

Abstract
Dysfunction of the type 1 cholecystokinin (CCK) receptor (CCK1R) as a result of increased gallbladder muscularis membrane cholesterol has been implicated in the pathogenesis of cholesterol gallstones. Administration of ursodeoxycholic acid, which is structurally related to cholesterol, has been shown to have beneficial effects on gallstone formation. Our aims were to explore the possible direct effects and mechanism of action of bile acids on CCK receptor function. We studied the effects of structurally related hydrophobic chenodeoxycholic acid and hydrophilic ursodeoxycholic acid in vitro on CCK receptor function in the setting of normal and elevated membrane cholesterol. We also examined their effects on a cholesterol-insensitive CCK1R mutant (Y140A) disrupting a key site of cholesterol action. The results show that, similar to the impact of cholesterol on CCK receptors, bile acid effects were limited to CCK1R, with no effects on CCK2R. Chenodeoxycholic acid had a negative impact on CCK1R function, while ursodeoxycholic acid had no effect on CCK1R function in normal membranes but was protective against the negative impact of elevated cholesterol on this receptor. The cholesterol-insensitive CCK1R mutant Y140A was resistant to effects of both bile acids. These data suggest that bile acids compete with the action of cholesterol on CCK1R, probably by interacting at the same site, although the conformational impact of each bile acid appears to be different, with ursodeoxycholic acid capable of correcting the abnormal conformation of CCK1R in a high-cholesterol environment. This mechanism may contribute to the beneficial effect of ursodeoxycholic acid in reducing cholesterol gallstone formation.
Keywords: cholecystokinin, gallstone disease, bile acids, cholesterol
cholesterol gallstones have long been recognized as the most common type of gallstones in Western populations (20). Formation of cholesterol gallstones has generally been attributed to lithogenic bile with elevated concentrations of cholesterol (14, 27, 31). Increased biliary cholesterol has also been observed in obesity (19, 39), and as a result of the increased global incidence of obesity (34), the incidence of cholesterol gallstone disease has also increased (45). Gallbladder stasis has also been implicated in the formation of gallstones, which, in the setting of lithogenic bile, is particularly problematic (36, 41).
The G protein-coupled type 1 cholecystokinin (CCK) receptor (CCK1R) is expressed on myocytes within the gallbladder muscularis and is the major physiological mediator of gallbladder contraction and emptying after a meal (26, 30). It is particularly interesting that this receptor has associations with obesity, cholesterol, and gallstone disease. The obesity connection relates to CCK being a satiety factor, with the vagal afferent CCK1R contributing to the physiological postcibal satiety response (29). The cholesterol connection relates to unique sensitivity of this receptor to membrane cholesterol (13, 23, 37), with elevated levels of this lipid, as have been observed in obese patients with metabolic syndrome (11, 35, 42), resulting in reduced biological responses to CCK (23, 48, 49, 52), whereas the closely related CCK2R is insensitive to cholesterol (37). The gallstone connection has largely been recognized in highly unusual kindreds, in which this receptor was found to be misprocessed and defective (32). Of note, the vast majority of patients with gallstones, including those with cholesterol gallstones, have normal CCK1R and CCK (33, 50).
In an attempt to better understand the possible role of CCK1R in cholesterol gallstone disease, we have extended the earlier work focused on the effect of cholesterol in which CCK responsiveness had been demonstrated to be abnormal (3, 46, 48), with the defect localized to the interface between CCK1R and its G (Gq) protein (48, 52). This had been demonstrated in animal models (51) and in patients with cholesterol gallstone disease (48, 49, 52). We showed a direct impact of cholesterol on the CCK1R, likely acting at a consensus site for cholesterol association with membrane proteins (12, 37).
Another fascinating set of observations includes the beneficial effect of the hydrophilic bile acid ursodeoxycholic acid on cholesterol gallstone formation (18, 47). Feeding this bile acid to enhance its presence in the bile acid pool has been reported to result in reduced formation of gallstones in animal models (47). There has also been a beneficial effect in patients treated with this bile acid (18). A series of effects of the ursodeoxycholic acid to reduce biliary cholesterol and, thereby, the lithogenicity of the bile have been described (40, 44), but it is not clear that these are the only effects contributing to this beneficial effect or how they contribute quantitatively to these clinical outcomes.
In the current study we have investigated the effect of bile acids on CCK receptor function to determine whether another mechanism for the beneficial effect of ursodeoxycholic acid might also be active. We postulated that the structural similarity between the bile acids and cholesterol could suggest that the bile acids might be working directly on the CCK1R at a site similar to the site at which cholesterol acts. The insights and tools previously developed to study the effects of cholesterol on the CCK1R have provided opportunities for such an investigation (12, 23, 37).
MATERIALS AND METHODS
Materials.
Synthetic CCK-(26–33) (CCK8) was purchased from Bachem (Torrance, CA). The benzodiazepine CCK1R antagonist BDZ-1 was provided by Professor Phillip Portoghese (1), the CCK1R antagonist T-0632 was prepared as we described previously (16), and the 1,5-benzodiazepine agonist GI181771X (2) was provided by GlaxoSmithKline Research Laboratories (Research Triangle Park, NC). Tauroursodeoxycholic acid (TUDC), taurochenodeoxycholic acid (TCDC), and 25-hydroxycholesterol were purchased from Sigma-Aldrich (St. Louis, MO), Quest fluo 8-AM from AAT Bioquest (Sunnyvale, CA), soybean trypsin inhibitor from Gibco Life Technologies (Grand Island, NY), bovine serum albumin from Equitech-Bio (Kerrville, TX), and lipoprotein-deficient serum from Intracel Resources (Frederick, MD). All other reagents were analytical grade.
Cell lines.
Chinese hamster ovary (CHO) cell lines expressing the CCK1R (CHO-CCK1R) and the type 2 CCK receptor (CHO-CCK2R) (7, 12), as well as the Y140A (CHO-CCK1R-Y140A) (12, 37), W166A (CHO-CCK1R-W166A) (37), and Y237A (CHO-CCK1R-Y237A) (37) mutants of the CCK1R, were used as sources of receptor for the current study. Cells were cultured at 37°C in an environment containing 5% CO2 on tissue culture plastic ware in Ham's F-12 medium (Gibco Life Technologies) supplemented with 5% FetalClone II (Hyclone, Logan, UT). An SRD15 cell line derived from CHO cells that expresses the CCK1R (SRD15-CCK1R) and was previously established and characterized (22) was cultured in similar conditions, except DMEM/F-12 medium (Gibco Life Technologies) was supplemented with 5% lipoprotein-deficient serum and 2.5 μM 25-hydroxycholesterol. Cells were passaged approximately twice a week.
Receptor-binding assays.
A radioligand competition-binding assay using whole cell binding in 24-well tissue culture plates was performed to determine the binding affinity of the natural agonist ligand CCK. This was applied to each of the cell lines in the absence and presence of bile acids. CHO-CCK1R, CHO-CCK1R-Y140A, CHO-CCK2R, or SRD15-CCK1R cells were plated and grown to ∼80% confluence prior to the assay. After they were washed once with PBS (pH 7.0), the cells were incubated for 15 min at 37°C in the absence or presence of TUDC and TCDC in Krebs-Ringer-HEPES (KRH) medium (25 mM HEPES, pH 7.4, 104 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM KH2PO4, and 1.2 mM MgSO4) containing 0.01% soybean trypsin inhibitor and 0.2% bovine serum albumin. A constant amount of the CCK-like radioligand 125I-d-Tyr-Gly-[(Nle28,31)CCK-(26–33)] (∼10 pM), prepared as described previously (38), and increasing concentrations (0–1 μM) of CCK were added to each well. After incubation for 1 h, the cells were washed twice with ice-cold KRH medium containing 0.01% soybean trypsin inhibitor and 0.2% bovine serum albumin to separate free from cell-bound radioligand before the cells were lysed with 0.5 M NaOH. Membrane-bound radioactivity was quantified with a γ-spectrometer. Nonspecific binding was determined in the presence of 1 μM CCK and represented <10% of total binding. The whole cell binding assay was also used for CCK1R antagonist T-0632 binding using 125I-T-0632, as we described previously (16).
The binding affinity of the small-molecule CCK1R antagonist BDZ-1 was determined in a standard radioligand competition-binding assay using receptor-bearing membranes prepared as described previously (7). Briefly, 4 μg of membranes were incubated with a constant amount of the radioligand 125I-BDZ-1 (∼28 pM) at room temperature for 1 h in 100 μl of KRH medium containing 0.2% bovine serum albumin and 0.01% soybean trypsin inhibitor in the absence or presence of increasing concentrations of nonradiolabeled BDZ-1. Membrane-bound and free radioligand were separated by filtration through a UniFilter-96 GF/B filter plate in a FilterMate harvester (PerkinElmer Life Sciences, Waltham, MA), as we described previously (7). Radioactivity in the plate was quantified using a TopCount NXT counter (PerkinElmer Life Sciences).
Intracellular calcium assays.
Effects of the bile acids on CCK biological activity were determined by measurement of the CCK-induced intracellular calcium responses in CCK receptor-bearing cell lines (12). Briefly, the cells were seeded in a 96-well black-walled clear-bottom plate and cultured for 24 h until they reached ∼80% confluence. The cells were washed once with KRH medium containing 1.2 mM MgCl2, 0.2% bovine serum albumin, and 2.5 mM probenecid and incubated with 0.75 μM fluo 8-AM at 37°C in the absence or presence of a constant amount of TCDC or TUDC. After incubation for 1 h, the cells were washed once with the KRH medium, and the calcium response assay was initiated by robotic addition of increasing concentrations (0–10−8 M) of agonist using a Flexstation 3 plate reader (Molecular Devices, Sunnyvale, CA) equipped with SoftMax Pro 5.4 software. Intracellular calcium responses were measured at 37°C by quantification of the fluorescence emission intensity at 525 nm after excitation of the samples at 485 nm, with data collection every 4 s over a 120-s period.
Data analysis.
All assays were performed in duplicate in a minimum of three independent experiments and are expressed as means ± SE. Radioligand binding and calcium concentration-response curves were analyzed and plotted using the nonlinear regression analysis program in the Prism software suite version 6 (GraphPad Software, San Diego, CA). Two-tailed t-tests were performed to evaluate the differences, and P < 0.05 was considered to be significant.
RESULTS
Effects of bile acids on CCK1R and CCK2R expressed in a normal membrane cholesterol environment.
The bile acids hydrophilic ursodeoxycholic acid and hydrophobic chenodeoxycholic acid are structurally similar to cholesterol, with differences only in positions 5–6, 7, and 24 (Fig. 1). The only difference between these two bile acids is orientation of the hydroxyl group in position 7: β-orientation in ursodeoxycholic acid and α-orientation in chenodeoxycholic acid (Fig. 1). Utilizing their taurine conjugates TUDC and TCDC to achieve better aqueous solubility than the unconjugated bile acids, we examined the impact of these bile acids on CCK binding and biological activity (Fig. 2). The binding data are displayed as percentage of saturable binding, as well as percentage of control levels of binding in the absence of the bile acids, since TCDC reduced the level of CCK binding. Concentration-response curves for TCDC to reduce CCK binding are also illustrated for each cell line (Fig. 2, insets). These data support the choice of 0.5 mM TCDC, which reduced the CCK binding to CCK1R by ∼50%, with enough saturable radioligand binding remaining to generate a meaningful CCK concentration-competition curve. TUDC was utilized at 1 mM, since, even at this high concentration, there was no reduction in CCK binding.
Fig. 1.

Chemical structures of cholesterol and the bile acids ursodeoxycholic (UDC) acid and chenodeoxycholic (CDC) acid. Structural similarity of UDC and CDC to cholesterol and differences between UDC and CDC and cholesterol are highlighted in gray circles. Taurine conjugates of UDC (TUDC) and CDC (TCDC) were used to enhance aqueous solubility.
Fig. 2.

Effects of bile acids on natural ligand binding to and biological activity at the CCK receptors CCK1R and CCK2R in a normal membrane environment. A, B, D, and E: CCK competition-binding curves for Chinese hamster ovary (CHO)-CCK1R (A and B) and CHO-CCK2R (D and E) cells in the absence (control) and presence of 1 mM TUDC and 0.5 mM TCDC. Insets: effects of increasing concentration of TCDC on specific CCK binding at the CCK1R (B) and CCK2R (E). Values (means ± SE of ≥3 independent experiments performed in duplicate) represent percentages of saturable binding for each curve (A and D) or percentage of control (B and E). C and F: typical concentration-dependent curves for intracellular calcium responses to CCK in CHO cells stably expressing CCK1R (C) and CCK2R (F) in the absence and presence of bile acids. Values (means ± SE of ≥8 independent experiments performed in duplicate) were normalized to maximal response to 0.1 mM ATP.
The hydrophilic bile acid TUDC at concentrations as high as 1 mM did not affect CCK binding or signaling at CCK1R expressed in CHO cells (Fig. 2, A–C). In contrast, the hydrophobic bile acid TCDC caused a concentration-dependent decrease in saturable binding of the CCK-like radioligand, with a ∼50% decrease in saturable binding at 0.5 mM (Fig. 2B, inset), along with a 3.5-fold rightward shift in the binding affinity at this concentration (Fig. 2A, Table 1). The decrease in binding affinity of CCK in the presence of 0.5 mM TCDC was accompanied by a significant 12-fold reduction in the potency of CCK-stimulated intracellular calcium responses (Fig. 2C, Table 1). These observations are different from the effects of excess cholesterol, which increases CCK binding affinity while reducing the potency of CCK-stimulated intracellular calcium responses (12, 22, 23, 37). It is interesting that CCK2R was significantly less sensitive to TCDC, exhibiting only a ∼20% decrease in specific binding of CCK under the same conditions, without a significant change in CCK binding affinity or potency (Fig. 2, D–F, Table 1). Similar to our findings at the CCK1R, TUDC had no effect on either CCK binding or signaling at this structurally related G protein-coupled receptor (Fig. 2, D–F, Table 1).
Table 1.
Impact of bile acids on ligand binding and biological activity
| Binding |
Biological Activity |
|||
|---|---|---|---|---|
| pIC50 | Specific binding, %control | pEC50 | Emax, % | |
| Effects on CCK on wild-type CCK receptors expressed in normal membrane or elevated cholesterol | ||||
| CCK1R | ||||
| Control | 8.62 ± 0.11 | 100 | 10.94 ± 0.07 | 70 ± 4 |
| TCDC, 0.5 mM | 7.90 ± 0.08** | 50 ± 4** | 10.14 ± 0.13** | 60 ± 6 |
| TUDC, 1 mM | 8.67 ± 0.10 | 101 ± 5 | 10.92 ± 0.17 | 80 ± 3 |
| CCK2R | ||||
| Control | 9.12 ± 0.10 | 100 | 9.54 ± 0.06 | 53 ± 5 |
| TCDC, 0.5 mM | 9.55 ± 0.28 | 85 ± 11 | 9.42 ± 0.07 | 43 ± 3 |
| TUDC, 1 mM | 8.70 ± 0.13 | 106 ± 3 | 9.40 ± 0.11 | 64 ± 6 |
| SRD15-CCK1R | ||||
| Control | 9.12 ± 0.19 | 100 | 9.59 ± 0.16 | 92 ± 4 |
| TCDC, 0.5 mM | 9.15 ± 0.23 | 104 ± 1 | 9.60 ± 0.35 | 70 ± 4** |
| TUDC, 0.1 mM | 10.69 ± 0.19** | 82 ± 8 | ||
| TUDC, 1 mM | 8.77 ± 0.08 | 97 ± 5 | 10.99 ± 0.18** | 79 ± 6 |
| Effects on allosteric ligand binding and action on wild-type CCK1R in normal membrane | ||||
| BDZ-1 | ||||
| Control | 8.82 ± 0.14 | 100 | ||
| TCDC, 0.5 mM | 9.03 ± 0.21 | 55 ± 5** | ||
| TUDC, 1 mM | 8.67 ± 0.06 | 96 ± 4 | ||
| T-0632 | ||||
| Control | 9.43 ± 0.02 | 100 | ||
| TCDC, 0.5 mM | 9.60 ± 0.06 | 66 ± 6** | ||
| TUDC, 1 mM | 9.50 ± 0.02 | 100 ± 5 | ||
| GI181771X | ||||
| Control | 9.43 ± 0.12 | 47 ± 5 | ||
| TCDC, 0.5 mM | 8.29 ± 0.10** | 31 ± 3* | ||
| TUDC, 1 mM | 9.27 ± 0.29 | 41 ± 3 | ||
| Effects on CCK binding and action on mutant CCK1R constructs in normal membrane | ||||
| CCK1R-Y140A | ||||
| Control | 9.68 ± 0.26 | 100 | 9.08 ± 0.08 | 71 ± 3 |
| TCDC, 0.5 mM | 9.78 ± 0.48 | 63 ± 7 | 8.88 ± 0.10 | 55 ± 9* |
| TUDC, 1 mM | 9.40 ± 0.05 | 104 ± 5 | 8.99 ± 0.16 | 64 ± 7 |
| CCK1R-Y237A | ||||
| Control | 7.78 ± 0.14 | 100 | 10.64 ± 0.20 | 79 ± 3 |
| TCDC, 0.5 mM | 7.06 ± 0.17* | 55 ± 6 | 9.45 ± 0.28** | 79 ± 9 |
| TUDC, 1 mM | ||||
| CCK1R-W166A | ||||
| Control | NDB | NDB | 9.01 ± 0.10 | 58 ± 6 |
| TCDC, 0.5 mM | NDB | NDB | >8.0** | 33 ± 2** |
| TUDC, 1 mM | NDB | NDB | ||
Values are means ± SE. 125I-BDZ-1 and 125I-T-0632 were utilized for binding BDZ-1 and T-0632, respectively; for all other binding, 125I-CCK radioligand was used.
CCK1R, CCK type 1 receptor; Emax, maximal response to 0.1 mM ATP; TCDC, taurochenodeoxycholic acid; TUDC, tauroursodeoxycholic acid.
P < 0.05,
P < 0.01 vs. control.
NDB, no detectable binding.
In addition, we tested the effects of TCDC and TUDC on the binding affinities and biological activity of allosteric ligands acting at the CCK1R. We utilized two allosteric antagonist radioligands, 125I-BDZ-1 (1) and 125I-T-0632 (16), in homologous competition-binding studies, and a benzodiazepine allosteric agonist, GI181771X (2), in intracellular calcium-signaling studies. The molecular basis of binding of allosteric ligands to the CCK1R has been extensively studied (7, 16, 21). These ligands bind exclusively to a docking cleft within the intramembranous helical bundle, which is distinct from the orthosteric CCK peptide-binding site of the CCK1R. Similar to 125I-CCK binding, ≤1 mM TUDC did not affect the function of the allosteric ligands at CCK1R (Fig. 3, Table 1). Also, TCDC decreased the total binding of 125I-BDZ-1 and 125I-T-0632 to the CCK1R in a concentration-dependent manner, with a ∼50% decrease at 0.5 mM (Fig. 3, B and D), but it did not affect binding affinities (Table 1). TCDC at 0.5 mM significantly affected the ability of the agonist GI181771X to stimulate intracellular calcium responses in wild-type CCK1R-bearing CHO cells, reducing its potency by 13.2-fold and its efficacy by 1.5-fold (Fig. 3E, Table 1).
Fig. 3.

Effects of bile acids on binding and biological activity of allosteric ligands at the CCK1R in a normal membrane environment. A–D: effects of 1 mM TUDC and 0.5 mM TCDC on homologous competitive binding of the allosteric antagonists BDZ-1 (A and B) and T-0632 (C and D). Insets: effects of increasing concentration of TCDC on specific binding of BDZ-1 (B) and T-0632 (D). E: effects of TUDC and TCDC on intracellular calcium responses stimulated by the allosteric agonist GI181771X. Values (means ± SE of ≥4 independent experiments performed in duplicate) represent percentage of saturable binding (A and C) or saturable binding as a percentage of control (B and D). Calcium responses were normalized to maximal response to 0.1 mM ATP.
Effects of bile acids on CCK1R expressed in an elevated membrane cholesterol environment.
Having established the effects of TCDC and TUDC in wild-type CCK1R in a normal membrane cholesterol environment, we examined the effect of TCDC and TUDC at the same wild-type CCK1R expressed in an elevated membrane cholesterol environment in the SRD15-CCK1R cell line (22). This cell line has been characterized as having an increased level of membrane cholesterol due to a genetic defect in the cholesterol biosynthetic pathway (28). The CCK1R expressed in this environment exhibits a higher binding affinity for CCK, which is nonproductive, associated with a significant reduction in potency to stimulate a biological response (12, 22, 23, 37). As shown in Fig. 4, A and B, TCDC and TUDC had no significant effects on CCK binding. Interestingly, 0.1 and 1 mM TUDC (concentrations below their critical micellar concentrations) improved the potency of CCK-induced intracellular calcium responses by fivefold (Fig. 4C, Table 1). The hydrophobic bile acid TCDC did not further modify the biological responses to CCK in this cell line (Fig. 4C, Table 1).
Fig. 4.
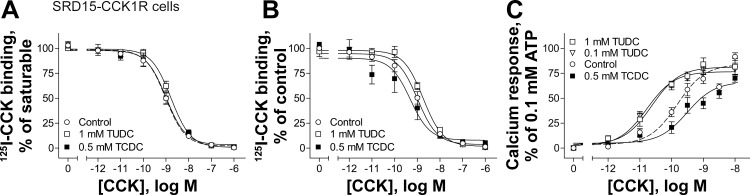
Effects of bile acids on CCK1R expressed in an elevated membrane cholesterol environment. A and B: CCK competition-binding curves at the CCK1R expressed in SRD15-CCK1R cells possessing increased membrane cholesterol in the absence (control) and presence of 1 mM TUDC and 0.5 mM TCDC. Values (means ± SE of ≥3 independent experiments performed in duplicate) represent percentage of saturable binding for each curve (A) and percentage of control (B). C: curves for concentration-dependent intracellular calcium responses to CCK in SRD15-CCK1R cells after treatments described in A and B. Values (means ± SE of ≥5 independent experiments performed in duplicate) were normalized to maximal response to 0.1 mM ATP.
Effects of bile acids on CCK1R constructs in which cholesterol-binding motifs were disrupted.
We previously studied the cholesterol recognition/interaction amino acid consensus pattern (CRAC) and the cholesterol consensus motif (CCM) by mutagenesis in CCK receptors (37). To test whether TUDC and TCDC differentially affect the CCK1R when these motifs are disrupted by mutation, we used the site mutant constructs Y140A and Y237A, disrupting the CRAC motifs in transmembrane domain 3 (TM3) and TM5, respectively, and W166A, disrupting the CCM motif in TM4 (37). We reported that, of these mutants, only the Y140A construct of the CCK1R exhibits insensitivity to a further decrease (37) or increase (12) in membrane cholesterol levels, while this mutant construct also mimics the behavior of the CCK1R in an elevated membrane cholesterol environment (12). Binding data for the effects of the bile acids on these CCK1R mutants are shown in Fig. 5 (Table 1), and biological activity data are shown in Fig. 6 (Table 1).
Fig. 5.

Effects of bile acids on CCK binding at the CCK1R with mutations in cholesterol recognition motifs. A–D: CCK competition-binding curves for CHO-CCK1R-Y140A (A and B) and CHO-CCK1R-Y237A (C and D) cells in the absence (control) and presence of TUDC and TCDC. Values (means ± SE of ≥3 independent experiments performed in duplicate) represent percentage of saturable binding for each curve (A and C) or percentage of control (B and D).
Fig. 6.

Effects of bile acids on CCK biological activity at the CCK1R with mutations in cholesterol-recognition motifs. A–C: curves illustrating CCK concentration-dependent intracellular calcium responses in CHO cells stably expressing the CCK1R mutants Y140A (A), Y237A (B), and W166A (C). Values (means ± SE of ≥5 independent experiments performed in duplicate) were normalized to maximal response to 0.1 mM ATP.
Data from homologous ligand competition-binding assays using the 125I-CCK radioligand and signaling assays show that the Y140A mutant of the CCK1R was not sensitive to TCDC treatment, with this bile acid having no effects on binding affinity or potency of CCK to stimulate intracellular calcium responses, in contrast to our findings in wild-type CCK1R (Figs. 5A and 6A). The total binding was reduced by ∼40% at the Y140A mutant (Fig. 5B) compared with ∼50% at the wild-type CCK1R (Fig. 2B). It is particularly interesting that the corrective effect of TUDC on signaling of the CCK1R that was observed at wild-type CCK1R expressed in an elevated membrane cholesterol environment (SRD15-CCK1R cells; Fig. 4C) was not observed at the Y140A mutant (Fig. 6A). An interpretation of this finding could be that although the Y140A mutant mimics the conformation of the CCK1R in a high-cholesterol environment, this mutation disrupts the ability of the bile acids to bind to that site.
In contrast, the Y237A (Fig. 5, C and D, and Fig. 6B) and W166A (Fig. 6C) mutants of the CCK1R remained sensitive to TCDC, similar to their effects on wild-type CCK1R. The W166A mutant was expressed only at a very low level on the cell surface, resulting in an inadequate level of saturable radioligand binding (data not shown). This likely reflects inappropriate folding of the receptor due to the mutation that resulted in intracellular trapping. However, there seem to be enough receptors on the cell surface to elicit a measurable calcium response to CCK stimulation in these cells (Fig. 6C).
DISCUSSION
CCK is a gastrointestinal peptide hormone synthesized in enteroendocrine I cells scattered along the mucosa of the proximal small intestine and secreted in response to luminal fat and protein. This hormone plays multiple roles relevant to nutritional homeostasis, acting via the CCK1R. These roles include stimulation of pancreatic exocrine secretion and gallbladder contraction to aid in digestion, slowing of gastric emptying and regulation of bowel transit to titrate the appropriate delivery of nutrients for absorption, and induction of postcibal satiety by acting on peripheral CCK receptors expressed on vagal afferent neurons (29, 43). Recognition of the latter role has supported the idea that this receptor might be an important target for the management of obesity (5, 6, 17, 25).
The CCK1R differs from the closely related CCK2R, in that the function of the CCK1R is affected by membrane cholesterol, whereas the function of the CCK2R is not (37). Lateral allosteric regulation of the CCK1R by cholesterol may be responsible for defective stimulus-activity coupling (23, 37), resulting in reduced contractile responses to CCK in cholesterol gallstone disease (49, 52). This defect is not observed in patients with pigment gallstones (10, 46, 49). A major difference between these two clinical settings is the presence of supersaturated cholesterol in lithogenic bile (14, 27, 31). Studies in humans and animals have shown that the abnormal gallbladder contractile response to CCK is correlated with elevated levels of cholesterol in the membrane of gallbladder smooth muscle cells in patients with cholesterol gallstones, and reduction of the cholesterol to normal levels by in vitro treatment of those cells with methyl-β-cyclodextrin has been shown to reverse this functional abnormality (9, 49, 51). Indeed, we have identified unique sensitivity of the CCK1R, and not the CCK2R, to increased membrane cholesterol in in vitro studies using model cell systems in which we explored the molecular basis of this action (12, 22, 23, 37).
Bile acids, another component of bile, are structurally related to cholesterol. Bile acids are present in high concentration in bile in the biliary tree and gallbladder, as well as the small bowel lumen, and are efficiently absorbed through bile acid transporters in the ileum (enterohepatic circulation). Bile acids can also be elevated in the circulation in cholestasis syndromes, but they are present in blood in much lower concentrations. The impact of bile acids on gallbladder function is quite instructive. In vitro and in vivo studies have shown that treatment with hydrophobic bile acids, such as chenodeoxycholic acid and cholic acid, has a negative impact on gallbladder emptying by reducing the gallbladder contractile response to CCK (47), whereas hydrophilic bile acids, such as ursodeoxycholic acid, are not inhibitory and may actually protect against the negative impact of the hydrophobic bile acids (and possibly cholesterol) (18). Ursodeoxycholic acid can improve gallbladder contraction and prevent gallbladder muscle dysfunction (18, 47). There have been suggestions that the beneficial effects of ursodeoxycholic in this setting relate to a decrease in the cholesterol content of bile and reduced impact on the gallbladder smooth muscle cells, as well as to the effect of this bile acid to reduce oxidative stress (18). It is not clear, however, that these are the only relevant effects of bile acids in this process.
Given the close structural similarity of bile acids to cholesterol, we postulate that there may exist an additional mechanism contributing to the reported beneficial clinical effects of ursodeoxycholic acid. Findings from the current study show that, similar to the effects of cholesterol on CCK receptors, bile acids had effects on the CCK1R, while they did not have recognizable effects on the structurally related CCK2R. This provided an important control for bile acid effects distal to the receptor in signaling pathways or on some other target in the membrane or the cell, since both CCK1R and CCK2R are believed to couple with the same G proteins and to induce similar signaling events in cells in which they are expressed. The differential impact of the bile acids on the same types of cells expressing these two types of CCK receptors is strongly suggestive that the bile acids work directly on the CCK1R.
Furthermore, the hydrophobic chenodeoxycholic acid had a negative effect on CCK1R function when present in a normal membrane environment, much like excess cholesterol. The absolute impact of chenodeoxycholic acid was distinct, negatively affecting both binding affinity and potency of biological activity (whereas cholesterol dissociates these two effects, increasing binding and reducing biological responses). Interestingly, there was no additional effect of this bile acid on the CCK1R in a high-cholesterol environment. In contrast, ursodeoxycholic acid did not affect CCK1R function in a normal-cholesterol environment and showed a significant protective effect against excess cholesterol, reversing the rightward shift of the CCK dose-response curve observed in the presence of high cholesterol. This is achieved without a change in the increased binding affinity typical of the high-cholesterol environment.
We previously studied a CCK1R with single amino acid mutations disrupting the CRAC and CCM cholesterol-association motifs (37) in an effort to localize the site of action of cholesterol. Of these CCK1R mutations, the Y140 residue in the CRAC motif at the bottom of TM3 near the DRY ionic lock mechanism was shown to be important in cholesterol binding to the CCK1R (37). Mutation of this residue yielded a CCK1R insensitive to a decrease (37) or an increase (12) in membrane cholesterol content, as demonstrated by a lack of change in CCK binding and signaling profiles, whereas mutations of other cholesterol-binding motifs within the CCK1R, Y237A (CRAC motif, TM5) and W166A (CCM, TM4), continued to be sensitive to cholesterol.
We have demonstrated that the Y140A mutant of the CCK1R mimics all the structural and functional effects typical of the wild-type CCK1R in a high-cholesterol environment (12). In the current study we show that, similar to cholesterol, the bile acid effects in wild-type CCK1R were absent in the Y140A mutant construct. Furthermore, mutations of Y237A and W166A had no impact on the bile acid effects. These data may be interpreted as follows: bile acids interact with the CCK1R at the same site as cholesterol and competitively inhibit the cholesterol interaction with the receptor. It is noteworthy that each bile acid affects the conformation of the CCK1R differently from cholesterol and differently from each other. On the basis of the current evidence, it can be postulated that chenodeoxycholic acid negatively affects the conformation of the CCK1R, which leads to G protein uncoupling, whereas ursodeoxycholic acid has a protective effect, possibly working to correct the abnormal receptor conformation induced by the high-cholesterol environment, which is reflected in significant improvement in CCK signaling in this setting.
To further understand the proposed mechanism, we consider that the CCK1R has three dominant domains: 1) the extracellular domain, where the natural peptide ligand binds, 2) the intracellular domain, where the heterotrimeric G protein interacts with the receptor, and 3) the intramembranous core helical bundle domain, in which a network of hydrogen bonds helps stabilize conformations and mediate conformational changes associated with agonist binding and activation (proposed mechanisms illustrated in Fig. 7). Clearly, the effect of cholesterol is a conformational change in the CCK1R that renders the helical bundle domain dysfunctional. In that situation, CCK binds with higher-than-normal affinity, yet the receptor-G protein coupling interaction is defective, resulting in reduced agonist-induced signaling. This suggests that the major defect when this domain is expressed in an elevated membrane cholesterol environment is in the transduction function of the core helical bundle of the CCK1R. It is probable that ursodeoxycholic acid can correct this defect to an extent that correlates with its clinical benefits in cholesterol gallstone disease. These data suggest that 1) bile acids compete with the action of cholesterol on the CCK1R, probably by interacting at the same site, although the conformational impact of this appears to be different with different bile acids, and 2) ursodeoxycholic acid is capable of correcting the abnormal conformation of the CCK1R in an elevated membrane cholesterol environment.
Fig. 7.

Summary of impact of bile acids on CCK1R function in normal and elevated membrane cholesterol environments: 7 transmembrane CCK1R in a normal membrane (light gray) or a membrane with elevated cholesterol level (dark gray), with CCK peptide docking to the receptor ectodomain and the heterotrimeric G protein (Gαq, β- and γ-subunits) associating at the receptor cytosolic face. Arrow illustrates transduction of the signal via conformational change in the helical bundle domain. CCK binding and signaling were reduced in response to TCDC, while TUDC had no effect on these CCK1R processes in a normal membrane. In a high-cholesterol environment, CCK1R binding was increased and signaling was decreased, with TCDC having no further effect, whereas TUDC corrected the signaling defect. It is proposed that these bile acid effects on the CCK1R reflect a direct interaction with the receptor, likely at the same site at which cholesterol binds and possibly competing with the detrimental effect of excess cholesterol. Each bile acid has its unique effects on CCK1R conformation and function.
In addition to cholesterol gallstone disease, the impact of ursodeoxycholic acid on CCK1R function might be beneficial in sphincter of Oddi dysfunction. This represents another site of the CCK1R that would be exposed to a high concentration of this bile acid, with its concentration elevated in the enterohepatic circulation after administration.
Given a key role of the CCK1R in obesity and metabolic syndrome (8, 15, 43, 49) and the failure of efforts to successfully develop a drug targeting this receptor (4, 5, 17, 24), there is still a need to develop alternate pharmacological therapeutic strategies to affect this target. The current observations that ursodeoxycholic acid has a positive effect on CCK1R function, perhaps acting through the site of negative impact of cholesterol, might provide a lead for a therapeutic approach to this problem. Because the efficient absorption of this bile acid occurs in the ileum, resulting in high concentrations only within the enterohepatic circulation, this (or any other) bile acid is unlikely to have therapeutic usefulness outside the gallbladder and biliary tree. However, the mechanism currently described to act on the CCK1R and proof-of-concept for this bile acid to correct the negative impact of cholesterol on the CCK1R may encourage the development of similar molecules that could have a high concentration in the systemic circulation to act on the vagal afferent CCK1R.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant DK-032878 and the Mayo Clinic.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.J.D., M.D., K.G.H., and L.J.M. developed the concept and designed the research; A.J.D., M.D., and K.G.H. performed the experiments; A.J.D., M.D., K.G.H., and L.J.M. analyzed the data; A.J.D., M.D., K.G.H., and L.J.M. interpreted the results of the experiments; A.J.D. and M.D. prepared the figures; A.J.D., M.D., and L.J.M. drafted the manuscript; A.J.D., M.D., and L.J.M. edited and revised the manuscript; L.J.M. approved the final version of the manuscript.
ACKNOWLEDGMENTS
The authors acknowledge the excellent technical assistance of M. L. Augustine and A. M. Ball.
REFERENCES
- 1.Akgun E, Korner M, Gao F, Harikumar KG, Waser B, Reubi JC, Portoghese PS, Miller LJ. Synthesis and in vitro characterization of radioiodinatable benzodiazepines selective for type 1 and type 2 cholecystokinin receptors. J Med Chem 52: 2138–2147, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aquino CJ, Armour DR, Berman JM, Birkemo LS, Carr RA, Croom DK, Dezube M, Dougherty RW Jr, Ervin GN, Grizzle MK, Head JE, Hirst GC, James MK, Johnson MF, Miller LJ, Queen KL, Rimele TJ, Smith DN, Sugg EE. Discovery of 1,5-benzodiazepines with peripheral cholecystokinin (CCK-A) receptor agonist activity. 1. Optimization of the agonist “trigger.” J Med Chem 39: 562–569, 1996. [DOI] [PubMed] [Google Scholar]
- 3.Behar J, Lee KY, Thompson WR, Biancani P. Gallbladder contraction in patients with pigment and cholesterol stones. Gastroenterology 97: 1479–1484, 1989. [DOI] [PubMed] [Google Scholar]
- 4.Berger R, Zhu C, Hansen AR, Harper B, Chen Z, Holt TG, Hubert J, Lee SJ, Pan J, Qian S, Reitman ML, Strack AM, Weingarth DT, Wolff M, Macneil DJ, Weber AE, Edmondson SD. 2-Substituted piperazine-derived imidazole carboxamides as potent and selective CCK1R agonists for the treatment of obesity. Bioorg Med Chem Lett 18: 4833–4837, 2008. [DOI] [PubMed] [Google Scholar]
- 5.Bignon E, Bachy A, Boigegrain R, Brodin R, Cottineau M, Gully D, Herbert JM, Keane P, Labie C, Molimard JC, Olliero D, Oury-Donat F, Petereau C, Prabonnaud V, Rockstroh MP, Schaeffer P, Servant O, Thurneyssen O, Soubrie P, Pascal M, Maffrand JP, Le Fur G. SR146131: a new potent, orally active, and selective nonpeptide cholecystokinin subtype 1 receptor agonist. I. In vitro studies. J Pharmacol Exp Ther 289: 742–751, 1999. [PubMed] [Google Scholar]
- 6.Castillo EJ, Delgado-Aros S, Camilleri M, Burton D, Stephens D, O'Connor-Semmes R, Walker A, Shachoy-Clark A, Zinsmeister AR. Effect of oral CCK-1 agonist GI181771X on fasting and postprandial gastric functions in healthy volunteers. Am J Physiol Gastrointest Liver Physiol 287: G363–G369, 2004. [DOI] [PubMed] [Google Scholar]
- 7.Cawston EE, Lam PC, Harikumar KG, Dong M, Ball AM, Augustine ML, Akgun E, Portoghese PS, Orry A, Abagyan R, Sexton PM, Miller LJ. Molecular basis for binding and subtype selectivity of 1,4-benzodiazepine antagonist ligands of the cholecystokinin receptor. J Biol Chem 287: 18618–18635, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chaudhri OB, Salem V, Murphy KG, Bloom SR. Gastrointestinal satiety signals. Annu Rev Physiol 70: 239–255, 2008. [DOI] [PubMed] [Google Scholar]
- 9.Chen Q, Amaral J, Biancani P, Behar J. Excess membrane cholesterol alters human gallbladder muscle contractility and membrane fluidity. Gastroenterology 116: 678–685, 1999. [DOI] [PubMed] [Google Scholar]
- 10.Chen Q, Amaral J, Oh S, Biancani P, Behar J. Gallbladder relaxation in patients with pigment and cholesterol stones. Gastroenterology 113: 930–937, 1997. [DOI] [PubMed] [Google Scholar]
- 11.Cofan M, Escurriol V, Garcia-Otin AL, Moreno-Iribas C, Larranaga N, Sanchez MJ, Tormo MJ, Redondo ML, Gonzalez CA, Corella D, Pocovi M, Civeira F, Ros E. Association of plasma markers of cholesterol homeostasis with metabolic syndrome components. A cross-sectional study. Nutr Metab Cardiovasc Dis 21: 651–657, 2011. [DOI] [PubMed] [Google Scholar]
- 12.Desai AJ, Harikumar KG, Miller LJ. A type 1 cholecystokinin receptor mutant that mimics the dysfunction observed for wild type receptor in a high cholesterol environment. J Biol Chem 289: 18314–18326, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Desai AJ, Miller LJ. Sensitivity of cholecystokinin receptors to membrane cholesterol content. Front Endocrinol 3: 123, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di Ciaula A, Wang DQ, Bonfrate L, Portincasa P. Current views on genetics and epigenetics of cholesterol gallstone disease. Cholesterol 2013: 298421, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dockray GJ. Cholecystokinin and gut-brain signalling. Regul Pept 155: 6–10, 2009. [DOI] [PubMed] [Google Scholar]
- 16.Dong M, Vattelana AM, Lam PC, Orry AJ, Abagyan R, Christopoulos A, Sexton PM, Haines DR, Miller LJ. Development of a highly selective allosteric antagonist radioligand for the type 1 cholecystokinin receptor and elucidation of its molecular basis of binding. Mol Pharmacol 87: 130–140, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elliott RL, Cameron KO, Chin JE, Bartlett JA, Beretta EE, Chen Y, Jardine Pda S, Dubins JS, Gillaspy ML, Hargrove DM, Kalgutkar AS, LaFlamme JA, Lame ME, Martin KA, Maurer TS, Nardone NA, Oliver RM, Scott DO, Sun D, Swick AG, Trebino CE, Zhang Y. Discovery of N-benzyl-2-[(4S)-4-(1H-indol-3-ylmethyl)-5-oxo-1-phenyl-4,5-dihydro-6H-[1,2,4]tri azolo[4,3-a][1,5]benzodiazepin-6-yl]-N-isopropylacetamide, an orally active, gut-selective CCK1 receptor agonist for the potential treatment of obesity. Bioorg Med Chem Lett 20: 6797–6801, 2010. [DOI] [PubMed] [Google Scholar]
- 18.Guarino MP, Cong P, Cicala M, Alloni R, Carotti S, Behar J. Ursodeoxycholic acid improves muscle contractility and inflammation in symptomatic gallbladders with cholesterol gallstones. Gut 56: 815–820, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gustafsson U, Sahlin S, Einarsson C. Biliary lipid composition in patients with cholesterol and pigment gallstones and gallstone-free subjects: deoxycholic acid does not contribute to formation of cholesterol gallstones. Eur J Clin Invest 30: 1099–1106, 2000. [DOI] [PubMed] [Google Scholar]
- 20.Hanauer SB. The burdens of digestive diseases. Nat Rev Gastroenterol Hepatol 6: 377, 2009. [DOI] [PubMed] [Google Scholar]
- 21.Harikumar KG, Cawston EE, Lam PC, Patil A, Orry A, Henke BR, Abagyan R, Christopoulos A, Sexton PM, Miller LJ. Molecular basis for benzodiazepine agonist action at the type 1 cholecystokinin receptor. J Biol Chem 288: 21082–21095, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harikumar KG, Potter RM, Patil A, Echeveste V, Miller LJ. Membrane cholesterol affects stimulus-activity coupling in type 1, but not type 2, CCK receptors: use of cell lines with elevated cholesterol. Lipids 48: 231–244, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harikumar KG, Puri V, Singh RD, Hanada K, Pagano RE, Miller LJ. Differential effects of modification of membrane cholesterol and sphingolipids on the conformation, function, and trafficking of the G protein-coupled cholecystokinin receptor. J Biol Chem 280: 2176–2185, 2005. [DOI] [PubMed] [Google Scholar]
- 24.Jordan J, Greenway FL, Leiter LA, Li Z, Jacobson P, Murphy K, Hill J, Kler L, Aftring RP. Stimulation of cholecystokinin-A receptors with GI181771X does not cause weight loss in overweight or obese patients. Clin Pharmacol Ther 83: 281–287, 2008. [DOI] [PubMed] [Google Scholar]
- 25.Kissileff HR, Pi-Sunyer FX, Thornton J, Smith GP. C-terminal octapeptide of cholecystokinin decreases food intake in man. Am J Clin Nutr 34: 154–160, 1981. [DOI] [PubMed] [Google Scholar]
- 26.Kreiss C, Schwizer W, Borovicka J, Jansen JB, Bouloux C, Pignol R, Bischof Delaloye A, Fried M. Effect of lintitript, a new CCK-A receptor antagonist, on gastric emptying of a solid-liquid meal in humans. Regul Pept 74: 143–149, 1998. [DOI] [PubMed] [Google Scholar]
- 27.Lamont JT, Carey MC. Cholesterol gallstone formation. 2. Pathobiology and pathomechanics. Prog Liver Dis 10: 165–191, 1992. [PubMed] [Google Scholar]
- 28.Lee PC, Sever N, Debose-Boyd RA. Isolation of sterol-resistant Chinese hamster ovary cells with genetic deficiencies in both Insig-1 and Insig-2. J Biol Chem 280: 25242–25249, 2005. [DOI] [PubMed] [Google Scholar]
- 29.Li Y, Owyang C. Endogenous cholecystokinin stimulates pancreatic enzyme secretion via vagal afferent pathway in rats. Gastroenterology 107: 525–531, 1994. [DOI] [PubMed] [Google Scholar]
- 30.Liddle RA, Gertz BJ, Kanayama S, Beccaria L, Coker LD, Turnbull TA, Morita ET. Effects of a novel cholecystokinin (CCK) receptor antagonist, MK-329, on gallbladder contraction and gastric emptying in humans. Implications for the physiology of CCK. J Clin Invest 84: 1220–1225, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McDougall RM, Walker K, Thurston OG. Prolonged secretion of lithogenic bile after cholecystectomy. Ann Surg 182: 150–153, 1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller LJ, Holicky EL, Ulrich CD, Wieben ED. Abnormal processing of the human cholecystokinin receptor gene in association with gallstones and obesity. Gastroenterology 109: 1375–1380, 1995. [DOI] [PubMed] [Google Scholar]
- 33.Nardone G, Ferber IA, Miller LJ. The integrity of the cholecystokinin receptor gene in gallbladder disease and obesity. Hepatology 22: 1751–1753, 1995. [PubMed] [Google Scholar]
- 34.Nguyen DM, El-Serag HB. The epidemiology of obesity. Gastroenterol Clin North Am 39: 1–7, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paragh G, Kovacs E, Seres I, Keresztes T, Balogh Z, Szabo J, Teichmann F, Foris G. Altered signal pathway in granulocytes from patients with hypercholesterolemia. J Lipid Res 40: 1728–1733, 1999. [PubMed] [Google Scholar]
- 36.Portincasa P, Di Ciaula A, Baldassarre G, Palmieri V, Gentile A, Cimmino A, Palasciano G. Gallbladder motor function in gallstone patients: sonographic and in vitro studies on the role of gallstones, smooth muscle function and gallbladder wall inflammation. J Hepatol 21: 430–440, 1994. [DOI] [PubMed] [Google Scholar]
- 37.Potter RM, Harikumar KG, Wu SV, Miller LJ. Differential sensitivity of types 1 and 2 cholecystokinin receptors to membrane cholesterol. J Lipid Res 53: 137–148, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Powers SP, Pinon DI, Miller LJ. Use of N,O-bis-Fmoc-d-Tyr-ONSu for introduction of an oxidative iodination site into cholecystokinin family peptides. Int J Pept Protein Res 31: 429–434, 1988. [DOI] [PubMed] [Google Scholar]
- 39.Reuben A, Qureshi Y, Murphy GM, Dowling RH. Effect of obesity and weight reduction on biliary cholesterol saturation and the response to chenodeoxycholic acid. Eur J Clin Invest 16: 133–142, 1986. [DOI] [PubMed] [Google Scholar]
- 40.Roma MG, Toledo FD, Boaglio AC, Basiglio CL, Crocenzi FA, Sanchez Pozzi EJ. Ursodeoxycholic acid in cholestasis: linking action mechanisms to therapeutic applications. Clin Sci (Lond) 121: 523–544, 2011. [DOI] [PubMed] [Google Scholar]
- 41.Roslyn JJ, Doty J, Pitt HA, Conter RL, Den Besten L. Enhanced gallbladder absorption during gallstone formation: the roles of cholesterol saturated bile and gallbladder stasis. Am J Med Sci 292: 75–80, 1986. [DOI] [PubMed] [Google Scholar]
- 42.Seres I, Foris G, Varga Z, Kosztaczky B, Kassai A, Balogh Z, Fulop P, Paragh G. The association between angiotensin II-induced free radical generation and membrane fluidity in neutrophils of patients with metabolic syndrome. J Membr Biol 214: 91–98, 2006. [DOI] [PubMed] [Google Scholar]
- 43.Smith GP, Gibbs J. The satiety effect of cholecystokinin. Recent progress and current problems. Ann NY Acad Sci 448: 417–423, 1985. [DOI] [PubMed] [Google Scholar]
- 44.Tint GS, Salen G, Shefer S. Effect of ursodeoxycholic acid and chenodeoxycholic acid on cholesterol and bile acid metabolism. Gastroenterology 91: 1007–1018, 1986. [DOI] [PubMed] [Google Scholar]
- 45.Tsai CJ, Leitzmann MF, Willett WC, Giovannucci EL. Prospective study of abdominal adiposity and gallstone disease in US men. Am J Clin Nutr 80: 38–44, 2004. [DOI] [PubMed] [Google Scholar]
- 46.Xiao ZL, Amaral J, Biancani P, Behar J. Impaired cytoprotective function of muscle in human gallbladders with cholesterol stones. Am J Physiol Gastrointest Liver Physiol 288: G525–G532, 2005. [DOI] [PubMed] [Google Scholar]
- 47.Xiao ZL, Biancani P, Carey MC, Behar J. Hydrophilic but not hydrophobic bile acids prevent gallbladder muscle dysfunction in acute cholecystitis. Hepatology 37: 1442–1450, 2003. [DOI] [PubMed] [Google Scholar]
- 48.Xiao ZL, Chen Q, Amaral J, Biancani P, Behar J. Defect of receptor-G protein coupling in human gallbladder with cholesterol stones. Am J Physiol Gastrointest Liver Physiol 278: G251–G258, 2000. [DOI] [PubMed] [Google Scholar]
- 49.Xiao ZL, Chen Q, Amaral J, Biancani P, Jensen RT, Behar J. CCK receptor dysfunction in muscle membranes from human gallbladders with cholesterol stones. Am J Physiol Gastrointest Liver Physiol 276: G1401–G1407, 1999. [DOI] [PubMed] [Google Scholar]
- 50.Xu HL, Hsing AW, Vogtmann E, Chu LW, Cheng JR, Gao J, Tan YT, Wang BS, Shen MC, Gao YT. Variants in CCK and CCKAR genes to susceptibility to biliary tract cancers and stones: a population-based study in Shanghai, China. J Gastroenterol Hepatol 28: 1476–1481, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu P, Chen Q, Biancani P, Behar J. Membrane cholesterol alters gallbladder muscle contractility in prairie dogs. Am J Physiol Gastrointest Liver Physiol 271: G56–G61, 1996. [DOI] [PubMed] [Google Scholar]
- 52.Yu P, Chen Q, Harnett KM, Amaral J, Biancani P, Behar J. Direct G protein activation reverses impaired CCK signaling in human gallbladders with cholesterol stones. Am J Physiol Gastrointest Liver Physiol 269: G659–G665, 1995. [DOI] [PubMed] [Google Scholar]