

Abstract
Phosphatidylinositol bisphosphate (PIP2) regulates epithelial sodium channel (ENaC) open probability. In turn, myristoylated alanine-rich C kinase substrate (MARCKS) protein or MARCKS-like protein 1 (MLP-1) at the plasma membrane regulates the delivery of PIP2 to ENaC. MARCKS and MLP-1 are regulated by changes in cytosolic calcium; increasing calcium promotes dissociation of MARCKS from the membrane, but the calcium-regulatory mechanisms are unclear. However, it is known that increased intracellular calcium can activate calmodulin and we show that inhibition of calmodulin with calmidazolium increases ENaC activity presumably by regulating MARCKS and MLP-1. Activated calmodulin can regulate MARCKS and MLP-1 in two ways. Calmodulin can bind to the effector domain of MARCKS or MLP-1, inactivating both proteins by causing their dissociation from the membrane. Mutations in MARCKS that prevent calmodulin association prevent dissociation of MARCKS from the membrane. Calmodulin also activates CaM kinase II (CaMKII). An inhibitor of CaMKII (KN93) increases ENaC activity, MARCKS association with ENaC, and promotes MARCKS movement to a membrane fraction. CaMKII phosphorylates filamin. Filamin is an essential component of the cytoskeleton and promotes association of ENaC, MARCKS, and MLP-1. Disruption of the cytoskeleton with cytochalasin E reduces ENaC activity. CaMKII phosphorylation of filamin disrupts the cytoskeleton and the association of MARCKS, MLP-1, and ENaC, thereby reducing ENaC open probability. Taken together, these findings suggest calmodulin and CaMKII modulate ENaC activity by destabilizing the association between the actin cytoskeleton, ENaC, and MARCKS, or MLP-1 at the apical membrane.
Keywords: calcium, CaMKII, ENaC, filamin, MARCKS
the amiloride-sensitive epithelial sodium channel (ENaC) plays a vital role in the regulation of total body fluid homeostasis and control of blood pressure; therefore, regulation of ENaC is critical for normal health. There are two modes of ENaC regulation: altering the density of channels in the membrane by changing membrane insertion or retrieval (40, 43) or altering the activity of individual channels by changing their open probability (1, 12, 21, 41). In particular, the open probability of ENaC is regulated by interaction with anionic lipids like phosphatidylinositol bisphosphate (PIP2) (1, 21, 41). The problem with understanding how this mode of regulation could occur is that PIP2 is a rare lipid in the membrane and ENaC is a relatively rare protein. If PIP2-ENaC interaction depended upon random lateral diffusion of the two molecules in the apical membrane, the channel would open only rarely. In fact, ENaC is typically open about half the time. This implies that the local concentration of PIP2 near ENaC is higher than would be expected based on the average density in the membrane generally. We have previously shown this can be accomplished by myristoylated alanine-rich C-kinase substrate (MARCKS) or MARCKS-like protein (MLP), which binds ENaC and electrostatically sequesters and concentrates PIP2 near ENaC (1). The effector domain of MARCKS contains multiple serine residues that are subject to phosphorylation by PKC, is responsible for binding anionic phospholipids, and harbors a calmodulin binding sequence (14, 35). MARCKS and MLP-1 are regulated by changes in cytosolic calcium; increasing calcium promotes dissociation of MARCKS from the membrane, but the mechanisms by which calcium promotes dissociation from the membrane are unclear (23). However, it is known that increased intracellular calcium can activate calmodulin. Activated calmodulin could regulate MARCKS and MLP-1 in two ways. Activated calmodulin can bind to the effector domains of MARCKS or MLP-1, inactivating both proteins, allowing their dissociation from the membrane (14). Mutations in MARCKS that prevent calmodulin association prevent dissociation of MARCKS from the membrane (14). Alternatively, calmodulin (CaM) also activates CaM kinase II (CaMKII). CaMKII is known to play an important role in the brain, but this multifunctional kinase is also expressed in other tissues, including the kidney. Timmins et al. (38) reported the major isoform of CaMKII expressed in murine kidneys was CaMKIIγ. Rothschild et al. (32) showed that only CaMKIIγ1 is expressed in zebrafish kidneys. The β-isoform of CaMKII has been best characterized for its role in actin cytoskeletal assembly. Hoffman and coworkers (16) recently determined isoform specificity of CaMKII, including the γ-isoform of CaMKII in mediating the inhibition of actin polymerization and modulation of the actin cytoskeleton (16). Disruption of the cytoskeleton with cytochalasin E reduces ENaC activity (29). Filamin is among the many different actin binding proteins that have been reported to bind and regulate the cell surface expression of transmembrane proteins. For example, filamin A has been shown to regulate the surface expression of BKCa channels (20), the CFTR (37), Kv4.2 potassium channels (27), calcium-sensing receptors (CaR) (42), the inwardly rectifying potassium channel Kir2.1 (33), and ENaC (39). Furthermore, filamins function as scaffolding proteins that are capable of connecting the actin cytoskeleton to various cytoplasmic signaling proteins. The association of ENaC with the cytoskeletal network is believed to help maintain its expression at the apical membrane and prevent its downregulation by endocytosis. There have been multiple studies performed to show the cytoskeleton is involved in the regulation of ENaC (8, 17, 19, 29). The present study corroborates our previous reports of ENaC being regulated by MARCKS and the actin cytoskeleton. Here, we show ENaC activity is sensitive to calcium-calmodulin and CaMKII. We focus on the effect of this pathway on MARCKS protein expression and posttranslational changes in the actin cytoskeletal protein filamin A to elucidate the mechanism by which ENaC is regulated.
MATERIALS AND METHODS
Cell culture and treatments.
Xenopus 2F3 cells were maintained as previously described (1, 4, 29). In some experiments, cells were cultured in media containing normal calcium (1.05 mM CaCl2) or high calcium (4 mM CaCl2) before being lysed in mammalian protein extraction reagent (MPER; Thermo Scientific, Rockford, IL) supplemented with Halt protease and phosphatase inhibitors (Thermo Scientific). All animal protocols are approved by the Emory University Institutional Care and Use Committee.
Site-directed mutagenesis.
Lysine residues 155 and 164 within the effector domain of MARCKS were mutated to glutamic acid residues by site-directed mutagenesis using a QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). PAGE purified oligonucleotides (Integrated DNA Technologies) used for site-directed mutagenesis were 5′-AAG CGT TTT TCA TTC GAA AAA TCT TTT GAG TTG AGT GGT TTC-3′ and its complement 3′-GAA ACC ACT CAA CTC AAA AGA TTT TTC GAA TGA AAA ACG CTT-5′. The underlined nucleotides correspond to the mutated bases. Constructs were confirmed by DNA sequencing.
Immunoprecipitation.
Cells were lysed in MPER and 200 μg of total protein freshly supplemented with protease and phosphatase inhibitors (Thermo Scientific) was incubated with a 1:250 dilution of MARCKS polyclonal antibody at 4°C for 4 h with end-over-end mixing. Complexes were incubated with a 1:10 dilution of prewashed 50% protein G agarose (Millipore, Billerica, MA) at 4°C for 6 h with end-over-end mixing. The beads were washed four times with ice-cold MPER, and the bound proteins were eluted in 1× SDS sample buffer and analyzed by SDS-PAGE, Western blotting, or Coomassie staining.
SDS-PAGE, Western blotting, and densitometric analysis.
Cells were washed once with 1× PBS before being scraped in MPER containing protease and phosphatase inhibitors (Thermo Scientific). The cell lysate was collected and then passed five times through a 23-gauge needle with a syringe before being incubated on ice for 1 h. The BCA protein assay (Thermo Scientific) was performed according to the manufacturer's instructions to determine protein concentration. Samples were prepared in Laemmli sample buffer (Bio-Rad, Hercules, CA) containing 5% β-mercaptoethanol. One hundred micrograms of protein was resolved on 4–20% Tris·HCl polyacrylamide precast gels using Tris glycine/SDS buffer (Bio-Rad) on a Criterion or Protean Western blotting system (Bio-Rad). The separated proteins were electrophoretically transferred onto Hybond C-extra nitrocellulose membranes (GE Healthcare, Piscataway, NJ) using prechilled Tris glycine buffer on a Criterion or Protean Western blotting system (Bio-Rad). The membranes were then blocked in 5% (wt/vol) dry milk in 1× TBS (Bio-Rad) for 1 h at room temperature. Western blotting was performed by first incubating the blocked membranes with rabbit polyclonal primary antibodies (anti-phospho-filamin A; phospho Ser2152, Cell Signaling Technology), anti-filamin A (Cell Signaling Technology), anti-CaMKII (Cell Signaling Technology), anti-PKC-α (Cell Signaling Technology), anti-MARCKS (Abcam), and anti-ENaC-α (1), -β (1), and -γ (22) prepared at a 1:1,000 dilution in 5% BSA-1× TBS for 8 h at 4°C. The membranes were washed three times at 5-min intervals with 1× TBS before being incubated with horseradish peroxidase-conjugated goat anti-rabbit secondary antibody at a dilution of 1:3,000 prepared in blocking solution. The membranes were washed four times at 4-min intervals with 1× TBS and incubated with SuperSignal Dura Chemiluminescent Substrate (Pierce) according to the manufacturer's instructions. The membranes were subjected to exposure using a Kodak Gel Logic 2200 Imager and Molecular Imaging software system (Carestream Health, Rochester, NY). Precision Plus Protein Standards (Bio-Rad) were used as markers to determine the relative molecular masses of the immunoreactive bands.
Recombinant protein production.
Recombinant proteins were expressed and purified as previously described (1–4).
Single-channel patch-clamp studies.
Micropipettes were pulled from filamented borosilicate glass capillaries (TW-150F, World Precision Instruments, Sarasota, FL) using a two-stage vertical puller (Narishige, Tokyo, Japan) and had a resistance of 6–10 MΩ. 2F3 cells were cultured to confluence on glutaraldehyde-fixed, collagen-coated Millipore-CM filters (Millipore) mounted onto the bottom of Lucite rings. At room temperature, cells were visualized with Hoffman modulation optics (Modulation Optics), and, after the pipette tip contacted the cell surface, negative pressure was applied to obtain a seal resistance of 10–20 GΩ. Physiological amphibian saline titrated with 0.1 N NaOH or HCl to a pH of 7.3–7.4 and consisting of (in mM) 95 NaCl, 3.4 KCl, 0.8 CaCl2, 0.8 MgCl2, and 10 HEPES was used for the patch pipette and extracellular bath solutions. A cell-attached patch configuration was used, and voltages are given as the negative of the patch pipette potential. Positive potentials represent depolarizations, while negative potentials represent hyperpolarizations of the cell membrane away from the resting potential. The product of the number of functional channels and the open probability was calculated using pCLAMP 10 software (Molecular Devices) and represents a measurement of ENaC activity within a patch.
Detergent extraction and sucrose density gradient centrifugation.
Cells were lysed in freshly prepared ice-cold 1% Brij 96/TNEV buffer (10 mM Tris·HCl, pH 7.5, 150 mM NaCl, 5 mM EDTA, 2 mM Na vanadate, and protease and phosphatase inhibitor cocktail). The cell lysate was collected and passed five times through a 23-gauge needle with a syringe before being incubated on ice for 1 h. Lysates were centrifuged at 10,000 rpm for 5 min at 4°C. Five hundred microliters of the supernatant was mixed with an equal volume of freshly prepared 80% sucrose in TNE (without Brij 96) and then transferred to a 13 × 23-mm Beckman centrifuge tube. Eighteen hundred microliters of freshly prepared 35% sucrose in TNE were carefully layered on top of the mixture followed by 500 μl of freshly prepared 5% sucrose. The sucrose gradient was then subject to ultracentrifugation in a SW50.1 rotor (Beckman) at 34,000 rpm for 16 h at 4°C. Thirteen fractions of 250-μl volume were carefully collected from the top to the bottom of the tube and then analyzed by Western blotting using flotilin and μ2 antibodies as protein markers.
Measurements of transepithelial voltage, resistance, and current.
Measurements were made from confluent cells using an epithelial voltmeter (EVOM, World Precision Instruments) under sterile conditions. Transepithelial current was calculated by Ohm's law and expressed as microamperes per square centimeter. To calculate amiloride-sensitive sodium current, the difference in total current and amiloride-insensitive current was determined after the addition of amiloride (0.5 μM) to the apical side of the inserts.
Statistical analysis.
A P value of <0.05 was considered significant. One-way ANOVA was performed for the comparison of more than one group.
RESULTS
Calmodulin inhibition augments endogenous ENaC activity in Xenopus 2F3 cells.
We previously demonstrated a role for MARCKS and the actin cytoskeleton in the regulation of ENaC activity in Xenopus 2F3 cells (1, 29). Calmodulin is a calcium binding protein that is known to regulate the function of MARCKS (14) and organization of the actin cytoskeleton (11). Here, we treated Xenopus 2F3 cells with a pharmacological inhibitor of calmodulin, calmidazolium, and performed single-channel patch-clamp recordings to determine whether ENaC activity is sensitive to calmodulin. Single-channel patch-clamp studies showed a significant increase in amiloride-sensitive ENaC current after application of calmidazolium, in which the activity was rescued after inducing calcium mobilization by application of ionomycin (Fig. 1A). Consistent with these results, analysis of single-channel patch-clamp data showed a statistically significant increase in the open probability of ENaC in response to calmidazolium and decrease in the open probability of the channels after ionomycin treatment (Fig. 1B).
Fig. 1.

Single-channel patch-clamp recordings measuring the effect of the calmodulin inhibitor calmidazolium on the open probability (Po) of the epithelial Na channel (ENaC). A: representative single-channel recordings of ENaC under control conditions (I), after application of 10 μM calmidazolium (II), and after application of 5 μM ionomycin (III). B: line graph showing a statistically significant increase in the Po of ENaC after application of calmidazolium and the rescue of ENaC activity after application of ionomycin. Values are means ± SE. *P < 0.05.
Calmodulin directly binds to MARCKS and CaMKII but not to ENaC in a calcium-dependent manner.
To determine whether the calmodulin effect on ENaC was in part due to direct binding between the two proteins and to confirm calmodulin binding to MARCKS and CaMKII in Xenopus 2F3 cells, we performed glutathione-S-transferase (GST) pulldown assays. Purified GST-calmodulin immobilized on a glutathione Sepharose support was used as the bait protein, and a crude cellular lysate from Xenopus 2F3 cells was used as the source of prey proteins, including ENaC, MARCKS, and CaMKII. GST pulldown assays showed calmodulin does not directly bind to ENaC, but does bind to MARCKS and CaMKII in a calcium-dependent manner (Fig. 2).
Fig. 2.

Glutathione-S-transferase (GST) pull-down assays showing the calcium-dependent interaction between recombinant GST-calmodulin (CaM), ENaC α-subunit, myristoylated alanine-rich C kinase substrate (MARCKS), and calmodulin kinase II (CaMKII). GST-CaM was expressed and purified from bacterial cells. GST-CaM fusion protein immobilized on a glutathione Sepharose support was used to pull down associated proteins from a crude Xenopus 2F3 cellular lysate in the presence of phosphatase and protease inhibitors and in the absence or presence of calcium (2 mM CaCl2). The ENaC α-subunit did not bind GST-calmodulin (A), but MARCKS (B) and CaMKII (C) bound in a calcium-dependent manner. Arrows indicate immunoreactive bands corresponding to each protein.
The calmodulin-dependent regulation of MARCKS involves lysine residues 155 and 164.
The myristoylation domain of MARCKS inserts itself hydrophobically within the inner leaflet of the plasma membrane, and the MARCKS effector domain interacts electrostatically with acidic lipids (5). Both domains of MARCKS are required for sufficient association with the cytosolic face of the plasma membrane (5). The effector domain of MARCKS plays multiple roles in the regulation of the protein because it also contains the calmodulin binding site. It is plausible that mutating the calmodulin binding site or introducing negative charges within the effector domain of MARCKS would alter calmodulin in the membrane. We made a single construct consisting of lysine residues 155 and 164 being mutated to glutamic acid residues by site-directed mutagenesis to evaluate both of these assumptions. First, we cotransfected green fluorescent protein (GFP)-calmodulin with MARCKS-cyan fluorescent protein (CFP) and investigated the effect of calcium augmentation on GFP-calmodulin translocation. We observed rapid translocation of GFP-calmodulin to the membrane after application of exogenous calcium (Fig. 3). Then, we monitored the ability of MARCKS-CFP to associate with the apical membrane before and after application of exogenous calcium. We observed less MARCKS associated with the membrane after application of exogenous calcium (Fig. 3). The release of MARCKS-CFP from the apical membrane in response to exogenous calcium was blocked in cells expressing mutant MARCKS-CFP compared with the wild-type control (Fig. 3).
Fig. 3.

Confocal microscopy and site-directed mutagenesis analysis of calcium-CaM-induced MARCKS translocation. A: Xenopus 2F3 cells were cotransfected with CaM-green fluorescent protein (GFP) and MARCK-cyan fluorescent protein (CFP). Forty-eight hours after transfection, cells were imaged for calmodulin-GFP and MARCKS-CFP. A moderate amount of calmodulin-GFP and MARCKS-CFP is expressed at the apical membrane in cells exposed to basal levels of calcium. An increase in CaM translocation to the apical membrane and MARCKS displacement from the membrane is shown for cells challenged with high levels of calcium (10 mM). B: Xenopus 2F3 cells were cotransfected with CaM-GFP and a mutant MARCKS-CFP (lysine residues 155 and 164 mutated to glutamic acid residues). Forty-eight hours after transfection, cells were imaged for calmodulin-GFP and MARCKS-CFP. Similar to A, a moderate amount of calmodulin-GFP and mutant MARCKS-CFP is expressed at the apical membrane in cells exposed to basal levels of calcium. An increase in CaM translocation to the apical membrane in response to calcium is shown, but the displacement of MARCKS from the membrane as seen in A is abrogated with the mutant MARCKS. All images shown represent the apical membrane and are the first of a series of images taken from the top to the bottom of the inset. Phase-contrast images show transfection efficiency and confluence of cells. In similar studies, using CaM-GFP and MARCKS-CFP, the cyan was pseudo colored red to shown colocalization and subcellular distribution between the plasma membrane and cytoplasm in the z-stack images (right).
CaMKII inhibition augments endogenous ENaC activity in Xenopus 2F3 cells.
The subcellular translocation of calmodulin and its effect on ENaC is dependent on calcium. The calcium-calmodulin complex regulates the activity of the serine/threonine-specific CaMKII. To determine whether CaMKII is involved in modulating ENaC activity, transepithelial resistance and voltage were recorded from Xenopus 2F3 cells and used to calculate amiloride-sensitive transepithelial current. Amiloride-sensitive transepithelial current increased in a time-dependent manner in response to application of the pharmacological inhibitor of CaMKII, KN93 (Fig. 4). Next, single-channel patch-clamp recordings were performed to corroborate a role for CaMKII in modulating ENaC activity and to determine whether CaMKII affects the open probability of ENaC. There was a statistically significant increase in the open probability of ENaC after application of the CaMKII inhibitor (Fig. 5).
Fig. 4.

Amiloride-sensitive transepithelial current measurements in Xenopus 2F3 cells after inhibition of CaMKII with KN93. Xenopus 2F3 cells grown on permeable supports were maintained in culture for 10 days to allow for the formation of tight junctions and the generation of measurable voltages and resistances across the monolayers. The pharmacological inhibitor of CaMKII, KN93 (0.5 μM) was applied to the apical side of Xenopus 2F3 cells at time 0 after recording of baseline voltage and resistance. Transepithelial voltages and resistances were used to calculate the current across the monolayers. A transient increase in transepithelial current was observed after 1 h of application of KN93. The increase in transepithelial current was sustained for an additional hour. At the end of the experiment, amiloride (0.5 μM) was applied to the apical side of the cells as a control (not shown).
Fig. 5.

Single-channel patch-clamp recordings measuring the effect of the CaMKII inhibitor KN93 on ENaC Po in Xenopus 2F3 cells. A: representative single-channel patch-clamp recordings illustrating inhibition of CaMKII with the pharmacological inhibitor KN93 (0.5 μM) increases the activity of ENaC. B: summary line graph showing the Po of ENaC increases after inhibiting CaMKII with KN93. Values are means ± SE. *P < 0.05.
MARCKS and MLP-1 protein expression is sensitive to CaMKII inhibition.
We previously reported a shift in MARCKS protein expression from light to heavy fractions with cytochalasin E treatment (29). In those studies, we used a recombinant antibody against the entire MARCKS protein to detect MARCKS and MLP-1 expression. To determine whether both MARCKS and MLP-1 protein expression are also sensitive to CaMKII, we performed similar studies in which we treated Xenopus 2F3 cells with the CaMKII inhibitor KN93 before performing sucrose density gradient and Western blot analysis of the different fractions. We found total protein expression levels of both MARCKS (Fig. 6, top bands) and MLP-1 (bottom bands) did not change significantly, but instead the expression of each protein shifted from heavy to light fractions in response to CaMKII inhibition (Fig. 6).
Fig. 6.

Sucrose density gradient and Western blot (WB) analysis of MARCKS and MARCKS-like protein 1 (MLP-1) in response to CaMKII inhibition in Xenopus 2F3 cells. Western blot analysis probing for MARCKS and MLP-1 shows the total amount of MARCKS and MLP-1 protein did not change, but the protein density shifted from heavy to light fractions. A greater amount of MARCKS and MLP-1 protein appear in lanes 1 and 4 with KN93 treatment (0.5 μM) compared with the vehicle (MOCK) treatment.
CaMKII inhibition and disruption of the cytoskeleton decrease phospho-filamin A.
Filamin A plays a role in linking the cytoskeleton to the plasma membrane. Wang et al. (39) showed filamin binds ENaC and decreases its activity. As indicated by studies by Bourguignon et al. (7), filamin may be a substrate of CaMKII. Ohta and Hartwig (25) showed filamin is phosphorylated in vitro at serine residues by CaMKII. Therefore, we wanted to determine whether CaMKII contributes to the phosphorylation of filamin in Xenopus 2F3 cells. We also wanted to determine whether disruption of the actin cytoskeleton affects filamin phosphorylation. We treated Xenopus 2F3 cells with either the cytoskeleton disrupter cytochalasin E or the CaMKII inhibitor KN93 and then performed SDS-PAGE and Western blot analysis, while probing for phospho- and total filamin protein. As shown in Fig. 7, both cytochalasin E and KN93 treatment attenuated the amount of phospho-filamin.
Fig. 7.
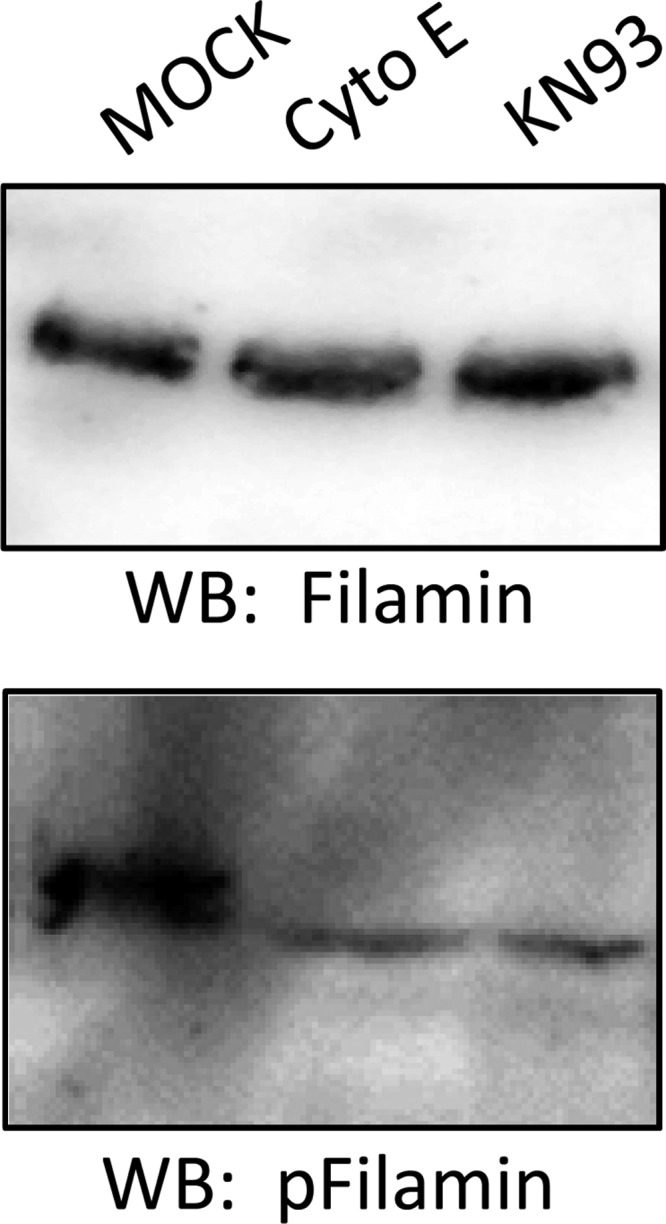
WB analysis of filamin and phospho-filamin in response to CaMKII inhibition and actin cytoskeleton disruption. Protein expression for phospho-filamin (bottom blot) decreased after CaMKII inhibition with KN93 or actin cytoskeleton disruption with cytochalasin E. Similar blots were probed for total filamin protein expression (top blot) as a control.
Calcium attenuates binding between ENaC subunits and filamin.
Although the interaction between ENaC and filamin has been reported, the molecular determinants governing the interaction are largely unknown. Here, we investigated the role of calcium in the regulation of ENaC activity. To elucidate the mechanism for this novel regulation, we investigated the effect of calcium on the association between ENaC subunits and filamin. There is a strong association between filamin and ENaC-α, -β, and -γ in Xenopus 2F3 cells cultured in basal levels of calcium (Fig. 8). The association between each ENaC subunit and filamin was attenuated in Xenopus 2F3 cells cultured in high levels of calcium (Fig. 8). We also investigated the association between PKC-α and filamin, since we previously showed MARCKS promotes the PIP2-dependent regulation of ENaC. PKC is known to regulate the function of MARCKS, and here we show PKC-α interacts with filamin in a calcium-dependent manner similar to ENaC and filamin (Fig. 8).
Fig. 8.

Immunoprecipitation (IP) and WB analysis showing the effect of calcium on the association between ENaC subunits and filamin. Xenopus 2F3 cells were cultured in either normal (1.05 mM CaCl2) or high calcium (4 mM CaCl2) for 8 h before being lysed in the presence of protease and phosphatase inhibitors. The lysate was used to immunoprecipitate ENaC subunits and PKC-α. All blots were probed for filamin. A strong association between filamin and ENaC α (A)-, β (B)-, γ (C)-subunits and PKC-α(D) is shown for cells treated with basal levels of calcium, and that association is attenuated for cells treated with high levels of calcium.
DISCUSSION
Multiple groups have shown sodium absorption in cortical collecting ducts is inhibited by increases in intracellular calcium (26, 31). Studies by Robins and Sandle (31) suggest increases in intracellular calcium inhibit wild-type ENaC but not Liddles-mutated ENaC, and this occurs by the removal of sodium channels from the apical membrane. This is consistent with observations by Plant et al. (28) which showed calcium can mobilize Nedd4 to the membrane. This would presumably signal ENaC for endocytosis and lysosomal degradation, thus reducing the density of the sodium channels at the membrane. Calmodulin has been suggested to play a role in calcium-dependent inhibition of ENaC activity, although the exact mechanism is unclear (31).
We recently reported a role for MARCKS (1) and the actin cytoskeleton (29) in the regulation of ENaC activity in Xenopus 2F3 cells. In this study, we show ENaC activity is modulated by calcium-calmodulin and CaMKII. It is likely that the actions of the calcium-calmodulin complex and CaMKII are linked, and the modulation in ENaC activity that is seen in response to pharmacological inhibition of either calmodulin or CaMKII involves other proteins including MARCKS/MLP-1 and other cytoskeletal-associated proteins. There are multiple mechanisms that could link the regulation of ENaC by calcium-calmodulin. Erk-1/2 phosphorylation is affected by CaMKII activity. Classic work by Illario et al. (18) showed inhibition of CaMKII activity by specific inhibitors inhibits Erk-1/2 phosphorylation by Ras-mediated activation of Raf-1. Accumulating evidence suggests Erk phosphorylation decreases ENaC cell surface expression by facilitating its interaction with the ubiquitin ligase Nedd4 (36).
The CaMKII-dependent activation of Erk1/2 is one mechanism for the regulation of ENaC activity. There is a large body of evidence that links ENaC to the actin cytoskeleton (19, 29, 44). CaMKII has been shown to participate in the reorganization of the actin cytoskeleton. Interestingly, Barros and Marshall (6) provided evidence for the Erk 1/2 MAP kinase pathway in the regulation of the actin cytoskeleton. This study, however, explores a different mechanism by which CaMKII can modulate ENaC activity through the regulation of the actin cytoskeleton. An intact cytoskeleton may stabilize ENaC at the membrane by multiple levels. First, it may potentiate ENaC interaction with MARCKS, which in turn promotes the interaction between ENaC and anionic phospholipids such as PIP2. We have recently shown MARCKS sequesters PIP2 and serves as a link between PIP2 and ENaC to increase channel open probability in Xenopus 2F3 cells (1). Second, the actin cytoskeleton may protect ENaC at the cell surface from clathrin-mediated endocytosis and degradation. The association between multiple cytoskeletal elements, such as MARCKS, spectrin, fodrin, actin, and filamin, may prevent binding of the ubiquitin ligase Nedd4-2 to ENaC if there is overlap between the binding sites. In this study, we investigated the ability of CaMKII to modulate the expression of several different proteins that are associated with the actin cytoskeleton. We compared different cellular fractions, including lipid rafts, cytoplasmic, and subplasma membrane by sucrose density gradient and Western blot analysis. We found that protein expression levels of MARCKS and MLP-1 between these different fractions was altered and the phosphorylation state of filamin was sensitive to CaMKII activity in Xenopus 2F3 cells. The shift in MARCKS/MLP-1 to fractions where ENaC is present in response to KN93 compared with MOCK treatment suggest endogenous CaMKII activity may alter the association between ENaC and MARCKS/MLP-1. The inhibition of endogenous CaMKII activity appears to cause a rearrangement and shift of actin cytoskeleton-associated proteins. Similarly, we observed a stronger association between ENaC subunits and MARCKS after inhibiting endogenous CaMKII activity (data not shown). These findings, in parallel with the rapid increase in ENaC activity we see after application of the CaMKII inhibitor KN93 are consistent with our hypothesis that MARCKS promotes ENaC activity when the actin cytoskeleton is intact. Additionally, the increased expression of MARCKS protein in light sucrose density gradient fractions containing lipid rafts after inhibition of CaMKII is also consistent with the observed increase in ENaC activity.
The effector domain of MARCKS and MLP-1 contains basic amino acid residues that are important for its association with the inner leaflet of the plasma membrane. We wanted to determine whether introducing negative charges within the effector domain would attenuate the association between MARCKS and the membrane. We mutated two lysines residues within the effector domain of MARCKS to glutamic acid residues, with one of these lysine residues residing within the calmodulin binding domain of MARCKS. Although the mutant MARCKS protein was able to translocate to and associate with the apical membrane of Xenopus 2F3 cells, its sensitivity to calcium-calmodulin was reduced. This indicates that introducing two negative charges is not enough to prevent MARCKS from interacting with plasma membrane and that either or both lysine 155 and lysine 164 are essential for the translocation of the protein.
We previously showed disruption of the actin cytoskeleton with cytochalasin E treatment reduces total fodrin protein expression and causes a change in its subcellular expression as demonstrated by sucrose density gradient and Western blot analysis (29). Our findings suggest disruption of the cytoskeletal complex reduces ENaC activity in these cells. In a different mechanism, Harris et al. (13) originally demonstrated fodrin proteolytic cleavage by calcium-dependent protease I is regulated by calmodulin binding. In our proposed model, either of these mechanisms that would presumably disrupt the association between ENaC and fodrin would affect ENaC activity. We did not observe any significant changes in the levels of fodrin protein expression in different sucrose density gradient fractions (data not shown) as we did for MARCKS and MLP-1. However, we did confirm a role for CaMKII in the phosphorylation of filamin in Xenopus 2F3 cells.
Here, we investigated whether proteins that link the cytoskeleton to the plasma membrane, such as filamin, are subject to regulation by CaMKII. Wang et al. (39) showed filamin directly binds ENaC and inhibits is activity. Filamin links actin filaments to membrane glycoproteins and potentiates the interaction between these proteins and various cytoplasmic signaling proteins. Bourguignon et al. (7) showed CaMKII upregulation leads to phosphorylation of filamin, and this reduces its interaction with filamentous actin to promote tumor cell migration. Nakamura et al. (24) provided the first direct evidence for the regulation of filamin by calcium-calmodulin. Biochemical studies revealed filamin is phosphorylated by CaMKII (7). Phosphorylation of filamin by CaMKII attenuates its binding to filamentous actin. Several different kinases have been shown to be able to phosphorylate filamin A and regulate its function as an actin cross-linking protein. Cukier and coworkers (10) showed cyclin B1/cyclin-dependent kinase 1 (Cdk1) binds and phosphorylates filamin A at a serine residue. The association between ENaC and filamin is interesting because this actin binding protein has been shown to link the actin cytoskeleton to β-arrestins (34). Chen et al. (9) reported influenza virus-mediated inhibition of ENaC depends on Src activation, which can be mediated by β-arrestin. This mechanism may involve the ability of Src to activate PLC, which in turn can subsequently lead to the activation of PKC. Our laboratory and others have shown PKC inhibits amiloride-sensitive sodium current.
Taken together, our results provide evidence for a calmodulin- and CaMKII-dependent mechanism of ENaC regulation in cells expressed in the distal nephron. This particular mechanism involves phosphorylation of filamin, destabilization of the actin cytoskeleton, and release of MARCKS/MLP-1 from the membrane (Fig. 9). Our work shows for the first time calmodulin and CaMKII modulate the activity of ENaC at the level of its open probability.
Fig. 9.

Proposed model depicting the role of calcium, CaM, and CaMKII on ENaC activity in Xenopus 2F3 cells. A: in the absence of calcium, filamin stabilizes the actin cytoskeleton including MARCKS/MLP-1 to allow for normal ENaC activity. B: in the presence of high calcium, the calcium-CaM complex activates CaMKII, which in turn phosphorylates filamin. The phosphorylation of filamin disrupts the association between ENaC and MARCKS/MLP-1 and causes reorganization of the actin cytoskeleton. Alternatively, calcium-CaM binds to the effector domain of MARCKS/MLP-1 and causes translocation of the protein from the membrane to the cytoplasm. Either the former or latter mechanisms result in loss of function of MARCKS/MLP-1 at the membrane and a decrease in ENaC activity.
GRANTS
This research was supported by National Institutes of Health Grants 1K01 DK099617 (to A. A. Alli), 5R01 DK067110 (to H.-P. Ma), and R37 DK037963 (to D. C. Eaton).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: A.A.A., H.-P.M., and D.C.E. provided conception and design of research; A.A.A., H.-F.B., B.-C.L., L.Y., S.A., and D.S.M. performed experiments; A.A.A., H.-F.B., B.-C.L., L.Y., S.A., and D.S.M. analyzed data; A.A.A. interpreted results of experiments; A.A.A., H.-F.B., B.-C.L., and L.Y. prepared figures; A.A.A. drafted manuscript; A.A.A. edited and revised manuscript; A.A.A., H.-F.B., B.-C.L., L.Y., S.A., D.S.M., H.-P.M., and D.C.E. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank B. J. Duke for maintaining cells in culture.
REFERENCES
- 1.Alli AA, Bao HF, Alli AA, Aldrugh Y, Song JZ, Ma HP, Yu L, Al-Khalili O, Eaton DC. Phosphatidylinositol phosphate-dependent regulation of Xenopus ENaC by MARCKS protein. Am J Physiol Renal Physiol 303: F800–F811, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alli AA, Gower WR Jr. The C type natriuretic peptide receptor tethers AHNAK1 at the plasma membrane to potentiate arachidonic acid-induced calcium mobilization. Am J Physiol Cell Physiol 297: C1157–C1167, 2009. [DOI] [PubMed] [Google Scholar]
- 3.Alli AA, Gower WR Jr. Molecular approaches to examine the phosphorylation state of the C type natriuretic peptide receptor. J Cell Biochem 110: 985–994, 2010. [DOI] [PubMed] [Google Scholar]
- 4.Alli AA, Song JZ, Al-Khalili O, Bao HF, Ma HP, Alli AA, Eaton DC. Cathepsin B is secreted apically from Xenopus 2F3 cells and cleaves the epithelial sodium channel (ENaC) to increase its activity. J Biol Chem 287: 30073–30083, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arbuzova A, Schmitz AA, Vergeres G. Cross-talk unfolded: MARCKS proteins. Biochem J 362: 1–12, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barros JC, Marshall CJ. Activation of either ERK1/2 or ERK5 MAP kinase pathways can lead to disruption of the actin cytoskeleton. J Cell Sci 118: 1663–1671, 2005. [DOI] [PubMed] [Google Scholar]
- 7.Bourguignon LY, Gilad E, Brightman A, Diedrich F, Singleton P. Hyaluronan-CD44 interaction with leukemia-associated RhoGEF and epidermal growth factor receptor promotes Rho/Ras co-activation, phospholipase C epsilon-Ca2+ signaling, and cytoskeleton modification in head and neck squamous cell carcinoma cells. J Biol Chem 281: 14026–14040, 2006. [DOI] [PubMed] [Google Scholar]
- 8.Cantiello HF, Stow JL, Prat AG, Ausiello DA. Actin filaments regulate epithelial Na+ channel activity. Am J Physiol Cell Physiol 261: C882–C888, 1991. [DOI] [PubMed] [Google Scholar]
- 9.Chen XJ, Seth S, Yue G, Kamat P, Compans RW, Guidot D, Brown LA, Eaton DC, Jain L. Influenza virus inhibits ENaC and lung fluid clearance. Am J Physiol Lung Cell Mol Physiol 287: L366–L373, 2004. [DOI] [PubMed] [Google Scholar]
- 10.Cukier IH, Li Y, Lee JM. Cyclin B1/Cdk1 binds and phosphorylates Filamin A and regulates its ability to cross-link actin. FEBS Lett 581: 1661–1672, 2007. [DOI] [PubMed] [Google Scholar]
- 11.Desrivieres S, Cooke FT, Morales-Johansson H, Parker PJ, Hall MN. Calmodulin controls organization of the actin cytoskeleton via regulation of phosphatidylinositol (4,5)-bisphosphate synthesis in Saccharomyces cerevisiae. Biochem J 366: 945–951, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo LJ, Alli AA, Eaton DC, Bao HF. ENaC is regulated by natriuretic peptide receptor-dependent cGMP signaling. Am J Physiol Renal Physiol 304: F930–F937, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harris AS, Croall DE, Morrow JS. Calmodulin regulates fodrin susceptibility to cleavage by calcium-dependent protease I. J Biol Chem 264: 17401–17408, 1989. [PubMed] [Google Scholar]
- 14.Hartwig JH, Thelen M, Rosen A, Janmey PA, Nairn AC, Aderem A. MARCKS is an actin filament crosslinking protein regulated by protein kinase C and calcium-calmodulin. Nature 356: 618–622, 1992. [DOI] [PubMed] [Google Scholar]
- 16.Hoffman L, Farley MM, Waxham MN. Calcium-calmodulin-dependent protein kinase II isoforms differentially impact the dynamics and structure of the actin cytoskeleton. Biochemistry 52: 1198–1207, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ilatovskaya DV, Pavlov TS, Levchenko V, Negulyaev YA, Staruschenko A. Cortical actin binding protein cortactin mediates ENaC activity via Arp2/3 complex. FASEB J 25: 2688–2699, 2011. [DOI] [PubMed] [Google Scholar]
- 18.Illario M, Cavallo AL, Bayer KU, Di MT, Fenzi G, Rossi G, Vitale M. Calcium/calmodulin-dependent protein kinase II binds to Raf-1 and modulates integrin-stimulated ERK activation. J Biol Chem 278: 45101–45108, 2003. [DOI] [PubMed] [Google Scholar]
- 19.Karpushev AV, Ilatovskaya DV, Pavlov TS, Negulyaev YA, Staruschenko A. Intact cytoskeleton is required for small G protein dependent activation of the epithelial Na+ channel. PLoS One 5: e8827, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim EY, Ridgway LD, Dryer SE. Interactions with filamin A stimulate surface expression of large-conductance Ca2+-activated K+ channels in the absence of direct actin binding. Mol Pharmacol 72: 622–630, 2007. [DOI] [PubMed] [Google Scholar]
- 21.Ma HP, Eaton DC. Acute regulation of epithelial sodium channel by anionic phospholipids. J Am Soc Nephrol 16: 3182–3187, 2005. [DOI] [PubMed] [Google Scholar]
- 22.Malik B, Schlanger L, Al-Khalili O, Bao HF, Yue G, Price SR, Mitch WE, Eaton DC. ENaC degradation in A6 cells by the ubiquitin-proteosome proteolytic pathway. J Biol Chem 276: 12903–12910, 2001. [DOI] [PubMed] [Google Scholar]
- 23.McLaughlin S, Murray D. Plasma membrane phosphoinositide organization by protein electrostatics. Nature 438: 605–611, 2005. [DOI] [PubMed] [Google Scholar]
- 24.Nakamura F, Hartwig JH, Stossel TP, Szymanski PT. Ca2+ and calmodulin regulate the binding of filamin A to actin filaments. J Biol Chem 280: 32426–32433, 2005. [DOI] [PubMed] [Google Scholar]
- 25.Ohta Y, Hartwig JH. Actin filament cross-linking by chicken gizzard filamin is regulated by phosphorylation in vitro. Biochemistry 34: 6745–6754, 1995. [DOI] [PubMed] [Google Scholar]
- 26.Palmer LG, Frindt G. Effects of cell Ca and pH on Na channels from rat cortical collecting tubule. Am J Physiol Renal Fluid Electrolyte Physiol 253: F333–F339, 1987. [DOI] [PubMed] [Google Scholar]
- 27.Petrecca K, Miller DM, Shrier A. Localization and enhanced current density of the Kv4.2 potassium channel by interaction with the actin-binding protein filamin. J Neurosci 20: 8736–8744, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Plant PJ, Yeger H, Staub O, Howard P, Rotin D. The C2 domain of the ubiquitin protein ligase Nedd4 mediates Ca2+-dependent plasma membrane localization. J Biol Chem 272: 32329–32336, 1997. [DOI] [PubMed] [Google Scholar]
- 29.Reifenberger MS, Yu L, Bao HF, Duke BJ, Liu BC, Ma HP, Alli AA, Eaton DC, Alli AA. Cytochalasin E alters the cytoskeleton and decreases ENaC activity in Xenopus 2F3 cells. Am J Physiol Renal Physiol 307: F86–F95, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robins GG, Sandle GI. Calcium rapidly down-regulates human renal epithelial sodium channels via a W-7-sensitive mechanism. J Membr Biol 247: 729–737, 2014. [DOI] [PubMed] [Google Scholar]
- 32.Rothschild SC, Francescatto L, Drummond IA, Tombes RM. CaMK-II is a PKD2 target that promotes pronephric kidney development and stabilizes cilia. Development 138: 3387–3397, 2011. [DOI] [PubMed] [Google Scholar]
- 33.Sampson LJ, Leyland ML, Dart C. Direct interaction between the actin-binding protein filamin-A and the inwardly rectifying potassium channel, Kir2.1. J Biol Chem 278: 41988–41997, 2003. [DOI] [PubMed] [Google Scholar]
- 34.Scott MG, Pierotti V, Storez H, Lindberg E, Thuret A, Muntaner O, Labbe-Jullie C, Pitcher JA, Marullo S. Cooperative regulation of extracellular signal-regulated kinase activation and cell shape change by filamin A and beta-arrestins. Mol Cell Biol 26: 3432–3445, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seykora JT, Myat MM, Allen LA, Ravetch JV, Aderem A. Molecular determinants of the myristoyl-electrostatic switch of MARCKS. J Biol Chem 271: 18797–18802, 1996. [DOI] [PubMed] [Google Scholar]
- 36.Shi H, Asher C, Chigaev A, Yung Y, Reuveny E, Seger R, Garty H. Interactions of beta and gamma ENaC with Nedd4 can be facilitated by an ERK-mediated phosphorylation. J Biol Chem 277: 13539–13547, 2002. [DOI] [PubMed] [Google Scholar]
- 37.Thelin WR, Chen Y, Gentzsch M, Kreda SM, Sallee JL, Scarlett CO, Borchers CH, Jacobson K, Stutts MJ, Milgram SL. Direct interaction with filamins modulates the stability and plasma membrane expression of CFTR. J Clin Invest 117: 364–374, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Timmins JM, Ozcan L, Seimon TA, Li G, Malagelada C, Backs J, Backs T, Bassel-Duby R, Olson EN, Anderson ME, Tabas I. Calcium/calmodulin-dependent protein kinase II links ER stress with Fas and mitochondrial apoptosis pathways. J Clin Invest 119: 2925–2941, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Q, Dai XQ, Li Q, Tuli J, Liang G, Li SS, Chen XZ. Filamin interacts with epithelial sodium channel and inhibits its channel function. J Biol Chem 288: 264–273, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu L, Cai H, Yue Q, Alli AA, Wang D, Al-Khalili O, Bao HF, Eaton DC. WNK4 inhibition of ENaC is independent of Nedd4-2-mediated ENaC ubiquitination. Am J Physiol Renal Physiol 305: F31–F41, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yue G, Malik B, Yue G, Eaton DC. Phosphatidylinositol 4,5-bisphosphate (PIP2) stimulates epithelial sodium channel activity in A6 cells. J Biol Chem 277: 11965–11969, 2002. [DOI] [PubMed] [Google Scholar]
- 42.Zhang M, Breitwieser GE. High affinity interaction with filamin A protects against calcium-sensing receptor degradation. J Biol Chem 280: 11140–11146, 2005. [DOI] [PubMed] [Google Scholar]
- 43.Zhou R, Patel SV, Snyder PM. Nedd4-2 catalyzes ubiquitination and degradation of cell surface ENaC. J Biol Chem 282: 20207–20212, 2007. [DOI] [PubMed] [Google Scholar]
- 44.Zuckerman JB, Chen X, Jacobs JD, Hu B, Kleyman TR, Smith PR. Association of the epithelial sodium channel with Apx and alpha-spectrin in A6 renal epithelial cells. J Biol Chem 274: 23286–23295, 1999. [DOI] [PubMed] [Google Scholar]