

Abstract
Cyclin-dependent kinase 1 (CDK1) is the only necessary CDK in cell proliferation and a novel target in the development of anticancer drugs. 8-Hydroxypiperidinemethyl-baicalein (BA-j) is a novel selective CDK1 inhibitor with broad spectrum anti-cancer activity (IC50 12.3 μM) and 2 tumor xenografts. Because of the differential mechanisms controlling redox-states in normal and cancer cells, BA-j can capture oxygen free radicals (·O2−) and selectively increase the level of H2O2 in cancer cells, thereby specifically oxidize and activate the intrinsic apoptosis pathway bypassing the extrinsic death receptor pathway, thus inducing apoptosis in cancer cells rather than in normal cells. BA-j is different from cytotoxic anticancer drugs which can activate both the intrinsic apoptosis pathway and the extrinsic death receptor pathway, and therefore harm normal cells while killing cancer cells. The molecular and biochemical mechanisms of reactive oxygen species (ROS) regulation suggest that BA-j may be developed into a novel anticancer agent.
The discovery of novel anticancer drugs that can selectively induce apoptosis in cancer cells is a challenging and significant task in pharmaceutical science. Cyclin-dependent protein kinases (CDKs) are key signaling molecules in the regulation of the cell cycle. CDKs are specific serine/threonine protein kinases in the cytoplasm and nucleus that act as mediators in signal transduction pathways. CDK1 is the only necessary CDK in cell proliferation, and a novel target in the development of anticancer drugs1,2,3. Recently, the global anticancer drug research community has turned its attention to CDK inhibitors, 20 of which have entered clinical trials1,4,5. However, the selectivity of most of the CDK inhibitors currently in clinical trials is unsatisfactory. Some showed inhibitory activity on CDK2 (i.e., acts on S phase and increases toxicity) and certain side effects because of their complex chemical structures. CDK inhibitors based on organic amine derivatives of flavonoid, such as Flavopiridol6,7 and P276-008,9,10, have attracted the most interest. However, because of their poor solubility and bioavailability, low blood concentration, difficulty in catabolism and rapid excretion by glucuronidation, the druggability of these molecules is unsatisfactory.
The most artificially cultivated medicinal species in China is Scutellaria baicalensis, a perennial herb whose dried root is commonly used in traditional Chinese medicine for the treatment of hyperlipomia, hypertension, hyperglycemia, inflammation, allergic reactions, viral infections, cancer and so on11. The major active ingredients in Scutellaria baicalensis are flavonoids and more than 40 flavonoid structures have been identified in this plant12,13. The most common flavonoid in Scutellaria baicalensis is Baicalin (9–21%), and its hydrolyzate, Baicalein (BA), possesses stronger potency. Natural flavonoids are selective CDK1 inhibitors, and BA is the most potent among them with the anti-proliferative activity IC50 25–75 μM14,15,16,17,18,19,20,21,22,23. Because of the differential mechanisms controlling redox-states in normal and cancer cells, by regulating reactive oxygen species (ROS) of BA24,25,26,27,28,29,30,31, ROS can specifically oxidize some enzymes with active site of cysteine. Such as, BA can inhibit CDK1 by oxidizing CDC25C, thus suppressing proliferation in cancer cells15,19,25,32. Further, BA can activate the intrinsic apoptotic pathways by oxidizing caspases15,16,19,21,22,24,25,26,27,28,29,30,33, bypassing the extrinsic death receptor pathway16,24,31, thus inducing apoptosis in cancer cells and activated lymphocytes rather than in normal cells17,21,23,30,31,33,34,35. However, the exact biochemical mechanism of BA by the regulation of ROS is only partially understood and so is the way BA regulating ROS.
Oral Baicalin can not be directly absorbed until it has been hydrolyzed into BA by intestinal microflora, yet “enterohepatic efflux effects” inactivate and excrete 95% of BA via glucuronidation and sulfation. Therefore, the level of BA in blood is very low (Cmax 0.26 μM) with poor bioavailability by oral BA36,37,38,39,40,41. In addition, BA is easily oxidized and practically insoluble in water, making it difficult to administer intravenously. Because of its poor bioavailability and undesirable traits as a drug, BA does not meet the requirements for the clinical treatment of cancer24.
Therefore, attempts have been made to increase the effectiveness of BA by structural modifications. The most effective structural modifications are likely to be BA Mannich base derivatives42,43,44. In our previous work, dozens of natural flavonoids were used as lead compounds to create hundreds of Mannich base derivatives of flavonoids. Using CDK1/Cyclin B inhibitory activity screening and structure-activity relationship studies, 8-hydroxypiperidine-methyl-baicalein (BA-j) was identified as the most effective flavonoid Mannich base derivative45. BA-j is a selective CDK1 inhibitor with a novel chemical structure45.
In this paper, the molecular and biological mechanism of BA-j specifically inducing apoptosis in cancer cells was studied and the way BA-j regulating ROS was explored by using a PF1 fluorescent probe to selectively determine the level of intracellular H2O2. These data provide evidence that BA-j could be developed into a novel anticancer agent for clinical use.
Results
Preparation and characterization of BA-j
The major active ingredient in the root of Scutellaria baicalensis Georgi is Baicalin. Baicalein (BA, Sigma, 98% pure) hydrolyzed from Baicalin was used as a lead compound and reacted with 4-hydroxy-piperidin and formaldehyde in methanol by Mannich reaction to obtain BA-j, with >90% yield, 99.0% purity and <2.0% total related substances (Fig. 1a)45.
Figure 1. Preparation and Targets screening of 8-Hydroxypiperidinemethyl-baicalein (BA-j).
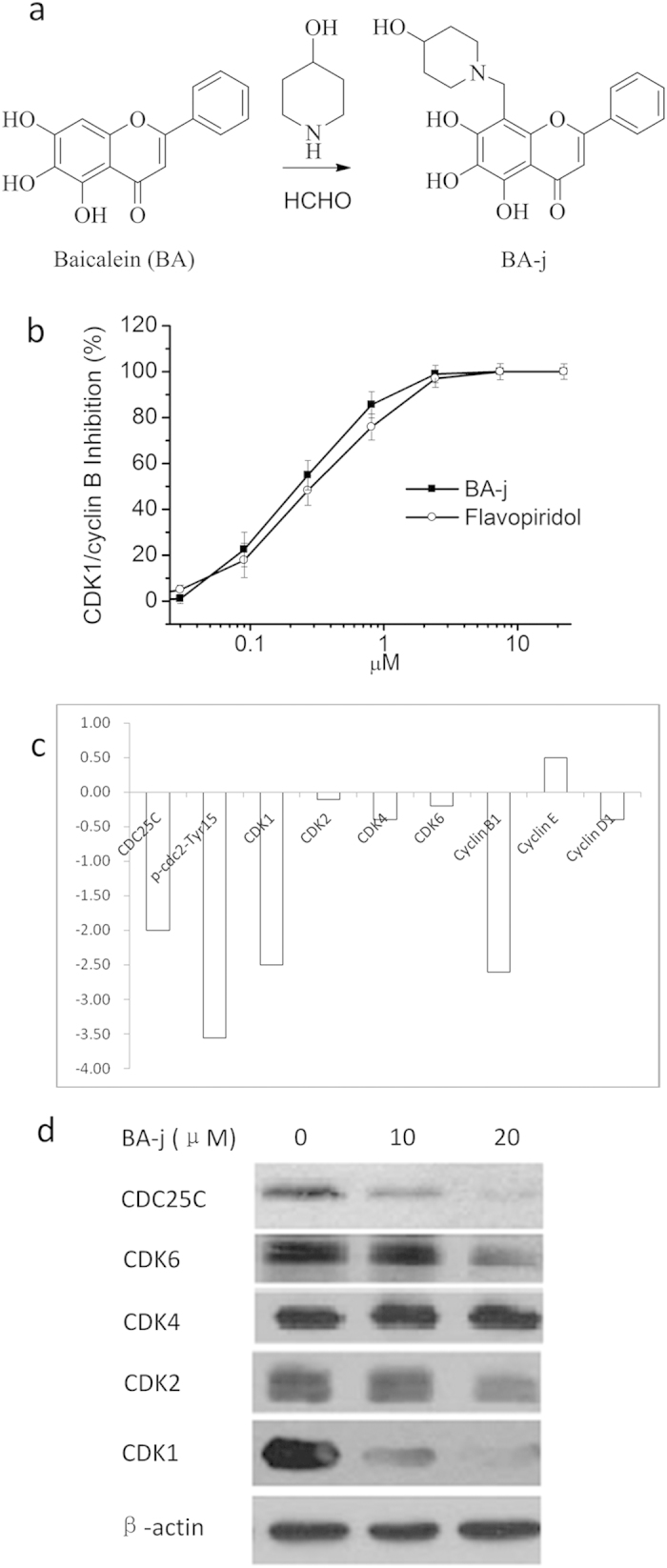
(a) Preparation of BA-j. (b) Inhibitory activity against CDK1 of 8-Hydroxypiperidinemethyl-baicalein (BA-j). (c) Hep G2 cells were treated with 10 μM BA-j for 24 h, and the assay of protein level in HepG2 cell signaling pathways were determined by Western blot for conjugated secondary antibody (goat-antimouse IgG/FITC) using densitometric scanning and normalized using internal standards (β-actin). (d) Hep G2 cells were treated with 10 and 20 μM BA-j for 24 h, and the expressions of CDKs and CDC25C proteins in HepG2 cell signaling pathways were determined by Western blot for the appropriate secondary antibody (horseradish peroxidase-conjugated goat-antimouse IgG).
BA-j is a yellow, solid, crystalline powder, containing 99.1% 5,6,7-Trihydroxy-8-(4-hydroxy-piperidin-1-ylmethyl)-2-phenyl-chromen. Dried under reduced pressure at 80 °C for 4 h, a single melting endothermic peak was shown at 215 °C by DSC. UV (methanol): λmax 277 nm, 322 nm. HRMS (TOF ES+) m/z: 384.1441[M+H]+ (theoretical value 384.1447), molecular weight (C21H21NO6): 383.14. IR (KBr, cm−1): 3059 (νArC-H), 2961, 2822, 1679 (νC=O), 1574 (νArC=C), 1549, 770, 696. BA-j CH3SO3H, 13C-NMR (DMSO-d6, 400 MHz) δ: 182.24 (C-4), 163.25 (C-2), 153.62 (C-9), 149.31 (C-7), 148.34 (C-5), 132.07 (C-1′), 130.81 (C-4′), 129.27 (C-3′,5′), 128.83 (C-6), 126.69 (C-2′,6′), 104.91 (C-8), 104.08 (C-10), 95.90 (C-3), 50.72 (C-4″), 47.60 (C-2″, 6″), 39.50 (C-1″, CH3SO3H), 29.35 (C-3″,5″). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.17-8.15 (m, 2H, Ar-2′, 6′-H), 7.65-7.59 (m, 3H, Ar-3′,4′,5′-H), 7.09 (s, 1H, 3-H), 4.98 (s,1H, 4″-OH), 4.51 (s, 2H, C8-CH2), 3.67 (m, 1H, 4″-CH), 3.41-3.40 (m, 4H, 2″, 6″-CH2), 2.30 (s, 3H, CH3SO3H), 1.85-1.59 (m, 4H, 3″, 5″-CH2). The characteristic peak of BA C8-H at δ6.60 disappears and is replaced by a BA-j C8-CH2 substitute characteristic peak at δ4.17. BA-j is 5, 6, 7-Trihydroxy- 8-(4-hydroxy-piperidin-1- ylmethyl)-2- phenyl-chromen.
The pKa of BA-j is 5.56, logP 0.68 (Octanol/Water), and its solubility is 58 μM in water and 376 μM in PBS. BA-j is stable in PBS in dark conditions. However, at room temperature under light, BA-j can capture. ·O2−, release H2O2, and be oxidized and hydrolyzed into 8-hydroxymethyl-5, 6-dehydrogenation-baicalein.
Targets of BA-j screening
Using natural flavonoids as lead compounds, hundreds of flavonoid Mannich base derivatives were semi-synthesized. These derivatives were screened for their inhibitory activity against CDK1/Cyclin B by FRET, and their structure-activity relationship analyzed44,45. The inhibitory activity of BA-j was the strongest (CC50 0.30 ± 0.11 μM), similar to Flavopiridol (CC50 0.33 ± 0.14 μM), and approximately 20-fold stronger than the lead compound BA (CC50 6.53 ± 0.21 μM), and approximately 50-fold stronger than Baicalin (CC50 14.36 ± 0.23 μM) (Fig. 1b). These results suggest that BA-j as a substrate compound for ATP can directly inhibit the activity of CDK1/ cyclin B.
To confirm the target of BA-j, Hep G2 cells were treated with 10 and 20 μM BA-j for 24 h, then the protein levels of signaling pathways in HepG2 cell were determined by Western blot for conjugated secondary antibody (goat-antimouse IgG/FITC) using densitometric scanning and normalized using internal standards (β-actin) , as shown in Fig. 1c. Among them, the expressions of CDKs and CDC25C proteins in HepG2 cell were determined by Western blot for the appropriate secondary antibody (horseradish peroxidase-conjugated goat-antimouse IgG), as shown in Fig. 1d. These results showed that 10 μM BA-j can inhibit the expression of CDC25C, CDK1 (p-cdc2-Tyr15-Thr14), CDK1 (p-cdc2-Tyr15) and Cyclin B1, rather than CDK2, CDK4, CDK6, Cyclin D and Cyclin E.
Because BA-j can capture ·O2− and selectively increase the level of H2O2 in Hep G2 cells, the activation of CDK1 was inhibited by H2O2 oxidizing CDC25C. These results suggest that BA-j can inhibit CDK1/cyclin B1 by oxidizing CDC25C, rather than other CDKs.
Anticancer activity
In vitro anti-proliferative effects of BA-j on cancer cells
The anti-proliferation activity of the BA Mannich base derivatives was screened, and BA-j was the most potent. BA-j had anti-proliferative effects on 12 human cancer cell lines in vitro and its IC50 was 12.3 μM (4.7 μg/ml, determined via MTT assay), approximately 4-fold more potent than BA (IC50 approximately 50 μM)11, as shown in Table 1. The average rate of proliferation inhibition in 60 types of human cancer cells treated with 10 μM BA-j was 40.6% (U.S. National Cancer and AIDS Research Center, by SRB). The SFDA “guidelines for antitumor drug research” provide that when the IC50 values of compounds or natural drugs inhibiting cancer cell proliferation are less than 10 μg/ml or 20 μg/ml, respectively, they are considered as having anticancer activity in vitro. When the tumor inhibitory rate is higher than 40%, the drug is considered to have antitumor activity in vivo. Our results indicate that BA-j has significant, broad-spectrum anti-proliferation activity in cancer, with no selectivity for a specific type of cancer. Furthermore, unlike cytotoxic anticancer drugs, BA-j can even reduce the growth of drug-resistant cancer cells.
Table 1. Anti-proliferation IC50 of BA-j in 12 cancer cell lines in vitro (48 h, determined by MTT assay, n = 4).
| No. | Cancer Cell Line | IC50 μM | ±μM |
|---|---|---|---|
| 1 | MCF-7 | 10.6 | 0.3 |
| 2 | Hela | 8.2 | 1.54 |
| 3 | SMMC-7721 | 11.1 | 3.02 |
| 4 | SGC-7901 | 16.8 | 1.83 |
| 5 | HL-60 | 12.5 | 1.63 |
| 6 | BGC-823 | 7.2 | 1.73 |
| 7 | PC-3 | 12.3 | 2.06 |
| 8 | H-460 | 12.7 | 2.62 |
| 9 | Hep G2 | 10.1 | 0.30 |
| 10 | K562 | 15.5 | 1.25 |
| 11 | HCT-116 | 15.8 | 3.21 |
| 12 | MDR-MB-231 | 14.8 | 0.84 |
| AV | 12.3 | 1.69 |
BA-j selectively inhibits proliferation in cancer cells rather than normal cells
To examine the effects of BA-j on normal cells, normal human primary hepatocyte cells (Liver) and normal human peripheral blood mononuclear cells (PBMCs) were cultured in the presence of BA-j for 48 h. The anti-proliferative activity of BA-j in normal cells was compared with that in Hep G2 and HL-60 cancer cells, shown in Fig. 2a–c. These results showed that BA-j exclusively inhibited proliferation in Hep G2 and HL-60 cells rather than normal Liver cells or PBMCs. These results indicate that BA-j has anti-proliferative activity in cancer cells rather than to normal cells, while the cytotoxic drug HCPT was active in both cancer cells and normal cells.
Figure 2. BA-j Anti-proliferation Activity in human cancer cells and 2 tumor xenografts.

Cells were cultured in the presence of BA-j for 48 h and an MTT assay was used to determine the anti-proliferation activity. (a) Anti-proliferation activity of BA-j in 12 human cancer cell lines compared with BA and HCPT treatment. (b) BA-j anti-proliferation activity comparison between Hep G2 cells and normal Liver cells. (c) BA-j anti-proliferation activity comparison between MCF-7 cells and normal PBMCs. (d,e) Tumor xenograft models of SGC-7901 and H-460 cells in male BALB/c nude mice treated with p.o. 10 mg/kg BA-j twice daily for 10 days.
In addition, when using BA-j with Acetyi-cysteine (as H2O2 scavenger) as combined treatment to Hep G2 and HL-60, the anti-proliferation activity of BA-j was inhibited determined by MTT, and the level of H2O2 reduced measured by PF1. Because BA-j can capture ·O2− and release H2O2 with oxidizing Acetyl-cysteine into cystine, BA-j inducing apoptosis was inhibited by Acetyl-cysteine. These results showed that Acetyl-cysteine can relieve the anti-proliferation ability of BA-j in cancer cells by reducing H2O246. (See Table 2) Thus it is suggested that CDC25C can be oxidized by H2O2 that is released by BA-j capturing ·O2−, resulting in anti-proliferation eventually.
Table 2. Acetyl-cysteine inhibiting anti-proliferation and H2O2 level of BA- j in Hep G2 and HL-60 by MTT(48 h) or by PF1(4 h).
| Cancer cells | Hep G2 | HL-60 | Hep G2 | HL-60 |
|---|---|---|---|---|
| BA-j μM | 10 | 10 | 10 | 10 |
| Acetyl-cysteine μM | 0 | 0 | 50 | 50 |
| Proliferation % | 55 | 60 | 96 | 95 |
| H2O2 μM | 40 | 35 | 6 | 5 |
In vivo anti-tumor efficacy of BA-j
The antitumor efficacy of BA-j in xenograft models of the human gastric carcinoma (SGC-7901) and non–small cell lung carcinoma (H-460) in BALB/c nude mice was further evaluated. Oral administration of 10 mg/kg BA-j twice daily for 10 days resulted in a significant reduction in tumor growth in mice bearing SGC-7901 and H-460 xenografts and an average weight loss of 10–15% compared with the control group. After 10 days of BA-j treatment, the tumor volume was decreased by >50% of the original tumor size in mice bearing SGC-7901 and H-460 xenografts (P < 0.05). These results indicate that the administration of BA-j can reduce the volume of SGC-7901 and H-460 tumors. The growth inhibition in mice bearing SGC-7901 and H-460 tumors on the 30th day after randomization was 76.7 ± 8.2% and 82.1 ± 10.3%, respectively, compared with the control group (n = 9, P < 0.01), as shown in Fig. 2d,e. These results indicate that BA-j (10 mg/kg) can reduce the growth of SGC-7901 and H-460 xenografts in vivo by >75%.
LD50 of injection administration of BA-j in mice is 1g/kg. To examine the effects of BA-j on normal mice, oral administration of 10 mg/kg BA-j twice daily for 10 days was conducted, and it showed almost no toxicity or abnormality by tissue sections. These results indicated that BA-j had no effects on normal mice.
Molecular mechanism of inducing apoptosis in cancer cells
Morphological observation of apoptosis induction in MCF-7 cells
Hoechst 33258 is an active fluorochrome that specifically binds DNA, mainly at A-T rich areas, emitting a blue fluorescence under UV excitation. The nuclei of living cells emit a uniform fluorescence, while apoptotic cells emit a bright blue fluorescence because of chromatin condensation and dense staining of the compact nuclei. After being treated with BA-j for 48 h, MCF-7 cells were stained by Hoechst 33258. Effects on the cellular morphology of these cells are shown in Fig. 3a. Increasing level of BA-j resulted in a sharp decrease in the number of living cells, as well as dense fluorescence emission from nuclei and the appearance of apoptotic bodies around the karyotheca margin, in a remarkably dose-dependent manner. These results indicate that BA-j can significantly induce apoptosis in MCF-7 breast cancer cells.
Figure 3. Apoptosis Effects Assays of BA-j.

(a) Fluorescent images of Hoechst 33258 stained MCF-7 cells showing the induction of apoptosis following a 48 h BA-j treatment (200×). (b) Effects of BA-j treatment (48 h) on the cell cycle in MCF-7 and HL-60 cells were determined using PI staining. (c) Treating MCF-7 and HL-60 for 48 h with BA-j induces apoptosis. Cell apoptosis was demonstrated using the Annexin V-FITC and PI method. (d) Inhibition of Bcl-2 expression after a 48 h BA-j treatment in MCF-7 and HL-60 cells. (e–g) The MMP is decreased and caspases are activated in MCF-7 and HL-60 cells treated with BA-j for 48 h. (e) BA-j treatment decreased the MMP in MCF-7 and HL-60 cells. (f) BA-j treatment activated caspase-8 and caspase-3 in MCF-7 cells. (g) BA-j treatment activated caspase-8 and caspase-3 in HL-60 cells.
Effects on the cell cycle
PI can specifically bind to DNA, with a good linear relationship between the fluorescence intensity and binding. After being fixed and stained with PI, the DNA content in cells can be quantitatively analyzed using flow cytometry. Because different cell cycle phases have varying levels of DNA content, the effects of a drug on progression through the cell phases can thus being determined. There are many distinct changes at the cellular and molecular level that occur during apoptosis, among the most characteristic of which are changes to the nucleus. Activated endonuclease results in DNA degradation, a decrease in the number of apoptotic cells, and the appearance of a hypodiploid apoptosis peak (sub-G1) before the G0/G1 peak. The results of PI fluorescence staining of MCF-7 and HL-60 cells treated with BA-j for 48 h are shown in Fig. 3b. BA-j treatment led to dose-dependent arrests in the G1 and G2 phase in MCF-7 and HL-60 cells, and a dose-dependent growth in the proportion of the apoptotic peak (sub-G1). Additionally, a high level of the drug resulted in a reduction in the number of cells in S phase. There was no DNA <185 Da in sub-G1. These results indicate that BA-j can induce G1 and G2/M arrest, reduce the number of cells in S phase, and induce apoptosis, rather than inflammation and necrosis, in MCF-7 and HL-60 cells.
Apoptosis induction assay
A series of morphological changes occur during apoptosis; changes in the plasma membrane are characteristic of early apoptosis. When cells begin to undergo apoptosis, the cell membrane phospholipid phthalein serine (PS) turns from the inside to the outside of the membrane, becoming exposed on the cell surface. Annexin V is a Ca2+-dependent phospholipid-binding protein, which possesses a high affinity to PS and can specifically bind to the PS exposed on the outside of the plasma membrane. However, PS transfers to the outside of the membrane not only during apoptosis but also during cell necrosis. The two mechanisms of death can be distinguished by the fact that the membrane is intact during the initial phases of apoptosis, but broken in cells undergoing necrosis. PI is a dye for nucleic acid that can permeate the membrane and stain the nuclear content in advanced stages of apoptosis and necrosis, but cannot penetrate the intact membrane of living and early-apoptotic cells. Thus, using Annexin V-FITC together with PI, the ratio of apoptotic and necrotic cells can be determined by flow cytometry. The results of co-staining with Annexin V-FITC and PI fluorescent dyes in MCF-7 and HL-60 cells treated with BA-j for 48 h are shown in Fig. 3c. BA-j dose-dependently induced apoptosis in MCF-7 and HL-60 cells. When MCF-7 and HL-60 cells were treated with 40 μM BA-j for 48 h, the percentage of early necrotic cells did not change, whereas the percentage of early apoptotic cells increased to 34.2% and 29.6%, respectively, and that of late apoptotic cells to 16.9% and 9.8%; therefore, total apoptotic cells increased to 51.1% and 39.4%. These data show that the amount of early apoptotic cells remarkably increased with higher drug level, while the increase in late apoptotic cells at this level was more modest. In contrast, the cytotoxic anticancer drug HCPT induced apoptosis at a low level, but inflammation and necrosis at a high level. These results suggest that unlike cytotoxic anticancer drugs, the anti-proliferative activity of BA-j in MCF-7 and HL-60 cells is associated with exclusively inducing apoptosis rather than directly killing cells or causing cell inflammation and necrosis.
BA-j decreases the expression of Bcl-2
The endoplasmic reticulum plays an important role in cell apoptosis; the anti-apoptotic protein Bcl-2 and the pro-apoptotic protein Bax reside on it. The balance of Bcl-2/Bax regulates the promotion and inhibition of cell apoptosis. Inhibiting the expression of Bcl-2 can result in the activation of caspases, leading to a cascade reaction, ultimately resulting in cell apoptosis. The expression levels of Bcl-2 in MCF-7 and HL-60 cells treated with BA-j for 48 h are shown in Fig. 3d. The expression of Bcl-2 showed a dose-dependent decrease following BA-j treatment, while there was no difference in Bax expression. These results suggest that BA-j inhibits the expression of Bcl-2, and this inhibition is associated with activation of the intracellular endoplasmic reticulum apoptosis pathway.
BA-j decreases the mitochondrial membrane potential
Mitochondria play a key role in cell apoptosis. The asymmetric distribution of protons and other ions on either side of the mitochondrial membrane creates the mitochondrial membrane potential (MMP). Decreasing the MMP are results in the opening of mitochondria permeability transition pores, and the release of pro-apoptotic active substances, such as cytochrome C, from the mitochondrial matrix into the cytoplasm. This results in the activation of caspase-9, which is the start of the mitochondrial apoptosis pathway, and causes a cascade reaction leading to further activation of caspase-3, ultimately resulting in cell apoptosis. Rhodamine 123 is an MMP indicator that can enter the mitochondrial matrix of living cells depending on the current MMP. When Rhodamine 123 is in the mitochondrial matrix, its fluorescence is diminished. During apoptosis, the integrity of the membrane is undermined and mitochondria permeability transition pores open, causing a collapse of the MMP and the release of Rhodamine 123 from mitochondria. This results in a strong, yellow-green fluorescence, whose intensity can be used to determine changes in the MMP. MCF-7 and HL-60 cells were treated with BA-j for 48 h and their Rhodamine123 fluorescence staining results are shown in Fig. 3e. The MMP was decreased with BA-j treatment in a dose-dependent manner. These results suggest that BA-j decreases the MMP, and this decrease is associated with activation of the intracellular mitochondria apoptosis pathway.
Activation of caspase-3 and caspase-8
Caspases (cysteine aspartic specific proteases) are a family of aspartic proteases containing cysteine, which possess similar structures and exist in the cytoplasm. Caspases are closely linked to apoptosis and are also involved in the regulation of cell growth and differentiation. Caspases are key enzymes in the process of apoptosis. Caspases are involved in almost all of the signal transduction pathways that mediate apoptosis. Caspases 8, 9, and 12 are early stage initiators of cell apoptosis, and caspases 3, 6, and 7 are late-stage executors. The active site of the caspases is a sulfydryl of cysteine, which exists in the inactive pro-caspase state and is sensitive to H2O2. When a pro-caspase is oxidized by H2O2 into a -S-S- dimer, it transforms into an active caspase-S-S-caspase, thus inducing apoptosis. If the sulfydryl of pro-caspase-9 is free, it will combine with phosphorylated survivin, which inhibits apoptosis and promotes cell division. Conversely, if the sulfydryl of pro-caspase-9 is oxidized by H2O2, it will separate from the phosphorylated survivin, which inhibits cell division and promotes apoptosis. MCF-7 and HL-60 cells were treated with BA-j for 48 h, and the activities of caspase-3 and caspase-8 were determined, as shown in Fig. 3f,g. The activity of caspase-3 and caspase-8 increased in a dose-dependent manner. These results indicate that the induction of apoptosis by BA-j in MCF-7 and HL-60 cells is associated with the activation of caspase-8 and caspase-3, via direct H2O2 oxidation, rather than death receptor-mediation.
BA-j inactivates Fas/FasL
Death receptors are transmembrane proteins that have numerous extracellular cysteine residues and possess intracellular proteolytic activity that further transmits the death signal. Death receptors belong to the tumor necrosis factor receptor family that includes Fas/FasL, TNFR-1/TNFR-α and TRAILR/TRAIL. Tumor necrosis factor receptors cannot be oxidized and activated by ·O2− or H2O2, but can be oxidized and activated by NaOCl, which has a stronger oxidation power. Cytotoxic agents can stimulate mitochondria to release NaOCl and activate death factor receptors, thus causing cell inflammation and necrosis.
Hep G2 cells were treated with BA-j for 48 h, and the expression of Fas/FasL was determined. As shown in Fig. 4, there was no dose-dependent effect of BA-j on Fas levels, whereas 2 μM of HCPT, a cytotoxic anticancer drug used as a positive control, showed an obvious induction of Fas/FasL expression.
Figure 4. Expression of Fas/FasL in Hep G2 cells treated with BA-j for 48 h compared with HCPT treatment.

2 μM HCPT was used.
These results suggest that the cell inflammation and necrosis caused by cytotoxic anticancer drugs is closely associated with extrinsic death receptors, which results in damage to both cancer cells and normal cells. These data also suggest that BA-j’s bypassing of the extrinsic death receptor pathway is associated with its resistance to NaOCl oxidation, which allows BA-j to protect normal cells from inflammation and necrosis.
Biochemical mechanism of inducing apoptosis in cancer cells by regulating ROS
Resistance to NaOCl oxidation is essential for protecting cells from inflammation and necrosis
BA-j (E0 = 0.80 ev) can be oxidized into 8-hydroxymethyl-5, 6-dehydrogenation-baicalein by capturing ·O2− and releasing H2O2 (determined by PF1 oxidizing into fluorescein), but BA-j cannot be oxidized by H2O2. BA-j can also be oxidized into 8-hydroxymethyl-5, 6-dehydrogenation-baicalein by NaOCl without releasing H2O2 (thus, PF1 is not oxidized into fluorescein), hydrogenating NaOCl into NaCl and H2O. ·O2− has a short survival, and it cannot directly oxidize a sulfhydryl of cysteine. H2O2 (E0 = 0.68 ev) can oxidize a sulfhydryl of cysteine into a disulfide bond rather than a sulfur-oxygen group. NaOCl (E0 = 1.49 ev) can oxidize a sulfhydryl of cysteine into both disulfide bonds and sulfur-oxygen groups. These results suggest that the resistance of BA-j to NaOCl oxidation is associated with protecting cells from inflammation and necrosis.
Increased intracellular H2O2 level is essential for selectively inducing apoptosis in cancer cells
We established a method of measuring the intracellular H2O2 level using PF1 selective fluorescent probes. To investigate the mechanism of the selective induction of apoptosis in cancer cells and the regulation of intracellular reactive oxygen species (ROS) by BA-j, an equal amount of primary human hepatocytes (Liver) and human peripheral blood mononuclear cells (PBMCs) were prepared and treated with BA-j for 48 h and compared with Hep G2 and MCF-7 cancer cells in intracellular H2O2 tests (PF1 method). Fig. 5a shows the fluorescence in Hep G2 and MCF-7 cancer cells treated with 12.5 μM BA-j for 4 h. Treating cells with a lower level of BA-j resulted in a weaker fluorescent emission, whereas higher levels of BA-j made most of the cells become apoptotic and increased the number of fluorescent bodies.
Figure 5. Comparison of BA-j effects on cancer cells and normal cells.

(a) Fluorescent imaging of intracellular H2O2 (detected by PF1) in Hep G2 and MCF-7 cancer cells compared with normal Liver cells and PBMCs. All cells were treated with 12.5 μM BA-j for 4 h. (Top: bright field, Bottom: excitation wavelength 488 nm, emission wavelength 525 nm, 64×). (b) Assay of intracellular H2O2 following drug treatment in Hep G2 and MCF-7 cancer cells compared with normal Liver cells and PBMCs. The cells were cultured in the presence of drug for 4 h and the level of intracellular H2O2 was detected by PF1.
Hep G2, Liver, MCF-7 and PBMCs were incubated with BA-j, BA, or HCPT for 4 h and the level of H2O2 was detected using PF1 fluorescence probes, as shown in Fig. 5b. When Hep G2 and MCF-7 cancer cells were incubated with BA-j, H2O2 levels exhibited a remarkable dose-dependent increase, whereas Liver cells and PBMCs did not. BA-j can selectively increase the H2O2 level in Hep G2 and MCF-7 cancer cells rather than in normal Liver cells and PBMCs by capturing ·O2− and releasing H2O2. BA-j is also associated with selectively inducing apoptosis in cancer cells but not in normal cells. By contrast, the results indicated that the cytotoxic drug HCPT had no selectivity in increasing H2O2 levels; it would harm normal cells and kill cancer cells. BA-j also was resistant to NaOCl oxidation, which is associated with preventing inflammation and necrosis in cells.
Metabolic pathway capturing ·O2 − and releasing H2O2
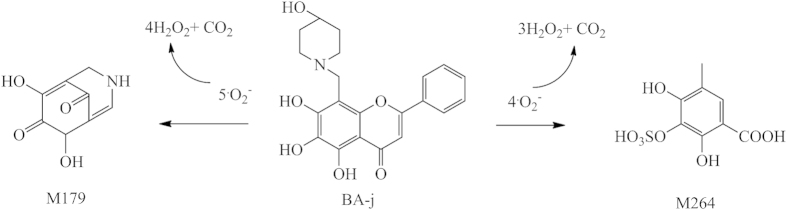
The major metabolic pathway of BA-j in macaques was identified by LC-MS-MS. The major metabolic pathway involved BA-j capturing 4-5 molecules of ·O2− and releasing 3-4 molecules of H2O2 based on the data given in Fig. 5b, with itself degrading into active intermediate metabolite dihydroflavonol, then while being oxidized and degraded mainly into the metabolites M179 and M264, as shown in Fig. 6. 95% of the BA that entered the body was excreted after glucuronidation or sulfation, and only a few of the BA molecules captured 1 molecule of ·O2− and released 1 molecule of H2O2 each, thus becoming oxidized. 4-hydroxylpiperidine possessed a strong activity against oxygen radicals because it captured multi-molecular ·O2−, and released multi-molecular H2O2 as it became oxidized and degraded. Introduction of 4-hydroxylpiperidine methylene into BA-activated flavonoid rings resulted in the easy capture of ·O2− and self-degradation. Meanwhile, the “enterohepatic efflux effect” by direct glucuronidation and sulfation was obviously inhibited. These results suggest that BA-j captures ·O2− in vivo, is degraded into M179 and M264, and releases H2O2, which is associated with increasing the intracellular H2O2 level in cancer cells. This metabolic pathway can inhibit the BA-j “efflux effect” caused by direct glucuronidation and sulfation. The pharmacokinetic nature of BA-j was better than that of BA.
Figure 6. The major metabolic pathway of BA-j in macaques.
Discussion
David Santamarıá et al. indicated in his 2007 article in Nature that CDK1 is the only essential CDK in cell proliferation. Moreover, in the absence of interphase CDKs, CDK1 can execute all the events that are required to drive cell1. When CDK1 is phosphorylated into CDK1 (p-cdc2-Tyr15-Thr14) by Weel and Mytl protein kinase, CDC25C dephosphorylate it into active CDK1 (p-cdc2-Tyr15), which combines Cyclin B1 and phosphorylate the substrate Survivin into Survivin (p-Thr14), thus promoting the cells into mitosis phase47. In 2010, Jorrit M Enserink and Richard D Kolodner posed that CDK1 could be used as the latest target for anti-proliferation drug research48.
BA is known to have selective inhibitory ability against CDK1, and inhibitory activity is weak against other kinases32. We found that BA-j as a substrate compound for ATP can directly inhibit the activity of CDK1 studied by FRET which is the enzyme activity determination method of the CDK1 in non-cancer cells, and the inhibitory activity of BA-j (CC50 0.30 μM) against CDK1/Cyclin B1 is approximately 20-fold and 50-fold stronger than BA and Baicalin. Because 4-hydroxy-piperidin as a potent antioxidant can capture multi-molecules of ·O2− and release multi-molecules of H2O2, BA was structurally modified into BA-j by Mannich with 4-hydroxy-piperidin, and the druggbility of BA-j is more excellent. It is worth noting that the former is the activity determination result of the CDK1 enzyme of non-cancer cells using FRET method, while the latter is the determination result in cancer cells in vitro by MTT. Principles are different between these two methods. Therefore, the results can only be compared qualitatively, but not quantitatively.
In Hep G2 cells, we founded that BA-j can selectively inhibit CDK1/Cyclin B1, rather than CDK2/Cyclin E or CDK4, 6/Cyclin D. BA-j can inhibit CDC25C too with dephosphorylating CDK1 (p-cdc2-Tyr15-Thr14) into active CDK1 (p-cdc2-Tyr15). Because BA-j can capture ·O2− and selectively increase the level of H2O2 in cancer cells rather than normal cells, which can oxidize CDC25C, and the dephosphorylation of CDK1is inhibited, but not other CDKs, thus inhibiting cell proliferation.
These results suggest that BA-j as a substrate compound for ATP is not only a direct inhibitor of CDK1/cyclin B, but also an indirect inhibitor of CDK1/cyclin B1 by H2O2 oxidizing CDC25C rather than of other CDKs. Therefore, BA-j is a novel selective CDK1 inhibitor.
BA-j showed broad-spectrum anti-cancer activity (IC50 12.3 μM, 4.7 μg/ml) and approximately 4-fold stronger than BA (IC50 50 μM) measured by MTT11. The anti proliferative activity of BA and Baicalin is not significant in vivo, because of their poor bioavailability by “enterohepatic efflux effects”. Because druggbility of BA-j is more excellent than BA, BA-j can significantly reduce the growth of SGC-7901 and H-460 xenografts (10 mg/kg, 10 day, >75%). The SFDA “guidelines for antitumor drug research” provide that when the IC50 values of compounds or natural drugs inhibiting cancer cell proliferation are less 10 μg/ml or 20 μg/ml, respectively, they are considered as having anticancer activity in vitro. When the tumor growth inhibitory rate is more than 40%, a drug is considered as having significant anti-tumor activity in vivo. BA-j showed no significant side effects within the therapeutic dosage range. Accordingly, the data suggest that BA-j has significant anti-tumor activity with high effect and lower toxicity. ROS are important messengers in cell signaling processes and form the biochemical basis of many physiological and pathological responses. ROS include oxygen free radicals (·O2−), hydroxyl radicals (·OH−), hydrogen peroxide (H2O2), sodium hypochlorite (NaOCl), nitric oxide (NO), lipid peroxides (ROOH), over oxygen nitrite (·ONOO−) and so on. H2O2 has a high diffusivity and can pass through the membrane. High levels of H2O2 can oxidize NaCl into NaOCl by catalysis with catalase (CAT).
The mechanisms of redox states are different between normal cells and cancer cells. In normal cells, where the activity of superoxide dismutase (SOD) is relatively high, over 95% of the ·O2− generated can be captured and composed into H2O2 by SOD. Then, excessive H2O2 is catalyzed and decomposed into H2O and O2 by glutathione enzyme (GPX), which oxidizes glutathione (GSH) into oxidized glutathione (GSSG); thus SOD and GPX maintain relatively low levels of ·O2− and H2O2, and the cells remain in a non-proliferative state. Whereas in tumor cells, the activity of SOD and CAT is weaker than in normal cells, leading to the accumulation of ·O2−, which promotes cell proliferation and increases the sensitivity of these cells to H2O2. Compared with normal cells, tumor cells are more likely to be induced to undergo apoptosis by the same level of H2O2.
Carole Nicco’s research group from Faculté de Médecine, University Paris V demonstrated for the first time that among the antioxidant enzymes that detoxify H2O2, the glutathione pathway is only weakly involved in the control of H2O2 production by tumor cells, but plays a major role in the control of H2O2 production in non-transformed cells. Thus, in normal cells, ROS are kept at low levels because the activity of NADPH oxidase and the H2O2 levels are regulated by the glutathione system. By contrast, in tumor cells, high levels of ROS, close to the threshold of cytotoxicity, are produced through the mitochondrial respiratory chain, and the H2O2 level is controlled by CAT. This discovery suggested for the first time that any agent that increases intracellular H2O2 levels could slow down cancer proliferation and lead to cell apoptosis. Conversely, any agent that decreases the intracellular H2O2 level could enhance tumor growth. H2O2 plays a pivotal role in controlling the fate of normal and tumor cells. Therefore, drugs that specifically increase intracellular H2O2 level in cancer cells can selectively induce cancer apoptosis46.
The effects of ROS on cell proliferation depend on their nature, sub-cellular origin and intracellular level. The intracellular level of H2O2 is critical because it easily reaches the threshold of toxicity in tumor cells. When some stimulation causes the H2O2 level in tumor cells to climb high enough, kinases with their active site of cysteine can become oxidized, for instance, H2O2 can cause depletion of the GSH content in cancer cells, target oxidation of CDC25, and inhibit activation of CDKs/Cyclins, suppressing proliferation47. H2O2 also can specifically oxidize caspases, thus activating the apoptotic pathway of mitochondria and endoplasmic reticulum and selectively inducing apoptosis in tumor cells. At low levels, cytotoxic drugs can improve the intracellular H2O2 level in cancer cells, oxidizing and activating the intrinsic apoptosis pathway. However, at high levels, they can stimulate mitochondria to produce NaOCl by H2O2, oxidizing NaCl with CAT catalysis, thus activating the extrinsic death receptors pathway and causing cell inflammation and necrosis. Therefore, cytotoxic drugs damage normal cells while killing cancer cells49.
The molecular mechanism responsible for the selectivity of BA-j is a result of the differential regulation of ROS between normal cells and cancer cells, which allows BA-j to selectively induce apoptosis in cancer cells while protecting normal cells from inflammation and necrosis. In normal cells, the activity of SOD and GPX is high, and ·O2− levels are low. BA-j captures ·O2− and produces small amounts of H2O2 that can be maintained at a very low level by GPX control, so the cells remain in a non-proliferative state. Whereas in cancer cells which have lower activity of SOD and CAT than normal cells, ·O2− is kept at a higher level. BA-j, similar to SOD, can capture ·O2− but produces more amounts of H2O2, which is maintained at a higher level.
The higher level of H2O2 can specifically oxidize kinases with active site of cysteine. For example, in cancer cells, H2O2 can cause depletion of GSH content50, target oxidation of CDC25, and inactivate CDKs/Cyclins, thus suppressing cancer proliferation15,19,25,32,47. H2O2 can specifically oxidize and activate the intrinsic apoptosis pathway (i.e., reduce the mitochondrial membrane potential, inhibit expression of Bcl-2, and activate caspase-8 and caspase-3, and therefore inducing apoptosis in cancer cells.
BA is known to have ROS-generating ability. However, the exact biochemical mechanism of BA regulating ROS is not understood. Because of their using of DCFH-DA, a non selective a probe, only the total ROS in cells was determined, but the differences among ·O2−, H2O2 and NaOCl were not distinguished. Therefore, questions such as why BA is different from cytotoxic anti-cancer drugs which harm normal cells while killing cancer cells could not be explained33,51.
In this paper, a novel method for assaying intracellular H2O2 was designed by using the H2O2 selective fluorescent probe PF152. We First confirmed that BA-j can regulate ROS level (reducing ·O2−, increasing H2O2, reducing NaOCl), but cytotoxic anti-cancer drugs can increase the ROS level (increasing ·O2−, H2O2, and NaOCl). Because of the differential mechanisms controlling redox states in normal and cancer cells, BA-j can capture oxygen free radicals (·O2−) and selectively increase the level of H2O2 in cancer cells, thereby specifically oxidize some enzymes with active site of cysteine, specifically oxidize and activate the intrinsic apoptosis pathway. BA-j can also resist oxidation by NaOCl, bypassing the extrinsic death receptor pathway (i.e., inactivate tumor necrosis factor receptor Fas/Fasl), which allows it to protect cells from inflammation and necrosis16,31. BA-j showed a higher activity but similar molecular mechanism to BA; see Fig. 7. This mechanism of BA-j is different from cytotoxic anticancer drugs, which can activate both the intrinsic apoptosis pathway and the extrinsic death receptors pathway, and therefore harm normal cells while killing cancer cells.
Figure 7. Mechanisms of ROS regulations.

(a) Molecular mechanisms of H2O2–regulated cell proliferation and apoptosis. (b) Mechanisms of ROS regulation by BA-j. BA-j can selectively induce apoptosis in cancer cells and protect normal cells from inflammation and necrosis.
Conclusion
The molecular and biochemical mechanisms of BA-j is the direct inhibition of CDK1 and capturing ·O2− and increasing level of H2O2 in cancer cells rather than in normal cells. Higher level of H2O2 is capable of specifically oxidizing kinases with cysteine at their active sites, such as oxidizing CDC25 with inactivating CDK1, and thus inhibiting proliferation cells, and oxidizing Caspases and activating the intrinsic apoptosis pathway thus inducing apoptosis in cancer cells rather than in normal cells. BA-j can also resist NaOCl oxidation, bypassing the extrinsic death receptor pathway, which allows it to protect cells from inflammation and necrosis. BA-j has broad-spectrum anticancer activity and can significantly reduce the growth of SGC-7901 and H-460 xenografts. These data provide evidence that BA-j may be developed into a novel anticancer drug.
Materials and Methods
Ethical Statements
Animal experiments were conducted in accordance with the guidelines issued by the State Food and Drug Administration (SFDA of China). The research and protocol were approved by the Animal Care and Use Committee of Dalian Medical University in China. The animals were housed and cared for in accordance with the guidelines established by the National Science Council of Republic China. The present study was performed in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council, NIH Publication No. 85–23, 1996). Briefly, male (6–8 week-old, 18–22 g, mixed-sex) BALB/c nude mice were provided by the Animal Care and Use Committee of Dalian Medical University in China.
Assay of intracellular H2O2 by PF1
PF1 was designed as an intracellular H2O2 selective fluorescent probe by Christopher J. Chang from the University of California-Berkeley. This probe is permeable through the cell membrane, stable and selective. PF1 is not fluorescent on its own, but it can be oxidized into yellow-green fluorescein FITC selectively by intracellular H2O2, rather than by ·O2− or NaOCl. PF1 is currently the only truly effective tool for assessing intracellular H2O2 levels induced by a drug52. A novel method to assay intracellular H2O2 was designed using the selective H2O2 fluorescent probe PF1 (synthesized by us), which was better than the non-selective ROS fluorescent probe DCFH-DA (2′, 7′-dichlorofluorescin diacetate)49.
Determination method: cancer cells were seeded in a 96-well black-transparent micro-plate (1 × 105 cells/ml and 100 μl/well) and incubated at 37 °C in an atmosphere of 5% CO2 for 30 h, or for 6 h in the case of normal cells. Then, 10 μl of a PBS-drug solution (drug was prepared as 4 mg/ml in DMSO and diluted with PBS to 500, 250, 125, 62.5, 31.25 and 0 μM) was added to 4 replicates. Cells were incubated for 3.5 h, then 10 μl of a PF1 fluorescent probes solution was added (a stock solution of PF1 was prepared as 1 mg/ml in DMSO and diluted with PBS to a 100 μM working solution), and cells were further incubated at 37 °C for 0.5 h while the micro-plate was shielded by foil. The fluorescence intensity was detected using fluorescence ELISA (excitation wavelength 488 nm, emission wavelength 525 nm), and the H2O2 concentration was calculated. For adherent cells, the foil and the media were removed, and the cells were washed with 200 μl PBS before being subjected to intracellular H2O2 fluorescence imaging via the fluorescence microscope. For suspended cells, 200 μl PBS was added to the media, and after 5 min, cells from the bottom of each well were collected and plated on glass coverslips and subjected to intracellular H2O2 fluorescence imaging via the fluorescence microscope.
Standard curve and quantitative limits: 90 μl of plasma culture media was added to each well, and then 10 μl of a PBS-H2O2 solution (H2O2 solution was diluted with PBS to 500, 250, 125, 62.5, 31.25 and 0 μM) was added to 4 replicates. Next, 10 μl of a 100 μM PF1 fluorescence probes solution was added, and micro-plates were shaken for 0.5 h at 37 °C. Then, the fluorescence intensity was measured and the standard curve was drawn.
The concentration of H2O2 in the plasma media ranged from 3 to 100 μM, and each well was treated with 10 μM PF1. With the square root of the blank correcting fluorescence OD differences as the abscissa (X), and the concentration of H2O2 (μM) as the ordinate (Y), Linear regression equation: Y = 0.592X (R2 = 0.995, n = 6) was derived, and a good linear relationship was observed.
CDK1/Cyclin B1 inhibition assay by FRET
The quantitative analysis technique of Fluorescence Resonance Energy Transfer (FRET) was adopted for screening the CDK1/Cyclin B inhibitory activity of the flavonoid Mannich base derivatives. Generally, the test compounds were prepared as 1 mg/ml DMSO solutions and diluted with DMSO into 8 geometric concentrations (3-fold). CDK1/Cyclin B inhibitory activities were measured by a fluorescence kinetic assay and CC50 values were calculated according to the instructions of the CDK1/Cyclin B kit (Invitrogen, USA)44.
Cell culture
Breast (MCF-7 [TCHu 74] and MDA-MB-231 [TCHu104]), cervical (Hela [TCHu187]), hepatoma (SMMC-7721 [TCHu 52] and Hep G2 [TCHu 72]), gastric (SGC-7901 [TCHu 46] and BGC-823 [TCHu 11]), prostate (PC-3 [TCHu158]), leukemia (HL-60 [TCHu 23] and K562 [TCHu191]), non–small cell lung carcinoma (H-460 [TCHu205]), and colon (HCT-116 [TCHu 99]) cancer cell lines were purchased from the Cell Bank of Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, China). The catalogue number of each cell line is indicated in the bracket following its name.
The cell lines were seeded in 4 × 6 cm flasks and cultured in DMEM (Sigma) supplemented with 10% heat-inactivated fetal bovine serum (Gibco), 100 U/ml Penicillin and 100 μg/ml streptomycin in a water-saturated atmosphere of 5% CO2 at 37 °C. The culture medium was changed every 48 h and cells were sub-cultured approximately once every 96 h. For sub-culturing, the medium was discarded, and the appropriate amount of 0.25% trypsin was added for a 2–3 min digestion at 37 °C. Observed under the microscope, these cells tended to become round. 10% FBS was added to terminate the digestion, and the cell solution was gently beaten with a straw several times so that cells were detached from the culture bottle wall and completely dispersed. These cells were divided into cell culture flasks and the appropriate amount of medium was added for further culture. For HL-60 cells, part of the medium was discarded leaving the appropriate amount of cells and medium, and fresh medium was added. For plating experiments, cells were observed and counted under the microscope, and cells in the logarithmic growth phase were used. RPMI 1640 medium was added for dilution, and cells were counted under the microscope and adjusted to 1 × 105 cells/ml.
Normal human primary hepatocytes (Liver) were purchased from the Dalian Institute of Physical Chemistry (Dalian, China). Liver was cut into small pieces and digested with the appropriate amount of 0.25% trypsin at 37 °C for 2–3 min. Observed under the microscope, these cells tended to become round. 10% FBS was added to terminate the digestion, and the cell solution was gently beaten with a straw several times so that cells were detached from the culture bottle wall and completely dispersed. RPMI 1640 medium was added for dilution, and the cells were counted under the microscope and adjusted to 1 × 105 cells/ml.
Normal human peripheral blood was purchased from the Dalian city blood bank and approved by the human blood use committee of the Dalian city blood bank in China. Normal human peripheral blood mononuclear cells (PBMCs) were isolated from heparinized venous blood of healthy volunteers by Ficoll–Paque density gradient centrifugation. After anticoagulation by heparin, 1 ml of fresh human peripheral blood was washed with PBS twice, and then 1 ml of Ficoll-PM400 was carefully added into the bottom of the test tube, forming a single layer. After 1000 r/min centrifugation for 7 min, the intermediate buffy coat layer was separated. RPMI 1640 medium was added for dilution, and the cells were counted under the microscope and adjusted to 1 × 105 cells/ml.
Proliferation assay by MTT
Cells were seeded in 96-well micro-plates (5 × 103 cells/well, HL-60 1 × 104 cells/well) and incubated at 37 °C for 24 h. In dark conditions, BA-j-DMSO solutions were diluted with PBS and added to the appropriate wells with final concentrations of 0, 5, 10, 20, and 40 μM, with 4 replicates of each condition. For the control, PBS containing 0.1% DMSO was added instead of the BA-j solution. The micro-plates were incubated at 37 °C for 48 h, and then the media were removed. 100 μl of a 0.5 mg/ml MTT solution diluted with PBS was added to the wells, and the micro-plates were further incubated for 3 h. After incubation, the MTT solution was removed, and 100 μl of DMSO was added to each well to dissolve the formazan. Micro-plates were read at 570 nm (630 nm as a reference) by a micro-plate reader (Sunrise, TECAN, Austria), according to Tim Mosmann’s method.
Tumor xenograft model
Human gastric carcinoma (SGC-7901) cells were chopped into fragments (approximately 1.5 mm), each of which was transplanted s.c. into the right axillary fossa of BALB/c nude mice. Human non–small carcinoma (H-460) cells were grown in RPMI 1640 containing 10% fetal bovine serum and harvested. Cells were resuspended in saline at 2.5 × 107 cells/ml and placed on ice. Severe combined immunodeficient mice were injected with a 0.2 ml cell suspension s.c. on the right flank and observed daily for tumor appearance. When the tumors attained a diameter of 5 mm, tumor xenograft models were randomized into two groups (n = 9). For 10 days, a 10 mg/kg BA-j mesylate injection was orally administered twice daily in the test group, and water was administered in the control group. Animals in both models were observed daily for signs of health deterioration and mortality for 30 days. Tumor size was measured once every 5 days in two perpendicular dimensions with vernier calipers and converted to tumor volume (TV) using the formula: (ab2)/2, where a and b refer to the longer and shorter dimensions, respectively. The body weight of the animals was measured once every 5 days, concurrently with the tumor dimension measurements. After observation for 30 days, mice were euthanized by cervical dislocation (this method of animal sacrifice is in accordance with the criteria set up by the Animal Care and Use Committee of Dalian Medical University in China) and weighed, and the tumor was segregated and weighed. No signs of ill health, pain, or suffering were observed, and therefore no action was taken to minimize suffering. There were no obvious adverse effects or mortality during the experiment.
Morphological observation of nuclear change
Hoechst 33258 staining method was adopted for analysis under dark conditions. Cells were seeded in 96-well micro-plates (1 × 106 cells/ml, 100 μl/well), and pre-incubated at 37 °C. Then, different concentrations (10, 20 and 40 μM) of BA-j were added, and the micro-plates were further incubated for 48 h. For staining, media were removed and cells were washed with PBS 3 times before being fixed in 0.5 ml of cold, 4% paraformaldehyde for 30 min at room temperature. Then, fixative was removed, and cells were washed with PBS 3 times, stained with 0.5 ml of 10 μg/ml Hoechst 33258 (Sigma) at 37 °C for 10 min, and incubated at 37 °C in 5% CO2 for 10 min. The presence of nuclear morphological changes in the cells was observed with an Olympus fluorescence microscope fitted with a UV excitation filter (Tokyo, Japan).
Cell cycle analysis
Propidium iodide (PI), which binds DNA, was used for staining analysis. Cells were seeded in flasks (5 × 104 cells/ml, 3 ml/flask), and cultured for 24 h in the dark. Then, different concentrations (10, 20 and 40 μM) of BA-j were added, and the micro-plates were further incubated for 48 h. After culturing, cells were collected (approximately 1 × 104), centrifuged at 1000 r/min for 5 min, and washed with cold PBS 3 times. Next, cells were fixed in 70% cold ethanol at 4 °C overnight. Then, fixative was removed, cells were washed with PBS twice, and incubated with DNase-free RNase (100 μg/ml) at 37 °C for 30 min. After incubation, the cells were re-suspended in 10 μg/ml PI (Sigma) solution and incubated at 4 °C for another 30 min in the dark, and then subjected to cell cycle analysis using flow cytometry. Flow cytometry (BD FACS cantoTM, USA) was used to detect cells in different phases of the cell cycle, and the percentages of cells in G1, S, and G2 phases were calculated using ModFit LT 3.0 software.
Apoptosis assay
Annexin V-FITC antibody immunofluorescence combined with PI/DNA binding was adopted for fluorescent analysis of apoptosis. Cells (2 × 105) were collected and subjected to quantitative flow cytometry analysis according to the instructions of the Annexin V-FITC kit (BioVision, USA).
Assay of Bcl-2 and Bax expression
Immunofluorescence was used to analyze Bcl-2 and Bax protein expression. Cells (1 × 106) were washed with cold PBS 3 times, fixed with 4% paraformaldehyde at 4 °C for 20 min, and then incubated with 0.5% TritonX-100 and 1% bovine serum albumin for 10 min. The cells were then washed with PBS twice, followed by the addition of primary antibodies against Bcl-2 or Bax (Santa Cruz) and further incubation at 37 °C for 1 h. Next, the cells were washed with PBS twice and incubated with the corresponding fluorescein isothiocyanate (FITC)-conjugated secondary antibodies at room temperature for 45 min in the dark. The cells were washed with PBS again, and the antigen density was analyzed by flow cytometry as described with minor modifications.
Assay of mitochondrial membrane potential
Mitochondrial membrane potential (MMP) changes were detected and analyzed with the Rhodamine 123 detection kit (Rh123, Nanjing Keygen Biotech., China). Cells (1 × 106) were incubated with 10 μg/ml Rh123 solution for 10 min at 37 °C in an atmosphere of 5% CO2. After incubation, the cells were washed twice with PBS to remove free Rh123, and then incubated in PBS for 60 min at 37 °C in an atmosphere of 5% CO2. The fluorescence intensity of Rh123 was measured immediately with Thermo Scientific Varioskan Flash (Thermo Scientific, USA) using excitation and emission wavelengths of 500 nm and 530 nm, respectively.
Caspase activity assay
Because the emission spectrum of free p-nitroaniline can be detected by a micro-plate reader at 405 nm, caspase activity can also be determined. A caspase-8 and caspase-3 colorimetric assay kit (Nanjing KeyGen Biotech., China) was used to determine caspase activity. Cells (2 × 106) were collected in a 1.5 ml centrifuge tube and lysed by 10 min incubation with 50 μl lysis buffer on ice. After 1000 r/min centrifugation at 4 °C for 1 min, the supernatant (i.e., cytoplasmic extract) was carefully pipetted into another tube and kept on ice. The protein concentration of each sample was determined using the Bradford method. Then, according to the protein level of each sample, certain amounts of protein (50–200 μg) were diluted in 50 μl cell lysis buffer for the next step of the experiment. Next, 50 μl of 2× reaction buffer (containing 10 mM DTT) and 5 μl of 4 mM DEVD p-nitroaniline (final concentration 200 μM) were added, and lysates were incubated at 37 °C for 4 h, then transferred into a 96-well micro-plate and read at 40 nm by a micro-plate reader.
Western blot analysis
Hep G2 cells (2 × 106) were lysed, and their protein content was separated by SDS-PAGE and electrophoretically transferred onto a PVDF membrane (Millipore). The membrane was blocked with 5% nonfat milk in TBST buffer and incubated overnight at 4 °C with specific primary antibodies, including anti-human Fas and FasL antibodies (Transduction Laboratory, USA). The membrane was washed with TBST buffer and incubated with the appropriate secondary antibody (horseradish peroxidase-conjugated goat-antimouse IgG). Determinations were performed using enhanced chemiluminescence kits (Amersham Biosciences, USA).
The membrane was blocked with 5% nonfat milk in TBST buffer and incubated overnight at 4 °C with specific primary antibodies, including anti-human CDC25C, CDK1 (p-cdc2-Tyr15-Thr14), CDK1 (p-cdc2-Tyr15), CDK2, CDK4, CDK6, Cyclin B1, Cyclin D, Cyclin E antibodies. The membrane was washed with TBST buffer, incubated with the appropriate secondary antibody (goat-antimouse IgG/ FITC), and the membrane was washed with TBST buffer. Differences in protein level were determined for conjugated goat-antimouse IgG/FITC by densitometric scanning (Quantity One software package, Biorad) and normalized using internal standards (i.e., β-actin)32.
Statistical analysis
Data are expressed as the mean ± S.E.M. from 4 repeated experiments and were evaluated using a one-way ANOVA. Differences are considered significant when P < 0.05.
Additional Information
How to cite this article: Zhang, S. et al. BA-j as a novel CDK1 inhibitor selectively induces apoptosis in cancer cells by regulating ROS. Sci. Rep. 5, 13626; doi: 10.1038/srep13626 (2015).
Acknowledgments
This work part is supported by U.S. National Institutes of Health, Cancer and AIDS Research Center (NCI, D749677-Y) and China Dalian Science and Technology Planning Project Fund (2008E11SF167, 2010E12SF063).
Footnotes
Author Contributions S.X.Z., Y.M.B. and X.L.J. designed the research. S.X.Z., X.L.J., K.J.L., H.Y.S., L.S.H., Y.Q., L.Z., X.D.S., J.L., Q.R.W. and Q.Y.F. conducted the experiments. S.X.Z., X.L.J., H.Y.S. and Y.Q. performed data analysis. S.X.Z., L.S.H., K.J.L. and H.Y.S. wrote or contributed to the writing of the manuscript. And all authors reviewed the manuscript.
References
- Santamaria D. et al. Cdk1 is sufficient to drive the mammalian cell cycle. Nature 448, 811–815 (2007). [DOI] [PubMed] [Google Scholar]
- Malumbres M. & Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 9, 153–166 (2009). [DOI] [PubMed] [Google Scholar]
- Diaz-Padilla I., Siu L. L. & Duran I. Cyclin-dependent kinase inhibitors as potential targeted anticancer agents. Invest New Drug 27, 586–594 (2009). [DOI] [PubMed] [Google Scholar]
- Rizzolio F., Tuccinardi T., Caligiuri I., Lucchetti C. & Giordano A. CDK inhibitors: from the bench to clinical trials. Curr Drug Targets 11, 279–290 (2010). [DOI] [PubMed] [Google Scholar]
- Wesierska-Gadek J., Maurer M., Zulehner N. & Komina O. Whether to target single or multiple CDKs for therapy? That is the question. J Cell Physiol 226, 341–349 (2011). [DOI] [PubMed] [Google Scholar]
- Phelps M. A. et al. Clinical response and pharmacokinetics from a phase 1 study of an active dosing schedule of flavopiridol in relapsed chronic lymphocytic leukemia. Blood 113, 2637–2645 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy B. et al. A dose-finding, pharmacokinetic and pharmacodynamic study of a novel schedule of flavopiridol in patients with advanced solid tumors. Invest New Drug 30, 629–638 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi K. S. et al. in vitro antitumor properties of a novel cyclin-dependent kinase inhibitor, P276-00. Mol Cancer Ther 6, 918–925 (2007). [DOI] [PubMed] [Google Scholar]
- Joshi K. S. et al. P276-00, a novel cyclin-dependent inhibitor induces G1-G2 arrest, shows antitumor activity on cisplatin-resistant cells and significant in vivo efficacy in tumor models. Mol Cancer Ther 6, 926–934 (2007). [DOI] [PubMed] [Google Scholar]
- Raje N. et al. Preclinical activity of P276-00, a novel small-molecule cyclin-dependent kinase inhibitor in the therapy of multiple myeloma. Leukemia 23, 961–970 (2009). [DOI] [PubMed] [Google Scholar]
- Li-Weber M. New therapeutic aspects of flavones: the anticancer properties of Scutellaria and its main active constituents Wogonin, Baicalein and Baicalin. Cancer Treat Rev 35, 57–68 (2009). [DOI] [PubMed] [Google Scholar]
- Horvath C. R., Martos P. A. & Saxena P. K. Identification and quantification of eight flavones in root and shoot tissues of the medicinal plant Huang-qin (Scutellaria baicalensis Georgi) using high-performance liquid chromatography with diode array and mass spectrometric detection. J Chromatogr A 1062, 199–207 (2005). [DOI] [PubMed] [Google Scholar]
- Liu G. et al. Investigation of flavonoid profile of Scutellaria bacalensis Georgi by high performance liquid chromatography with diode array detection and electrospray ion trap mass spectrometry. J Chromatogr A 1216, 4809–4814 (2009). [DOI] [PubMed] [Google Scholar]
- Bonham M. et al. Characterization of chemical constituents in Scutellaria baicalensis with antiandrogenic and growth-inhibitory activities toward prostate carcinoma. Clin Cancer Res 11, 3905–3914 (2005). [DOI] [PubMed] [Google Scholar]
- Chao J. I., Su W. C. & Liu H. F. Baicalein induces cancer cell death and proliferation retardation by the inhibition of CDC2 kinase and survivin associated with opposite role of p38 mitogen-activated protein kinase and AKT. Mol Cancer Ther 6, 3039–3048 (2007). [DOI] [PubMed] [Google Scholar]
- Chen Y. N. et al. Involvement of Intrinsic and Extrinsic Apoptotic Pathways in Baicalein-Induced Apoptosis in Human Hepatoma Hep3B Cells. J Cancer Mol 1, 37–45 (2005). [Google Scholar]
- Hsu S. L., Hsieh Y. C., Hsieh W. C. & Chou C. J. Baicalein induces a dual growth arrest by modulating multiple cell cycle regulatory molecules. Eur J Pharmacol 425, 165–171 (2001). [DOI] [PubMed] [Google Scholar]
- Ikemoto S. et al. Antitumor effects of Scutellariae radix and its components baicalein, baicalin, and wogonin on bladder cancer cell lines. Urology 55, 951–955 (2000). [DOI] [PubMed] [Google Scholar]
- Leung H. W. C., Yang W. H., Lai M. Y., Lin C. J. & Lee H. Z. Inhibition of 12-lipoxygenase during baicalein-induced human lung nonsmall carcinoma H460 cell apoptosis. Food Chem Toxicol 45, 403–411 (2007). [DOI] [PubMed] [Google Scholar]
- Li B. Q. et al. Flavonoid baicalin inhibits HIV-1 infection at the level of viral entry. Biochem Bioph Res Co 276, 534–538 (2000). [DOI] [PubMed] [Google Scholar]
- Ma Z. et al. Baicalein, a component of Scutellaria radix from Huang-Lian-Jie-Du-Tang (HLJDT), leads to suppression of proliferation and induction of apoptosis in human myeloma cells. Blood 105, 3312–3318 (2005). [DOI] [PubMed] [Google Scholar]
- Takahashi H. et al. Baicalein, a component of Scutellaria baicalensis, induces apoptosis by Mcl-1 down-regulation in human pancreatic cancer cells. BBA-Mol Cell Res 1813, 1465–1474 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D. Y. et al. Inhibition of cancer cell proliferation and prostaglandin E2 synthesis by Scutellaria baicalensis. Cancer Res 63, 4037–4043 (2003). [PubMed] [Google Scholar]
- Chen Y. J. et al. Baicalein Triggers Mitochondria-Mediated Apoptosis and Enhances the Antileukemic Effect of Vincristine in Childhood Acute Lymphoblastic Leukemia CCRF-CEM Cells. Evid-Based Compl Alt 2013, 124747 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo H. M. et al. Mitochondrial-dependent caspase activation pathway is involved in baicalein-induced apoptosis in human hepatoma J5 cells. Int J Oncol 35, (2009). [DOI] [PubMed] [Google Scholar]
- Lee J. H. et al. The role of Ca2+ in baicalein-induced apoptosis in human breast MDA-MB-231 cancer cells through mitochondria- and caspase-3-dependent pathway. Anticancer Res 28, 1701–1711 (2008). [PubMed] [Google Scholar]
- Li Y. C. et al. Baicalein-induced apoptosis via endoplasmic reticulum stress through elevations of reactive oxygen species and mitochondria dependent pathway in mouse-rat hybrid retina ganglion cells (N18). Neurochem Res 34, 418–429 (2009). [DOI] [PubMed] [Google Scholar]
- Lin Y. T. et al. Baicalein induces apoptosis in SCC-4 human tongue cancer cells via a Ca2+-dependent mitochondrial pathway. In Vivo 21, 1053–1058 (2007). [PubMed] [Google Scholar]
- Lu H. F. et al. ROS mediates baicalin-induced apoptosis in human promyelocytic leukemia HL-60 cells through the expression of the Gadd153 and mitochondrial-dependent pathway. Anticancer Res 27, 117–125 (2007). [PubMed] [Google Scholar]
- Ueda S. et al. Baicalin induces apoptosis via mitochondrial pathway as prooxidant. Mol Immunol 38, 781–791 (2002). [DOI] [PubMed] [Google Scholar]
- Zhang Y. et al. Baicalein selectively induces apoptosis in activated lymphocytes and ameliorates concanavalin a-induced hepatitis in mice. Plos One 8, e69592 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye F. et al. The effect of Scutellaria baicalensis on the signaling network in hepatocellular carcinoma cells. Nutr Cancer 61, 530–537 (2009). [DOI] [PubMed] [Google Scholar]
- Taniguchi H. et al. Baicalein overcomes tumor necrosis factor-related apoptosis-inducing ligand resistance via two different cell-specific pathways in cancer cells but not in normal cells. Cancer Res 68, 8918–8927 (2008). [DOI] [PubMed] [Google Scholar]
- Chen C. H. et al. Baicalein, a novel apoptotic agent for hepatoma cell lines: a potential medicine for hepatoma. Nutr Cancer 38, 287–295 (2000). [DOI] [PubMed] [Google Scholar]
- Parajuli P., Joshee N., Rimando A. M., Mittal S. & Yadav A. K. In vitro antitumor mechanisms of various Scutellaria extracts and constituent flavonoids. Planta Med 75, 41–48 (2009). [DOI] [PubMed] [Google Scholar]
- Akao T. et al. Efflux of baicalin, a flavone glucuronide of Scutellariae Radix, on Caco-2 cells through multidrug resistance-associated protein 2. J Pharm Pharmacol 59, 87–93 (2007). [DOI] [PubMed] [Google Scholar]
- Feng N. P., Di B. & Liu W. Y. Comparison of the metabolism of baicalin in rats orally administered with Radix scutellariae extract and Shuang-Huang-Lian extract. Chem Pharm Bull 53, 978–983 (2005). [DOI] [PubMed] [Google Scholar]
- Lai M. Y., Hsiu S. L., Chen C. C., Hou Y. C. & Chao P. D. Urinary pharmacokinetics of baicalein, wogonin and their glycosides after oral administration of Scutellariae Radix in humans. Biol Pharm Bull 26, 79–83 (2003). [DOI] [PubMed] [Google Scholar]
- Muto R. et al. The chemical structure of new substance as the metabolite of baicalin and time profiles for the plasma concentration after oral administration of Sho-Saiko-To in human. Yakugaku Zasshi 118, 79–87 (1998). [DOI] [PubMed] [Google Scholar]
- Tian S. et al. Pharmacokinetic study of baicalein after oral administration in monkeys. Fitoterapia 83, 532–540 (2012). [DOI] [PubMed] [Google Scholar]
- Zhang L., Lin G., Chang Q. & Zuo Z. Role of intestinal first-pass metabolism of baicalein in its absorption process. Pharmaceut Res 22, 1050–1058 (2005). [DOI] [PubMed] [Google Scholar]
- Gao H., Nishioka T., Kawabata J. & Kasai T. Structure-activity relationships for alpha-glucosidase inhibition of baicalein, 5,6,7-trihydroxyflavone: the effect of A-ring substitution. Biosci Biotech Bioch(BBB) 68, 369–375 (2004). [DOI] [PubMed] [Google Scholar]
- Sun X., Hu C. Q., Huang X. D. & Dong J. C. Mannich reaction of Baicalein. Chinese J Org Chem 23, 81–85 (2003). [Google Scholar]
- Zhang S. et al. Nitrogen-containing flavonoid analogues as CDK1/cyclin B inhibitors: synthesis, SAR analysis, and biological activity. Bioorgan Med Chem 16, 7128–7133 (2008). [DOI] [PubMed] [Google Scholar]
- Zhang S, Bo y, Inventors; Cyclin-dependent protein kinase inhibitors of scutellaria flavonoid organic amine derivatives, synthesis and use thereof. United States patent US 8377895B2. 2013 Feb 19 and China patent ZL200910140275.4. 2013 August 21.
- Nicco C., Laurent A., Chereau C., Weill B. & Batteux F. Differential modulation of normal and tumor cell proliferation by reactive oxygen species. Biomed Pharmacother 59, 169–174 (2005). [DOI] [PubMed] [Google Scholar]
- Contour-Galcera M. O., Sidhu A., Prevost G., Bigg D. & Ducommun B. What’s new on CDC25 phosphatase inhibitors. Pharmacol Therapeut 115, 1–12 (2007). [DOI] [PubMed] [Google Scholar]
- Enserink J. M. & Kolodner R. D. An overview of Cdk1-controlled targets and processes. Cell Div 5, 11 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S. et al. Cyclane-aminol 10-hydroxycamptothecin analogs as novel DNA topoisomerase I inhibitors induce apoptosis selectively in tumor cells. Anti-Cancer Drug 25, 614–623 (2014). [DOI] [PubMed] [Google Scholar]
- Chang W. H., Chen C. H. & Lu F. J. Different effects of baicalein, baicalin and wogonin on mitochondrial function, glutathione content and cell cycle progression in human hepatoma cell lines. Planta Med 68, 128–132 (2002). [DOI] [PubMed] [Google Scholar]
- Lin H. Y., Shen S. C., Lin C. W., Yang L. Y. & Chen Y. C. Baicalein inhibition of hydrogen peroxide-induced apoptosis via ROS-dependent heme oxygenase 1 gene expression. BBA-Mol Cell Res 1773, 1073–1086 (2007). [DOI] [PubMed] [Google Scholar]
- Miller E. W., Albers A. E., Pralle A., Isacoff E. Y. & Chang C. J. Boronate-based fluorescent probes for imaging cellular hydrogen peroxide. J Am Chem Soc 127, 16652–16659 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]