

ABSTRACT
Phage shock protein A (PspA), which is responsible for maintaining inner membrane integrity under stress in enterobacteria, and vesicle-inducting protein in plastids 1 (Vipp1), which functions for membrane maintenance and thylakoid biogenesis in cyanobacteria and plants, are similar peripheral membrane-binding proteins. Their homologous N-terminal amphipathic helices are required for membrane binding; however, the membrane features recognized and required for expressing their functionalities have remained largely uncharacterized. Rigorously controlled, in vitro methodologies with lipid vesicles and purified proteins were used in this study and provided the first biochemical and biophysical characterizations of membrane binding by PspA and Vipp1. Both proteins are found to sense stored curvature elastic (SCE) stress and anionic lipids within the membrane. PspA has an enhanced sensitivity for SCE stress and a higher affinity for the membrane than Vipp1. These variations in binding may be crucial for some of the proteins’ differing roles in vivo. Assays probing the transcriptional regulatory function of PspA in the presence of vesicles showed that a relief of transcription inhibition occurs in an SCE stress-specific manner. This in vitro recapitulation of membrane stress-dependent transcription control suggests that the Psp response may be mounted in vivo when a cell’s inner membrane experiences increased SCE stress.
IMPORTANCE
All cell types maintain the integrity of their membrane systems. One widely distributed membrane stress response system in bacteria is the phage shock protein (Psp) system. The central component, peripheral membrane protein PspA, which mitigates inner membrane stress in bacteria, has a counterpart, Vipp1, which functions for membrane maintenance and thylakoid biogenesis in plants and photosynthetic bacteria. Membrane association of both these proteins is accepted as playing a pivotal role in their functions. Here we show that direct membrane binding by PspA and Vipp1 is driven by two physio-chemical signals, one of which is membrane stress specific. Our work points to alleviation of membrane stored curvature elastic stress by amphipathic helix insertions as an attractive mechanism for membrane maintenance by PspA and Vipp1. Furthermore, the identification of a physical, stress-related membrane signal suggests a unilateral mechanism that promotes both binding of PspA and induction of the Psp response.
INTRODUCTION
Stress response systems are prevalent throughout all cell types, and several important stress responses function to maintain the cell envelope. The page shock protein (Psp) response is one such membrane-targeted stress response system that is widely distributed in bacterial, archaea, and higher plants (1, 2). It is induced under a number of conditions proposed to result in inner membrane (IM) stress, including the mislocalization of outer membrane secretins (such as filamentous phage pIV protein), extreme temperature, and hyperosmotic shock. Involved in pathogenicity, biofilm formation, and multidrug-resistant persister cell formation, the Psp response appears to stabilize the IM and prevent dissipation of the proton motive force (PMF) (3).
A minimal Psp system in Proteobacteria is proposed to consist of PspA and the transcriptional activator PspF, and in enterobacteria it is supported by the IM proteins PspB and PspC, which are implicated in IM stress sensing and signal transduction (1, 2). PspA functions as both a negative regulator and effector of the system. In an uninduced state, PspA forms a 6:6 inhibitory cocomplex with PspF and prevents σ54-dependent expression of the PspA promoter (4). Upon IM stress, the PspA-PspF complex is disturbed, allowing PspF to activate transcription of the psp operon (5). PspA expression is upregulated and a switch from negative regulator to effector function is observed, resulting in the formation of higher-order PspA oligomers that bind to the IM to maintain its integrity (6).
Induction of the Psp response and the regulatory role of PspA have been well characterized (5, 7–9); however, insights into its effector function are relatively sparse. Although PspA is known to bind the IM of Escherichia coli and Yersinia enterocolitica (6, 10), recognition of features within the membrane and the mechanism by which membrane stress is alleviated are only just beginning to emerge. For example, the E. coli PspA protein is reported to preferentially bind to anionic lipids in vitro (3), and cardiolipin (CL) has been implicated in signaling (11). PspA exhibits significant similarity with Vipp1, a protein involved in biogenesis of photosystems, thylakoid membrane biogenesis, and protection in cyanobacteria and higher plants (12–15), but again, the binding determinants in the target membranes are unclear.
Recent studies have described putative N-terminal amphipathic helices (AHs) in both PspA and Vipp1 (14, 16). Membrane-binding AHs are a common motif encountered in many peripheral membrane-binding proteins and, although as yet incompletely characterized, the putative N-terminal AH regions of PspA and Vipp1 have been shown to play a role in bilayer association. Jovanovic et al. (16) identified two putative AH sequences at the N-terminus of PspA, residues 2 to 19 (here referred to as AHa) and residues 25 to 42 (AHb), separated by a P25 helix-breaker proline residue. An AHa deletion mutant in E. coli loses effector function and IM localization in vivo, while an AHb deletion mutant retains effector function but loses regulatory function. Vipp1 has a conserved P25 residue preceded by a putative AH sequence, deletion of which in the Arabidopsis thaliana protein results in the loss of the protein’s ability to associate with pea chloroplast vesicles (14).
The role played by membrane composition as a targeting factor of PspA and Vipp1 has yet to be thoroughly examined. Previous in vitro studies of the interaction between PspA and Vipp1 with phospholipid bilayers were limited to qualitative studies with native E. coli anionic lipids (3, 14). There is, however, a significant body of evidence showing that the functions of peripheral membrane proteins in cells are often modulated by the physical properties of biological membranes. This includes stored curvature elastic (SCE) stress and electrostatic-mediated membrane associations, which are particularly relevant in AH-mediated membrane associations. SCE stress has been identified as a membrane-binding determinant for a number of peripheral membrane-binding proteins (17, 18). This physical torque stress occurs within a bilayer when lipids in the constituent monolayers are forced to adopt an unfavorable packing conformation, such as when type II conical lipids (e.g., phosphatidylethanolamine [PE] and diacylglycerols [DAG]) form a major component of the membrane. Sustaining SCE stress levels within a critical range is important for biological membranes to prevent phase transition into a porous state (17). SCE stress is thought to result in hydrophobic cavities within the membrane, which are known as lipid-packing defects, a phenomenon also linked to increased membrane curvature and shown to promote AH membrane binding in a number of cases (19, 20).
Here, we establish a simple and quantitative technique employing the use of native PAGE supported by classical sucrose gradient centrifugation to provide insights into the physical determinants that promote membrane binding of PspA and Vipp1. We show that both proteins possess at least two distinct sensing mechanisms for direct membrane binding, an anionic lipid-binding determinant and preferential binding to membranes with increased SCE stress. Establishment of a direct protein-membrane interaction that is stress specific yet nonspecific to particular phospholipid head groups suggests that a conserved membrane stress-sensing feature may be recognized in all organisms that use PspA and Vipp1. Additionally, by using in vitro transcription assays, we show relief of PspA’s transcription repression function upon its exposure to membranes with high levels of SCE stress. We infer that SCE stress likely plays a role in IM stress signaling and induction of the Psp response in vivo. We propose that PspA and Vipp1 target areas of biological membranes possessing increased lipid-packing defects arising from SCE stress. At these sites, PspA or Vipp1 higher-order oligomers may then stabilize the stressed membrane by multiple AH insertions, alleviating SCE stress and imparting a scaffold effect that prevents the membrane phase transition into a porous state.
RESULTS
PspA-membrane binding quantitatively measured in a native PAGE-based assay.
Wild-type (WT) E. coli PspA was purified as described previously (7, 8), and native functionality was confirmed via PspA-PspF interaction assays (see Fig. S1 in the supplemental material). In order to quantify the binding of PspA to phospholipid vesicles, a native PAGE-based assay was developed (Fig. 1a). Results with purified PspA and 100 nm E. coli total lipid extract (TLE) vesicles are shown in Fig. 1. The intensity of the PspA band significantly decreased with samples containing TLE vesicles (Fig. 1b) and titrated down to around 10% that of the free PspA standard. The percentage of vesicles that bound PspA was calculated based on the decrease in band intensity of the positive vesicle samples compared to the PspA input. Figure 1c shows the calculated amount of vesicle-bound PspA positively correlates with increasing TLE concentration. Controls using non-membrane-binding PspF1–275 established that protein-membrane binding was the cause of the decreased protein signal for PspA. Incubation with vesicles resulted in no significant decrease in the free PspF1–275 band (see Fig. S1). PspA-vesicle cocomplexes migrating within the gel were evident when we performed the assay with small vesicles (diameters, <100 nm), providing evidence of a direct protein-membrane interaction (see Fig. S2 in the supplemental material). To further confirm a direct PspA-membrane interaction, sucrose density gradient centrifugation was undertaken. PspA alone was observed only in dense fractions; however, when incubated with vesicles, PspA was then found in lower-density fractions, where TLE vesicles fractionate (Fig. 1d). It is clear from these results that our purified PspA is able to bind the membrane of vesicles with lipid compositions similar to those found in E. coli.
FIG 1 .

Native PAGE-based binding assay results, which provide a quantitative measure of PspA-membrane binding. (a) Schematic for the assay. (b) Image of a typical vesicle titration, Sypro Ruby-stained native gel (image shown is 10 µM PspA titrated with 100 nm TLE vesicles). Boxed areas signify signal-integrated regions, and the percent band intensity, compared to results without vesicles (−, lane 1), is shown for each lane. (c) Binding isotherm of vesicle-bound PspA as a function of lipid concentration for 100 nm TLE vesicles derived from the relative intensity of the PspA band versus lipid concentration (insert). Error bars show standard deviations, calculated from three independent experiments using different sets of vesicles. (d) Fractions from sucrose gradient centrifugation were analyzed via SDS-PAGE and Sypro Ruby protein staining of PspA with and without TLE vesicles. Starred fractions indicate those where NBD-PE-labeled vesicles were found to reside.
E. coli lipid extracts deficient in anionic lipids still bind PspA.
To probe the contributions of native E. coli phospholipids to PspA-membrane binding, vesicles produced from cell extracts were used. Zwitterionic PE, anionic phosphatidylglycerol (PG), and dianionic CL make up around 75%, 20%, and 5% of the IM phospholipid content of E. coli, respectively (21). Strains of E. coli lacking the CL synthase gene (cls) have greatly diminished amounts of CL, while strains lacking the PG synthase gene (pgsA) have significantly reduced PG and CL (22). Lipids were extracted from E. coli WT cells and from cls and pgsA deletion mutants. Mass spectrometry of the extracts confirmed a great reduction in PG for the pgsA mutant (data not shown). PspA bound vesicles of the three lipid extracts with a similar affinity (Fig. 2a), as the slight differences in binding between the extracts were statistically insignificant at any of the lipid concentrations tested (for all comparisons, P > 0.05, repeated-measures analysis of variance [ANOVA]). Our results imply that membranes deficient in anionic lipids can still bind PspA. Although low levels of anionic lipids within the pgsA vesicles and <1% phosphatidylserine (PS) are present in membranes (23), our results suggest that a high anionic lipid content in membranes is not a requirement for PspA binding.
FIG 2 .

Membrane binding of PspA as a function of lipid composition. (a) Binding of PspA to vesicles (100 nm) composed of E. coli lipids extracted from WT, cls (deficient in CL), and pgsA (deficient in PG and CL) at increasing lipid concentrations. (b) Membrane binding of PspA to zwitterionic vesicles compositions of increasing SCE stress (from DMPC/DOPC 4:6 to DOPE/DOPC 4:6) at 0.5 and 1.5 mM lipid concentrations. (c) PspA binding as a function of SCE stress within vesicles at increasing lipid concentrations. Differences in binding between vesicle compositions were statistically significant at all lipid concentrations (P < 0.05). (d) Binding of PspA to DMPC/DOPC 4:6 (low-SCE stress) vesicles containing different percentages of the anionic lipids PG (DOPG), CL (14:0 CL), and PS (DOPS). All experiments (in this and remaining figures unless otherwise stated) were carried out in triplicate with different vesicle preparations, and error bars represent standard errors. (e) Binding of PspA as a function of anionic lipid content and SCE stress. Binding levels to vesicles with low (DMPC/DOPC 4:6) and high (DOPE/DOPC 4:6) SCE stress with and without 10% anionic DOPG are shown.
Membrane stored curvature elastic stress-dependent binding.
Investigations using synthetic phospholipid vesicles were undertaken. This approach allows for enhanced control over the biophysical properties of the membrane compared with use of lipid extracts, as both lipid head and tail groups can be precisely selected. Here, 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) vesicles were used to assess binding to neutral (zwitterionic) lipids, and this resulted in a pronounced membrane interaction (see Fig. S3a and b in the supplemental material). As suggested by the data, anionic lipids cannot be the sole membrane-binding determinant. Although DOPC is not a native E. coli lipid, binding may be due to a physical, nonspecific feature also present in native membranes; we note that SCE stress has been shown to modulate binding of proteins containing AHs to vesicles (17, 18). Binary lipid vesicles consisting of DMPC with DOPC, or DOPC with DOPE, were used to assess the binding of PspA as a function of the SCE stress profile of the bilayer. These chosen vesicle compositions result in a monotonic increase in SCE stress from DMPC/DOPC to DOPE/DOPC mixtures and have been employed in a number of protein-membrane interaction studies of the effects of SCE stress (17, 18). As illustrated in Fig. 2b, from DMPC/DOPC 4:6 (molar ratio) to DOPE/DOPC 4:6, increasing PspA binding was observed in concert with an increase in SCE stress at both lipid concentrations. At a 0.5 mM lipid concentration (50% of binding saturation for TLE vesicles), PspA exhibited a 4-fold increase in binding for DMPC/DOPC 4:6 versus DOPE/DOPC 4:6 vesicles. The effect of increasing vesicle concentrations from 0.25 to 2.5 mM (Fig. 2c) resulted in an initial linear increase in binding up to saturation for all lipid compositions while preserving the SCE stress-modulated binding trend. Binding saturation with DOPC/DOPE (6:4 or 8:2) and DOPC vesicles at lipid concentrations of 2.5 mM showed that SCE stress is sufficient to completely sequester all of the PspA to the membrane.
Anionic lipids play a role in PspA-membrane binding.
Electrostatic binding interactions were evaluated by addition of anionic lipids to the system. Inclusion of PG, CL, and PS in DMPC/DOPC 4:6 vesicles (chosen because these lipid compositions exhibited minimal SCE stress-specific binding) at concentrations of 0.5, 2, and 10 mol% were studied. Despite higher anionic lipid concentrations in the native E. coli IM, concentrations were kept low to minimize their contribution to the SCE stress profile of the vesicles. Preparations of DMPC/DOPC with as little as 0.5% anionic lipid increased binding of all species (Fig. 2d). Cardiolipin, which carries two negative charges and often resides in curved regions within the bacterial membrane, was found to have the largest effect at a low concentration, with 0.5% giving a >2-fold binding increase. The anionic lipid contribution to membrane binding peaked at 2%, with 10% leading to similar amounts of PspA binding. Combining the two binding determinants by using 10% anionic lipids in vesicles with elevated SCE stress (DOPE/DOPC 4:6) increased membrane associations of PspA over that with 10% anionic lipids under low-SCE stress (DMPC/DOPC 4:6) vesicles (Fig. 2e). The binding affinity was similar to that of neutral vesicles with high SCE stress, and so it appears that anionic lipids reduce but do not eliminate the SCE stress-sensing ability of PspA.
PspA1–186 and PspAΔAHa provide insights into structure-specific lipid binding.
PspA variants were investigated to study the effects of oligomerization and the putative N-terminal AHa on membrane binding. PspA1–186, which lacks α-helical domain HD4 (Fig. 3a), retains negative regulatory function and purifies as a monomer/dimer unable to form higher-order oligomers (4) but carries AHa and is able to bind the IM when overproduced (11). PspA1–186 binds TLE vesicles, and increased membrane SCE stress correlates with additional binding (Fig. 3b). As such, PspA1–186 behaves similarly to wild-type PspA for membrane binding. Apparently, the oligomeric state (and so HD4) observed in solution does not play an essential role for PspA in binding to vesicles.
FIG 3 .
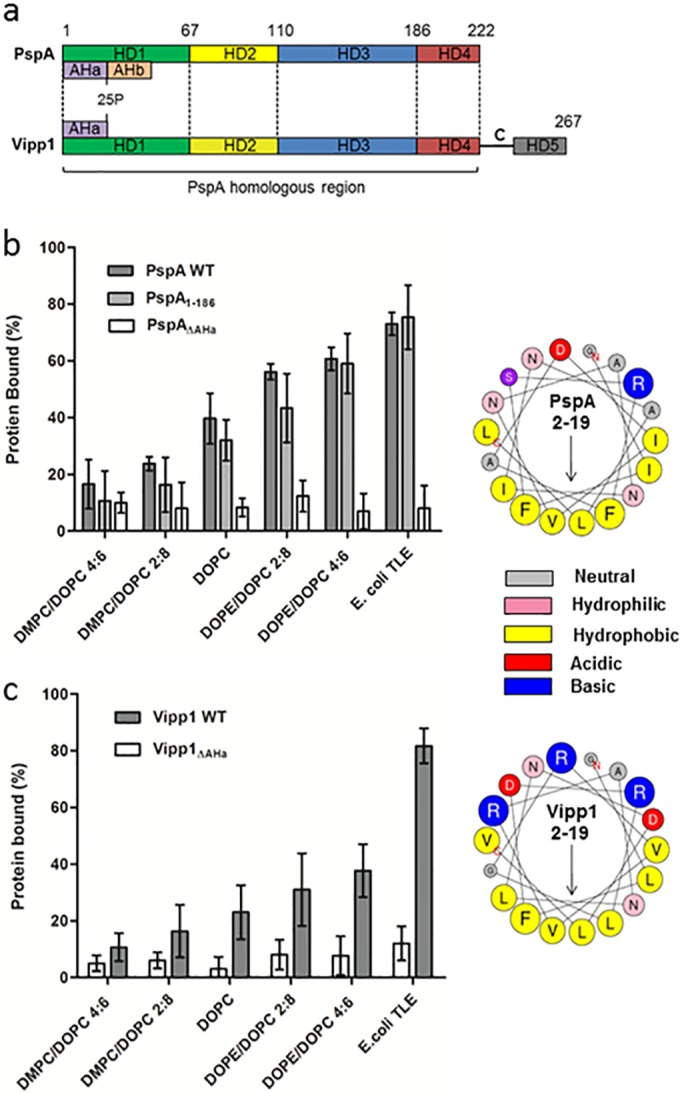
Membrane binding of PspA variants and the PspA homologue Vipp1. (a) Schematic of the E. coli PspA and Synechocystis Vipp1 protein sequences, with the positions of the helical domains (HD1 to 4 according to regions defined for PspA and HD5 of Vipp1) and AH regions. (b) Affinity of PspA, PspA1–186, and PspAΔAHa for vesicles under increasing SCE stress and E. coli TLE (1 mM lipid). (c) Affinities of Vipp1 and Vipp1ΔAHa within the same assay but at a 2 mM lipid concentration. Concentrations for all proteins were 10 µM within the assay. ANOVA showed differences in binding between lipid composition to be statistical significant (P < 0.05) for all proteins apart from the ΔAHa mutants of PspA and Vipp1. (b and c) Right sides show helical wheel projections of the N-terminal AHa (residues 2 to 19) of PspA and Vipp1, with arrows showing the direction of the hydrophobic moment. Residue sizes are proportional to the amino acid side chain volumes.
PspAΔAHa, a variant of PspA that lacks residues 2 to 19 (AHa) (Fig. 3a), did not bind the IM in vivo, and only residual binding was observed upon IM stress (16). Unlike PspA and PspA1–186, no PspAΔAHa was found in the membrane fraction during its purification. Purified soluble PspAΔAHa exhibited no significant vesicle binding, irrespective of lipid concentration, anionic lipid composition (see Fig. S4c in the supplemental material), or SCE stress (Fig. 3b). Apparently, residues 2 to 19, encoding AHa, are required for a direct PspA-lipid-binding interaction. To confirm that binding is a direct interaction between the PspA N-terminal AHa and the membrane, residues 2 to 19 of PspA were fused to the hydrophilic protein enhanced green fluorescent protein (eGFP; an approach utilized by Salje et al. to show direct membrane binding of the N-terminal AH of EcMreB [24]). Similar to the findings for EcMreB reported by Salje et al., membrane binding of the fusion protein could not be detected with a single copy of AHa. However, when present in duplicate or triplicate (mimicking the multiple AHas present in a PspA oligomer), binding was observed following IM stress via cell fractionation, single-molecule imaging studies, and in vitro native PAGE-based membrane-binding assays (see Fig. S5 in the supplemental material). These results strengthen the evidence for direct binding of PspA AHa to the stressed membrane and also suggest that multiple AHa-membrane interactions in higher-order oligomer PspA enhances the strength of the bilayer association.
Vipp1-membrane binding.
Synechocystis sp. PCC 6308 Vipp1 was expressed and purified by using a protocol similar to that for PspA. Circular dichroism studies on the purified Vipp1 gave a predominantly α-helical structure (see Fig. S5a in the supplemental material), similar to the secondary structure reported and predicted in silico (25), and indicated a native fold. When analyzed via native PAGE, Vipp1 yields a diffuse, slow-running band similar to that of PspA. Vipp1 can bind E. coli IM vesicles (14) and, in agreement, the native PAGE and sucrose gradient centrifugation assays showed clear binding of Vipp1 to E. coli TLE vesicles (see Fig. S5b). Affinity of Vipp1 for the TLE vesicles was less than half that of PspA when assessed using the native PAGE system (Fig. 3b); however, general binding trends were conserved between the two, as Vipp1 exhibited both anionic and SCE stress-dependent binding. While increased SCE stress still correlated with Vipp1 membrane binding, the effect was less marked than that with PspA (Fig. 3b and c; see also Fig. S5c). Incorporation of Vipp1 in anionic lipid assays showed a slight increase in binding with an additional membrane negative charge; however, the effects were again less marked than those with PspA (see Fig. S5e). Combining the two determinants using 10% anionic lipids in vesicles of high SCE stress gave a binding affinity similar to that of neutral vesicles of high SCE stress, suggesting a preference for SCE stress-specific binding with low levels of anionic lipids (see Fig. S5f). Membrane binding of Vipp1 to vesicles composed of the major lipid species within cyanobacterial membranes, the galactolipids monogalactosyldiacylglycerol (MGDG) and digalactosyldiacylglycerol (DGDG) and phospholipid PG, was also observed (see Fig. S6 in the supplemental material). This showed a direct bilayer association of Vipp1 to lipid compositions similar to those seen in native Synechocystis thylakoid and cytoplasmic membranes.
Similar to the AHa of PspA, Vipp1 contains a putative N-terminal AH region important for membrane association (14) (see also Fig. 3a in the supplemental material). For a direct comparison with PspA, residues 2 to 19 were designated Vipp1AHa, and purification of a Vipp1 mutant lacking these residues (Vipp1ΔAHa) was also undertaken. Within the native PAGE and sucrose gradient centrifugation assays, no binding of Vipp1ΔAHa to vesicles, irrespective of lipid composition, was observed (Fig. 3c; see also Fig. S5d in the supplemental material). It is apparent that the presence of AHa of Vipp1 is required for its direct bilayer association.
Vesicle binding by PspA is sufficient to derepress transcription in vitro.
To recapitulate the transcription regulatory role PspA plays within the Psp response in the presence of lipid bilayers, a small primed RNA (spRNA) in vitro transcription assay was performed. Using the Sinorhizobium meliloti nifH test promoter and RNA polymerase holoenzyme in the presence of dinucleotide primer UpG, radiolabeled GTP, and cold dATP for PspF1–275 action results in production of a small spRNA product, UpGGG (Fig. 4a) (26). Upon addition of PspA, production of the spRNA was repressed (Fig. 4a) via formation of the PspA-PspF1–275 inhibitory cocomplex (A-F complex). When TLE vesicles were included, spRNA production recovered to a level similar to that seen when PspA was absent (Fig. 4a). Controls established that vesicles do not have a PspA- or PspF1–275-independent stimulatory effect on spRNA formation (see Fig. S7a in the supplemental material). These data are indicative of PspA-PspF complex disruption upon membrane exposure, presumably by PspA being sequestered to the vesicle surface. Native PAGE analysis of a PspA/PspF protein mixture with and without vesicles supported this view (see Fig. S7a).
FIG 4 .
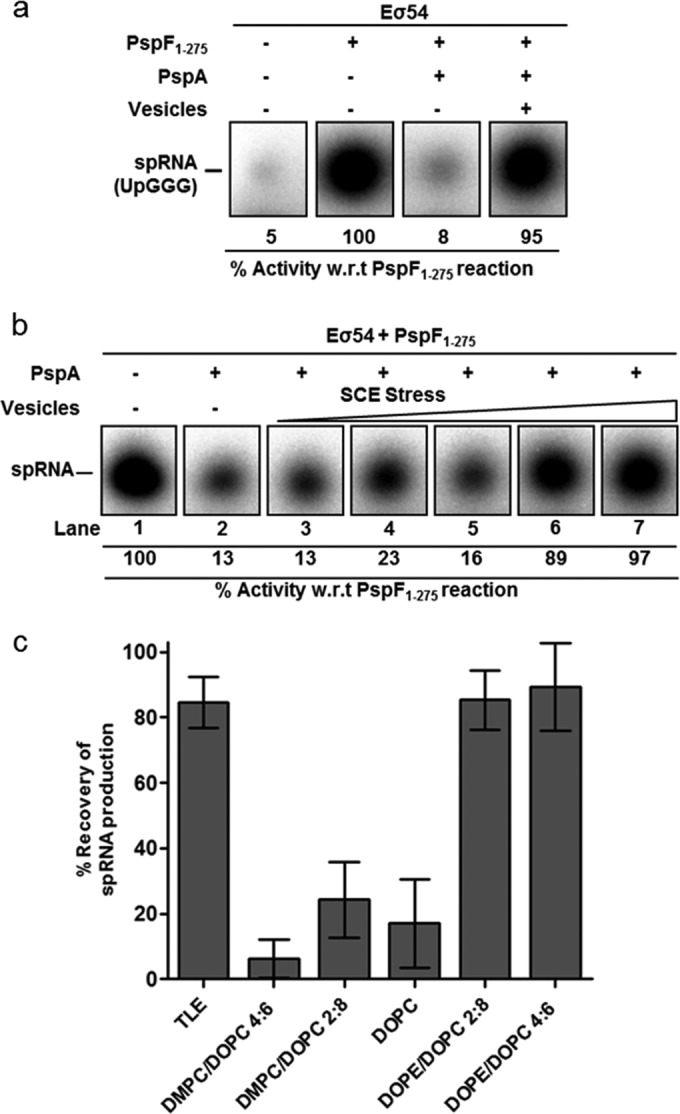
Lipid vesicles are able to derepress transcription and disrupt a PspA-PspF inhibitory complex. (a) Denaturing gels, showing that production of the spRNA product (UpGGG) in the presence of the transcriptional activator PspF (lane 2) is inhibited upon addition of PspA (lane 3). Repression was relieved with the addition of TLE vesicles (lane 4). (b) spRNA production with incorporation of an increasing SCE stress vesicle set. Increasing membrane stored energy composition levels are shown, from left to right: DMPC/DOPC 4:6, DMPC/DOPC 2:8, DOPC, DOPE/DOPC 2:8, DOPE/DOPC 4:6. (c) Recovery of spRNA production plotted for each vesicle composition at 1 mM lipid concentration.
Other vesicle compositions were assayed to assess their effects on transcription derepression. The increases in spRNA production following release of a PspA negative control arising from the inclusion of SCE stress vesicles (same lipid compositions as used in the native PAGE assays described above) are shown in Fig. 4b and c (see also Fig. S7b in the supplemental material). Significant recovery of transcription was evident for the two vesicles with the highest levels of SCE stress (DOPE/DOPC 4:6 and 2:8), while the three other compositions present limited transcriptional recovery. When PspA1–186 instead of full-length PspA was incorporated into the assay mixture, no vesicle compositions were able to relieve transcription inhibition (see Fig. S7c in the supplemental material), suggesting a higher affinity for PspF1–275 than the membrane. Native PAGE analysis also showed vesicles were unable to disrupt the PspA1–186/PspF1–275 inhibitory complex (see Fig. S7d). These results revealed that exposure of the PspA/PspF1–275 inhibitory cocomplex to bilayers of certain lipid compositions is sufficient to disrupt the transcription-inhibiting properties of PspA but apparently only if HD4 of PspA, responsible for higher-order oligomerization of PspA, is present (compare the activities of full-length PspA and PspA1–186).
DISCUSSION
The results obtained revealed detailed insights into the membrane-binding determinants of PspA and Vipp1 and indicated possible mechanisms of plasma membrane stress signaling and the actions of these proteins for membrane maintenance. SCE stress within the membrane was found to be a major membrane-binding determinant for PspA and Vipp1. Lipid compositions used to test this were chosen to specifically probe for increased membrane SCE stress. For the large unilamellar vesicles (diameter, >100 nm), membrane curvature was minimal in comparison to the effect of lipid-packing parameters. The bilayers can therefore essentially be considered flat (27). Dynamic light scattering confirmed the same distribution of vesicle diameters in all samples (see Fig. S8 in the supplemental material), and previous studies of DMPC/DOPC and DOPE/DOPC binary systems found they exhibit no phase separation (17). The use of zwitterionic lipids eliminated the possibility of electrostatic-mediated protein-membrane interactions. With DMPC/DOPC vesicles lipid head groups are net uncharged, and with DOPE/DOPC vesicles chains are unaffected; thus, any phospholipid head or chain group chemistry is not important. Lipid molecular area dependence for binding was also inconsistent with the data, as the average cross-sectional area per molecule initially increased from 67 Å2 for 100 mol% DMPC to 76 Å2 at 100% DOPC, while it was reduced to 69 Å2 for 60 mol% DOPE (18). Crucially, the spontaneous curvature changes monotonically from DMPC to DOPC to DOPE and, consequently, so too does the SCE stress. From DMPC to DOPC, unsaturation increases the propensity of chains to splay, causing curvature toward the polar head. DOPC to DOPE results in a further preference for negative curvature due to the lower hydrophilicity of the PE head group (18).
It is vitally important for biological membranes to maintain their SCE stress within a critical range to prevent the phase transition into a porous state (17). An obvious detrimental effect of this phase transition within the Gram-negative IM is dissipation of the PMF. PspA is strongly implicated in preventing dissipation of the PMF both in vivo and in vitro (2, 3), and this could be through controlling SCE stress levels to maintain an impermeable lamellar bilayer. We observed that for PspA and Vipp1, membrane binding increased with SCE stress. This behavior has been shown with other peripheral proteins, such as CCT and Rab (17, 18), for which insertion into the membrane interface allows the chains of nearby lipids to splay and alleviate some of the SCE stress. It appears plausible that PspA and Vipp1 could exploit this mechanism to both target and alleviate areas of the membrane exhibiting high levels of SCE stress, in order to stabilize the membrane.
Higher affinities for the vesicles when anionic lipids were included were observed for both PspA and Vipp1. Binding mediated by electrostatic interactions plays a key role in membrane association of many proteins via interactions between anionic lipids and positively charged amino acids (28, 29). Previous studies with PspA highlighted interactions between the anionic lipids PG and PS, enabling membrane binding without CL determinacy (3). We showed here that the presence of anionic lipids at low concentrations promotes a PspA-membrane-binding interaction with vesicles of low SCE stress. As all three anionic lipids appear to have a similar effect on PspA binding despite having different headgroup chemical structures, it is unlikely that the increase in binding is due to recognition of any specific lipid functional group. Nonspecific electrostatic interactions between positively charged amino acid residues on PspA and anionic lipids in the bilayer could be attributable to the enhanced membrane binding.
The conserved N-terminal AH regions of both PspA and Vipp1 (Fig. 3a) appear to be vital for membrane association in both SCE stress and anionic lipid-determined binding, as shown from the loss of function of ΔAHa mutants. In addition, the ability of PspA’s AHa to recruit eGFP to the membrane (see Fig. S4 in the supplemental material) provides strong evidence for a direct N-terminal AH-mediated membrane association mechanism for both PspA and Vipp1. AHs have been shown to bind the membrane in an SCE stress-dependent manner both in vitro (17) and in silico (30). We propose for PspA and Vipp1 that upon membrane association, the hydrophobic face of the AH lies parallel to the bilayer with its central axis positioned at the interface between the phospholipid polar heads and alkyl chains, akin to a model AH (19). Hydrophobic residue insertion within the fatty acyl chains can act as a wedge to alleviate SCE stress (17). This association may be complemented by an electrostatic interaction between anionic lipids within the membrane and the positively charged residues located on the hydrophilic face of the PspA and Vipp1 AHas.
The ability of AHs to sense membrane curvature by binding to hydrophobic lipid-packing defects enriched on curved surfaces has been reported (31, 32). This effect goes hand in hand with SCE stress-dependent binding, as both phenomena result from membranes being forced to adopt unfavorable lipid-packing conformations. Packing defects expose the hydrophobic membrane interior, providing sites for AH insertion and alleviating SCE stress once inserted (33). With the SCE stress assay, the increasing stored membrane tension creates more packing defects, likely providing more binding sites for PspA and Vipp1 and therefore increasing their membrane binding. Recent work has shown that the curvature sensing ability of AHs is modulated by variations in charge on the nonpolar face and residues surrounding the AH (32). Also, dampening of the AH curvature-sensing ability is observed with increased membrane negative charge (20, 32). This model is consistent with the observations made in this study, where PspA’s SCE stress sensing ability was mitigated by an increase in the anionic lipid content (though SCE stress sensing was still observed in the presence of anionic lipids).
The compositions and lipid species within the Synechocystis thylakoid and cytoplasmic membranes with which Vipp1 associates are very different from those found within the IM of E. coli, to which PspA binds. This study showed that membrane binding of PspA and Vipp1 is driven by two physical membrane signals of SCE stress and lipid headgroup charge. The nonspecific nature of these signals means that they can be exhibited across biological membranes irrespective of specific lipid species and suggests that conservation of an AH-driven membrane-binding mechanism by PspA and Vipp1 in vivo is possible throughout all species in which corresponding homologues are found. Indeed this work shows that Vipp1 directly binds to membranes consisting predominantly of both phospholipids and galactolipids. This may explain why Vipp1 from cyanobacteria can substitute for the PspA effector function in E. coli and, in direct correspondence, PspA from E. coli can substitute for Vipp1 and act in cyanobacteria as an effector to resolve protein translocation defects (34). The specific function of Vipp1 in thylakoid membrane biogenesis might be attributed to a different substructure of Vipp1 compared to PspA (Fig. 3a and see below). Though PspA and Vipp1 proteins each possess N-terminal AHs, the amino acid sequence within them is not strictly conserved. Analysis of both AHas may explain some of the differences observed in membrane binding of the proteins. PspA’s increased membrane-binding ability (for all membrane compositions) could in part be attributable to a higher average AH hydrophobicity (H) of 0.606 H, compared with 0.377 H for Vipp1 (calculated with the Fauchere and Pliska hydrophobicity scale via the method of Heliquest [35]) increasing its affinity for lipids. Less-charged residues on the hydrophilic face of PspA (6R, 9D; net charge [Z] of 0) could explain its enhanced sensitivity for SCE stress-dependent binding compared with Vipp1 (5D, 6R, 9R, 12R, 17D; Z = 1), as increasing the charged residues on the polar face has been shown to dampen SCE stress sensing in other AHs (36, 37).
While many bilayer-associating AHs, such as SCE stress-sensing ALPS motifs, do not induce membrane curvature or vesicle remodeling, other AHs possess this ability and participate in vesicle formation (19). Therefore, although we saw shared binding determinants, the distinguishing effector function of the PspA and Vipp1 proteins may arise from variation within the N-terminal AHs as well as Vipp1’s extra C-terminal domain. In E. coli, the peripheral membrane-binding proteins MreB (shown to interact with PspA [10]) and MinD also associate with the IM via an AH (24, 38). Overall, it seems highly likely that PspA and Vipp1 bind the membrane by insertion of their N-terminal AHs into the lipid bilayer.
Many inducing agents and conditions have been identified for the Psp response (2). While a unifying signal manifested by all inducers has been suggested to be due to variations in IM properties (1, 2), such a specific chemical or physical change has not been identified or proposed. The results from this work now suggest for the first time that SCE stress or the resulting lipid-packing defects within the IM could well be this unifying signal. Unfortunately, directly quantifying the SCE stress in biological membranes has yet to be achieved, so the effect that membrane stressors have on the precise magnitude of SCE stress in vivo cannot yet be ascertained. However, many (if not all) Psp-inducing conditions will likely result in local or global changes in SCE stress within the IM. For example, defects in protein translocation or the mislocalization of outer membrane proteins (including secretin pIV) could cause a hydrophobic mismatch, which can cause large changes in the lateral stress profile of the surrounding bilayer (39). Extreme temperatures, which induce the Psp response, could increase IM SCE stress through increased acyl chain trans-gauche rotation, and similarly, hyperosmotic shock could cause increased lipid-packing defects through bilayer expansion. Another inducer of the Psp response, free fatty acids (for which the Psp response has shown to be highly upregulated) have also been shown to increase the SCE stress within membranes (40). SCE stress may also play a role in Vipp1’s ability to prevent the formation of balloon-like swollen chloroplasts in Arabidopsis thaliana under osmotic stress (12). Membrane swelling will increase the lateral stress in the bilayer and most likely induce lipid-packing defects favoring membrane association of Vipp1. Future studies correlating the levels of SCE stress (and the resultant protein binding) seen in these in vitro studies to those seen under stressed and nonstressed conditions in vivo should help to confirm that an SCE stress-sensing mechanism drives the effector functions of PspA and Vipp1.
Our study also provides insights into PspA’s regulatory role and suggests a new unilateral mode of IM stress-dependent induction of the Psp response. We showed that the PspA-PspF complex is unable to survive when exposed to the membranes of TLE vesicles and that this results in relief of transcription repression. Under certain growth conditions, PspA’s ability to sense the state of the membrane could result in direct signaling to PspA, release of PspF, and induction of the Psp response, effectively bypassing any stress sensing by PspB and PspC. Increased transcription, which is seen with high-SCE stress vesicles, indicates that increased lipid-packing defects coupled with SCE stress provide energetically favorable binding sites for insertion of PspA’s AH that overcome the repressive PspA-PspF interaction. This probably results from conformational changes in PspA upon membrane binding, as a competitive binding site for PspF seems unlikely, since PspAΔAHa can still interact with PspF (23). The Psp response is induced under conditions that impair the IM integrity (2). As described above, stimuli such as protein translocation defects and mislocalization of outer membrane secretins into the IM require PspBC to release the PspA-PspF inhibitory complex and so induce the Psp response in enterobacteria. However, under certain severe stress conditions (extreme temperature, hyperosmotic shock [2], when secretins are produced in cells with disturbed cell wall synthesis [11] or under anaerobic conditions [41]), the induction of psp is partially or completely PspBC independent. Therefore, the induction of psp upon stress may include both a PspA-PspF interaction with PspBC and a direct PspA interaction with the stressed IM. It is possible that severe stresses provide sufficient increases in SCE stress to overcome the affinity of PspA for PspF and so, in concert with PspBC, induce the Psp response (Fig. 5). Comparative studies of genome/operon organization within Proteobacteria have revealed that PspA and PspF are the most conserved Psp proteins and constitute the Psp minimal system (42). This supports our findings that the PspA-PspF complex can be directly regulated by stressed membranes.
FIG 5 .

Working model of the Psp response of E. coli in vivo, based on findings of this study. The PspA-PspF inhibitory complex detaches from the nucleoid and diffuses to the IM. PspA can sense the state of the IM either via assistance from PspBC sensors or via its N-terminal AH and binds directly to the IM at locations of SCE stress and membrane-packing defects. This results in disruption of the PspA-PspF complex, allowing PspF to initiate transcription of the psp operon and thus expression of the Psp response. The upregulated PspA forms higher-order oligomers directly bound to the IM in areas of high SCE stress. Multiple helix insertions into the IM from PspA oligomers reduce some of the SCE stress to stabilize the membrane and so may help yield higher-order self-assemblies of PspA to reduce their dissociation from the IM under stress.
Based on the findings of this study, we propose an updated model of the induction and effector function of the Psp response that takes into account the effects of SCE stress that could manifest in the IM as a result of Psp-inducing conditions (Fig. 5).
MATERIALS AND METHODS
Bacterial strains and plasmids.
Bacterial strains, plasmids their sources and constructions are described in Text S1 and listed in Table S1 in the supplemental material.
Protein purification.
Proteins were overexpressed in E. coli BL21 and purified using Ni2+-nitrilotriacetic acid affinity purification (see Text S1 in the supplemental material for the protocol).
Preparation of lipid vesicles.
Lipids were purchased from Avanti Polar Lipids (Alabastar, AL, USA). Buffer was added {25 mM Tris-HCl (pH 7.5), 200 mM NaCl, 75 mM NaSCN, and 0.005% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS)} to a 5 mM final phospholipid concentration and left at 25°C for 1 h. The suspension was then subject to five freeze-thaw-vortex cycles. Vesicles were produced by extruding the suspension through polycarbonate filters of 100-nm and 400-nm pore sizes using a miniextruder (Avanti Polar Lipids). Vesicle sizes were characterized via dynamic light scattering (DLS) to ensure homogeneity.
Native PAGE protein-vesicle binding assay.
Purified protein (5 to 10 µM) in 20 mM Tris-HCl (pH 7.8), 150 mM NaCl, 75 mM NaSCN, 5% glycerol, 0.005% CHAPS was incubated with increasing amounts of lipid vesicles for 15 min at 25°C in a total reaction volume of 20 µl. Four microliters of 5× native PAGE loading dye (250 mM Tris-HCl [pH 7.4], 50% [vol/vol] glycerol, 0.5 mg/ml bromophenol blue) was added to the samples before loading onto a polyacrylamide native gel (5% acrylamide concentration; OmniPAGE system; Geneflow). The gels were run in a Tris-glycine buffer (National Diagnostics; 25 mM Tris, 192 mM glycine [pH 8.3]) at 90 V for 80 min. Gels were stained with Sypro Ruby protein stain (Invitrogen), and bands were quantified using a Fujifilm FLA-5000 PhosphorImager and Aida image analyzer. The percentage of protein membrane bound was calculated from the relative intensity of the free protein band compared with that of a protein-only control.
Sucrose gradient centrifugation.
A 100-µl (20 µM) sample of protein was incubated with 50 µl (3.5 mM) vesicles for 15 min at 25°C. Samples were then carefully layered on top of a 4.9-ml 0 to 30% sucrose gradient in PspA buffer (pH 7.8) in centrifuge tubes. After centrifugation (160,000 × g, 16 h, 4°C) with a swingout rotor, fractions of 0.5 ml were collected and analyzed by SDS-PAGE and Sypro Ruby protein staining. The location of vesicles after centrifugation was monitored using vesicles containing 0.5% fluorescent (N-(7-nitroben-2-oxa-1,3-diazol-4-yl)-1,2-dihexadecanoyl-sn-glycero-3–PE (NBD-PE), with the fluorescence of collected fractions measured by using a BMG Omega plate reader.
Small primed RNA synthesis assay.
An spRNA synthesis assay was performed in 10 µl of STA buffer (25 mM Tris-acetate [pH 8.0], 8 mM Mg-acetate, 10 mM KCl, 3.5% [wt/vol] polyethylene glycol 6000). One hundred nanomoles of Eσ54 (reconstituted using a 1:5 E:σ54 ratio) and 20 nM promoter nifH probe were incubated at 25°C for 5 min to allow Eσ54-DNA complex formation. Addition of 4 mM dATP (for PspF ATPase) and a further 5-min incubation were followed by addition of protein mix (containing the desired amounts of PspA, PspF, and vesicles) and a final 30-min incubation at 37°C. The spRNA (UpGGG) synthesis was initiated by addition of a mix containing 4 µCi [α-32P]GTP, 0.5 mM UpG, and 100 µg/ml heparin followed by 37°C incubation for 20 min. The reaction was quenched via addition of loading buffer and analyzed on a 20% denaturing gel, visualized, and quantified using a Fuji FLA-5000 PhosphorImager.
SUPPLEMENTAL MATERIAL
Supplementary methods. Download
Controls undertaken for the native PAGE-based protein-membrane-binding assay. (A) Controls showing functionality of purified PspA. Native PAGE band shift assay results with PspA and PspF1–275, showing formation of a PspA-PspF1–275 cocomplex (left) and the resulting ATPase inhibition of PspF1–275 at a 1:1 PspA-to-PspF1–275 molar ratio, monitored via an NADH-coupled assay (right). (B) Five percent native PAGE gel with decreasing amounts of PspA stained via Sypro Ruby protein stain. Intensity of the integrated PspA bands plotted against protein used produced a linear trend depicted in the adjacent graph. (C) Native PAGE assay with PspF1–275 and 100 nm TLE vesicles with integrated band intensities with respect to (w.r.t) a PspF1–275 ΔV, showing no decrease on addition of vesicles. (D) Gel from native PAGE protein-membrane binding (SCE stress binding assay) undertaken with reduced running buffer pH (pH 7.5, compared to the typical 8.2) with binding trends for PspA preserved. Vesicles of increasing SCE stress compositions correspond to; DMPC/DOPC 4:6 (lane 1), DMPC/DOPC 2:8 (lane 2), DOPC (lane 3), DOPE/DOPC 2:8 (lane 4), and DOPE/DOPC 4:6 (lane 5). Lane 6 contains E. coli TLE vesicles incubated with PspA, and lane 7 contains E. coli TLE vesicles alone. Download
Visualization of a protein-vesicle complex in the native gel matrix, obtained using small unilamellar vesicles (SUVs). (A) Fluorescence image of sonicated SUVs (composed of DOPC plus 0.2% NBD-PE and DOPC/DOPG 8:2 plus 0.2% NBD-PE) run on a 4.5% native PAGE gel for 75 min at 100 V. (B) Fluorescence image before and after SYPRO Ruby staining of sonicated DOPC/DOPG 8:2 plus 0.2% NBD-PE SUVs run with and without 10 µM PspA WT. (C) Sypro-stained 4.5% native gel of Vipp1 WT (10 µM) incubated with increasing concentrations of DOPC/DOPG 8:2 SUVs. Download
Native PAGE-based protein-membrane-binding assays for PspA. (A) Gels from SCE stress assay at 0.5 and 1 mM lipid concentrations. Ten micromoles of PspA was used with integrated intensities of each band with respect to the PspA ΔV sample shown. Vesicles of increasing SCE stress contained the same composition and are presented in the same order as described for Fig. S1D. (B) Gels from anionic lipid-binding assays with PspA. Vesicles were extruded through 400 nm filters and composed of DMPC/DOPC 4:6 with the addition of anionic lipids PG (DOPG), CL (14:0 CL), or PS (DOPS), as specified. Integrated intensities of each band with respect to the PspA ΔV sample are shown. (C) Gels from the SCE stress assay with PspAΔAHa, with intensities of each band with respect to (w.r.t). to the PspAΔAHa ΔV sample shown. Increasing SCE stress vesicles were the same lipid compositions and are presented in the same order as described for Fig. S1D. Download
Direct interaction of multiple copies of PspA AHa with the membrane. (A) Schematic presentation of constructs expressing His-tagged eGFP-(L−) PspA (full length; pGJ92), eGFP-(L−) with 2 (2AHa; pGJ100) or 3 (3AHa; pGJ101) copies of PspA N-terminal AHas fused or with eGFP alone (pGJ102). (B) SMI (TIRF mode) of live cells, showing that in the absence of membrane stress (-pIV), eGFP-PspA foci localize in the membrane close to the pole (white arrow), while eGFP and eGFP-3AHa are distributed in the cytoplasm. Under stress (+pIV), eGFP-PspA foci localize in the polar and lateral membranes (white arrows), eGFP is located in the cytoplasm, and eGFP-3AHa is found mainly in the lateral membrane (white arrows). Bar, 1 µm (C) Membrane localization of eGFP-PspA, eGFP-2AHa, and eGFP-3AHa proteins following Triton X100-based fractionation of cells into soluble (Sol) and inner membrane (IM) fractions and Western blotting using GFP antibodies. Numbers represent total intensities of protein bands, where the maximal intensity was set as 1. (D) Vesicle binding of purified eGFP-3AHa, eGFP-PspA and eGFP determined from the native PAGE-based protein-membrane-binding assay. Vesicles of 100 nm were used at a 1 mM lipid concentration, and proteins were used at a concentration of 5 µM. Download
Protein-membrane-binding assays for Vipp1. (A) Circular dichroism spectra of purified Vipp1 and PspA1–186 between 180 and 260 nm. Spectra were normalized to the mean residue elipticity and are averages of 3 scans at 25°C. (B) Collected fractions from sucrose gradient centrifugation were analyzed via SDS-PAGE and Sypro Ruby protein staining of Vipp1, with and without 100 nm E. coli TLE vesicles. Starred fractions indicate those found to contain NBD-PE-labeled vesicles (monitored via fluorescence emission at 515 nm). (C) Gels from a SCE stress assay with 10 µM Vipp1 and a 2.5 mM lipid concentration. Integrated intensities of each band with respect to the Vipp1 ΔV sample are shown. Vesicles of increasing SCE stress had the same composition and were used in the same order as described for Fig. S1. (D) SCE stress assay with 10 µM Vipp1ΔAHa. Intensities of each band were compared to that for the Vipp1ΔAHa ΔV sample shown. Vesicles of increasing SCE stress had the same composition and are presented in the same order as for Fig. S1D. (E) Gels from anionic lipid-binding assays. All lanes contain 10 µM Vipp1. Vesicles were extruded through 400 nm filters and composed of DMPC/DOPC 4:6 with the addition of the anionic lipids PG (DOPG), CA (14:0 CA), and PS (DOPS) as indicated. The lipid concentration was 2.5 mM. Integrated intensities of each band with respect to the Vipp1 ΔV sample are shown. (F) Membrane binding of Vipp1 as a function of anionic lipids and SCE stress combined. A native PAGE gel (left) with 10 µM Vipp1 was incubated with DMPC/DOPC 4:6 vesicles and DOPE/DOPC 4:6 vesicles with or without 10% DOPG, with the calculated membrane-binding levels plotted in the bar graph (right). Download
Vipp1 binding to vesicles composed of the major lipid species within cyanobacterial thylakoid membranes, the galactolipids monogalactosyldiacylglycerol (MGDG) and digalactosyldiacylglycerol (DGDG) and phospholipid PG. (A) Gel (4.5%) from the native PAGE-based binding assay. Each lane contained 10 µM Vipp1 WT, either alone or with 1 mM or 2.5 mM of the specified vesicles. Vesicles were extruded through 400-nm filters with the lipid composition of each as follows: DGDG/DOPG, 3:1; DGDG/MGDG/DOPG, 2:1:1; DOPC/DOPG, 3:1. The intensity of each band with respect to the Vipp1 WT-alone sample on each gel is shown. (B) A bar graph showing the membrane binding of Vipp1 to each set of vesicles at 1 mM and 2.5 mM lipid concentrations. Data were obtained from three separate Vipp1 preparations. PG was used in all vesicle compositions to prevent aggregation. Download
Denaturing gels showing the spRNA (UpGG*G*) product and free GTP for transcription assays probing regulation of PspF1–275 by PspA upon membrane exposure. (A) Synthesis of the spRNA product occurred in the presence of PspF1–275 (lanes 2 and 3) was inhibited upon addition of PspA (lanes 5 and 6) and restored again with addition of vesicles (lanes 7 and 8). Controls for PspF dependency of UpGGG formation are in lanes 1 and 4. Txn components refer to inclusion of the reagents and the nifH promoter probe from which in vitro transcription occurred. (B) Denaturing gel showing the effect on spRNA production using vesicles of increasing SCE stress. Vesicles of increasing SCE stress were the same composition and used in the same order as described for Fig. S1. (C) PspA1–186 inhibition of transcription cannot be recovered by addition of vesicles. (D) Native gel image of the PspA-PspF inhibitory complex (lane 3) formed after incubation of 10 µM PspA WT with 10 µM PspF1–275, followed by its disruption and reappearance of the PspF band upon addition of 1 mM vesicles (lane 4). The PspA1–186-PspF1–275 inhibitory complex (lane 6) formed after incubation of 10 µM PspA1–186 with 10 µM PspF1–275 was not disrupted upon addition of 1 mM vesicles (lane 7). Download
Dynamic light scattering data for vesicles used within this study, with the number of particles plotted against particle size. (A) E. coli TLE vesicles. (B) Vesicles made from cls pgsA and WT cell extracts. (C) SCE stress assay vesicle set. (D) DMPC/DOPC 4:6 vesicles with inclusion of anionic lipids. Download
Strains and plasmids used.
ACKNOWLEDGMENTS
This work was funded by a Leverhulme Trust project grant (RPG-2012-705), the EPSRC via grant EP/G00465X/1 and by an EPSRC Centre for Doctoral Training Studentship from the Institute of Chemical Biology (Imperial College London) awarded to C.M.
We acknowledge L. Ying, D. Jach, P. Mehta, C. Hogg, and N. S. F. Mohd Suhaimi (Imperial College London) for technical help, C. Mullineaux and G. Bennardo (Queen Mary University of London) for plasmid-borne Synechocystis Vipp1, and W. Dowhan (University of Texas) for strain UE54.
Footnotes
Citation McDonald C, Jovanovic G, Ces O, Buck M. 2015. Membrane stored curvature elastic stress modulates recruitment of maintenance proteins PspA and Vipp1. mBio 6(5):e01188-15. doi:10.1128/mBio.01188-15.
REFERENCES
- 1.Darwin AJ. 2005. The phage-shock-protein response. Mol Microbiol 57:621–628. doi: 10.1111/j.1365-2958.2005.04694.x. [DOI] [PubMed] [Google Scholar]
- 2.Joly N, Engl C, Jovanovic G, Huvet M, Toni T, Sheng X, Stumpf MP, Buck M. 2010. Managing membrane stress: the phage shock protein (psp) response, from molecular mechanisms to physiology. FEMS Microbiol Rev 34:797–827. doi: 10.1111/j.1574-6976.2010.00240.x. [DOI] [PubMed] [Google Scholar]
- 3.Kobayashi R, Suzuki T, Yoshida M. 2007. Escherichia coli phage-shock protein A (PspA) binds to membrane phospholipids and repairs proton leakage of the damaged membranes. Mol Microbiol 66:100–109. doi: 10.1111/j.1365-2958.2007.05893.x. [DOI] [PubMed] [Google Scholar]
- 4.Joly N, Burrows PC, Engl C, Jovanovic G, Buck M. 2009. A lower-order oligomer form of phage shock protein A (PspA) stably associates with the hexameric AAA(+) transcription activator protein PspF for negative regulation. J Mol Biol 394:764–775. doi: 10.1016/j.jmb.2009.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jovanovic G, Engl C, Mayhew AJ, Burrows PC, Buck M. 2010. Properties of the phage-shock-protein (psp) regulatory complex that govern signal transduction and induction of the psp response in Escherichia coli. Microbiology 156:2920–2932. doi: 10.1099/mic.0.040055-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamaguchi S, Gueguen E, Horstman NK, Darwin AJ. 2010. Membrane association of PspA depends on activation of the phage-shock-protein response in Yersinia enterocolitica. Mol Microbiol 78:429–443. doi: 10.1111/j.1365-2958.2010.07344.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elderkin S, Jones S, Schumacher J, Studholme D, Buck M. 2002. Mechanism of action of the Escherichia coli phage shock protein PspA in repression of the AAA family transcription factor PspF. J Mol Biol 320:23–37. doi: 10.1016/S0022-2836(02)00404-7. [DOI] [PubMed] [Google Scholar]
- 8.Elderkin S, Bordes P, Jones S, Rappas M, Buck M. 2005. Molecular determinants for PspA-mediated repression of the AAA transcriptional activator PspF. J Bacteriol 187:3238–3248. doi: 10.1128/JB.187.9.3238-3248.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Model P, Jovanovic G, Dworkin J. 1997. The Escherichia coli phage-shock-protein (psp) operon. Mol Microbiol 24:255–261. doi: 10.1046/j.1365-2958.1997.3481712.x. [DOI] [PubMed] [Google Scholar]
- 10.Engl C, Jovanovic G, Lloyd LJ, Murray H, Spitaler M, Ying L, Errington J, Buck M. 2009. In vivo localizations of membrane stress controllers PspA and PspG in Escherichia coli. Mol Microbiol 73:382–396. doi: 10.1111/j.1365-2958.2009.06776.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jovanovic G, Mehta P, Ying L, Buck M. 2014. Anionic lipids and the cytoskeletal proteins MreB and RodZ define the spatio-temporal distribution and function of membrane stress controller PspA in Escherichia coli. Microbiology 160:2374–2386. doi: 10.1099/mic.0.078527-0. [DOI] [PubMed] [Google Scholar]
- 12.Zhang L, Sakamoto W. 2012. Possible function of VIPP1 in thylakoids: protection but not formation? Plant Signal Behav 8:1–3. doi: 10.4161/psb.22860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nordhues A, Schöttler MA, Unger AK, Geimer S, Schönfelder S, Schmollinger S, Rütgers M, Finazzi G, Soppa B, Sommer F, Mühlhaus T, Roach T, Krieger-Liszkay A, Lokstein H, Crespo JL, Schroda M. 2012. Evidence for a role of VIPP1 in the structural organization of the photosynthetic apparatus in Chlamydomonas. Plant Cell 24:637–659. doi: 10.1105/tpc.111.092692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Otters S, Braun P, Hubner J, Wanner G, Vothknecht UC, Chigri F. 2013. The first alpha-helical domain of the vesicle-inducing protein in plastids 1 promotes oligomerization and lipid binding. Planta 237:529–540. doi: 10.1007/s00425-012-1772-1. [DOI] [PubMed] [Google Scholar]
- 15.Zhang S, Shen G, Li Z, Golbeck JH, Bryant DA. 2014. Vipp1 is essential for the biogenesis of photosystem I but not thylakoid membranes in Synechococcus sp. PCC 7002. J Biol Chem 289:15904–15914. doi: 10.1074/jbc.M114.555631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jovanovic G, Mehta P, McDonald C, Davidson AC, Uzdavinys P, Ying L, Buck M. 2014. The N-terminal amphipathic helices determine regulatory and effector functions of phage shock protein A (PspA) in Escherichia coli. J Mol Biol 426:1498–1511. doi: 10.1016/j.jmb.2013.12.016. [DOI] [PubMed] [Google Scholar]
- 17.Attard GS, Templer RH, Smith WS, Hunt AN, Jackowski S. 2000. Modulation of CTP: phosphocholine cytidylyltransferase by membrane curvature elastic stress. Proc Natl Acad Sci U S A 97:9032–9036. doi: 10.1073/pnas.160260697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kirsten ML, Baron RA, Seabra MC, Ces O. 2013. Rab1a and Rab5a preferentially bind to binary lipid compositions with higher stored curvature elastic energy. Mol Membr Biol 30:303–314. doi: 10.3109/09687688.2013.818725. [DOI] [PubMed] [Google Scholar]
- 19.Drin G, Antonny B. 2010. Amphipathic helices and membrane curvature. FEBS Lett 584:1840–1847. doi: 10.1016/j.febslet.2009.10.022. [DOI] [PubMed] [Google Scholar]
- 20.Drin G, Casella JF, Gautier R, Boehmer T, Schwartz TU, Antonny B. 2007. A general amphipathic alpha-helical motif for sensing membrane curvature. Nat Struct Mol Biol 14:138–146. doi: 10.1038/nsmb1194. [DOI] [PubMed] [Google Scholar]
- 21.Foss MH, Eun YJ, Weibel DB. 2011. Chemical-biological studies of subcellular organization in bacteria. Biochemistry 50:7719–7734. doi: 10.1021/bi200940d. [DOI] [PubMed] [Google Scholar]
- 22.Cronan JE. 2003. Bacterial membrane lipids: where do we stand? Annu Rev Microbiol 57:203–224. doi: 10.1146/annurev.micro.57.030502.090851. [DOI] [PubMed] [Google Scholar]
- 23.Raetz CR. 1976. Phosphatidylserine synthetase mutants of Escherichia coli—genetic-mapping and membrane phospholipid composition. J Biol Chem 251:3242–3249. [PubMed] [Google Scholar]
- 24.Salje J, van den Ent F, de Boer P, Löwe J. 2011. Direct membrane binding by bacterial actin MreB. Mol Cell 43:478–487. doi: 10.1016/j.molcel.2011.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fuhrmann E, Bultema JB, Kahmann U, Rupprecht E, Boekema EJ, Schneider D. 2009. The vesicle-inducing protein 1 from Synechocystis sp. PCC 6803 organizes into diverse higher-ordered ring structures. Mol Biol Cell 20:4620–4628. doi: 10.1091/mbc.E09-04-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burrows PC, Joly N, Buck M. 2010. A prehydrolysis state of an AAA+ ATPase supports transcription activation of an enhancer-dependent RNA polymerase. Proc Natl Acad Sci U S A 107:9376–9381. doi: 10.1073/pnas.1001188107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goto K, Hozumi Y, Nakano T, Martelli AM, Kondo H. 2010. Methods for measuring the activity and expression of diacylglycerol kinase, p 77–109. In Murphy E, Rosenberger T (ed), Lipid-mediated signalling. CRC Press, Boca Raton, FL. [Google Scholar]
- 28.Zhou W, Parent LJ, Wills JW, Resh MD. 1994. Identification of a membrane-binding domain within the amino-terminal region of human immunodeficiency virus type 1 gag protein which interacts with acidic phospholipids. J Virol 68:2556–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McLaughlin S, Aderem A. 1995. The myristoyl-electrostatic switch: a modulator of reversible protein-membrane interactions. Trends Biochem Sci 20:272–276. doi: 10.1016/S0968-0004(00)89042-8. [DOI] [PubMed] [Google Scholar]
- 30.Zhan H, Lazaridis T. 2013. Inclusion of lateral pressure/curvature stress effects in implicit membrane models. Biophys J 104:643–654. doi: 10.1016/j.bpj.2012.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hatzakis NS, Bhatia VK, Larsen J, Madsen KL, Bolinger PY, Kunding AH, Castillo J, Gether U, Hedegård P, Stamou D. 2009. How curved membranes recruit amphipathic helices and protein anchoring motifs. Nat Chem Biol 5:835–841. doi: 10.1038/nchembio.213. [DOI] [PubMed] [Google Scholar]
- 32.Chong SS, Taneva SG, Lee JM, Cornell RB. 2014. The curvature sensitivity of a membrane-binding amphipathic helix can be modulated by the charge on a flanking region. Biochemistry 53:450–461. doi: 10.1021/bi401457r. [DOI] [PubMed] [Google Scholar]
- 33.Davies SM, Epand RM, Kraayenhof R, Cornell RB. 2001. Regulation of CTP: phosphocholine cytidylyltransferase activity by the physical properties of lipid membranes: an important role for stored curvature strain energy. Biochemistry 40:10522–10531. doi: 10.1021/bi010904c. [DOI] [PubMed] [Google Scholar]
- 34.DeLisa MP, Lee P, Palmer T, Georgiou G. 2004. Phage shock protein PspA of Escherichia coli relieves saturation of protein export via the Tat pathway. J Bacteriol 186:366–373. doi: 10.1128/JB.186.2.366-373.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eisenberg D, Weiss RM, Terwilliger TC. 1982. The helical hydrophobic moment—a measure of the amphiphilicity of a helix. Nature 299:371–374. doi: 10.1038/299371a0. [DOI] [PubMed] [Google Scholar]
- 36.Mishra VK, Palgunachari MN. 1996. Interaction of model class A1, class A2, and class Y amphipathic helical peptides with membranes. Biochemistry 35:11210–11220. doi: 10.1021/bi960760f. [DOI] [PubMed] [Google Scholar]
- 37.Jensen M, Bhatia VK, Jao CC, Rasmussen JE, Pedersen SL, Jensen KJ, Langen R, Stamou D. 2011. Membrane curvature sensing by amphipathic helixes: a single liposome study using alpha-synuclein and annexin B12. J Biol Chem 286:42603–42614. doi: 10.1074/jbc.M111.271130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Szeto TH, Rowland SL, Habrukowich CL, King GF. 2003. The MinD membrane targeting sequence is a transplantable lipid-binding helix. J Biol Chem 278:40050–40056. doi: 10.1074/jbc.M306876200. [DOI] [PubMed] [Google Scholar]
- 39.Nielsen C, Goulian M, Andersen OS. 1998. Energetics on inclusion-induced bilayer deformations. Biophys J 74:1966–1983. doi: 10.1016/S0006-3495(98)77904-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seddon JM, Templer RH, Warrender NA, Huang Z, Cevc G, Marsh D. 1997. Phosphatidylcholine fatty acid membranes: effects of headgroup hydration on the phase behavior and structural parameters of the gel and inverse hexagonal (H II) phases. Biochim Biophys Acta 1327:131–147. doi: 10.1016/S0005-2736(97)00047-3. [DOI] [PubMed] [Google Scholar]
- 41.Jovanovic G, Engl C, Buck M. 2009. Physical, functional and conditional interactions between ArcAB and phage shock proteins upon secretion-induced stress in Escherichia coli. Mol Microbiol 74:16–28. doi: 10.1111/j.1365-2958.2009.06809.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huvet M, Toni T, Sheng X, Thorne T, Jovanovic G, Engl C, Buck M, Pinney JW, Stumpf MP. 2011. The evolution of the phage shock protein response system: interplay between protein function, genomic organization, and system function. Mol Biol Evol 28:1141–1155. doi: 10.1093/molbev/msq301. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary methods. Download
Controls undertaken for the native PAGE-based protein-membrane-binding assay. (A) Controls showing functionality of purified PspA. Native PAGE band shift assay results with PspA and PspF1–275, showing formation of a PspA-PspF1–275 cocomplex (left) and the resulting ATPase inhibition of PspF1–275 at a 1:1 PspA-to-PspF1–275 molar ratio, monitored via an NADH-coupled assay (right). (B) Five percent native PAGE gel with decreasing amounts of PspA stained via Sypro Ruby protein stain. Intensity of the integrated PspA bands plotted against protein used produced a linear trend depicted in the adjacent graph. (C) Native PAGE assay with PspF1–275 and 100 nm TLE vesicles with integrated band intensities with respect to (w.r.t) a PspF1–275 ΔV, showing no decrease on addition of vesicles. (D) Gel from native PAGE protein-membrane binding (SCE stress binding assay) undertaken with reduced running buffer pH (pH 7.5, compared to the typical 8.2) with binding trends for PspA preserved. Vesicles of increasing SCE stress compositions correspond to; DMPC/DOPC 4:6 (lane 1), DMPC/DOPC 2:8 (lane 2), DOPC (lane 3), DOPE/DOPC 2:8 (lane 4), and DOPE/DOPC 4:6 (lane 5). Lane 6 contains E. coli TLE vesicles incubated with PspA, and lane 7 contains E. coli TLE vesicles alone. Download
Visualization of a protein-vesicle complex in the native gel matrix, obtained using small unilamellar vesicles (SUVs). (A) Fluorescence image of sonicated SUVs (composed of DOPC plus 0.2% NBD-PE and DOPC/DOPG 8:2 plus 0.2% NBD-PE) run on a 4.5% native PAGE gel for 75 min at 100 V. (B) Fluorescence image before and after SYPRO Ruby staining of sonicated DOPC/DOPG 8:2 plus 0.2% NBD-PE SUVs run with and without 10 µM PspA WT. (C) Sypro-stained 4.5% native gel of Vipp1 WT (10 µM) incubated with increasing concentrations of DOPC/DOPG 8:2 SUVs. Download
Native PAGE-based protein-membrane-binding assays for PspA. (A) Gels from SCE stress assay at 0.5 and 1 mM lipid concentrations. Ten micromoles of PspA was used with integrated intensities of each band with respect to the PspA ΔV sample shown. Vesicles of increasing SCE stress contained the same composition and are presented in the same order as described for Fig. S1D. (B) Gels from anionic lipid-binding assays with PspA. Vesicles were extruded through 400 nm filters and composed of DMPC/DOPC 4:6 with the addition of anionic lipids PG (DOPG), CL (14:0 CL), or PS (DOPS), as specified. Integrated intensities of each band with respect to the PspA ΔV sample are shown. (C) Gels from the SCE stress assay with PspAΔAHa, with intensities of each band with respect to (w.r.t). to the PspAΔAHa ΔV sample shown. Increasing SCE stress vesicles were the same lipid compositions and are presented in the same order as described for Fig. S1D. Download
Direct interaction of multiple copies of PspA AHa with the membrane. (A) Schematic presentation of constructs expressing His-tagged eGFP-(L−) PspA (full length; pGJ92), eGFP-(L−) with 2 (2AHa; pGJ100) or 3 (3AHa; pGJ101) copies of PspA N-terminal AHas fused or with eGFP alone (pGJ102). (B) SMI (TIRF mode) of live cells, showing that in the absence of membrane stress (-pIV), eGFP-PspA foci localize in the membrane close to the pole (white arrow), while eGFP and eGFP-3AHa are distributed in the cytoplasm. Under stress (+pIV), eGFP-PspA foci localize in the polar and lateral membranes (white arrows), eGFP is located in the cytoplasm, and eGFP-3AHa is found mainly in the lateral membrane (white arrows). Bar, 1 µm (C) Membrane localization of eGFP-PspA, eGFP-2AHa, and eGFP-3AHa proteins following Triton X100-based fractionation of cells into soluble (Sol) and inner membrane (IM) fractions and Western blotting using GFP antibodies. Numbers represent total intensities of protein bands, where the maximal intensity was set as 1. (D) Vesicle binding of purified eGFP-3AHa, eGFP-PspA and eGFP determined from the native PAGE-based protein-membrane-binding assay. Vesicles of 100 nm were used at a 1 mM lipid concentration, and proteins were used at a concentration of 5 µM. Download
Protein-membrane-binding assays for Vipp1. (A) Circular dichroism spectra of purified Vipp1 and PspA1–186 between 180 and 260 nm. Spectra were normalized to the mean residue elipticity and are averages of 3 scans at 25°C. (B) Collected fractions from sucrose gradient centrifugation were analyzed via SDS-PAGE and Sypro Ruby protein staining of Vipp1, with and without 100 nm E. coli TLE vesicles. Starred fractions indicate those found to contain NBD-PE-labeled vesicles (monitored via fluorescence emission at 515 nm). (C) Gels from a SCE stress assay with 10 µM Vipp1 and a 2.5 mM lipid concentration. Integrated intensities of each band with respect to the Vipp1 ΔV sample are shown. Vesicles of increasing SCE stress had the same composition and were used in the same order as described for Fig. S1. (D) SCE stress assay with 10 µM Vipp1ΔAHa. Intensities of each band were compared to that for the Vipp1ΔAHa ΔV sample shown. Vesicles of increasing SCE stress had the same composition and are presented in the same order as for Fig. S1D. (E) Gels from anionic lipid-binding assays. All lanes contain 10 µM Vipp1. Vesicles were extruded through 400 nm filters and composed of DMPC/DOPC 4:6 with the addition of the anionic lipids PG (DOPG), CA (14:0 CA), and PS (DOPS) as indicated. The lipid concentration was 2.5 mM. Integrated intensities of each band with respect to the Vipp1 ΔV sample are shown. (F) Membrane binding of Vipp1 as a function of anionic lipids and SCE stress combined. A native PAGE gel (left) with 10 µM Vipp1 was incubated with DMPC/DOPC 4:6 vesicles and DOPE/DOPC 4:6 vesicles with or without 10% DOPG, with the calculated membrane-binding levels plotted in the bar graph (right). Download
Vipp1 binding to vesicles composed of the major lipid species within cyanobacterial thylakoid membranes, the galactolipids monogalactosyldiacylglycerol (MGDG) and digalactosyldiacylglycerol (DGDG) and phospholipid PG. (A) Gel (4.5%) from the native PAGE-based binding assay. Each lane contained 10 µM Vipp1 WT, either alone or with 1 mM or 2.5 mM of the specified vesicles. Vesicles were extruded through 400-nm filters with the lipid composition of each as follows: DGDG/DOPG, 3:1; DGDG/MGDG/DOPG, 2:1:1; DOPC/DOPG, 3:1. The intensity of each band with respect to the Vipp1 WT-alone sample on each gel is shown. (B) A bar graph showing the membrane binding of Vipp1 to each set of vesicles at 1 mM and 2.5 mM lipid concentrations. Data were obtained from three separate Vipp1 preparations. PG was used in all vesicle compositions to prevent aggregation. Download
Denaturing gels showing the spRNA (UpGG*G*) product and free GTP for transcription assays probing regulation of PspF1–275 by PspA upon membrane exposure. (A) Synthesis of the spRNA product occurred in the presence of PspF1–275 (lanes 2 and 3) was inhibited upon addition of PspA (lanes 5 and 6) and restored again with addition of vesicles (lanes 7 and 8). Controls for PspF dependency of UpGGG formation are in lanes 1 and 4. Txn components refer to inclusion of the reagents and the nifH promoter probe from which in vitro transcription occurred. (B) Denaturing gel showing the effect on spRNA production using vesicles of increasing SCE stress. Vesicles of increasing SCE stress were the same composition and used in the same order as described for Fig. S1. (C) PspA1–186 inhibition of transcription cannot be recovered by addition of vesicles. (D) Native gel image of the PspA-PspF inhibitory complex (lane 3) formed after incubation of 10 µM PspA WT with 10 µM PspF1–275, followed by its disruption and reappearance of the PspF band upon addition of 1 mM vesicles (lane 4). The PspA1–186-PspF1–275 inhibitory complex (lane 6) formed after incubation of 10 µM PspA1–186 with 10 µM PspF1–275 was not disrupted upon addition of 1 mM vesicles (lane 7). Download
Dynamic light scattering data for vesicles used within this study, with the number of particles plotted against particle size. (A) E. coli TLE vesicles. (B) Vesicles made from cls pgsA and WT cell extracts. (C) SCE stress assay vesicle set. (D) DMPC/DOPC 4:6 vesicles with inclusion of anionic lipids. Download
Strains and plasmids used.