect the dependency of persistence on different parameters without the complicating factors of growth heterogeneity and low persister levels (20, 31, 32). On the basis of the prevailing model of persistence, which attributes cell survival to transient dormancy and antibiotic primary target inactivity (14), the expectation was that tolerance toward ofloxacin should result from reduced DNA gyrase activity and manifest from lower levels of ofloxacin-induced DNA damage than in cells that lose their culturability from treatment, either by death or entry into the viable-but-nonculturable (VBNC) state (32–34). To assess the accuracy of this model, we used transcriptional reporters of DNA damage, flow cytometry, and FACS to measure SOS induction in persisters relative to that in nonpersisters within a stationary-phase population. The SOS response is triggered by DNA damage, and it is induced by numerous DNA-damaging agents, including FQs (26, 35). During this process, RecA is involved in a variety of functions (36). One key function, the stimulation of LexA repressor self-cleavage and dissociation (37), leads to the induction of more than 50 genes, approximately 30 of which have been associated with the SOS response and DNA damage repair (38, 39). Thus, SOS induction has been used as an indicator of DNA damage in E. coli in numerous studies (28–30), and we used induction of SOS genes as a reporter for ofloxacin-induced DNA damage here.
Ofloxacin induces a heterogeneous SOS response in stationary-phase E. coli.
With transcriptional fusions of the PrecA, PrecN, PsulA, and PtisB promoters to gfp, we observed induction of the SOS response, indicating DNA damage, in ofloxacin-treated stationary-phase cultures of E. coli (Fig. 1A). We note that FQs do not (i) rapidly destroy bacterial cell walls to allow release of cytoplasmic contents or (ii) target the translational apparatus, which enabled the use of GFP as a reporter in ofloxacin-treated samples. Induction of GFP in response to ofloxacin was confirmed to be SOS dependent with the use of the ΔrecA mutant and an uncleavable lexA mutant, lexA3 (see Fig. S1 in the supplemental material) (40). In addition, the killing dynamics of all of the reporter strains under these treatment conditions were confirmed to be biphasic, which is a necessary condition for persister quantification (see Fig. S2 and S3 in the supplemental material). Interestingly, SOS induction was found to be heterogeneous, with only 20 to 30% of the total population exhibiting increased fluorescence from the SOS transcriptional reporters (Fig. 1A). Because we used induction of a fluorescent protein to indicate DNA damage, we considered the possibility that the heterogeneity observed arose from a differential ability of cells to synthesize new protein. To determine if this was the case, we employed a synthetic construct with mCherry under the control of an isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible promoter, the PrecA, PrecN, PsulA, and PtisB reporters, and two-color flow cytometry. As depicted in Fig. 1B, mCherry induction produced a single, well-defined, high-fluorescence population, in both the R and nR subpopulations. Controls that confirm mCherry was produced in the R and nR subpopulations during ofloxacin treatment are provided in Fig. S4 in the supplemental material. These data showed that although only a subpopulation (20 to 30%) of the stationary-phase cells responded to ofloxacin-induced DNA damage, the general protein synthesis capabilities of the R and nR subpopulations were equivalent during drug treatment. This indicated that stationary-phase E. coli exhibited differential induction of the SOS response upon ofloxacin treatment. Given the prevailing model of persister tolerance, we anticipated that the subpopulations that failed to induce the SOS response should be enriched with persisters because of a lesser extent of ofloxacin-induced DNA damage.
FIG 1 .
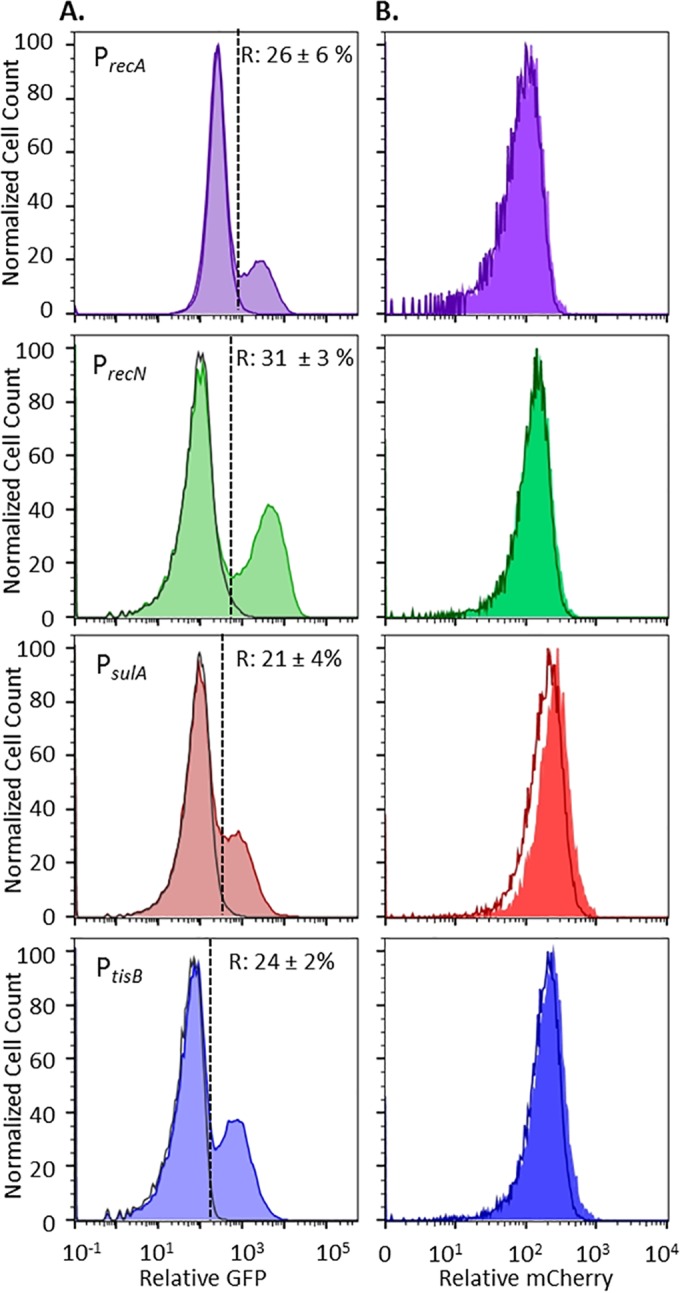
Stationary-phase E. coli responds to ofloxacin by SOS response induction. (A) GFP induction from SOS transcriptional reporters upon ofloxacin treatment (shaded colored curves). R and nR subpopulations were delineated with respect to untreated controls (unshaded gray curves). nR cells are to the left of the dashed lines, and R cells are to the right. The average percentages of the total populations classified as R ± the standard deviation of at least three replicates are indicated for each promoter. Controls that confirm SOS-dependent induction in response to ofloxacin under these treatment conditions are provided in Fig. S1 in the supplemental material. Data that demonstrate biphasic killing of these strains in response to the ofloxacin treatment conditions used here are provided in Fig. S2 and S3 in the supplemental material. (B) Induction of mCherry from PT5lacO occurs in both the R (shaded curves) and nR (unshaded curves) subpopulations. For each reporter, mCherry in R cells and that in nR cells largely overlap. Controls that confirm induction of mCherry during ofloxacin treatment as well as the gating strategies used are provided in Fig. S4 in the supplemental material.
Persisters arise with equivalent frequency from SOS-responding and nonresponding subpopulations.
To assess persister abundance in the subpopulations that did and did not elicit an ofloxacin-induced SOS response, we used FACS to segregate ofloxacin-treated cultures into four subpopulations on the basis of their induction of SOS promoters. Gates A and B were designed to capture the nR subpopulation, since ≥98% of the events from ofloxacin-treated ΔrecA mutant controls fell within these gates. The nR subpopulation was further separated such that quantile A contained a minimum number of events with the lowest GFP expression and quantile B contained the remainder. Gates C and D were designed to capture the R subpopulation, since ≤2% of the events from ofloxacin-treated ΔrecA mutant controls fell within these gates. The R subpopulation was further segregated such that quantile D included the top 10% of the cells with the highest GFP expression and quantile C contained the remainder of the R subpopulation. Surprisingly, and contrary to what was expected, we observed that persisters were equally distributed among all of the gates for each SOS reporter; in other words, survival was independent of SOS induction (Fig. 2). To confirm that these results were not due to imprecision in the sorting experiments, we reanalyzed the sorted fractions (A, B, C, and D) of each reporter (see Fig. S5 in the supplemental material). As anticipated, subpopulation A had the highest composition of nR cells upon reanalysis (~93% when averaged across all four reporters), whereas subpopulation D had the highest composition of R cells upon reanalysis (~78% when averaged across all four reporters). When this degree of imprecision was accounted for (see Text S1 in the supplemental material for details), the average frequencies of persisters in the nR and R subpopulations did not deviate by more than 13% from what was measured for the A and D subpopulations for each reporter (see Fig. S5 in the supplemental material), which confirmed that persisters arose from the nR and R subpopulations with approximately equal likelihood. These data suggested that persisters and nonpersisters experienced and responded to DNA damage equivalently. Further, these data implied that (i) the majority of stationary-phase E. coli cells, irrespective of whether they induced the SOS response, experienced a lethal level of DNA damage from ofloxacin and (ii) induction of the SOS response during ofloxacin treatment was inconsequential to persistence. The first of these implications is based on the fact that the majority of the cells in both the nR and R subpopulations died or lost their culturability during ofloxacin treatment, whereas the second is a direct corollary of equivalent persister frequencies in the nR and R subpopulations.
FIG 2 .
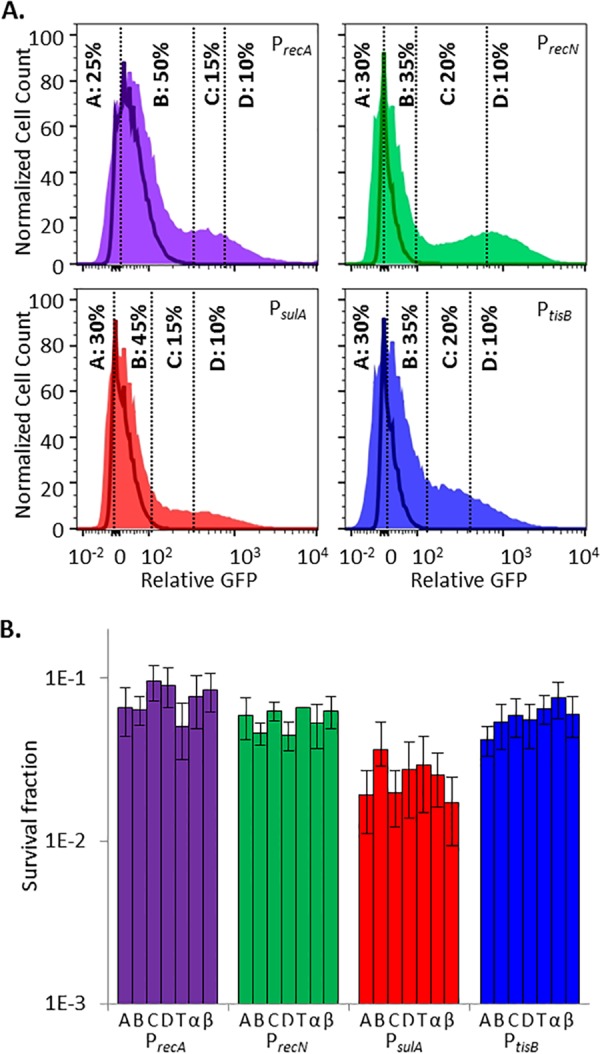
Induction of the SOS response in stationary-phase persisters and nonpersisters is equivalent. (A) GFP induction from SOS transcriptional reporters with ofloxacin. Unshaded curves, ΔrecA mutant controls treated for 5 h with ofloxacin; shaded curves, samples treated for 5 h with ofloxacin. Events in gates C and D were considered to have responded to ofloxacin because ≤2% of the events from ofloxacin-treated ΔrecA mutant controls fell within those gates. Controls for each reporter are provided in Fig. S1 and S4 in the supplemental material. (B) Survival fractions of ofloxacin-treated cells from gates designated in panel A. T, total-population control passed through the sorter (all gates); α and β, samples diluted to postsorting cell density before and after sorting that did not themselves travel through the sorter. Sorting did not significantly reduce sample culturability. This was determined by a lack of significant difference in survival between the total sorted population (T) and the presorting (α) or postsorting (β) controls (by Student’s t test, two tails with unequal variance) for any of the SOS reporters tested. Survival fractions were calculated relative to the cell density of sorted samples, 3 × 105 cells/ml. Differences between the survival of PsulA (fractions or controls) and the other three SOS reporters were not significant for the majority of the comparisons, as determined by Student’s t test (two tails with unequal variance). Biphasic killing was observed for all strains (see Fig. S2 in the supplemental material), and reanalysis of FACS-segregated populations is provided in Fig. S5 in the supplemental material.
Persisters and nonpersisters dramatically induce the SOS response during recovery.
To provide evidence beyond culturability measurements (numbers of CFU per milliliter) that the majority of stationary-phase E. coli cells experienced considerable DNA damage upon treatment with ofloxacin, we reasoned that most of the cells would exhibit filamentation and SOS induction, hallmarks of stress from DNA damage (41), upon the removal of ofloxacin and exposure to fresh nutrients (recovery). We note that filamentation was not observed while treating stationary-phase cells with ofloxacin because they were in a nongrowing state. To determine whether the above hypothesis was correct, ofloxacin was removed from stationary-phase cultures of PrecA-gfp and the cells were then transferred to fresh liquid LB medium. After 2 h of recovery, >90% of the cells showed an impressive SOS response, with expression from PrecA reaching >100 times that achieved with the ΔrecA mutant control (Fig. 3A). In addition, microscopic inspection of these cultures demonstrated that most of the cells had undergone filamentation (Fig. 3D). These data provide further evidence that most of the stationary-phase E. coli cells experienced ofloxacin-induced DNA damage, regardless of the induction of the SOS response during the treatment period. Interestingly, during the 2-h recovery period, the number of CFU per milliliter was found to be stable (Fig. 3B), suggesting that persisters had yet to initiate any measurable replication. Therefore, we used FACS to segregate the culture after 2 h of recovery and determine whether persisters were enriched in any subpopulation (e.g., low-SOS-responding fraction, high-SOS-responding fraction) or equally distributed throughout the population. As depicted in Fig. 3C, persisters were found at almost equal frequencies along the entire distribution of SOS induction. This provided additional compelling evidence that ofloxacin-induced DNA damage within persisters and nonpersisters of stationary cultures was equivalent. To further explore this discovery, we reasoned that if persisters in nongrowing populations suffered extensive DNA damage, most of them should require DNA repair machinery for survival.
FIG 3 .

Persisters and nonpersisters induce an impressive SOS response within 2 h of recovery in liquid medium. (A) Shaded purple curve, WT containing PrecA-gfp treated in stationary phase for 5 h with ofloxacin, followed by 2 h of recovery in antibiotic-free LB medium. Unshaded curve, same as purple curve, except that the ΔrecA mutant containing PrecA-gfp was used. Vertical lines indicate the FACS gating strategy used. Additional controls are provided in Fig. S6 in the supplemental material. (B) The number of CFU per milliliter remained unchanged through 2 h of recovery in fresh LB medium. Numbers of CFU per milliliter are reported as average values ± the minimum and maximum values of two biological replicates. (C) Survival fractions from the A, B, C, and D gates designated in panel A. T, total population (all gates); α, pre-FACS control; β, post-FACS control. Survival fractions were calculated relative to the cell density of sorted samples, 3 × 105 cells/ml. Data are average values ± standard errors from at least three biological replicates. (D) WT morphology and induction of PrecA in cells subjected to ofloxacin treatment (OFL tx) for 5 h in stationary phase after transfer to antibiotic-free LB medium for 0 and 2 h, along with untreated controls. Controls demonstrating that PrecA induction and filamentation are RecA dependent are provided in Fig. S6 in the supplemental material. All images are 87.36 by 66.56 µm.
DNA break repair machinery is required for ofloxacin persistence in stationary phase.
To test the above hypothesis, we examined the importance of different DNA repair machinery for persister survival by using genetic mutants. We measured persister abundance in strains lacking key DNA break repair enzymes (ΔrecA, ΔrecB, ΔrecF, ΔrecN, ΔdksA, and ΔruvA deletion mutants), and the TisB toxin (ΔtisAB deletion mutant). RecA is central to recombination events and DNA break repair, as it catalyzes DNA strand exchange reactions in homologous recombination, recruitment of enzymes to the site of DNA damage, LexA cleavage, and induction of the SOS response upon DNA damage (37). RecB interacts with RecA (42, 43), possesses DNA-dependent ATPase activity (44), and is essential for homologous recombination and DNA break repair by RecBCD (45). RecF stabilizes RecA–single-stranded DNA complexes (46), RecN is essential for the repair of two or more double-strand breaks (47), DksA is involved in RecN-DNA assembly (48), RuvA is a known DNA-binding protein involved in RecBCD-dependent DNA repair (49), and TisB is a membrane-associated peptide implicated in persister formation in exponentially growing populations (29).
Upon the treatment of stationary-phase cultures of the mutants with ofloxacin, we observed biphasic killing and various levels of survival (Fig. 4). The ΔrecF, ΔtisAB, and ΔdksA mutants exhibited persistence similar to that of wild-type (WT) cells, whereas survival was significantly reduced (P values of <0.05) in the ΔrecN and ΔruvA mutants by approximately 50-fold, in the ΔrecB mutant by >1,000-fold, and in the ΔrecA mutant by >10,000-fold. These data provide evidence that persisters in stationary phase suffer ofloxacin-induced DNA damage that needs to be repaired to support survival. In consideration of the FACS data (Fig. 2), which suggested that SOS induction during ofloxacin treatment was inconsequential to persistence in stationary phase, the requirement of different DNA repair systems for persister survival raised several interesting questions. These included whether basal levels of DNA repair machinery are sufficient for persistence and, if not, when SOS induction, which activates the expression of RecN, RuvA, RecB, and RecA, is required for the phenotype.
FIG 4 .
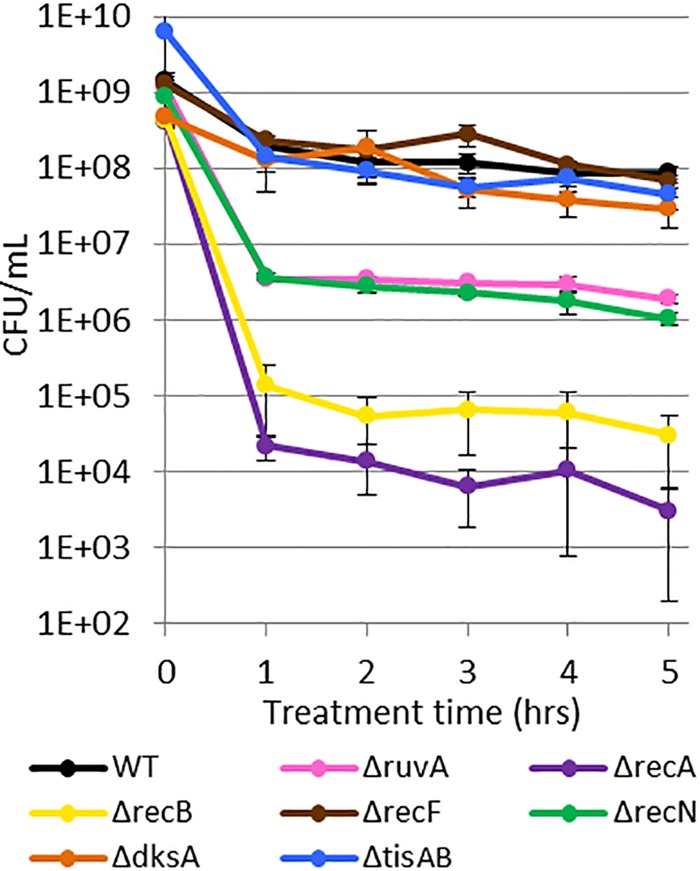
DNA damage repair system mutants have reduced survival after ofloxacin treatment in stationary phase. Ofloxacin persisters decreased approximately 104-fold in ΔrecA mutants, 103-fold in ΔrecB mutants, and 50-fold in ΔruvA and ΔrecN mutants. ΔrecF, ΔtisAB, and ΔdksA resulted in persister levels comparable to those of the WT. Data are average values ± standard errors from at least three biological replicates.
Transcription and translation, including induction of the SOS response, during ofloxacin treatment are not required for persistence.
To begin to answer the above questions, we sought to provide evidence, in addition to the FACS analysis, that demonstrated that induction of the SOS response during ofloxacin treatment was inconsequential to persistence in stationary-phase cultures. To do this, we treated PrecN reporters with 100 µg/ml rifampin (RIF) or 50 µg/ml chloramphenicol (CM) to stop RNA or protein synthesis prior to and during ofloxacin treatment. Both RIF and CM, at these concentrations, prevented GFP induction by ofloxacin, confirming that the treatments used were sufficient to prevent RNA and protein synthesis (Fig. 5A). When RIF- or CM-treated cultures were assayed for persistence to ofloxacin, survival fractions were indistinguishable from the samples treated with ofloxacin only (Fig. 5B). This illustrated that transcription and translation during ofloxacin treatment, including but not limited to induction of the SOS response, are not required for persistence to ofloxacin in nongrowing populations.
FIG 5 .

Induction of transcription and translation during ofloxacin (OFL) treatment is not required for persister survival. (A) Treatment with CM (shaded blue curve) or RIF (shaded red curve) inhibits ofloxacin-induced translation or transcription, respectively, from PrecN-gfp. Unshaded gray curves, non-ofloxacin-treated control. (B) Treatment with CM or RIF does not alter ofloxacin persister levels or culturability; all ofloxacin-treated samples displayed biphasic killing. CM and RIF were dissolved in dimethyl sulfoxide (DMSO). (C) Ofloxacin persisters decreased 1,000-fold in the presence of LexA3 (gray curve) relative to the WT (black curve). Kill curve data are average values ± standard errors from at least three biological replicates.
As a complement to the RIF and CM treatment assays, we measured persistence to ofloxacin in stationary-phase cultures of the lexA3 mutant, which is an uncleavable mutant that prevents SOS induction in response to DNA damage. Interestingly, the lexA3 mutant had a statistically significant ~1,000-fold reduction in persisters compared to the WT (Fig. 5C). An important difference between the RIF and CM experiments and those performed with lexA3 is that RIF and CM were removed after ofloxacin treatment, which relieved suppression of transcription and translation during the postantibiotic recovery period, whereas lexA3 continuously prohibited SOS induction both during and after ofloxacin treatment. In fact, a parallel can be drawn for the DNA repair system mutants, which are devoid of the systems both during and after ofloxacin treatment.
Collectively, the data presented in Fig. 1 to 5 demonstrate that persisters induce the SOS response both during and after ofloxacin treatment and that DNA repair systems and SOS induction are required for the phenotype. Interestingly, SOS induction during treatment was found to be dispensable. These results inspired us to further examine when persisters in nongrowing populations require DNA repair systems and SOS induction to survive ofloxacin treatment.
Persisters require DNA damage repair systems and the SOS response only during recovery.
To determine when stationary-phase ofloxacin persisters require DNA repair machinery, we enumerated persisters from samples with RecA expression induced prior to ofloxacin treatment, during treatment, and during recovery only. We found that while persisters were 10,000-fold less abundant in recA-deficient cells than in WT cells (Fig. 4), induction of RecA only during recovery was sufficient to restore persister levels to those with RecA expressed prior to and during treatment in stationary-phase cultures (Fig. 6A). RecA was used in these experiments because it was found to be the most important DNA repair system for persister survival from ofloxacin treatment (Fig. 4).
FIG 6 .

RecA and the SOS response are critical to persistence only during recovery from ofloxacin treatment (OFL tx). (A) Ofloxacin persisters decreased >1,000-fold in the absence of RecA expression (dashed purple) relative to when RecA was expressed from 2 h pretreatment with ofloxacin through recovery (dashed blue). RecA induction on LB plates during recovery only (red) was sufficient to restore persisters to WT levels. (B) Ofloxacin persisters decreased 25-fold with LexA3 (blue) relative to the WT (black). LexA3 induction on LB plates only (red) maintained this reduced survival. We note that LexA3 induction only during recovery reduced the overall viability in both ofloxacin-treated and untreated samples, and this was accounted for by plotting the surviving fraction. Data are average values ± standard errors from at least three biological replicates. Numbers of CFU per milliliter for all samples and controls are provided in Fig. S7 in the supplemental material.
Because RecA is involved in both DNA damage repair and SOS induction, we tested persistence in cells harboring inducible lexA3 to more precisely examine the requirement of the SOS response. LexA3 is a dominant negative mutation (40), and therefore, in this system, lexA remained on the chromosome, which enabled normal SOS induction in the absence of lexA3 expression. We enumerated persisters from samples with lexA3 expression induced prior to ofloxacin treatment, during treatment, and during recovery only. With impaired SOS induction, which was achieved with lexA3 expression from 2 h before treatment through recovery, we observed a significant (P value of <0.05) 25-fold decrease in survival compared to that of the uninduced control. This held true with lexA3 induction during recovery only (Fig. 6B). Thus, induction of the SOS response only during recovery from ofloxacin treatment is critical for persister survival.
To probe the generality of this phenomenon, we performed similar recA expression experiments with stationary-phase cultures treated with mitomycin C (MMC), a non-FQ DNA-damaging agent (50). As depicted in Fig. S7B in the supplemental material, induction of RecA only during recovery was sufficient to restore persister levels to those with RecA present prior to and during MMC treatment, which mirrors the results obtained with ofloxacin.
DISCUSSION
The prevailing model of persistence asserts that persisters survive antibiotic treatment by entering a transient state of dormancy, in which they sustain substantially less antibiotic-induced damage than cells that die (6, 8, 11, 13, 14). In addition to the studies already mentioned that analyzed persistence to β-lactams by time-lapse microscopy (10, 18), evidence for an analogous dependence between lack of primary target corruption and persistence to other drug classes has been obtained for growing cultures (28, 51). Dorr and colleagues used a λ-CI reporter, which is activated by massive DNA damage and strong SOS induction, to examine persistence to ciprofloxacin in exponential-phase populations and found that few persisters sustained extensive DNA damage relative to the bulk of growing cells that died (28). Using flow cytometry and confocal microscopy, Kim and colleagues observed that persisters to norfloxacin in growing populations are less likely to undergo filamentation in response to antibiotic treatment than are their dying kin, which also lent support to the prevailing model (51). In fact, even recent amendments to the collective understanding of persistence, which include the participation of multidrug efflux pumps (52, 53) and prodrug-activating enzymes (54), conform to a model where tolerance is achieved with a reduction in corruption of the antibiotic’s primary target. However, these lines of evidence were derived largely from growing populations, and it remained unknown, for antibiotics that retain activity against nongrowing bacteria, whether persisters from nongrowing populations sustained less antibiotic-induced damage than their kin that were also nongrowing but succumbed to the treatment. Therefore, we examined the responses of stationary-phase persisters and nonpersisters (dead cells and those that have lost their culturability [VBNC cells]) to ofloxacin. Ofloxacin binds DNA-bound DNA gyrase A and allows its endonuclease activity to continue while inhibiting its religation activity, thereby producing DNA breaks and stimulating the SOS response (27). By employing flow cytometry, FACS, and persister assays, we showed that the distribution of antibiotic-induced damage in persisters from nongrowing populations was indistinguishable from that in their nonpersister kin (Fig. 2 and 3). Consistent with these results, we found that persister levels in stationary-phase cultures were significantly reduced by several mutations to DNA damage repair systems, which included RecA, RecB, RuvA, and RecN (Fig. 4). These data, from nongrowing cultures, complement previous work that implied that DNA damage repair is important for persistence (28, 29) and more recent work that found DinG, UvrD, and RuvA to be important for ciprofloxacin persistence in stationary-phase cells cultured under different conditions (55). In fact, we postulate that the strong parallels between the mutations that reduce FQ persister levels in exponential- and stationary-phase cultures may exist because the majority of persisters in exponentially growing cultures arise from the small fraction of nongrowing bacteria within those populations (10, 18, 20, 32).
Interestingly, even though ofloxacin-induced DNA damage required repair in stationary-phase persisters (Fig. 4), induction of the SOS response during treatment did not impact persister survival (Fig. 2 and 5). However, lexA3 did reduce persister levels by 1,000-fold (Fig. 5C). This outcome directed us to probe when SOS induction and DNA damage repair are important for ofloxacin persister survival. Employing inducible recA and lexA3 expression systems, we illustrate that persistence to ofloxacin in stationary phase requires SOS induction and DNA damage repair machinery only during recovery (Fig. 6). While persister frequencies drop 10,000-fold relative to those of the WT in the absence of RecA, WT persister frequencies are robustly restored upon recA induction only during recovery on LB agar plates (Fig. 6A). Analogously, quenching the SOS response with LexA3 only during recovery reduced persistence to levels that were equivalent to those seen in cultures where inhibition of the SOS response began before ofloxacin treatment and continued throughout recovery (Fig. 6B). These data imply that both SOS induction and DNA damage repair by RecA during recovery participate in defining whether or not a nongrowing cell damaged by ofloxacin will survive as a persister.
In conclusion, the findings presented here complement the current understanding of the persister phenotype but suggest that an amendment of the prevailing dormancy model is in order. In particular, the results presented indicate that the extent of damage by FQs in nongrowing populations is not the key parameter that differentiates persisters and nonpersisters. Although the differentiating characteristic(s) was not explicitly identified in this study, on the basis of the work presented here, we postulate that it may be the location of DNA damage and/or the nature of that damage (e.g., single- versus double-strand DNA breaks [56]) that delineates ofloxacin persisters from nonpersisters in nongrowing populations. Further, this work demonstrates that, in addition to the fact that stationary-phase ofloxacin persisters sustain DNA damage, they do not require repair machinery prior to or during treatment (Fig. 6). This differs from findings on exponentially growing cultures that imply that what happens during treatment is important for persister formation (28, 29). Alternatively, we have discovered that recovery, an element of antibiotic tolerance assays that is frequently overlooked, is pivotal for persister survival. Indeed, we found this to be true also for the DNA-damaging agent MMC (see Fig. S7B in the supplemental material), which suggests that the importance of recovery periods for survival is not a phenomenon specific to FQs but one that could be quite general. Building upon this knowledge, we postulate that focusing antibacterial discovery efforts on inhibition of damage repair systems (57) will facilitate the identification of therapies to eliminate persisters from nongrowing populations and thereby improve the treatment outcomes for recalcitrant infections.
MATERIALS AND METHODS
Bacterial strains and plasmids.
All of the bacterial strains used in this study (see Table S1A in the supplemental material) were derived from MG1655, and their construction is described in Text S1 in the supplemental material. The construction of all of the plasmids used in this study (see Table S1A) is also detailed in Text S1 in the supplemental material.
Chemicals, media, and culture conditions.
All chemicals, unless otherwise noted, were purchased from Fisher Scientific or Sigma-Aldrich. IPTG was purchased from Gold Biotechnology (St. Louis, MO).
Liquid LB medium and LB agar plates were used for planktonic starter cultures and enumeration of CFU, respectively. M9 medium supplemented with 10 mM glucose as the sole carbon source (M9-glucose) was used for planktonic growth. Antibiotics were used at the following concentrations for selection, transcription, or translational inhibition, respectively: 50 µg/ml kanamycin (KAN), 100 µg/ml RIF, or 50 µg/ml CM. For persister assays, 5 µg/ml ofloxacin (16, 19, 20, 58) or 5 µg/ml MMC was used (59).
Unless otherwise noted, overnight cultures were prepared as follows. One milliliter of LB medium was inoculated from a 25% glycerol −80°C stock and grown for 4 h at 37°C with shaking (250 rpm), and then 20 µl of this starter culture was used to inoculate 2 ml of M9-glucose and subsequently grown at 37°C with shaking for another 16 h. To prepare cultures for ofloxacin or MMC treatment, 250-ml baffled flasks containing 25 ml of M9-glucose were inoculated with overnight cultures to an optical density at 600 nm (OD600) of 0.01. Cultures were then propagated for 20 h at 37°C with shaking (250 rpm) unless otherwise noted. All cultures of plasmid-bearing strains were supplemented with 50 µg/ml KAN for plasmid retention.
Inhibition of transcription and translation with RIF and CM.
Transcription and translation were inhibited in cells harboring the PrecN-gfp reporter plasmid by treatment with 100 µg/ml RIF and 50 µg/ml CM, respectively, for 30 min prior to and during ofloxacin treatment.
Persistence measurements.
Persistence was measured by enumerating CFU after exposure to 5 µg/ml ofloxacin or MMC for up to 5 h, which was sufficient to be in the second phase of the biphasic killing curve. Five-hundred-microliter samples were removed pretreatment, and 500-µl samples were collected hourly for 5 h. Cells were pelleted (3 min, 15,000 rpm), resuspended in an equal volume of phosphate-buffered saline (PBS), and serially diluted in PBS. Ten microliters per dilution of sample was spotted onto LB agar plates with or without 1 mM IPTG and incubated for 16 h at 37°C, and 10 to 100 colonies were counted per sample to determine persister levels (16). Survival fractions in sorted samples (Fig. 2B and 3C) were calculated relative to the cell density of sorted samples, 3 × 105 cells/ml. All reported colony counts and survival fractions are average values of three or more replicates, and error bars indicate standard errors.
To ensure that late-waking persisters were not a significant source of CFU after 16 h of recovery, plates were imaged and CFU were enumerated after 16 and 48 h of recovery at 37°C (see Fig. S8 in the supplemental material). To ensure that ofloxacin carryover was not impacting CFU measurements, WT and ΔrecA mutant samples were enumerated after plating on LB medium with or without 20 mM MgSO4 and incubated for 16 h at 37°C (see Fig. S7C in the supplemental material).
Flow cytometry of reporter strains.
Transcriptional SOS response reporters (see Table S1A in the supplemental material) were tested by inducing DNA damage by treatment with 5 µg/ml ofloxacin, while general protein production ability was tested by inducing mCherry expression from MO001 with 1 mM IPTG.
Reporters were induced for 5 h at 37°C and 250 rpm. Reporter samples were diluted to 107 cells/ml in PBS plus 50 µg/ml CM (to halt protein synthesis) from which single E. coli cells were identified by forward scatter (FSC) and side scatter (SSC) on an LSRII (BD Biosciences, San Jose, CA). GFP and mCherry fluorescence values were acquired on a per-cell basis by interrogating each cell with a 488- or 561-nm laser and collecting fluorescence with a 525/25- or 610/10-nm band-pass filter (respectively). Data were acquired on a FACSDiVa (BD Biosciences, San Jose, CA) and analyzed by FlowJo software (TreeStar, Ashland, OR). All collected events were gated as either R or nR by relative GFP expression, with the boundary set such that >99% of the untreated control events were nonresponders (Fig. 1). R and nR subpopulations were then plotted by relative mCherry expression. The GFP and mCherry expression distributions of all reporter strains were measured by flow cytometry prior to any follow-up sorting experiments. All flow cytometry experiments were repeated at least three times, and representative curves are presented. MG1655, MG1655 plus pUA66 (promoterless), and uninduced MG1655 plus reporter (e.g., without ofloxacin, without IPTG) were run as negative controls, whereas MG1655 plus PT5lac-gfp and MO001, both induced with IPTG, were used as GFP-positive and mCherry-positive controls, respectively.
Additionally, the PrecA-gfp SOS response reporter was treated with ofloxacin as described above and allowed to recover after ofloxacin treatment in fresh liquid LB medium (inoculated to an OD600 of 0.05 after ofloxacin removal by cell centrifugation and resuspension in fresh medium) for 2 h at 37°C and 250 rpm prior to analysis by flow cytometry and imaging. This liquid recovery was devised to resemble the nutrient environment experienced by cells when they were plated for CFU measurements, which were always conducted on LB agar, and thus, LB was used in this assay.
FACS.
Transcriptional SOS response and general protein production ability reporters (see Table S1A in the supplemental material) were cultured as described above, and FACS analysis of ofloxacin-treated samples was performed (Fig. 2 and 3). Samples were prepared for sorting by diluting cells to 2.5 × 107 cells/ml in warm, spent culture medium sterilized with a 0.22-µm filter. For pre- and postsorting controls, cells were further diluted to 3 × 105/ml in PBS, which was the postsorting cell concentration, before (alpha sample) and after (beta sample) sorting experiments. Additionally, the total unsegregated population (T sample) was collected after passage through the sorter. These controls were included to confirm that neither passage through the cell sorter nor the time between the conclusion of ofloxacin treatment and the conclusion of FACS was altering persister levels.
Cells were sorted with a Vantage cell sorter (Sony-iCyt Mission Technology, Champaign, IL) with a 70-µm nozzle in PBS sheath fluid at 30 lb/in2. Single E. coli cells were identified and gated by using FSC and SSC, and four physiologically distinct subpopulations (A to D) were segregated on the basis of GFP fluorescence. Fluorescence was measured with a 488-nm laser and a 530/15-nm band-pass filter.
FACS gating for all reporters is detailed in Text S1 in the supplemental material. All FACS experiments were repeated at least three times, and representative curves are presented. All reported survival fractions are average values of three or more replicates, and error bars indicate standard errors.
Microscopy.
Samples treated with ofloxacin and allowed to recover in fresh liquid LB medium as described above were imaged with a Nikon 90i epifluorescence microscope equipped with a 100× 1.4 numerical aperture objective (Nikon) and a Hamamatsu ORCA-R2 charge-coupled device camera. Five-hundred-microliter samples were collected upon inoculation (t = 0 h) and, after 2 h, pelleted (3 min, 15,000 rpm), resuspended in an equal volume of 4% paraformaldehyde, and incubated at room temperature for 25 min. Cells were then pelleted again, resuspended in an equal volume of PBS, and repelleted to a 10-fold concentration in PBS. Samples were spotted onto 1% LB low-melting-point agar pads and imaged for phase-contrast and GFP fluorescence microscopy. NIS Elements software (Nikon) was used to automate image acquisition for phase-contrast and fluorescence channels.
Statistical analysis.
The statistical significances of chemical treatments, genetic mutations, and inducible gene expression for persister formation were determined with two-tailed Student t tests with unequal variances. These tests are detailed in Text S1 in the supplemental material.
SUPPLEMENTAL MATERIAL
SOS reporter controls. (A) GFP expression from PrecA, PrecN, PsulA, and PtisB in ofloxacin-treated ΔrecA (unshaded colored curves) and untreated ΔrecA (unshaded gray curves) mutant cells. Empty-vector (pUA66) in ofloxacin-treated MG1655 (light-gray-shaded curves) and GFP-positive controls (MG1655 with PT5lacO-gfp harbored by pUA66, induced with IPTG since inoculation) (dark-gray-shaded curves) are included for reference. (B) GFP expression from PrecA, PrecN, PsulA, and PtisB in ofloxacin-treated lexA3 mutant cells (shaded colored curves) and untreated lexA3 mutant cells (unshaded gray curves). The empty vector (pUA66) in ofloxacin-treated lexA3 mutant cells (light-gray-shaded curves) and GFP-positive controls (MG1655 with PT5lacO-gfp harbored by pUA66, induced with IPTG since inoculation) (dark-gray-shaded curves) are included for reference. Download
SOS transcriptional reporter kill curves. WT (A) and MO001 (B) reporters and empty-vector pUA66 control kill curves display biphasic killing within 5 h of ofloxacin treatment. Mathematical quantification of biphasic killing is provided in Fig. S3. Data are average values ± standard errors from at least three biological replicates. Download
Mathematical analysis and quantification of biphasic killing kinetics. Experimental survival data (see Fig. S2, WT) (black dots) were captured by using a heterogeneous-population model (black line) that included two populations (CN, CP) with different constant death rates (kN, kP) and the assumption that growth during stationary phase is negligible. A homogeneous-population model (red line) that included a single population (CN), a single constant killing rate (kN), and the assumption that growth during stationary phase is negligible failed to capture the experimental data. The decimal reduction time, tdec, which is the time required for the number of CFU per milliliter of the population to drop by 90%, was calculated for each population considered. We note that the tdec for the persister subpopulation was ~25 times longer than that calculated for normal cells in the heterogeneous-population model. Models and parameter optimization are described in Text S1. Download
Gating schema and controls for SOS and protein production reporters. (A) Entire sample population displaying characteristic FSC and SSC areas. (B) GFP distribution of entire SOS reporter populations in ofloxacin-treated (shaded colored curves) and untreated samples (unshaded gray curves). Empty-vector (pUA66) negative controls in ofloxacin-treated samples (light-gray-shaded curves) and GFP-positive controls (MG1655 with PT5lacO-gfp harbored by pUA66, induced with IPTG since inoculation) (dark-gray-shaded curves) are included for reference. R and nR subpopulation boundaries were defined by untreated controls and are approximated by the dashed vertical lines. (C) mCherry distribution of R (shaded colored curves) and nR (unshaded color curves) subpopulations after 5 h of concurrent 1 mM IPTG induction and ofloxacin treatment. Empty vector (pUA66) in uninduced MO001 treated with ofloxacin (light-gray-shaded curves) and mCherry-positive controls (MO001 with 1 mM IPTG throughout growth) (dark-gray-shaded curves) are provided for reference. Download
Reanalysis of FACS-processed subpopulations. (A to D) Cells from subpopulations A, B, C, and D were reanalyzed as described in Text S1. To quantify the sorting precision with regard to the segregation of the nR and R subpopulations, the abundances of each sorted fraction (A, B, C, D) within the nR and R gates were measured during reanalysis. The nR/R threshold was based on the ΔrecA mutant control from the primary sorting. Percentages are reported as average values ± the minimum and maximum values of two biological replicates. nR and R boundaries are indicated by the dashed lines. (E) Average persister frequencies in subpopulations A and D compared to those calculated for the nR and R subpopulations, respectively, given the sorting imprecision quantified in panels A to D. Detailed calculation steps for fnR and fR are provided in Text S1. Download
Additional controls associated with the recovery experiments described in Fig. 3. (A) Shaded dark purple curve, WT containing PrecA-gfp that was treated in stationary phase for 5 h with ofloxacin and then allowed to recover for 2 h in antibiotic-free LB medium. Shaded light purple curve, same as the dark purple curve, except without the 2-h recovery period. Unshaded curve, same as the dark purple curve, except that the ΔrecA mutant containing PrecA-gfp was used. Shaded light gray curve, same as the dark purple curve, except that the WT containing the empty vector (pUA66) was used. Dark-gray-shaded curve, GFP-positive control (MG1655 with PT5lacO-gfp on pUA66, induced with IPTG since inoculation). (B) Reanalysis of sorted fractions from the gating strategy used for Fig. 3A. (C) ΔrecA mutant morphology and induction of PrecA in cells treated with ofloxacin (OFL) for 5 h in stationary phase after transfer to antibiotic-free LB medium for 0 and 2 h, along with untreated controls. All images are 87.36 by 66.56 µm. Download
Persister CFU-per-milliliter data. (A) Numbers of CFU per milliliter for ΔrecA mutant and WT cells containing different inducible systems or empty-vector controls. Error bars for MG1655 plus PT5lacO-lexA3 are depicted in blue to highlight the variability observed in the initial measurement, which would have been masked if left in black. Data are average values ± the standard errors from at least three biological replicates. (B) Numbers of CFU per milliliter for ΔrecA strains and empty-vector pUA66 negative controls upon treatment with the DNA-damaging agent MMC. The limit of detection is indicated by the red line at 10 CFU/ml. The ΔrecA mutant plus PT5lacO-recA was treated with 5 µg/ml MMC and RecA induction as indicated. WT (solid black curve with open markers) and ΔrecA mutant (dashed purple curve with open markers) cells plus empty vector pUA66 were treated with 5 µg/ml MMC. Data are average values ± standard errors of at least three biological replicates. (C) WT MG1655 and ΔrecA mutant ofloxacin persister survival is equivalent upon recovery on LB and LB plus 20 mM MgSO4 plates. Data are average values of two biological replicates with minimum and maximum error bars. Download
Persister quantification after 16 and 48 h. Numbers of CFU per milliliter are equivalent when determined after 16 and 48 h of incubation after ofloxacin (A) and MMC (B) treatments in the WT and ΔrecA mutant strains. Note that because of excessive growth at 48 h, the persister levels reported and analyzed were obtained after 16 h of postantibiotic incubation. Download
Bacterial strains, plasmids (A), and primers (B) used in this study.
Supplemental information. Download
ACKNOWLEDGMENTS
We are grateful to Christina DeCoste, John Grady, Albert Siryaporn, Hsin-Jung Li, Theresa Henry, Stephanie Amato, Mehmet Orman, Zemer Gitai, Thomas Silhavy, and Ned Wingreen for technical assistance and fruitful discussions. We thank the National BioResource Project (NIG, Japan) for distribution of the Keio collection.
This research was supported by the Department of the Army (W81XWH-12-2-0138), the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (R21AI115075), and Princeton University. The content is solely our responsibility and does not necessarily represent the official views of the Department of the Army, the National Institutes of Health, or Princeton University. The funders had no role in study design, data collection and analysis, the decision to publish, or preparation of the manuscript. We have no competing interests to declare.
Footnotes
Citation Völzing KG, Brynildsen MP. 2015. Stationary-phase persisters to ofloxacin sustain DNA damage and require repair systems only during recovery. mBio 6(5):e00731-15. doi:10.1128/mBio.00731-15.
REFERENCES
- 1.Klevens RM, Edwards JR, Richards CL, Horan TC, Gaynes RP, Pollock DA, Cardo DM. 2007. Estimating health care-associated infections and deaths in U.S. hospitals, 2002. Public Health Rep 122:160–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scott RD., II 2009. The direct medical costs of health care-associated infections in U.S. hospitals and the benefits of prevention. Centers for Disease Control and Prevention, Atlanta, GA: http://www.cdc.gov/HAI/pdfs/hai/Scott_CostPaper.pdf. [Google Scholar]
- 3.Costerton JW, Stewart PS, Greenberg EP. 1999. Bacterial biofilms: a common cause of persistent infections. Science 284:1318–1322. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- 4.Allison KR, Brynildsen MP, Collins JJ. 2011. Heterogeneous bacterial persisters and engineering approaches to eliminate them. Curr Opin Microbiol 14:593–598. doi: 10.1016/j.mib.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fauvart M, De Groote VN, Michiels J. 2011. Role of persister cells in chronic infections: clinical relevance and perspectives on anti-persister therapies. J Med Microbiol 60:699–709. doi: 10.1099/jmm.0.030932-0. [DOI] [PubMed] [Google Scholar]
- 6.Gefen O, Balaban NQ. 2009. The importance of being persistent: heterogeneity of bacterial populations under antibiotic stress. FEMS Microbiol Rev 33:704–717. doi: 10.1111/j.1574-6976.2008.00156.x. [DOI] [PubMed] [Google Scholar]
- 7.Levin BR, Concepción-Acevedo J, Udekwu KI. 2014. Persistence: a copacetic and parsimonious hypothesis for the existence of non-inherited resistance to antibiotics. Curr Opin Microbiol 21:18–21. doi: 10.1016/j.mib.2014.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lewis K. 2007. Persister cells, dormancy and infectious disease. Nat Rev Microbiol 5:48–56. doi: 10.1038/nrmicro1557. [DOI] [PubMed] [Google Scholar]
- 9.Wiuff C, Zappala RM, Regoes RR, Garner KN, Baquero F, Levin BR. 2005. Phenotypic tolerance: antibiotic enrichment of noninherited resistance in bacterial populations. Antimicrob Agents Chemother 49:1483–1494. doi: 10.1128/AAC.49.4.1483-1494.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S. 2004. Bacterial persistence as a phenotypic switch. Science 305:1622–1625. doi: 10.1126/science.1099390. [DOI] [PubMed] [Google Scholar]
- 11.Lewis K. 2010. Persister cells. Annu Rev Microbiol 64:357–372. doi: 10.1146/annurev.micro.112408.134306. [DOI] [PubMed] [Google Scholar]
- 12.Amato SM, Fazen CH, Henry TC, Mok WW, Orman MA, Sandvik EL, Volzing KG, Brynildsen MP. 2014. The role of metabolism in bacterial persistence. Front Microbiol 5:70. doi: 10.3389/fmicb.2014.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Balaban NQ. 2011. Persistence: mechanisms for triggering and enhancing phenotypic variability. Curr Opin Genet Dev 21:768–775. doi: 10.1016/j.gde.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 14.Balaban NQ, Gerdes K, Lewis K, McKinney JD. 2013. A problem of persistence: still more questions than answers? Nat Rev Microbiol 11:587–591. doi: 10.1038/nrmicro3076. [DOI] [PubMed] [Google Scholar]
- 15.Grant SS, Hung DT. 2013. Persistent bacterial infections, antibiotic tolerance, and the oxidative stress response. Virulence 4:273–283. doi: 10.4161/viru.23987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amato SM, Orman MA, Brynildsen MP. 2013. Metabolic control of persister formation in Escherichia coli. Mol Cell 50:475–487. doi: 10.1016/j.molcel.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 17.Maisonneuve E, Gerdes K. 2014. Molecular mechanisms underlying bacterial persisters. Cell 157:539–548. doi: 10.1016/j.cell.2014.02.050. [DOI] [PubMed] [Google Scholar]
- 18.Maisonneuve E, Castro-Camargo M, Gerdes K. 2013. (p)ppGpp controls bacterial persistence by stochastic induction of toxin-antitoxin activity. Cell 154:1140–1150. doi: 10.1016/j.cell.2013.07.048. [DOI] [PubMed] [Google Scholar]
- 19.Shah D, Zhang Z, Khodursky A, Kaldalu N, Kurg K, Lewis K. 2006. Persisters: a distinct physiological state of E. coli. BMC Microbiol 6:53. doi: 10.1186/1471-2180-6-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Orman MA, Brynildsen MP. 2013. Dormancy is not necessary or sufficient for bacterial persistence. Antimicrob Agents Chemother 57:3230–3239. doi: 10.1128/AAC.00243-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hansen S, Lewis K, Vulić M. 2008. Role of global regulators and nucleotide metabolism in antibiotic tolerance in Escherichia coli. Antimicrob Agents Chemother 52:2718–2726. doi: 10.1128/AAC.00144-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y, Zhang Y. 2007. PhoU is a persistence switch involved in persister formation and tolerance to multiple antibiotics and stresses in Escherichia coli. Antimicrob Agents Chemother 51:2092–2099. doi: 10.1128/AAC.00052-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luidalepp H, Jõers A, Kaldalu N, Tenson T. 2011. Age of inoculum strongly influences persister frequency and can mask effects of mutations implicated in altered persistence. J Bacteriol 193:3598–3605. doi: 10.1128/JB.00085-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Modai J. 1999. High-dose intravenous fluoroquinolones in the treatment of severe infections. J Chemother 11:478–485. doi: 10.1179/joc.1999.11.6.478. [DOI] [PubMed] [Google Scholar]
- 25.Drlica K, Zhao X. 1997. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol Mol Biol Rev 61:377–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kohanski MA, Dwyer DJ, Collins JJ. 2010. How antibiotics kill bacteria: from targets to networks. Nat Rev Microbiol 8:423–435. doi: 10.1038/nrmicro2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sato K, Inoue Y, Fujii T, Aoyama H, Mitsuhashi S. 1986. Antibacterial activity of ofloxacin and its mode of action. Infection 14(Suppl 4):S226–S230. doi: 10.1007/BF01661277. [DOI] [PubMed] [Google Scholar]
- 28.Dörr T, Lewis K, Vulić M. 2009. SOS response induces persistence to fluoroquinolones in Escherichia coli. PLoS Genet 5:e1000760. doi: 10.1371/journal.pgen.1000760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dörr T, Vulić M, Lewis K. 2010. Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLoS Biol 8:e1000317. doi: 10.1371/journal.pbio.1000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pennington JM, Rosenberg SM. 2007. Spontaneous DNA breakage in single living Escherichia coli cells. Nat Genet 39:797–802. doi: 10.1038/ng2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keren I, Kaldalu N, Spoering A, Wang Y, Lewis K. 2004. Persister cells and tolerance to antimicrobials. FEMS Microbiol Lett 230:13–18. doi: 10.1016/S0378-1097(03)00856-5. [DOI] [PubMed] [Google Scholar]
- 32.Roostalu J, Jõers A, Luidalepp H, Kaldalu N, Tenson T. 2008. Cell division in Escherichia coli cultures monitored at single cell resolution. BMC Microbiol 8:68. doi: 10.1186/1471-2180-8-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oliver JD. 2005. The viable but nonculturable state in bacteria. J Microbiol 43:93–100. [PubMed] [Google Scholar]
- 34.Orman MA, Brynildsen MP. 2013. Establishment of a method to rapidly assay bacterial persister metabolism. Antimicrob Agents Chemother 57:4398–4409. doi: 10.1128/AAC.00372-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baharoglu Z, Mazel D. 2014. SOS, the formidable strategy of bacteria against aggressions. FEMS Microbiol Rev 38:1126–1145. doi: 10.1111/1574-6976.12077. [DOI] [PubMed] [Google Scholar]
- 36.Keseler IM, Mackie A, Peralta-Gil M, Santos-Zavaleta A, Gama-Castro S, Bonavides-Martínez C, Fulcher C, Huerta AM, Kothari A, Krummenacker M, Latendresse M, Muñiz-Rascado L, Ong Q, Paley S, Schröder I, Shearer AG, Subhraveti P, Travers M, Weerasinghe D, Weiss V. 2013. EcoCyc: fusing model organism databases with systems biology. Nucleic Acids Res 41:D605–D612. doi: 10.1093/nar/gks1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.d’Ari R. 1985. The SOS system. Biochimie 67:343–347. doi: 10.1016/S0300-9084(85)80077-8. [DOI] [PubMed] [Google Scholar]
- 38.Keseler IM, Mackie A, Peralta-Gil M, Santos-Zavaleta A, Gama-Castro S, Bonavides-Martínez C, Fulcher C, Huerta AM, Kothari A, Krummenacker M, Latendresse M, Muñiz-Rascado L, Ong Q, Paley S, Schröder I, Shearer AG, Subhraveti P, Travers M, Weerasinghe D, Weiss V. 2013. EcoCyc: fusing model organism databases with systems biology. Nucleic Acids Res 41:D605–D612. doi: 10.1093/nar/gks1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salgado H, Peralta-Gil M, Gama-Castro S, Santos-Zavaleta A, Muñiz-Rascado L, García-Sotelo JS, Weiss V, Solano-Lira H, Martínez-Flores I, Medina-Rivera A, Salgado-Osorio G, Alquicira-Hernández S, Alquicira-Hernández K, López-Fuentes A, Porrón-Sotelo L, Huerta AM, Bonavides-Martínez C, Balderas-Martínez YI, Pannier L, Olvera M. 2013. RegulonDB v8.0: omics data sets, evolutionary conservation, regulatory phrases, cross-validated gold standards and more. Nucleic Acids Res 41:D203–D213. doi: 10.1093/nar/gks1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mount DW. 1977. A mutant of Escherichia coli showing constitutive expression of the lysogenic induction and error-prone DNA repair pathways. Proc Natl Acad Sci U S A 74:300–304. doi: 10.1073/pnas.74.1.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hill TM, Sharma B, Valjavec-Gratian M, Smith J. 1997. sfi-independent filamentation in Escherichia coli is lexA dependent and requires DNA damage for induction. J Bacteriol 179:1931–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu M, Souaya J, Julin DA. 1998. The 30-kDa C-terminal domain of the RecB protein is critical for the nuclease activity, but not the helicase activity, of the RecBCD enzyme from Escherichia coli. Proc Natl Acad Sci U S A 95:981–986. doi: 10.1073/pnas.95.3.981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu M, Souaya J, Julin DA. 1998. Identification of the nuclease active site in the multifunctional RecBCD enzyme by creation of a chimeric enzyme. J Mol Biol 283:797–808. doi: 10.1006/jmbi.1998.2127. [DOI] [PubMed] [Google Scholar]
- 44.Hickson ID, Robson CN, Atkinson KE, Hutton L, Emmerson PT. 1985. Reconstitution of RecBC DNase activity from purified Escherichia coli RecB and RecC proteins. J Biol Chem 260:1224–1229. [PubMed] [Google Scholar]
- 45.Courcelle J, Hanawalt PC. 2003. RecA-dependent recovery of arrested DNA replication forks. Annu Rev Genet 37:611–646. doi: 10.1146/annurev.genet.37.110801.142616. [DOI] [PubMed] [Google Scholar]
- 46.Whitby MC, Lloyd RG. 1995. Altered SOS induction associated with mutations in recF, recO and recR. Mol Gen Genet 246:174–179. doi: 10.1007/BF00294680. [DOI] [PubMed] [Google Scholar]
- 47.Finch PW, Finch PW, Chambers P, Emmerson PT. 1985. Identification of the Escherichia coli recN gene product as a major SOS protein. J Bacteriol 164:653–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meddows TR, Savory AP, Grove JI, Moore T, Lloyd RG. 2005. RecN protein and transcription factor DksA combine to promote faithful recombinational repair of DNA double-strand breaks. Mol Microbiol 57:97–110. doi: 10.1111/j.1365-2958.2005.04677.x. [DOI] [PubMed] [Google Scholar]
- 49.Shinagawa H, Makino K, Amemura M, Kimura S, Iwasaki H, Nakata A. 1988. Structure and regulation of the Escherichia coli ruv operon involved in DNA repair and recombination. J Bacteriol 170:4322–4329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weng MW, Zheng Y, Jasti VP, Champeil E, Tomasz M, Wang Y, Basu AK, Tang MS. 2010. Repair of mitomycin C mono- and interstrand cross-linked DNA adducts by UvrABC: a new model. Nucleic Acids Res 38:6976–6984. doi: 10.1093/nar/gkq576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim J-S, Heo P, Yang T-J, Lee K-S, Jin Y-S, Kim S-K, Shin D, Kweon D-H. 2011. Bacterial persisters tolerate antibiotics by not producing hydroxyl radicals. Biochem Biophys Res Commun 413:105–110. doi: 10.1016/j.bbrc.2011.08.063. [DOI] [PubMed] [Google Scholar]
- 52.Wu Y, Vulić M, Keren I, Lewis K. 2012. Role of oxidative stress in persister tolerance. Antimicrob Agents Chemother 56:4922–4926. doi: 10.1128/AAC.00921-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adams KN, Takaki K, Connolly LE, Wiedenhoft H, Winglee K, Humbert O, Edelstein PH, Cosma CL, Ramakrishnan L. 2011. Drug tolerance in replicating mycobacteria mediated by a macrophage-induced efflux mechanism. Cell 145:39–53. doi: 10.1016/j.cell.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wakamoto Y, Dhar N, Chait R, Schneider K, Signorino-Gelo F, Leibler S, McKinney JD. 2013. Dynamic persistence of antibiotic-stressed mycobacteria. Science 339:91–95. doi: 10.1126/science.1229858. [DOI] [PubMed] [Google Scholar]
- 55.Theodore A, Lewis K, Vulic M. 2013. Tolerance of Escherichia coli to fluoroquinolone antibiotics depends on specific components of the SOS response pathway. Genetics 195:1265–1276. doi: 10.1534/genetics.113.152306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Snyder M, Drlica K. 1979. DNA gyrase on the bacterial chromosome: DNA cleavage induced by oxolinic acid. J Mol Biol 131:287–302. doi: 10.1016/0022-2836(79)90077-9. [DOI] [PubMed] [Google Scholar]
- 57.Lu TK, Collins JJ. 2009. Engineered bacteriophage targeting gene networks as adjuvants for antibiotic therapy. Proc Natl Acad Sci U S A 106:4629–4634. doi: 10.1073/pnas.0800442106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Allison KR, Brynildsen MP, Collins JJ. 2011. Metabolite-enabled eradication of bacterial persisters by aminoglycosides. Nature 473:216–220. doi: 10.1038/nature10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Keren I, Shah D, Spoering A, Kaldalu N, Lewis K. 2004. Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. J Bacteriol 186:8172–8180. doi: 10.1128/JB.186.24.8172-8180.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SOS reporter controls. (A) GFP expression from PrecA, PrecN, PsulA, and PtisB in ofloxacin-treated ΔrecA (unshaded colored curves) and untreated ΔrecA (unshaded gray curves) mutant cells. Empty-vector (pUA66) in ofloxacin-treated MG1655 (light-gray-shaded curves) and GFP-positive controls (MG1655 with PT5lacO-gfp harbored by pUA66, induced with IPTG since inoculation) (dark-gray-shaded curves) are included for reference. (B) GFP expression from PrecA, PrecN, PsulA, and PtisB in ofloxacin-treated lexA3 mutant cells (shaded colored curves) and untreated lexA3 mutant cells (unshaded gray curves). The empty vector (pUA66) in ofloxacin-treated lexA3 mutant cells (light-gray-shaded curves) and GFP-positive controls (MG1655 with PT5lacO-gfp harbored by pUA66, induced with IPTG since inoculation) (dark-gray-shaded curves) are included for reference. Download
SOS transcriptional reporter kill curves. WT (A) and MO001 (B) reporters and empty-vector pUA66 control kill curves display biphasic killing within 5 h of ofloxacin treatment. Mathematical quantification of biphasic killing is provided in Fig. S3. Data are average values ± standard errors from at least three biological replicates. Download
Mathematical analysis and quantification of biphasic killing kinetics. Experimental survival data (see Fig. S2, WT) (black dots) were captured by using a heterogeneous-population model (black line) that included two populations (CN, CP) with different constant death rates (kN, kP) and the assumption that growth during stationary phase is negligible. A homogeneous-population model (red line) that included a single population (CN), a single constant killing rate (kN), and the assumption that growth during stationary phase is negligible failed to capture the experimental data. The decimal reduction time, tdec, which is the time required for the number of CFU per milliliter of the population to drop by 90%, was calculated for each population considered. We note that the tdec for the persister subpopulation was ~25 times longer than that calculated for normal cells in the heterogeneous-population model. Models and parameter optimization are described in Text S1. Download
Gating schema and controls for SOS and protein production reporters. (A) Entire sample population displaying characteristic FSC and SSC areas. (B) GFP distribution of entire SOS reporter populations in ofloxacin-treated (shaded colored curves) and untreated samples (unshaded gray curves). Empty-vector (pUA66) negative controls in ofloxacin-treated samples (light-gray-shaded curves) and GFP-positive controls (MG1655 with PT5lacO-gfp harbored by pUA66, induced with IPTG since inoculation) (dark-gray-shaded curves) are included for reference. R and nR subpopulation boundaries were defined by untreated controls and are approximated by the dashed vertical lines. (C) mCherry distribution of R (shaded colored curves) and nR (unshaded color curves) subpopulations after 5 h of concurrent 1 mM IPTG induction and ofloxacin treatment. Empty vector (pUA66) in uninduced MO001 treated with ofloxacin (light-gray-shaded curves) and mCherry-positive controls (MO001 with 1 mM IPTG throughout growth) (dark-gray-shaded curves) are provided for reference. Download
Reanalysis of FACS-processed subpopulations. (A to D) Cells from subpopulations A, B, C, and D were reanalyzed as described in Text S1. To quantify the sorting precision with regard to the segregation of the nR and R subpopulations, the abundances of each sorted fraction (A, B, C, D) within the nR and R gates were measured during reanalysis. The nR/R threshold was based on the ΔrecA mutant control from the primary sorting. Percentages are reported as average values ± the minimum and maximum values of two biological replicates. nR and R boundaries are indicated by the dashed lines. (E) Average persister frequencies in subpopulations A and D compared to those calculated for the nR and R subpopulations, respectively, given the sorting imprecision quantified in panels A to D. Detailed calculation steps for fnR and fR are provided in Text S1. Download
Additional controls associated with the recovery experiments described in Fig. 3. (A) Shaded dark purple curve, WT containing PrecA-gfp that was treated in stationary phase for 5 h with ofloxacin and then allowed to recover for 2 h in antibiotic-free LB medium. Shaded light purple curve, same as the dark purple curve, except without the 2-h recovery period. Unshaded curve, same as the dark purple curve, except that the ΔrecA mutant containing PrecA-gfp was used. Shaded light gray curve, same as the dark purple curve, except that the WT containing the empty vector (pUA66) was used. Dark-gray-shaded curve, GFP-positive control (MG1655 with PT5lacO-gfp on pUA66, induced with IPTG since inoculation). (B) Reanalysis of sorted fractions from the gating strategy used for Fig. 3A. (C) ΔrecA mutant morphology and induction of PrecA in cells treated with ofloxacin (OFL) for 5 h in stationary phase after transfer to antibiotic-free LB medium for 0 and 2 h, along with untreated controls. All images are 87.36 by 66.56 µm. Download
Persister CFU-per-milliliter data. (A) Numbers of CFU per milliliter for ΔrecA mutant and WT cells containing different inducible systems or empty-vector controls. Error bars for MG1655 plus PT5lacO-lexA3 are depicted in blue to highlight the variability observed in the initial measurement, which would have been masked if left in black. Data are average values ± the standard errors from at least three biological replicates. (B) Numbers of CFU per milliliter for ΔrecA strains and empty-vector pUA66 negative controls upon treatment with the DNA-damaging agent MMC. The limit of detection is indicated by the red line at 10 CFU/ml. The ΔrecA mutant plus PT5lacO-recA was treated with 5 µg/ml MMC and RecA induction as indicated. WT (solid black curve with open markers) and ΔrecA mutant (dashed purple curve with open markers) cells plus empty vector pUA66 were treated with 5 µg/ml MMC. Data are average values ± standard errors of at least three biological replicates. (C) WT MG1655 and ΔrecA mutant ofloxacin persister survival is equivalent upon recovery on LB and LB plus 20 mM MgSO4 plates. Data are average values of two biological replicates with minimum and maximum error bars. Download
Persister quantification after 16 and 48 h. Numbers of CFU per milliliter are equivalent when determined after 16 and 48 h of incubation after ofloxacin (A) and MMC (B) treatments in the WT and ΔrecA mutant strains. Note that because of excessive growth at 48 h, the persister levels reported and analyzed were obtained after 16 h of postantibiotic incubation. Download
Bacterial strains, plasmids (A), and primers (B) used in this study.
Supplemental information. Download