

Abstract
Acute kidney injury (AKI) is a common hospital complication. There are no effective treatments to minimize kidney injury or limit associated morbidity and mortality. Currently, serum creatinine and urine output remain the gold standard used clinically in the diagnosis of AKI. Several novel biomarkers can diagnose AKI earlier than elevations of serum creatinine and changes in urine output. Recent long-term observational studies have elucidated a subgroup of patients who have positive biomarkers of AKI but do not meet criteria for AKI by serum creatinine or urine output, termed subclinical AKI. These patients with subclinical AKI have increased risk of both short- and long-term mortality. In this review, we will highlight the implications of what these patients may represent and the need for better phenotyping of AKI by etiology, severity of injury, and ability to recover. We will discuss two AKI biomarkers, neutrophil gelatinase-associated lipocalin (NGAL) and breast regression protein-39 (BRP-39)/YKL-40, that exemplify the need to characterize the complexity of the biological meaning behind the biomarker, beyond elevated levels reporting on tissue injury. Ultimately, careful phenotyping of AKI will lead to identification of therapeutic targets and appropriate patient populations for clinical trials.
Keywords: subclinical injury, heterogeneity, outcomes, NGAL, chitinase 3-like 1
acute kidney injury (AKI) is a common complication that is increasing in incidence among hospitalized patients (26, 56). Epidemiological data suggest that an episode of AKI portends an increased risk of both short- and long-term mortality (12, 16). Even though the limitations of serum creatinine in accurately estimating glomerular filtration rate (GFR) and diagnosing AKI are well accepted, serum creatinine remains the gold standard used to define AKI in both clinical settings as well as in basic science research using animal models of AKI. The recent discovery of novel renal injury biomarkers has identified plasma and urinary biomarkers that can identify AKI earlier than serum creatinine and differentiate between hemodynamic changes in kidney function with structural injury.
This review will highlight the observational data demonstrating poor outcomes associated with subclinical AKI (biomarker positive, serum creatinine negative) as an example of the need to better phenotype AKI (functional vs. structural), characterize meaningful AKI (AKI associated with poor clinical outcomes), and identify pathways of injury and repair leading to potential therapeutic targets. Current AKI biomarker research is still in its infancy in predicting recovery from AKI and identification of pathways of recovery. Our ability to accurately characterize meaningful AKI will better identify at-risk patient populations for enrollment in clinical trials based on AKI phenotype as well as guide clinical management of the post-AKI patient to mitigate long-term morbidity and mortality.
Serum Creatinine as a Poor Marker for AKI
Serum creatinine and decreased urine output have long been used to define AKI. Multiple classification systems (AKIN, RIFLE, KDIGO) that incorporate these two metrics have been derived to assist in research with the hopes of improving clinical practice, although the incidence of AKI differs depending on the definition used (59). The limitations of serum creatinine as a biomarker for AKI are well accepted (Table 1). Increases in serum creatinine are delayed 48–72 h in the setting of acute injury. Serum creatinine levels can be highly variable in the setting of low muscle mass, hypercatabolism in critical illness, the dilutional effect of fluid overload, decreased production in sepsis, and drug-induced inhibition of tubular secretion (17, 20, 35, 36). Furthermore, studies in animal models have shown development of renal fibrosis and abnormal gene expression profiles in kidneys after an episode of acute ischemic kidney injury despite recovery of serum creatinine back to baseline levels (4, 5).
Table 1.
Domains of kidney disease to be assessed
| Domains of Kidney Disease to be Assessed | Serum Creatinine | Role of Biomarkers/Examples and Limitations |
|---|---|---|
| Renal reserve | Serum creatinine may not rise until >50% decrease in kidney functioning units or kidney mass. | Cut-offs for AKI biomarkers in patients with and without baseline CKD not established. |
| Use of novel CKD biomarkers such as collagen PIIINP in conjunction with AKI biomarkers not yet established in assessing AKI on CKD. | ||
| Delay in diagnosis of reduced GFR | Delayed diagnosis by 48–72 h in settings of acute injury. | Earlier diagnosis at the time of insult. |
| NGAL, KIM-1, IL-18, IGFBP7/TIMP-2 within 12 h postop or presentation with critical illness. | ||
| Phenotyping | Cannot distinguish between functional vs. structural kidney injury within etiologies of kidney disease. | Can distinguish between functional (prerenal) vs. structural kidney injury. |
| Current biomarkers (i.e., NGAL, KIM-1) unable to subtype structural AKI in settings such as ischemic vs. septic. | ||
| Specificity (location of kidney injury) | Cannot distinguish between glomerular injury or tubular injury along the nephron. | Biomarkers can help localize injury within the kidney. |
| Glomerular: Albumin | ||
| Proximal tubule: NGAL, KIM-1, IL-18, Albumin | ||
| Distal tubule: NGAL, L-FABP | ||
| Recovery | Estimations of kinetic GFR possible, currently not used clinically. | Decreasing levels of injury biomarkers may predict recovery. |
| “Recovery” biomarker yet to be determined. | ||
| Overall kinetics of biomarkers in initial injury and persistent injury vs. recovery not fully established. | ||
| Differentiation of normal repair from maladaptive healing promoting fibrosis with repair biomarkers such as NGAL and YKL-40 needs to be established. |
AKI, acute kidney injury; GFR, glomerular filtration rate; CKD, chronic kidney disease; NGAL, neutrophil gelatinase-associated lipocalin; KIM-1, kidney injury molecule-1; TIMP-2, tissue inhibitor of metalloproteinases-2; L-FABP, liver-type fatty acid-binding protein.
Novel AKI Biomarkers
The limitations of serum creatinine have fueled research in the identification of better biomarkers, such as neutrophil gelatinase-associated lipocalin (NGAL), IL-18, kidney injury molecule-1 (KIM-1), L-type fatty acid-binding protein (L-FABP), tissue inhibitor of metalloproteinases-2 (TIMP-2), and IGFBP7. These novel AKI biomarkers allow for earlier diagnosis of acute injury within 12 h of presentation of critical illness or after surgery (both cardiac and noncardiac). Another important advance is the ability of AKI biomarkers to distinguish decreases in GFR due to prerenal, hemodynamic conditions with concomitant rise in serum creatinine without tubular injury from decreases in GFR resulting from intrinsic renal tubular damage. The characterization of biomarker expression along the nephron allows for the localization of acute injury. Several recent reviews have summarized AKI-specific biomarker characteristics and performance, which will not be discussed here (2, 10, 17, 45).
Limitations of novel AKI biomarkers.
While AKI biomarkers can address diagnostic delay in AKI and differentiate between prerenal (functional) vs. structural injury, there are still domains of AKI that require further research. Three areas that require more definition include our ability to estimate renal reserve, subphenotype the etiologies of AKI, and predict recovery (outlined in Table 1). The current data have not reached the granularity needed to carefully phenotype the type of AKI by etiology and risk for long-term mortality, both of which have important implications in the development of effective therapeutic targets. For example, the pathophysiology of septic AKI vs. sterile postoperative hypoperfusion may have overlapping characteristics and biomarkers, but may have very different pathways that dictate injury and normal repair processes. These elements of AKI that remain to be defined expand the classic 2 × 2 table differentiating hemodynamic changes in kidney function from structural injury to reflect the complexity of AKI syndromes (Fig. 1).
Fig. 1.

Identification of clinically relevant structural kidney damage using biomarkers. Concurrent use of intrinsic kidney injury biomarkers and serum creatinine-based estimation of glomerular filtration rate (GFR) identifies at least 4 clinically relevant categories differentiating prerenal declines in GFR without structural injury and structural injury by biomarker positivity with or without changes in serum creatinine. Establishing acute kidney injury (AKI) biomarker signatures that can further refine and subtype AKI on multiple dimensions has the potential to better identify treatment targets and therapeutic windows. CKD, chronic kidney disease; CVD, cardiovascular disease; RAAS, renin-angiotensin-aldosterone system; SCr. serum creatinine.
Subclinical AKI
One recent observation identifies a subgroup of patients with subclinical AKI, a group that does not meet serum creatinine-based definitions of AKI, but is biomarker positive, highlighting one of the four core scenarios differentiating between changes in GFR and true structural kidney injury (Fig. 1). In a pooled analysis of single and multicenter studies of outcomes in studies reporting serum and/or urine NGAL- and serum creatinine-based definitions of AKI, patients with elevated NGAL levels without elevated serum creatinine [NGAL(+)/SCr(−); SCr, serum creatinine], falling into the category depicted in the gray box in Fig. 1, were at increased risk of needing renal replacement therapy, prolonged ICU and hospital length of stay, and more importantly, increased in-hospital mortality compared with the NGAL(−)/SCr(−) group (23). This subset of biomarker-positive, serum creatinine-negative patients is not only at risk for short-term morbidity and mortality but also for long-term adverse outcomes. A three-year follow-up of adults enrolled in the Translational Research in Biomarker Endpoints in AKI (TRIBE-AKI) cohort showed that the highest tertiles of peak urinary IL-18 and KIM-1 within 3 days after cardiac surgery in patients without clinical AKI were independently associated with increased long-term mortality (15). Similarly, renal transplant recipients who did not develop delayed graft function but had high perioperative urinary biomarker levels (NGAL and IL-18) had higher rates of poor graft outcomes at 1 yr than even those who developed delayed graft function but had below median levels of urinary biomarkers (24).
The significance of this subgroup of subclinical AKI with increased long-term mortality has not been fully characterized. Knowing the limitations of serum creatinine, the potential for false negative serum creatinine is possible. In this case, a “missed” AKI occurred, in which true intrinsic structural renal injury was not detected by serum creatinine-based definitions. This could have resulted from several causes such as adequate renal reserve which absorbed the minor changes in GFR associated with injury, dilution with intravenous fluids, or concomitant reduction in creatinine production in a critically ill patient. Alternatively, elevations in these biomarkers may reflect systemic disease and reduced host fitness without overt decreases in GFR or tubular damage. As biomarker levels are not yet established in chronic kidney disease (CKD), one could consider the possibility that low levels of elevated biomarkers could reflect underlying CKD without an acute injury or change in serum creatinine.
The observed increased risk of mortality based on injury biomarker positivity further highlights our need to differentiate functional AKI (prerenal, hemodynamic decrease in GFR, not associated with increased poor short- and long-term outcomes) from intrinsic kidney damage (associated with long-term risk of CKD and mortality) in patients with elevated levels of serum creatinine. Presenting levels of urinary NGAL in the emergency room department is superior in predicting adverse events than serum creatinine and can augment our ability to distinguish patients with prerenal AKI vs. those with intrinsic AKI (43, 44). In patients with liver cirrhosis, a patient population with a high incidence of AKI in which the use of fractional excretion rate of sodium in differential diagnosis is limited, urinary biomarkers can distinguish patients with functional AKI from those with intrinsic AKI at risk for AKI progression and death (6, 7). Physiological increases in serum creatinine associated with hemodynamic changes without actual structural damage often underlie the effects of antagonists of the renin-angiotensin-aldosterone system (RAAS). RAAS inhibitors are often discontinued in the setting of an acute increase in serum creatinine. While appropriate in certain clinical contexts (such as hypovolemic hypotension), it is unclear whether discontinuation of RAAS inhibitors is necessary in all contexts of increased serum creatinine, especially in patients who benefit from RAAS inhibition, such as patients with heart failure and diabetes mellitus. In a study of patients undergoing cardiac surgery, preoperative use of angiotensin-converting enzyme inhibitors and/or angiotensin receptor blockers was associated with an increased risk of functional AKI, as defined by increased serum creatinine (14). However, this increase in serum creatinine was not coupled with an increased risk of intrinsic AKI as defined by urinary injury biomarkers. The ability to discriminate between GFR reduction due to purely RAAS inhibition vs. that due to structural AKI will be critically important in the clinical decision-making process of holding and/or reinitiating anti-RAAS agents in patients who can benefit from both short- and long-term RAAS inhibition.
The current available clinical data provide only support for the association of long-term mortality after clinical and subclinical AKI and cannot determine causality. As proposed previously (13), the episode of AKI could have identified a group of patients who failed a “renal stress test” in the setting multiple comorbidities and were thus predestined to worse outcomes. If the associations are causal, current clinical and animal data would suggest that even after recovery of serum creatinine levels after an episode of AKI, residual injury and fibrosis within the kidney lead to advancing CKD with concomitant increased morbidity and mortality related to CKD. As our clinical goal is to attenuate the risk of morbidity and mortality in our patients, the increased risk of mortality, regardless of whether the AKI was causal, remains an important clinical outcome. The identification of a subgroup of patients with subclinical AKI but biomarker positivity further supports the notion of an underlying disease mechanism that predisposes to increased mortality.
These observations press the need to expand our current classification of AKI from a four-core scenario model (Fig. 1) to subtyping functional AKI, subclinical AKI, and clinical AKI. This expanded model will help inform future research in elucidating the underlying disease mechanisms driving injury, progressive disease, and mortality.
Predicting Recovery
Current analysis of AKI biomarkers has shown associations of higher biomarker levels with worse outcomes: need for dialysis, in-hospital mortality, and long-term mortality (15, 32, 44, 48). Higher levels of biomarkers have also been shown to predict progression of AKI in the setting of liver cirrhosis and postoperative cardiac surgery (6, 31). Current prediction tools proposed for clinical use include primarily kinetic GFR equations based on creatinine changes over unit time (11) or by urinary creatinine excretion rate (50). While both have potential to be helpful in acute clinical management, both are limited by the dependence on estimated creatinine production rates which not only vary among individuals but are often dynamically altered in the setting of acute illness.
A few studies have assessed the ability of biomarker kinetics to predict renal recovery. In most cases, lower levels and/or decreasing levels of biomarkers were predictive of renal recovery. In AKI associated with community-acquired pneumonia, lower plasma NGAL levels were associated with renal recovery (54). In critically ill patients who developed AKI and required renal replacement therapy, decreasing urinary NGAL and urinary hepatocyte growth factor (HGF) in the first 14 days was associated with increased odds of renal recovery (55). Similarly, the evaluation of TIMP-2 and IGFBP7, two G1 cell cycle arrest proteins and novel biomarkers of AKI (30), in the prediction of AKI in postoperative cardiac surgery patients, showed that a decline of urinary [TIMP-2]*[IGFBP7] levels between 4 and 24 h after surgery was most predictive of renal recovery, compared with the difference in NGAL levels (40). Recent investigation of early postoperative serum cytokine levels in cardiac surgery patients with AKI showed that high serum IL-10 levels among postoperative AKI patients with elevated urinary NGAL was associated with decreased risk of all-cause mortality after cardiac surgery, suggesting a potential role in the repair pathway of kidney injury (60).
The biological significance and meaning of biomarker levels with respect to renal recovery and effect on long-term survival has not yet been fully defined. Whether higher levels of a biomarker solely reflect severity of injury or whether the biomarker itself is pathogenic, or conversely, assisting with renal repair is not completely clear in most cases. Two biomarkers in particular, NGAL and YKL-40, are examples of biomarkers in which their roles in renal injury, repair, fibrosis, and infection are multifaceted, thus their actual concentrations in the plasma or urine and even their kinetics will be context dependent.
NGAL.
NGAL, or lipocalin 2 (LCN2), is the most widely studied biomarker for AKI. Through binding bacterial siderophores and sequestering iron, NGAL acts to inhibit bacterial growth. Thus NGAL is critically important in host defense against pathogens that require siderophores for iron acquisition and survival. NGAL is expressed by neutrophils as well as epithelial cells in the liver and kidney in response to injury, inflammation, and neoplastic transformation (52). Mouse models of intraperitoneal and urinary tract infections with Escherichia coli highlight the bacteriostatic role of NGAL in promoting bacterial clearance and survival (8, 46). However, not all bacteria require siderophores for iron acquisition, and some do not require iron for survival (42). Streptococcus pneumoniae, one of the most common respiratory pathogens, does not require siderophores for iron acquisition. In a mouse model of Pneumococcal pneumonia, NGAL-induced IL-10 formation in macrophages impairs bacterial clearance and increases mortality as Lcn2 knockout animals had less bacterial burden in the lungs, less bacteremia, and improved survival (58). In critically ill patients, pneumonia caused by gram-positive bacteria, but not gram-negative bacteria, elevated NGAL levels correlated with mortality (58).
Similarly, the role of NGAL in renoprotection and renal repair after ischemic injury also appears to be context dependent. Administration of recombinant NGAL either before, during, or after ischemia-reperfusion (I/R) injury was protective (41). NGAL expression in innate immune cells is also implicated in limiting inflammation in nephrotoxic serum nephritis as well as mediating the protective effect of IL-10 overexpression in macrophages in renal I/R injury (29). However, it is unclear why there was no difference in renal outcomes after renal I/R injury in Lcn2 knockout mice compared with wild-type mice (8). In CKD models, EGFR-dependent expression of NGAL is thought to contribute to progression of disease. Lcn2 knockout animals were protected from hyperproliferation and cyst formation in CKD models of nephron loss and polycystic disease, respectively (57). As both the nephron loss and cystic disease models have an element of chronic injury, persistent NGAL expression is likely to be representative of a chronic repair response in these models, ultimately resulting in hyperproliferation and fibrosis. Urine NGAL levels in patients with polycystic kidney disease, however, do not correlate with progression of CKD (47). Taken together, the current data would suggest that NGAL expression has temporal, disease process, and cell-type specificity as well as differences in mouse and human disease processes. These characteristics may indeed be able to explain the wide variability of performance of NGAL in human clinical studies covering a wide array of patient populations and types of AKI.
BRP-39/YKL-40.
Mouse BRP-39 and the human homologue YKL-40 (also known as chitinase 3-like-1) are chitinase-like proteins, which are evolutionarily conserved 18-glycosyl hydrolase proteins that lack enzymatic activity and cannot cleave chitin. BRP-39 and YKL-40 are produced in many cell types including neutrophils, monocytes/macrophages, synovial cells, muscle cells, smooth muscle cells, endothelial cells, tumor cells, and colonic, ductal, and airway epithelial cells (33). The role of BRP-39/YKL-40 appears to be diverse, and, like NGAL, dependent on disease context. Similar to NGAL, BRP-39 is important in host defense and repair responses. Brp39 null mice infected with S. pneumoniae developed heightened lung injury, decreased bacterial clearance, and increased mortality (18). In hypoxic lung injury, BRP-39/YKL-40 limits lung injury, inflammation, and epithelial apoptosis (53). On the other hand, BRP-39/YKL-40 mediates allergen-induced Th2 inflammation and fibroproliferative lung disease (34, 61). In the case of lung fibrosis modeled by bleomycin-treated mice, BRP-39 plays an important role in limiting lung inflammation and epithelial cell death while promoting repair early in the injury phase of the disease process (61). However, late in the repair phase, BRP-39 promotes lung fibrosis through alternative activation of macrophages, fibroblast proliferation, and matrix deposition.
BRP-39/YKL-40 was recently discovered to be a biomarker for AKI and delayed graft function in deceased donor renal transplant patients (25, 51). Studies of the Brp39 knockout mouse revealed that macrophage-derived BRP-39 was critical in limiting tubular apoptosis via activation of Akt, thereby improving survival after renal I/R injury (51). Although BRP-39/YKL-40 is an important mediator of the reparative response after ischemic kidney injury, in both AKI and renal transplantation, high levels of urinary and circulating YKL-40 were associated with AKI progression and in-hospital death as well as delayed graft function, respectively. As the animal model would suggest that BRP-39 is required for normal repair responses, higher levels of YKL-40 in patients with AKI or delayed graft function more likely reflect severity and/or persistence of injury. It remains to be determined whether there is an entity of BRP-39/YKL-40 resistance, in which high levels reflect the inability to utilize the reparative capacity of BRP-39/YKL-40.
Based on their characteristics, NGAL and BRP-39/YKL-40 are representative of evolutionarily conserved pathways of host defense, critical for mounting antimicrobial defenses in certain infections while limiting tissue injury in the setting of inflammation required for pathogen killing and promoting tissue repair after clearance of the pathogen. In both cases, prolonged expression in the setting of chronic injury promotes the development of fibrosis. As the effects of these molecular pathways are diverse, depending on the type and acuity of the injury, it is clear that biomarker signatures that can clarify the type of injury (infectious vs. sterile injury), the acuity of disease (transient vs. persistent), and the temporality of disease (early injury vs. repair phase) are needed. Ultimately, biomarker signatures that can report on the overall short- and long-term fitness of the patient will be critically important in delineating the appropriate therapeutic windows for effective treatments to maximally limit injury, promote repair, and prevent fibrosis without hindering an appropriate host defense.
Mortality After AKI
While the epidemiological association of both short- and long-term mortality with AKI has been well established, the ultimate cause of mortality is difficult to determine. Major proposed pathways mediating the increased risk of mortality after an episode of AKI are summarized in Fig. 2 and discussed below.
Fig. 2.
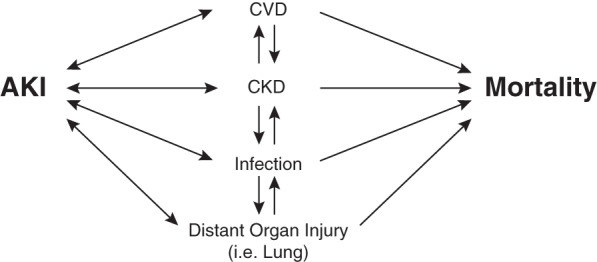
Mortality after AKI. Multiple interconnected pathways are proposed to mediate increased risk of mortality after an episode of AKI.
There is increasing appreciation of the cross talk that occurs with the kidney and other organ systems in the setting of acute illness. Distant organ injury in the setting of AKI, as well as the kidney as the distant organ injured in the setting of acute lung injury (ALI) can both contribute to the increased mortality in patients with subclinical AKI, especially short-term in-hospital mortality. Both epidemiological data and studies in animal models have suggested that AKI and ALI often occur together, one causing and/or predisposing the other (35).
A likely clinical scenario that can lead to increased long-term mortality in patients with subclinical AKI is undetected CKD based on serum creatinine estimation. Association of increased cardiovascular disease and mortality with CKD is well established (3, 22). More recently, clinical observational studies have shown an increased risk of community-acquired infections (39) and an increased risk of mortality in pneumonia (28) and septic shock (37). Animal models have confirmed that prior kidney injury and CKD models incur an increased risk of mortality after sepsis induced by sublethal cecal ligation and puncture associated with increased bacteremia, septic shock, and splenocyte apoptosis (19). As serum creatinine can normalize after AKI, despite the development of focal renal fibrosis, persistent changes in gene expression, and altered renal hemodynamics and sodium handling, (4, 5, 49), these observations of increased mortality in patients with subclinical AKI suggest that we are likely misclassifying a significant cohort of patients with true CKD despite “normalization” of serum creatinine. Ultimately, identification of biomarkers for CKD (21) will need to include patients with subclinical AKI. Questions to be considered during the identification and analysis of CKD biomarkers is biological relevance to pathways that can inform us, not only on progression of kidney disease but also on systemic effects such as cardiovascular disease, immunosuppression, and the risk of mortality.
Future Directions
It has become increasingly clear that AKI syndromes are complex and diverse. Our ability to subphenotype AKI syndromes remains limited. While the identification of novel AKI biomarkers has allowed for earlier diagnosis of AKI and differentiation between functional and structural kidney injury, we are still limited in our ability to identify effective therapeutic targets and windows. The implications of poor short- and long-term outcomes in patients with subclinical AKI highlight the need to further subphenotype AKI in several dimensions, including etiology of injury, kinetics of injury and repair, and disease determinants for poor outcomes. Future research goals include utilizing currently available data and biomarkers as well as unbiased studies to identify unique biomarkers specific to unique subtypes of AKI and the biological processes that mediate disease (summarized in Table 2).
Table 2.
Future directions
| Future Research Goals |
|---|
| • Improve characterization of subclinical AKI. |
| • Broaden the phenotyping of patients with AKI based on biological mechanisms of injury. |
| Applying Biomarker Characteristics |
| -Kinetics and reference range for severity of injury and chance of recovery/survival |
| -Reference range in CKD |
| Unbiased studies in AKI to identify unique biomarkers specific to different subtypes of AKI and respective biological pathways driving injury. |
| • Expand statistical methods |
| Combine biomarkers and/or clinical parameters with biomarkers. |
| Cluster methods for improved phenotyping. |
| Better approaches (i.e., Bayesian methods) to improve biomarker combinations. |
| Combine existing biomarker studies to harness knowledge using meta-analytic techniques. |
| • Extrapolate findings from clinical studies to precisely predict risk in an individual |
| • Develop and institute point-of-care biomarker assays for rapid turnaround time for diagnosis, clinical trial enrollment, and risk assessment for clinical decision-making. |
| • Advanced biomarkers as drug development tools through FDA biomarker qualification pathways (prognostic, predictive, surrogate, and safety biomarkers). |
| • Improve AKI animal models to incorporate comorbidities and aging. |
Key areas of future research include defining the kinetics and reference ranges for current AKI biomarkers including biomarker levels in the setting of baseline CKD. Incorporating emerging CKD biomarkers such as collagen PIIINP (27) with AKI markers may clarify CKD with or without acute injury. AKI biomarkers should be studied in combination with each other as well as clinical parameters to better define AKI subphenotypes. Examples of the power of Bayesian methods and cluster analyses in identifying subphenotypes of disease include the subclassification of patients with acute respiratory distress syndrome (ARDS) (9) and congestive heart failure (CHF) (1). The use of these methods in ARDS and CHF has identified distinct subphenotypes that have distinct prognoses and response profiles to therapy within these diseases. Application of these methods to AKI has the potential to help clarify the heterogeneity of AKI, improve our ability to select appropriate patient populations for clinical trials, and, in the future, individualize AKI diagnosis and management. Current use of AKI biomarkers remains in the realm of clinical research. Development and institution of point-of-care biomarker assays for rapid turnaround for diagnosis and risk assessment will not only enhance clinical trial enrollment but also improve clinical decision-making. AKI biomarkers should also be advanced as drug development tools through the Food and Drug Administration biomarker qualification pathway.
Current kidney injury biomarkers have identified several important biological injury response pathways in AKI. Some biomarkers, such as NGAL and YKL-40, have roles in repair as well as progressive maladaptive healing, highlighting the need to better understand mechanisms that dictate normal repair vs. fibrosis. Furthermore, our current understanding of these biomarkers is limited in our ability to differentiate the types of AKI in patients, such as ischemic, septic, and drug-induced nephrotoxicity. The identification of unique biological pathways driving injury in these specific types of AKI can enhance our ability to individualize therapeutic targets based on the cause of AKI. An unbiased approach to identify biomarkers that can differentiate subphenotypes is needed. A coordinated effort will be required to work through several barriers in AKI research, such as the lack of kidney tissue to correlate molecular signatures and histological injury as well as the lack of standardized high-throughput proteomic analyses.
Phenotyping different animal AKI models that reflect diverse modes of injury such as sepsis and ischemia may shed light on unique biological pathways that mediate injury and repair. As comorbidities have been proposed to contribute to the lack of translatability of animal models (38), the incorporation of common comorbidities such as diabetes and aging into animal models of AKI can further shed light on the spectrum of AKI disease in patients. Experimentally, it will be important to develop methods to limit the incremental variability with added variables into animal models.
Conclusion
In conclusion, currently available biomarkers have improved our ability to diagnose intrinsic kidney injury. We have yet to fully characterize biological pathways that promote kidney repair and long-term survival. Novel biomarkers NGAL and BRP-39/YKL-40 are examples of important factors that mediate cellular protection early after injury, but are likely to promote maladaptive repair and fibrosis in the setting of severe and persistent injury. Furthermore, the diverse roles they play in the inflammatory response in infection and sterile injury highlight the need to fully understand the time and contextual considerations when interpreting biomarker levels. The implications of carefully phenotyping AKI are wide-ranging, including the development of effective therapeutic strategies depending on the disease context and identification of the appropriate patient populations for enrollment in clinical trials. Clinically, identification of at-risk post-AKI patients, including those with subclinical AKI and underlying CKD without abnormal estimated GFR based on serum creatinine, will be important for establishing appropriate follow-up to improve long-term outcomes.
GRANTS
C. R. Parikh is supported by National Institutes of Health Grants R01HL085757 and K24DK090203. S. C. Huen is supported by American Heart Association Grant 13FTF17070000.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: S.C.H. and C.R.P. provided conception and design of research; S.C.H. and C.R.P. prepared figures; S.C.H. and C.R.P. drafted manuscript; S.C.H. and C.R.P. edited and revised manuscript; S.C.H. and C.R.P. approved final version of manuscript; C.R.P. performed experiments; C.R.P. interpreted results of experiments.
REFERENCES
- 1.Ahmad T, Pencina MJ, Schulte PJ, O'Brien E, Whellan DJ, Pina IL, Kitzman DW, Lee KL, O'Connor CM, Felker GM. Clinical implications of chronic heart failure phenotypes defined by cluster analysis. J Am Coll Cardiol 64: 1765–1774, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alge JL, Arthur JM. Biomarkers of AKI: a review of mechanistic relevance and potential therapeutic implications. Clin J Am Soc Nephrol 10: 147–155, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bakris G, Vassalotti J, Ritz E, Wanner C, Stergiou G, Molitch M, Nesto R, Kaysen GA, Sowers JR; CKD Consensus Working Group. National Kidney Foundation consensus conference on cardiovascular and kidney diseases and diabetes risk: an integrated therapeutic approach to reduce events. Kidney Int 78: 726–736, 2010. [DOI] [PubMed] [Google Scholar]
- 4.Basile DP, Donohoe D, Roethe K, Osborn JL. Renal ischemic injury results in permanent damage to peritubular capillaries and influences long-term function. Am J Physiol Renal Physiol 281: F887–F899, 2001. [DOI] [PubMed] [Google Scholar]
- 5.Basile DP, Fredrich K, Alausa M, Vio CP, Liang M, Rieder MR, Greene AS, Cowley AW. Identification of persistently altered gene expression in the kidney after functional recovery from ischemic acute renal failure. Am J Physiol Renal Physiol 288: F953–F963, 2005. [DOI] [PubMed] [Google Scholar]
- 6.Belcher JM, Garcia-Tsao G, Sanyal AJ, Thiessen-Philbrook H, Peixoto AJ, Perazella MA, Ansari N, Lim J, Coca SG, Parikh CR; TRIBE-AKI Consortium. Urinary biomarkers and progression of AKI in patients with cirrhosis. Clin J Am Soc Nephrol 9: 1857–1867, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belcher JM, Sanyal AJ, Peixoto AJ, Perazella MA, Lim J, Thiessen-Philbrook H, Ansari N, Coca SG, Garcia-Tsao G, Parikh CR, TRIBE-AKI Consortium. Kidney biomarkers and differential diagnosis of patients with cirrhosis and acute kidney injury. Hepatology 60: 622–632, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berger T, Togawa A, Duncan GS, Elia AJ, You-Ten A, Wakeham A, Fong HEH, Cheung CC, Mak TW. Lipocalin 2-deficient mice exhibit increased sensitivity to Escherichia coli infection but not to ischemia-reperfusion injury. Proc Natl Acad Sci USA 103: 1834–1839, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calfee CS, Delucchi K, Parsons PE, Thompson BT, Ware LB, Matthay MA. Subphenotypes in acute respiratory distress syndrome: latent class analysis of data from two randomised controlled trials. Lancet Respir Med 2: 611–620, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Charlton JR, Portilla D, Okusa MD. A basic science view of acute kidney injury biomarkers. Nephrol Dial Transplant 29: 1301–1311, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen S. Retooling the creatinine clearance equation to estimate kinetic GFR when the plasma creatinine is changing acutely. J Am Soc Nephrol 24: 877–888, 2013. [DOI] [PubMed] [Google Scholar]
- 12.Chertow GM, Burdick E, Honour M, Bonventre JV, Bates DW. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J Am Soc Nephrol 16: 3365–3370, 2005. [DOI] [PubMed] [Google Scholar]
- 13.Coca SG. Long-term outcomes of acute kidney injury. Curr Opin Nephrol Hypertens 19: 266–272, 2010. [DOI] [PubMed] [Google Scholar]
- 14.Coca SG, Garg AX, Swaminathan M, Garwood S, Hong K, Thiessen-Philbrook H, Passik C, Koyner JL, Parikh CR; TRIBE-AKI Consortium. Preoperative angiotensin-converting enzyme inhibitors and angiotensin receptor blocker use and acute kidney injury in patients undergoing cardiac surgery. Nephrol Dialysis Transplant 28: 2787–2799, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coca SG, Garg AX, Thiessen-Philbrook H, Koyner JL, Patel UD, Krumholz HM, Shlipak MG, Parikh CR, TRIBE-AKI Consortium. Urinary biomarkers of AKI and mortality 3 years after cardiac surgery. J Am Soc Nephrol 25: 1063–1071, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coca SG, Yusuf B, Shlipak MG, Garg AX, Parikh CR. Long-term risk of mortality and other adverse outcomes after acute kidney injury: a systematic review and meta-analysis. Am J Kidney Dis 53: 961–973, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Geus HRH, Betjes MG, Bakker J. Biomarkers for the prediction of acute kidney injury: a narrative review on current status and future challenges. Clin Kidney J 5: 102–108, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dela Cruz CS, Liu W, He CH, Jacoby A, Gornitzky A, Ma B, Flavell R, Lee CG, Elias JA. Chitinase 3-like-1 promotes Streptococcus pneumoniae killing and augments host tolerance to lung antibacterial responses. Cell Host Microbe 12: 34–46, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doi K, Leelahavanichkul A, Hu X, Sidransky KL, Zhou H, Qin Y, Eisner C, Schnermann J, Yuen PST, Star RA. Pre-existing renal disease promotes sepsis-induced acute kidney injury and worsens outcome. Kidney Int 74: 1017–1025, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doi K, Yuen PST, Eisner C, Hu X, Leelahavanichkul A, Schnermann J, Star RA. Reduced production of creatinine limits its use as marker of kidney injury in sepsis. J Am Soc Nephrol 20: 1217–1221, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fassett RG, Venuthurupalli SK, Gobe GC, Coombes JS, Cooper MA, Hoy WE. Biomarkers in chronic kidney disease: a review. Kidney Int 80: 806–821, 2011. [DOI] [PubMed] [Google Scholar]
- 22.Go AS, Chertow GM, Fan D, McCulloch CE, Hsu Cy. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med 351: 1296–1305, 2004. [DOI] [PubMed] [Google Scholar]
- 23.Haase M, Devarajan P, Haase-Fielitz A, Bellomo R, Cruz DN, Wagener G, Krawczeski CD, Koyner JL, Murray P, Zappitelli M, Goldstein SL, Makris K, Ronco C, Martensson J, Martling CR, Venge P, Siew E, Ware LB, Ikizler TA, Mertens PR. The outcome of neutrophil gelatinase-associated lipocalin-positive subclinical acute kidney injury: a multicenter pooled analysis of prospective studies. J Am Coll Cardiol 57: 1752–1761, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hall IE, Doshi MD, Reese PP, Marcus RJ, Thiessen-Philbrook H, Parikh CR. Association between peritransplant kidney injury biomarkers and 1-year allograft outcomes. Clin J Am Soc Nephrol 7: 1224–1233, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hall IE, Stern EP, Cantley LG, Elias JA, Parikh CR. Urine YKL-40 is associated with progressive acute kidney injury or death in hospitalized patients. BMC Nephrol 15: 133, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsu RK, McCulloch CE, Dudley RA, Lo LJ, Hsu Cy. Temporal changes in incidence of dialysis-requiring AKI. J Am Soc Nephrol 24: 37–42, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ix JH, Biggs ML, Mukamal K, Djousse L, Siscovick D, Tracy R, Katz R, Delaney JA, Chaves P, Rifkin DE, Hughes-Austin JM, Garimella PS, Sarnak MJ, Shlipak MG, Kizer JR. Urine collagen fragments and CKD progression—The Cardiovascular Health Study. J Am Soc Nephrol [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.James MT, Quan H, Tonelli M, Manns BJ, Faris P, Laupland KB, Hemmelgarn BR, Acute Kidney Disease Network. CKD and risk of hospitalization and death with pneumonia. Am J Kidney Dis 54: 24–32, 2009. [DOI] [PubMed] [Google Scholar]
- 29.Jung M, Sola A, Hughes J, Kluth DC, Vinuesa E, Viñas JL, Pérez-Ladaga A, Hotter G. Infusion of IL-10-expressing cells protects against renal ischemia through induction of lipocalin-2. Kidney Int 81: 969–982, 2012. [DOI] [PubMed] [Google Scholar]
- 30.Kashani K, Al-Khafaji A, Ardiles T, Artigas A, Bagshaw SM, Bell M, Bihorac A, Birkhahn R, Cely CM, Chawla LS, Davison DL, Feldkamp T, Forni LG, Gong MN, Gunnerson KJ, Haase M, Hackett J, Honore PM, Hoste EA, Joannes-Boyau O, Joannidis M, Kim P, Koyner JL, Laskowitz DT, Lissauer ME, Marx G, McCullough PA, Mullaney S, Ostermann M, Rimmelé T, Shapiro NI, Shaw AD, Shi J, Sprague AM, Vincent JL, Vinsonneau C, Wagner L, Walker MG, Wilkerson RG, Zacharowski K, Kellum JA. Discovery and validation of cell cycle arrest biomarkers in human acute kidney injury. Crit Care 17: R25, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koyner JL, Garg AX, Coca SG, Sint K, Thiessen-Philbrook H, Patel UD, Shlipak MG, Parikh CR; TRIBE-AKI Consortium. Biomarkers predict progression of acute kidney injury after cardiac surgery. J Am Soc Nephrol 23: 905–914, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koyner JL, Shaw AD, Chawla LS, Hoste EA, Bihorac A, Kashani K, Haase M, Shi J, Kellum JA; Sapphire Investigators. Tissue inhibitor metalloproteinase-2 (TIMP-2) IGF-binding protein-7 (IGFBP7) levels are associated with adverse long-term outcomes in patients with AKI. J Am Soc Nephrol 26: 1747–1754, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee CG, Da Silva CA, Dela Cruz CS, Ahangari F, Ma B, Kang MJ, He CH, Takyar S, Elias JA. Role of chitin and chitinase/chitinase-like proteins in inflammation, tissue remodeling, and injury. Annu Rev Physiol 73: 479–501, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee CG, Hartl D, Lee GR, Koller B, Matsuura H, Da Silva CA, Sohn MH, Cohn L, Homer RJ, Kozhich AA, Humbles A, Kearley J, Coyle A, Chupp G, Reed J, Flavell RA, Elias JA. Role of breast regression protein 39 (BRP-39)/chitinase 3-like-1 in Th2 and IL-13-induced tissue responses and apoptosis. J Exp Med 206: 1149–1166, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu KD, Thompson BT, Ancukiewicz M, Steingrub JS, Douglas IS, Matthay MA, Wright P, Peterson MW, Rock P, Hyzy RC, Anzueto A, Truwit JD; National Institutes of Health National Heart Lung, and Blood Institute Acute Respiratory Distress Syndrome Network. Acute kidney injury in patients with acute lung injury: impact of fluid accumulation on classification of acute kidney injury and associated outcomes. Crit Care Med 39: 2665–2671, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Macedo E, Bouchard J, Soroko SH, Chertow GM, Himmelfarb J, Ikizler TA, Paganini EP, Mehta RL; Program to Improve Care in Acute Renal Disease Study. Fluid accumulation, recognition and staging of acute kidney injury in critically-ill patients. Crit Care 14: R82, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maizel J, Deransy R, Dehedin B, Secq E, Zogheib E, Lewandowski E, Tribouilloy C, Massy ZA, Choukroun G, Slama M. Impact of non-dialysis chronic kidney disease on survival in patients with septic shock. BMC Nephrol 14: 77, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCafferty K, Forbes S, Thiemermann C, Yaqoob MM. The challenge of translating ischemic conditioning from animal models to humans: the role of comorbidities. Dis Model Mech 7: 1321–1333, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McDonald HI, Thomas SL, Nitsch D. Chronic kidney disease as a risk factor for acute community-acquired infections in high-income countries: a systematic review. BMJ Open 4: e004100, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meersch M, Schmidt C, Van Aken H, Martens S, Rossaint J, Singbartl K, Görlich D, Kellum JA, Zarbock A. Urinary TIMP-2 and IGFBP7 as early biomarkers of acute kidney injury and renal recovery following cardiac surgery. PLoS One 9: e93460, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mishra J, Mori K, Ma Q, Kelly C, Yang J, Mitsnefes M, Barasch J, Devarajan P. Amelioration of ischemic acute renal injury by neutrophil gelatinase-associated lipocalin. J Am Soc Nephrol 15: 3073–3082, 2004. [DOI] [PubMed] [Google Scholar]
- 42.Neilands JB. Siderophores: structure and function of microbial iron transport compounds. J Biol Chem 270: 26723–26726, 1995. [DOI] [PubMed] [Google Scholar]
- 43.Nickolas TL, O'Rourke MJ, Yang J, Sise ME, Canetta PA, Barasch N, Buchen C, Khan F, Mori K, Giglio J, Devarajan P, Barasch J. Sensitivity and specificity of a single emergency department measurement of urinary neutrophil gelatinase-associated lipocalin for diagnosing acute kidney injury. Ann Intern Med 148: 810–819, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nickolas TL, Schmidt-Ott KM, Canetta P, Forster C, Singer E, Sise M, Elger A, Maarouf O, Sola-Del Valle DA, O'Rourke M, Sherman E, Lee P, Geara A, Imus P, Guddati A, Polland A, Rahman W, Elitok S, Malik N, Giglio J, El-Sayegh S, Devarajan P, Hebbar S, Saggi SJ, Hahn B, Kettritz R, Luft FC, Barasch J. Diagnostic and prognostic stratification in the emergency department using urinary biomarkers of nephron damage: a multicenter prospective cohort study. J Am Coll Cardiol 59: 246–255, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ostermann M, Philips BJ, Forni LG. Clinical review: biomarkers of acute kidney injury: where are we now? Crit Care 16: 233, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Paragas N, Kulkarni R, Werth M, Schmidt-Ott KM, Forster C, Deng R, Zhang Q, Singer E, Klose AD, Shen TH, Francis KP, Ray S, Vijayakumar S, Seward S, Bovino ME, Xu K, Takabe Y, Amaral FE, Mohan S, Wax R, Corbin K, Sanna-Cherchi S, Mori K, Johnson L, Nickolas T, D'Agati V, Lin CS, Qiu A, Al-Awqati Q, Ratner AJ, Barasch J. α-Intercalated cells defend the urinary system from bacterial infection. J Clin Invest 124: 2963–2976, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parikh CR, Dahl NK, Chapman AB, Bost JE, Edelstein CL, Comer DM, Zeltner R, Tian X, Grantham JJ, Somlo S. Evaluation of urine biomarkers of kidney injury in polycystic kidney disease. Kidney Int 81: 784–790, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parikh CR, Devarajan P, Zappitelli M, Sint K, Thiessen-Philbrook H, Li S, Kim RW, Koyner JL, Coca SG, Edelstein CL, Shlipak MG, Garg AX, Krawczeski CD; TRIBE-AKI Consortium. Postoperative biomarkers predict acute kidney injury and poor outcomes after adult cardiac surgery. J Am Soc Nephrol 22: 1748–1757, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pechman KR, De Miguel C, Lund H, Leonard EC, Basile DP, Mattson DL. Recovery from renal ischemia-reperfusion injury is associated with altered renal hemodynamics, blunted pressure natriuresis, and sodium-sensitive hypertension. Am J Physiol Regul Integr Comp Physiol 297: R1358–R1363, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pickering JW, Mellas J. A simple method to detect recovery of glomerular filtration rate following acute kidney injury. Biomed Res Int 2014: 542069, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schmidt IM, Hall IE, Kale S, Lee S, He CH, Lee Y, Chupp GL, Moeckel GW, Lee CG, Elias JA, Parikh CR, Cantley LG. Chitinase-like protein Brp-39/YKL-40 modulates the renal response to ischemic injury and predicts delayed allograft function. J Am Soc Nephrol 24: 309–319, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Singer E, Marko L, Paragas N, Barasch J, Dragun D, Müller DN, Budde K, Schmidt-Ott KM. Neutrophil gelatinase-associated lipocalin: pathophysiology and clinical applications. Acta Physiol (Oxf) 207: 663–672, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sohn MH, Kang MJ, Matsuura H, Bhandari V, Chen NY, Lee CG, Elias JA. The chitinase-like proteins breast regression protein-39 and YKL-40 regulate hyperoxia-induced acute lung injury. Am J Respir Crit Care Med 182: 918–928, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Srisawat N, Murugan R, Lee M, Kong L, Carter M, Angus DC, Kellum JA; Genetic and Inflammatory Markers of Sepsis (GenIMS) Investigators. Plasma neutrophil gelatinase-associated lipocalin predicts recovery from acute kidney injury following community-acquired pneumonia. Kidney Int 80: 545–552, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Srisawat N, Wen X, Lee M, Kong L, Elder M, Carter M, Unruh M, Finkel K, Vijayan A, Ramkumar M, Paganini E, Singbartl K, Palevsky PM, Kellum JA. Urinary biomarkers and renal recovery in critically ill patients with renal support. Clin J Am Soc Nephrol 6: 1815–1823, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Susantitaphong P, Cruz DN, Cerda J, Abulfaraj M, Alqahtani F, Koulouridis I, Jaber BL; Acute Kiidney Injury Advisory Group of the American Society of Nephrology. World incidence of AKI: a meta-analysis. Clin J Am Soc Nephrol 8: 1482–1493, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Viau A, El Karoui K, Laouari D, Burtin M, Nguyen C, Mori K, Pillebout E, Berger T, Mak TW, Knebelmann B, Friedlander G, Barasch J, Terzi F. Lipocalin 2 is essential for chronic kidney disease progression in mice and humans. J Clin Invest 120: 4065–4076, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Warszawska JM, Gawish R, Sharif O, Sigel S, Doninger B, Lakovits K, Mesteri I, Nairz M, Boon L, Spiel A, Fuhrmann V, Strobl B, Müller M, Schenk P, Weiss G, Knapp S. Lipocalin 2 deactivates macrophages and worsens pneumococcal pneumonia outcomes. J Clin Invest 123: 3363–3372, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zeng X, McMahon GM, Brunelli SM, Bates DW, Waikar SS. Incidence, outcomes, and comparisons across definitions of AKI in hospitalized individuals. Clin J Am Soc Nephrol 9: 12–20, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang WR, Garg AX, Coca SG, Devereaux PJ, Eikelboom J, Kavsak P, McArthur E, Thiessen-Philbrook H, Shortt C, Shlipak M, Whitlock R, Parikh CR. Plasma IL-6 and IL-10 concentrations predict AKI and long-term mortality in adults after cardiac surgery. J Am Soc Nephrol [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou Y, Peng H, Sun H, Peng X, Tang C, Gan Y, Chen X, Mathur A, Hu B, Slade MD, Montgomery RR, Shaw AC, Homer RJ, White ES, Lee CM, Moore MW, Gulati M, Geun Lee C, Elias JA, Herzog EL. Chitinase 3-like 1 suppresses injury and promotes fibroproliferative responses in mammalian lung fibrosis. Sci Transl Med 6: 240–ra276., 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]