

Abstract
Lemurs, the living primates most distantly related to humans, demonstrate incredible diversity in behaviour, life history patterns and adaptive traits. Although many lemur species are endangered within their native Madagascar, there is no high-quality genome assembly from this taxon, limiting population and conservation genetic studies. One critically endangered lemur is the blue-eyed black lemur Eulemur flavifrons. This species is fixed for blue irises, a convergent trait that evolved at least four times in primates and was subject to positive selection in humans, where 5′ regulatory variation of OCA2 explains most of the brown/blue eye colour differences. We built a de novo genome assembly for E. flavifrons, providing the most complete lemur genome to date, and a high confidence consensus sequence for close sister species E. macaco, the (brown-eyed) black lemur. From diversity and divergence patterns across the genomes, we estimated a recent split time of the two species (160 Kya) and temporal fluctuations in effective population sizes that accord with known environmental changes. By looking for regions of unusually low diversity, we identified potential signals of directional selection in E. flavifrons at MITF, a melanocyte development gene that regulates OCA2 and has previously been associated with variation in human iris colour, as well as at several other genes involved in melanin biosynthesis in mammals. Our study thus illustrates how whole-genome sequencing of a few individuals can illuminate the demographic and selection history of nonmodel species.
Keywords: convergent evolution, de novo genome assembly, demographic inference, Eulemur, selection scan
Introduction
The fields of genetics and population genetics have traditionally focused on model systems for which extensive resources are available, notably reference genomes. Until recently, building a genome for a new species was prohibitively expensive and computationally intractable for individual laboratories, but this situation has been upended with the advent of next-generation sequencing technologies and assembly programs. These developments promise to transform the study of nonmodel organisms. The assembly of even a single diploid genome allows unprecedented investigations into evolutionary processes that are largely inaccessible by other approaches. For instance, data from one diploid genome can be used to infer linkage disequilibrium and recombination (Reich et al. 2002; Haubold et al. 2010), as well as demographic history (Miller et al. 2012; Zhan et al. 2013) and evolutionary relationships (Gnerre et al. 2011; Amemiya et al. 2013). Moreover, when closely related species differ in a positively selected trait with a simple genetic architecture, whole-genome sequencing may be used to evaluate candidate loci influencing the trait.
Here, we use a de novo assembly of the blue-eyed black lemur (Eulemur flavifrons) and a reference-based assembly of its close relative, the black lemur (E. macaco), to investigate the two species’ evolutionary history (Fig.1). The blue-eyed black lemur inhabits a narrow range of northwestern Madagascar (Andrianjakarivelo 2004; Randriatahina & Rabarivola 2004) and is one of at least 60 lemurs that are endangered or critically endangered (Schwitzer et al. 2013). Despite the lemurs’ diversity and conservation significance, there is still no high-quality genome assembly of a lemur species (Yoder 2013). Many lemurs are separated by large phylogenetic distances; for instance, the blue-eyed black lemur is 5.8% divergent (39 MY; based on 35 kb of sequence data from 54 nuclear genes; Perelman et al. 2011) from the mouse lemur (Microcebus murinus; Lindblad-Toh et al. 2011) and 6.5% (59 MY; Perelman et al. 2011) from the aye-aye (Daubentonia madagascariensis; Perry et al. 2012b), the only lemurs for which draft genome assemblies are available. Thus, as is the case for most nonmodel organisms, genomewide studies of any lemur species currently require a de novo assembly.
Fig. 1.
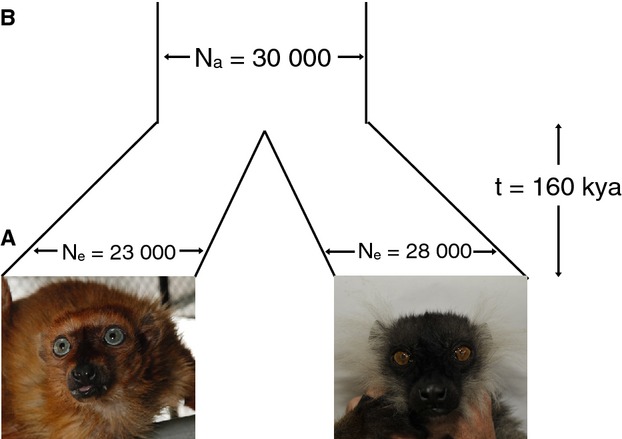
Samples and demographic parameters estimated. (A) Pictured at left is the blue-eyed black lemur (Harlow) sequenced to high coverage in this study; at right is the high-coverage black lemur (Harmonia); pictures are courtesy of David Haring (Duke Lemur Center). (B) This diagram displays the split time and effective population sizes inferred from diversity and divergence data from the two individuals (Appendix S14, Supporting information).
In addition to its conservation importance, the blue-eyed black lemur is of particular interest because it is fixed for blue iris pigmentation, whereas its sister species, the black lemur, is fixed for brown irises. Blue irises evolved independently in at least three other primate lineages, in which iris colour is currently polymorphic: humans (Homo sapiens; Eiberg et al. 2008), Japanese macaques (Macaca fuscata; Yamagiwa 1979) and spider monkeys (brown: Ateles hybridus, formerly Ateles belzebuth hybridus; and closely related Colombian black: Ateles geoffroyi or fusciceps, subspecies rufiventris; Hernandez-Camacho & Cooper 1976). Examples of convergent evolution such as this provide opportunities to query the extent of constraint on the evolution of a particular trait, by determining whether the same or different genetic mechanisms have been responsible for the acquisition of this trait in different lineages.
Because the blue iris phenotype is highly similar across primate species (Meyer et al. 2013) and human iris pigmentation loci are well characterized, these loci provide the natural focal points for testing whether the same genetic mechanisms are involved in lemurs. Several loci have been associated with natural iris pigmentation variation in humans, and most of these play known roles in the melanin synthesis pathway (Frudakis et al. 2003; Sulem et al. 2007; Sturm et al. 2008; Liu et al. 2010). Yet unlike many complex human traits, the majority of blue vs. brown variation in iris colour can be explained by a single locus. The human causal site is a regulatory variant of the OCA2 gene located within an intron of HERC2 (Eiberg et al. 2008; Sturm et al. 2008), and thus, previous studies aimed at identifying iris pigmentation-associated differences between the two lemur species have focused on the ortholog of this region. We found no fixed differences in 1.2 kb orthologous to the human causal site (Meyer et al. 2013; see also Bradley et al. 2009), although we were unable to investigate the full OCA2 region because of the size of the gene (344 kb, including introns, in humans, 297 kb in mouse). In order to extend the search for a causal locus beyond this region, as well as to query both coding and noncoding variation within it for potentially causal sites, genomewide data in large scaffolds are particularly helpful.
Materials and methods
We assembled the blue-eyed black lemur genome de novo, and we then assembled the black lemur genome using this de novo assembly as a reference. A schematic overview of our assembly and analysis pipeline, along with a brief glossary of assembly-related terms, may be found in Fig. S1 (Supporting information).
Samples
DNA was obtained from frozen blood or tissue for four blue-eyed black lemurs and four black lemurs from the Duke Lemur Center (Appendix S1, Supporting information). One blue-eyed black lemur and one black lemur were sequenced to high fold coverage to generate genome assemblies. All lemurs were outbred from wild-caught ancestors.
Short read sequencing, error correction and genome assembly
Library preparation and sequencing were performed using the recommended Illumina protocols at the University of Chicago, the core sequencing facility at Princeton University, and the Keck Center for Comparative Genomics, University of Illinois (Appendix S2, Supporting information). Redundant mate pair (MP) reads were removed, and paired end (PE) reads with called bases were stripped of adapters and trimmed using fastx (http://hannonlab.cshl.edu/fastx_toolkit/; Appendix S3, Supporting information). Trimmed reads were error-corrected using quake (Kelley et al. 2010) and assembled using soapdenovo v1.05 (Li et al. 2010) with a k-mer size of 33 (Appendices S4–S5, Supporting information). After estimating insert sizes for each library and resolving bimodal distributions (Appendix S6, Supporting information), PE and MP information was loaded onto the graph sequentially from shortest to longest estimated insert size to join unique contigs into scaffolds, and the ‘Gap Closure’ module was run to resolve repeats. Final contigs were extracted by breaking scaffolds at the remaining gaps. See Appendix S7 (Supporting information) for command line parameters and Appendix S8 and Fig. S2 (Supporting information) for memory consumption at each step.
Evaluation of assembly quality and accuracy
As a measure of assembly quality, we calculated N50, or the size of the largest contig or scaffold with the property that 50% of the assembly is contained in contigs or scaffolds of that size or greater. To assess scaffold accuracy, we calculated the proportion of PE reads that were correctly oriented and within three standard deviations of the mean insert size (Li & Durbin 2009; Li et al. 2009; Appendix S4, Supporting information). Additionally, we employed two other methods to assess assembly quality: (i) we aligned contigs from the blue-eyed black lemur genome assembly to previously sequenced BACs (Appendix S9, Supporting information), and (ii) we assessed the proportion of highly conserved, core eukaryotic genes covered by scaffolds in the assembly (Parra et al. 2009; Appendix S10, Supporting information).
Reference-based assembly of the black lemur genome
Following quality filtering using fastx (Appendix S3, Supporting information), reads were aligned to the blue-eyed black lemur assembly using BWA (Li & Durbin 2009) with trimming parameter 15 and all other parameters set to default values. Reads with mapping quality <10 were eliminated using samtools (Li et al. 2009), and duplicates were marked using picard (http://picard.sourceforge.net). Consensus sequence was generated using the output from the Genome Analysis Toolkit (GATK, DePristo et al. 2011, see below), requiring a minimum filtered depth of three and incorporating the higher coverage allele at polymorphic sites, to mimic the consensus generation for the blue-eyed black lemur from soapdenovo, using a custom script (Appendix S7, Supporting information).
Identification of polymorphic sites within and divergent sites between high-coverage samples
We called single nucleotide polymorphisms (SNPs) from the alignments of reads to the assembly using the recommended pipeline for the unifiedgenotyper tool of GATK (DePristo et al. 2011); we used the ‘emit all sites’ option to include nonvariable sites in the resulting Variant Call Format (VCF) file and allow generation of consensus files. We subsequently filtered both heterozygous and homozygous sites by base quality, read depth and mapping quality as recommended (Abecasis et al. 2010; Appendix S11, Supporting information). We resequenced approximately 9.9 kb (blue-eyed black lemur) and 15.1 kb (black lemur) of the high-coverage individuals by Sanger sequencing. Concordance between the GATK-derived sequence and Sanger sequence suggested that the SNP calling was highly reliable (Appendix S12, Supporting information).
We estimated heterozygosity by counting the number of high-quality polymorphic sites per high-quality base pair within the VCF file for each species and divergence between species by comparing high-quality sites between the VCF files, with a single allele chosen at random for each species at heterozygous sites. We used a modified version of the vcf2fq function from vcfutils.pl (Appendix S11, Supporting information) to generate SNP-inclusive consensuses, which were used for ancestral Ne inference.
Inference of demographic history
We used the pairwise sequentially Markovian coalescent (PSMC; Li & Durbin 2011) to infer demographic history separately in the blue-eyed black lemur and black lemur, using the sequence of confidently called (quality >30) homozygous and heterozygous sites within scaffolds >10 kb in length. We summarized heterozygosity over 75-bp windows, chosen so that ∼1% of windows would contain more than one polymorphic site; other window sizes yielded similar results (Fig. S3A, Supporting information). We obtained an estimate of confidence by resampling scaffolds, with replacement, to obtain sequence of comparable total length, splitting large scaffolds into 250-kb segments. Because PSMC was developed for fully assembled genome data, we performed simulations to determine the effect of missing data and nonindependence of scaffolds (Appendix S13; Fig. S3B, Supporting information).
Estimating species split time
Under a simple isolation model, the probability of individuals from two species sharing a neutral polymorphism at any given locus due to identity by descent (IBD) is a function of time since the species split. Specifically, this probability is the product of (i) the probability of no coalescent events occurring since the species split and (ii) the probability of the lineages coalescing within the ancestral population in an order that would give rise to a shared polymorphism.
These probabilities can be written in terms of τ (the split time scaled by 2Ne) and Na/Ne (the ratio of Ne in the ancestral branch to current Ne). The expected pairwise divergence between species can also be written as a function of τ and Na/Ne. We estimated τ and Na/Ne by substituting our estimates of genomewide divergence and the proportion of shared polymorphic sites from the two-sample data set into these two equations (i.e. assuming that any shared SNPs were due to IBD, which is reasonable given the species’ recent split) (Appendix S14, Supporting information). We then estimated the split time from τ under plausible estimates of mutation rate and generation time (Appendix S15, Supporting information).
Scan for selection in the two-sample data set
We identified initial candidate regions for recent positive selection in one lineage by searching for large regions with low within-species diversity relative to the total number of polymorphic sites. Specifically, we considered the statistic Ps1 = (hs1 + ss)/(hs1 + hs2 + ss + fd), where hs1 represents the number of sites heterozygous only within the focal species, hs2 the number of sites heterozygous only within the other species, ss the number of shared heterozygous sites and fd the number of sites at which the two species are homozygous for different alleles (Appendix S16, Supporting information). To determine the empirical genomewide significance for each region, we summarized Ps1 across 20-kb nonoverlapping windows.
Annotation of pigmentation genes
We identified exons of the lemur orthologs of ASIP, MITF, OCA2 and TYR by comparing the human coding sequence (downloaded from Ensembl GRCh37) to our draft assembly using blastn 2.2.26+ (Altschul et al. 1990) (Appendix S17, Supporting information).
Additional resequencing and regulatory annotation of scaffold containing the ortholog of OCA2
We resequenced 18 ∼1.1-kb regions from this scaffold in the two high-coverage-sequenced individuals, eight additional blue-eyed black lemurs and six additional black lemurs by Sanger sequencing (Appendix S12, Supporting information). We used these data to assess the reliability of our SNP calls and to estimate local genetic differentiation between the two species. We identified candidate differential transcription factor binding sites by assessing the position weight matrix (PWM) scores for the E-box (MITF), LEF1 and RUSH (HLTF) PWMs for each species at each site within the scaffold (Appendix S18, Supporting information). We focused on sites at which the black lemur PWM score was in the 10% tail scaffoldwide and was log(10) greater than the blue-eyed black lemur PWM score, and where the blue-eyed black lemur allele was inferred to be derived (Appendix S18, Supporting information). In the combined data set, we specifically considered sites with posterior probability ≥0.8 (from angsd and ngstools, see below) of being fixed differences between species.
Resequencing of additional individuals at low fold coverage
We sequenced three additional individuals of each species, chosen to minimize relatedness among samples (Appendix S1, Supporting information), to low-to-medium depth (4–15x). Following raw read quality control and read mapping as for the high-coverage black lemur sample, alignments were processed following the recommended pipeline for GATK (DePristo et al. 2011) and then input to angsd (http://popgen.dk/wiki/index.php/ANGSD) and ngstools (https://github.com/mfumagalli/ngsTools), which use an empirical Bayesian approach to estimate within-population allele frequencies, heterozygosity, divergence and the number of segregating sites from low-coverage sequencing data, accounting for uncertainty (Fumagalli 2013; Fumagalli et al. 2013; Korneliussen et al. 2014).
Estimation of FST and Ps1 from the combined sample
We estimated FST and Ps1 from the sample of all high- and low-coverage samples, using the output from angsd and ngstools. We calculated single species FST as 1 – Hw/HB, where Hw represents within-species heterozygosity and HB represents between-species heterozygosity (reported by ngstools as dXY). We calculated Ps1 as ss1/Pvar, where ss1 represents the probability of a segregating site in species 1, and Pvar represents the probability of a site being variable in the whole sample. We summarized FST as 1 – ∑Hw/∑HB, and Pva as ∑ss1/∑Pvar across regions of 20 kb, excluding sites that had a posterior probability <0.8 of being polymorphic within the whole sample (Appendix S19, Supporting information).
Assessment of admixture in the combined sample
We performed principal components analysis (PCA) and an estimation of admixture proportions by applying ngsCovar (Fumagalli 2013; Fumagalli et al. 2013) and NGSadmix (Skotte et al. 2013), respectively, to a subset of high confidence polymorphic sites in the combined sample (Appendix S20, Supporting information).
Empirical P-values for a region including multiple windows
For each pigmentation gene, we first calculated the maximum FST among all windows overlapping its exons (annotated as in Appendix S17, Supporting information). We then drew 10 000 regions with the same number of consecutive windows at random from the genome, excluding the gene region, and determined the maximum FST for each region. We considered the empirical P-value for the pigmentation gene to be the proportion of all randomly drawn regions with maximum FST greater than or equal to that observed for the gene region. We used the same procedure, focusing on the regional minimum statistic, to determine an empirical P-value for PS1.
Gene ontology enrichment analysis
We annotated genes overlapping the 1% blue-eyed FST tail from the combined sample using tblastn 2.2.22+, retaining only those genes whose best match for any subset of the sequence fell within the FST tail regions (Appendix S21, Supporting information). We used the functional annotation chart produced by the Database for Annotation, Visualization and Integrated Discovery (david) v6.7 (Huang et al. 2008, 2009) to determine fold enrichment and EASE score (adjusted Fisher's exact test P-value). To correct for gene lengths, we selected subsets of all genes whose medians matched those of the categories of interest (Appendix S21, Supporting information).
Results
We assembled the blue-eyed black lemur genome de novo from Illumina PE and MP read libraries using the de Bruijn graph assembler soapdenovo (Li et al. 2010) (see Appendix S1–S8 for details, Supporting information). We obtained 205 Gbp total raw sequence from 2.26 billion raw reads using 11 lanes of sequencing (Table S1, Supporting information). We used a range of target insert sizes (180, 500, and 1000-bp PE and 3- and 8-kb MP) to achieve a draft genome with large contiguous sequences (contigs) and scaffolds (Fig.2), representing approximately 79% of the 2.68 Gb genome (Appendix S22, Supporting information) at an average coverage of 52X.
Fig. 2.
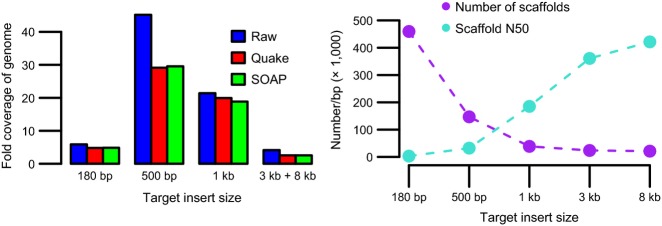
Libraries of multiple insert sizes contributed to overall assembly quality. (A) Coverage of raw reads and reads corrected by Quake or SOAP (Appendix S4, Supporting information), calculated using the genome size estimated from the k-mer distribution (Appendix S22, Supporting information). The number of low-quality raw reads in the 500-bp library far exceeded those in other libraries, as evidenced by the drop in coverage in the corrected reads. (B) Increase in scaffold N50 and decrease in total scaffold number with the addition of paired end (PE) and mate pair (MP) libraries of increasing insert size.
We generated a reference-based assembly for the black lemur genome by mapping one lane of 500-bp PE reads to the blue-eyed black lemur draft genome (Li & Durbin 2009; Li et al. 2009). The resulting genome (based on 65 Gbp of raw sequence data from 650 million raw reads) has 21X average coverage. Based on current prices for Illumina HiSeq2500 sequencing, obtaining the same total sequence length for these two individuals would cost around $10 000 today—within the reach of individual laboratories.
The draft assembly of the blue-eyed black lemur consists of large contigs and scaffolds, and several lines of evidence indicate that this assembly is of high quality and completeness. The contig N50 of the assembly was 16.3 kb and the scaffold N50 was 421.5 kb (Table S2, Supporting information), 96% of base pairs had at least 20X coverage (Fig. S4A, Supporting information), and 99.95% of corrected reads mapped with mapping quality at least 20. Previously sequenced black lemur bacterial artificial chromosomes (BACs) aligned to the assembly across 88% of their length, and alignable blocks shared on average 99.4% identity with the blue-eyed black lemur contigs (Appendix S9, Supporting information), as expected based on previous resequencing data (Perelman et al. 2011) and the divergence estimate from our own data. Moreover, of the 248 core single-copy genes that are highly conserved in a wide range of taxa (Parra et al. 2009), 94.7% were present in the assembly as full-length ortholog matches or fragments of genes (Appendix S10, Supporting information).
Genome assemblies of single diploid individuals provide an opportunity to estimate a key population genetic parameter: the nucleotide diversity or heterozygosity (Hartl & Clark 2007). This quantity is informative for conservation purposes, because an extremely low level of genetic diversity, as is sometimes observed in critically endangered species, may inhibit species’ response to novel selective pressures (Lynch & Lande 1998). Increased reporting of this information will also enable evaluation of possible determinants of species diversity levels (Leffler et al. 2012; Romiguier et al. 2014; Corbett-Detig et al. 2015). Few recent genome assembly papers have reported such estimates, however, possibly because of the difficulty of confidently assessing both heterozygous and homozygous sites. We estimated heterozygosity in the two species by calling heterozygous sites from the mapping of reads to the blue-eyed black lemur genome assembly; Sanger sequencing confirmed a low error rate for these calls (Materials and Methods; Appendix S11 and S12, Supporting information). We estimated a genomewide diversity (π) of 0.174% per bp in blue-eyed black lemurs and 0.219% in black lemurs (Fig.3).
Fig. 3.
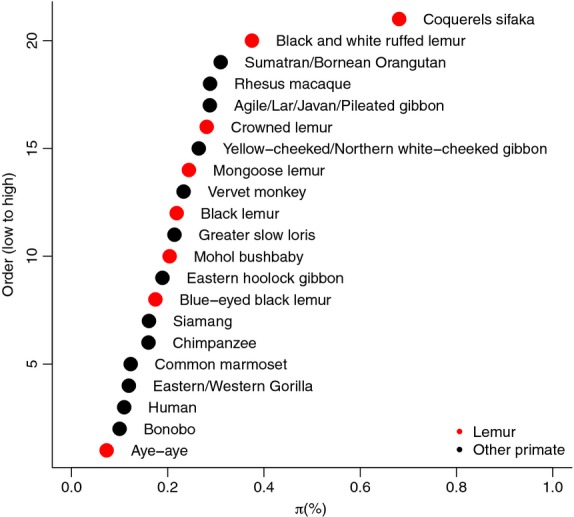
Intraspecific diversity (π) across primates. Values other than those calculated in this study are from Leffler et al. (2012) and represent average nuclear diversity estimates based on data from at least three autosomal regions. Red dots represent diversity estimates for lemurs. x-axis: π in %/base pair; y-axis: rank among primate species included.
Beyond average diversity levels, patterns of variation along the genome can be used to infer demographic history, notably using the PSMC (46), even when the assembly consists of scaffolds rather than a linear genome (Appendix S13, Supporting information). During the time interval within which demographic histories can be well-characterized by this approach (i.e. excluding very recent and ancient time periods within which the method is unreliable: Li & Durbin 2011; MacLeod et al. 2013), the estimated effective population size (Ne) of each species fluctuates between ∼20 000 and ∼60 000 (Fig.4). Such a range of Ne is comparable to that inferred for primates such as great apes, with a slightly higher minimum Ne (Prado-Martinez et al. 2013). The two lemur species share a similar trajectory from the distant past until approximately 250 kya, including a drop in population size around 1 Mya and subsequent population growth around 400 Kya. Some changes in Ne correspond to known environmental changes; for instance, the decrease approximately 1 Mya accords with the timing of a shift to a more arid climate and corresponding loss of vegetation in southern and eastern Africa, the locations with paleoclimate data nearest Madagascar (deMenocal 1995). Following the species split, the two demographic histories appear to be negatively correlated, although the sum of the two Nes remains nearly constant. This may indicate a limit to the carrying capacity of a shared habitat range.
Fig. 4.
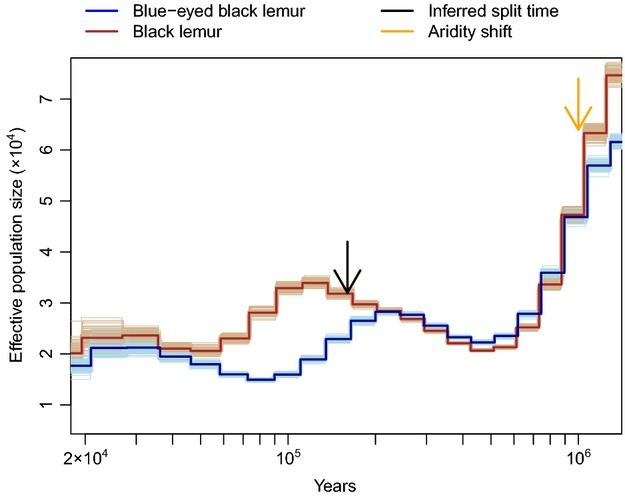
Inferred demographic history for blue-eyed black lemur and black lemur. The thick blue line and thick brown line represent the inferred trajectories for blue-eyed black lemur and black lemur, respectively, with light lines representing demographic histories inferred from 100 bootstrap resamplings. The split time (black arrow) was estimated from diversity and divergence between the species (Appendix S14, Supporting information). The orange arrow represents the time at which African paleoclimate records indicate a shift towards more arid, open conditions (deMenocal 1995). The generation time was taken to be 5 years and the mutation rate per base pair per generation 2 × 10−8 in blue-eyed black lemurs and 1.951 × 10−8 in brown-eyed black lemurs, with the reduction in black lemurs due to reduced power to call heterozygous sites in lower coverage data (Appendix S15, Supporting information).
Focusing on the divergence between the two species, we estimated a genomewide value of 0.366% per bp, less than two-fold higher than diversity within species (see Materials and methods). Assuming a simple demographic model, we used the number of shared SNPs and pairwise differences between the two genomes together with the within-species heterozygosities to estimate the split time and ancestral effective population size Na (see Materials and methods). Given a mutation rate of 2.0 × 10−8 substitutions per bp per generation and a generation time of 5 years (Appendix S15, Supporting information), the estimated date of the species split is approximately 160 kya, and the estimate of Na is 30 000 (Appendix S14, Supporting information; Fig.1B). Although this is somewhat more recent than the time at which the two species’ PSMC-inferred trajectories begin to overlap (Fig.4), this discrepancy may be explained by a tendency of PSMC to overestimate the time of population splits due to smoothing (Appendix S13, Supporting information). We note that gene flow since the species split would lead us to underestimate the true time at which the populations initially split and to overestimate the true Na (Wall 2003; Leman et al. 2005; Becquet & Przeworski 2007); however, these effects should not bias our subsequent analyses.
The recent split between the blue-eyed black lemur and black lemur suggests a natural approach to look for targets of recent positive selection within one of the two lineages. Shortly after the fixation of a beneficial substitution, neutral diversity at linked sites should be reduced but neutral divergence unaffected (Smith & Haigh 1974; Birky & Walsh 1988). If the substitution is caused by a selective sweep, the signature should persist for roughly Ne generations (Przeworski 2002), which is comparable to the estimated split time of the blue-eyed black lemur and black lemur, and, if the selection coefficient were as large as 1%, could affect as much as 0.1 cM (Appendix S16, Supporting information). In order to identify regions that may have been subject to recent selective pressures, we therefore located windows of at least 20 kb (corresponding to on average ∼0.02 cM in humans; Kong et al. 2004) in which one species had unusually few polymorphic sites relative to the other species and to divergence levels (Materials and Methods, Appendix S16, Supporting information).
We were particularly interested in using the results of this scan for selection to test whether the convergent evolution of blue irises in lemurs occurred via a similar genetic mechanism to that operating in humans. Similar selective pressures operating in distinct lineages can frequently lead to convergent evolution, and in humans, there is evidence for a partial selective sweep in Europeans at the causal locus for blue irises (Donnelly et al. 2012). Blue irises have fixed in the blue-eyed black lemurs since their recent split with black lemurs, and this fixation may also have been due to selection. Although an advantage of blue irises in lemurs has not been directly demonstrated, plausible hypotheses include sexual selection and species recognition (Bradley et al. 2009). We therefore looked for signals of selection in the lemur genome overlapping known human iris pigmentation genes. Our analysis identified the scaffold containing the orthologs of human genes OCA2 and HERC2 among regions with the most extreme signatures of selection in the blue-eyed black lemur lineage (π = 3.73 × 10−5, compared to 1.74 × 10−3 genomewide; Fig.5A). This region contained multiple windows with extreme statistics for FST and Ps1, the proportion of all polymorphisms that are heterozygous only in the blue-eyed black lemur (see Materials and Methods). Of 47 nonoverlapping 20-kb windows in the region, 26 had FST = 1 in blue-eyed black lemur (the upper 9.1%-tile of windows genomewide), and 34 had Ps1 = 0, that is no SNPs in the blue-eyed black lemur (the lower 10.7%-tile).
Fig. 5.

The region surrounding the ortholog of OCA2 displays a tentative signature of selection in the blue-eyed black lemur. (A) In the two-sample data set, diversity within the blue-eyed black lemur (blue) is low throughout the scaffold containing the ortholog of OCA2 and adjacent scaffolds (Appendix S23, Supporting information), in contrast to diversity within the black lemur (orange) and divergence (grey) or fixed differences (black) between species. Dashed lines show genomewide mean values for summary statistics. Regions Sanger sequenced in a larger sample (Appendix S12, Supporting information) are shown as points with colours representing the magnitude of blue-eyed black lemur FST from Sanger sequencing. (B) When incorporating both low-coverage and high-coverage whole-genome sequenced individuals, diversity within the blue-eyed black lemur sample is reduced relative to black lemur diversity and between-species divergence, yet the difference is less extreme and more localized. The region contains several candidate mutations inferred to influence the binding of transcription factors that regulate OCA2 in humans (Appendix S18, Supporting information); these are displayed as red lines, with those in the strongly differentiated region starred. Other fixed differences are displayed as black vertical lines. In all plots, exons of the orthologs of HERC2 and OCA2 are shown in dark green and purple, respectively.
Unusually low diversity in blue-eyed black lemurs relative to diversity within black lemurs and divergence between species is consistent with a recent selective sweep in this region. We examined whether such a sweep may have acted on coding changes by determining the amino acid sequence of OCA2 in both sequenced individuals (Materials and Methods, Appendix S17, Supporting information). Observing no fixed amino acid changes, we further annotated potential regulatory changes by computational inference of differential binding sites for the transcription factors that regulate OCA2 in humans (Appendix S18, Supporting information). These analyses, in combination with additional Sanger sequencing in a larger sample (Appendix S12, Supporting information), yielded a list of 26 candidate causal regulatory mutations within two regions of extremely reduced diversity totalling 140 kb in length.
To evaluate the potential signal of directional selection in the HERC2/OCA2 region further, we sequenced the genomes of three additional individuals per species to a mean depth of 7X and used likelihood-based methods to estimate summaries of genetic diversity from the reads (Fumagalli 2013; Fumagalli et al. 2013; Korneliussen et al. 2014) (Materials and Methods; Appendix S19, Supporting information). In this larger data set, the strongest signal within the region is in a 20-kb window overlapping OCA2 with blue-eyed FST = 0.90 (upper 4.2%-tile) and Ps1 = 0.16 (5.7%-tile). Given that there are as many as eight windows to consider, the evidence for selection in this region is highly tentative. Notably, if we correct for multiple testing using an empirical approach (see Materials and Methods), there is no longer good evidence for unusually reduced diversity in this region (FST, P = 0.211; Ps1 P = 0.283). Intriguingly, however, the peak of high differentiation in the larger data set lies above a promising functional candidate, namely a fixed difference in which the derived blue-eyed allele is predicted to reduce HLTF binding 34-fold and MITF binding 119-fold. Such a regulatory site is therefore a candidate for the substitution driving the local increase in population differentiation (Fig.5B).
Beyond OCA2, we investigated patterns of genetic variation at 16 genes that had been associated with iris pigmentation variation in humans or mice, reasoning that a priori these represent good candidate genes (Appendix S17, Supporting information). One window of nine overlapping MITF, one of the transcription factors directly regulating OCA2 (Sturm et al. 2008; Visser et al. 2012), had FST = 0.96 (upper 0.5%-tile) and Ps1 = 0.05 (lower 0.3%-tile). Moreover, at another direct binding target of MITF, TYR, one of five windows had FST = 0.84 (11.4%-tile) and Ps1 = 0.14 (3.7%-tile). Other than the signals at OCA2, MITF, and TYR, the only candidate iris pigmentation genes with unusual differentiation were ASIP, which lies farther upstream in the pathway (one of seven windows had FST = 0.88, 6.4%-tile; Ps1 = 0.17, 6.6%-tile), as well as LYST (of six windows, one had FST = 0.88, 6.1%-tile; Ps1 = 0.15, 4.3%-tile, and another FST = 0.90, 4.0%-tile, Ps1 = 0.16, 5.0%-tile) and NPLOC4 (one of three windows had FST = 0.92, 2.5%-tile, Ps1 = 0.15, 4.3%-tile), which reside within regions recently associated with quantitative iris colour but whose function in melanin biosynthesis is not yet understood (Liu et al. 2010). After our correction for multiple windows, however, diversity patterns at TYR, LYST and NPLOC4 were no longer unusual, whereas the signal at MITF (P = 0.029 for FST and P = 0.014 for Ps1) remained genomewide significant.
A further complication in this analysis is the number of candidate genes considered, as one of 16 genes displaying empirical significance is expected by chance. To address this, we performed a gene ontology (GO) enrichment analysis on the set of genes overlapping regions in the 1% tail of blue-eyed FST. Through this analysis, we found an enrichment of genes in two pigmentation-related categories, even after matching the background set for gene length to account for the possibility that pigmentation genes are simply longer (KEGG melanogenesis pathway: 7.3-fold enrichment, P = 0.056; Biocarta melanocyte development: 25.0-fold enrichment, P = 0.065) (Materials and Methods; Appendix S1, Supporting information). These enriched GO categories highlighted a few additional candidate genes: notably, signals at KITLG and TCF7 that are somewhat unlikely by chance, even after correcting for number of windows (FST P = 0.033 and Ps1 P = 0.052 for KITLG; FST P = 0.021 and Ps1 P = 0.020 for TCF7). We also observed tentative signals at two other loci recently implicated in melanogenesis but not included in the GO categories, namely SERPINB2 (FST P = 0.014 and Ps1 P = 0.007), which was demonstrated through a siRNA screen to influence melanogenesis (Ganesan et al. 2008) and FIG4 (FST P = 0.005 and Ps1 P = 0.006), mutations in which influence melanosome distribution in mouse hair follicles (Chow et al. 2007).
Two of the candidate genes with potential signatures of selection in the larger data set play well-understood and important roles in the development of melanocytes and the production of melanin. Specifically, MITF is the primary transcription factor regulating melanocyte differentiation, proliferation and survival (Levy et al. 2006), and binds the OCA2 enhancer (Sturm et al. 2008; Visser et al. 2012). MITF has been associated with Waardenburg and Tietz syndrome phenotypes in humans (Smith et al. 2000; Pingault et al. 2010). We found no fixed differences in amino acid sequences of MITF between the two lemur species (Appendix S17, Supporting information), indicating that if it is involved in lemur iris pigmentation, regulatory changes must be responsible. KITLG is a member of the MAPK signalling pathway, and interactions between KIT and KITLG are essential for melanocyte proliferation and melanin production (Picardo & Cardinali 2011). KITLG has been strongly associated with hair pigmentation variation and tentatively associated with skin pigmentation variation in European human populations (Sulem et al. 2007; Guenther et al. 2014).
Discussion
We provide the most complete assembly of a lemur genome to date, representing 79% of the total genome length with a scaffold N50 of 421 kb. These statistics compare favourably to several other de novo genome assemblies of nonmodel organisms obtained by consortia using similar coverage with Illumina and/or 454 technology (Atlantic cod: Star et al. 2011; Heliconius butterfly: The Heliconius Genome Consortium 2012; African coelacanth: Amemiya et al. 2013), even though the blue-eyed black lemur genome is substantially larger than several of these (Star et al. 2011; The Heliconius Genome Consortium 2012). We attribute the completeness and large scaffold length of the assembly in part to the use of multiple insert size libraries, particularly the inclusion of 3 and 8-kb MPs, which substantially increased our N50 (Fig.2B). Estimating the insert size of libraries from the alignment of reads to the assembly also enabled us to provide more accurate prior information about the insert size distribution, which improved contiguity (Table S2, Supporting information). The resulting high-quality draft genome is a first for the lemur clade and will provide a valuable resource for future studies within this taxon (Yoder 2013). We also provide a list of well-supported polymorphic and divergent sites within and between the blue-eyed black lemur and black lemur, which will assist in continued conservation efforts in these species.
We leveraged the high-quality diploid genome sequences for these two individuals to learn about the species’ demographic and selective history. In contrast to traditional methods relying on data from many individuals at a few loci, we used the information from many loci for a few individuals to perform similar evolutionary inferences. Because our assembly represents the majority of the species’ approximately 2.6 Gb genome, these sequences should represent the many distinct evolutionary histories of chromosomal segments within our sampled individuals. We first used these data to estimate genomewide genetic diversity, which, assuming the majority of the genome is neutrally evolving, provides a way of estimating effective population sizes. The estimated mean diversity levels for the blue-eyed black lemur and black lemur suggest current effective population sizes of approximately 23 000 and 28 000, respectively (Fig.1B).
Using variation in diversity levels along the genome, we were also able to infer with confidence changes in effective population size throughout most of the past million years. These values have fluctuated by approximately three-fold, with several notable periods of change corresponding to contemporaneous changes in environment (Fig.4). When considering the influence of more recent changes in environment on the lemurs’ genetic diversity, it is interesting to note that our estimates of current nucleotide diversity for both species fall in the middle of population-specific estimates for both lemurs and all primates (Fig.3). Moreover, the diversity of the two species is almost identical, despite the blue-eyed black lemur being categorized as ‘critically endangered’ and the black lemur as ‘vulnerable’ (Schwitzer et al. 2013). Such a discrepancy between census and effective population size may indicate a recent rapid decline in blue-eyed black lemurs; diversity in the wild may be further reduced if this species suffered an extreme bottleneck since the collection of samples in the 1960s–1980s. Studies of the two species in the wild highlight their tiny habitat distribution and decreasing numbers (Mittermeier et al. 2006; Volampeno et al. 2010), indicating that they will require intensive conservation efforts, and the residual diversity in blue-eyed black lemurs may be seen as a positive sign in that regard (Perry et al. 2012a).
In estimating demographic parameters from captive-bred individuals, we are assuming that these individuals are a representative sample of their respective species, an assumption that may be violated if, for example, the samples are unusually inbred. Alternatively, the ancestors of the sample may have experienced admixture due to hybridization with their sister species, a process that has been noted in the wild (Rabarivola et al. 1991). High levels of inbreeding would result in an underestimate of intraspecific genetic diversity, whereas extensive admixture would lead to an overestimate, as well as a corresponding overestimate of historic Ne. To minimize the effects of inbreeding, we used individuals indicated as outbred according to recorded pedigrees. The results of our PCA and admixture proportion estimation further indicated that our samples are not admixed (Fig. S5, Supporting information).
The recent split between these two species enabled us to use reduced heterozygosity in one lineage to identify signatures of recent positive selection. Unlike signatures such as increased dN/dS (Kimura 1977) or extreme McDonald–Kreitman test values (McDonald & Kreitman 1991), which rely on an enrichment of fixed noncoding substitutions in genes, our test can identify signatures of more recent selection in both genic and nongenic regions. Because of the large size of our assembly's scaffolds, we were able to summarize the evidence for selection across regions at least 20 kb in length and to identify the orthologs of human genes overlapping these regions. Notably, this genomewide scan in the two initially sequenced individuals identified a signal surrounding the ortholog of OCA2, the gene whose expression determines most of the blue-brown iris pigmentation variation in humans (Duffy et al. 2007; Eiberg et al. 2008; Sturm et al. 2008). This suggested that a similar genetic mechanism may have led to the convergent evolution of blue irises in lemurs and in humans. When we performed further resequencing of a larger sample to investigate this signal in more detail, however, the signal weakened, and instead the lowest diversity signal was seen at MITF, among loci known to contribute to iris pigmentation variation in humans. We also observed signals at FIG4, KITLG, SERPINB2 and TCF7, which play roles in melanogenesis but have not been previously associated specifically with iris pigmentation. Given the prior evidence for the role of these genes in influencing pigmentation and the tentative evidence for recent selection in the blue-eyed black lemur lineage, these regions are therefore candidates for the location of the mutation or mutations causing blue irises in blue-eyed black lemurs. Alternatively, strong differentiation at these genes may be due to an influence on coat colour, which also differs subtly between lemur species in the lighter coloured females (Mittermeier et al. 2008).
Neither changes in the ortholog of the human OCA2 enhancer (Bradley et al. 2009; Meyer et al. 2013) nor amino acid changes in OCA2, ASIP, MITF or TYR (Materials and Methods, Appendix S17, Supporting information) are fixed between the two lemur species, indicating that, if these genes also play a role in iris colour in lemurs, distinct regulatory changes are likely to be responsible. The repeated use of regulatory, rather than coding, changes, to achieve the same phenotype suggests that potential coding changes may be subject to deleterious pleiotropy (Stern 2011). Such negative pleiotropy is consistent with the association of other mutations leading to blue irises, particularly coding changes in ASIP, MITF, OCA2 and TYR, with disease phenotypes in humans (Oetting & King 1999; Smith et al. 2000; Pingault et al. 2010) and domesticated species (Bultman et al. 1992; Juraschko et al. 2003; Hauswirth et al. 2012).
Using our genome assembly together with results from targeted Sanger sequencing and computational identification of transcription factor binding site changes, we were able to identify a small number of candidate causal regulatory sites in the OCA2 region. Analysis of variation in the larger sample, including individuals sequenced to low depth, further enabled the identification of fixed differences in the sample of eight chromosomes in all pigmentation regions, as well as narrowing the candidate list at OCA2. This resulted in 14 candidate sites in the OCA2 region, three of which lie under peaks in FST (Fig.4B). Given the difficulty of functional studies in the appropriate tissue (developing iridial melanocytes) in a nonmodel organism, population genetic analyses of this kind can therefore provide a key step in narrowing down the genetic basis of adaptations in natural populations.
More generally, our study demonstrates the feasibility of building de novo assemblies of nonmodel organisms and illustrates how they can be used for population genetic inferences about demographic history and selection as well as for the more widespread analyses of molecular evolution. Thus, our study provides both a valuable resource for further research in the genetics of lemurs and a model for potential uses of genome sequence data in nonmodel organisms.
Acknowledgments
We thank Catelyn Michelini and George Perry, Jr. at the University of Chicago, Ana Faigon and Peter Andolfatto at Princeton University, and Alvaro Gonzalo Hernandez at the University of Illinois at Urbana-Champaign for library preparation and high-throughput sequencing. We are grateful to Erin Ehmke, Sarah Zehr and David Haring (Duke Lemur Center) for provision of samples and information about and photographs of the lemurs. We thank Doris Bachtrog, Colleen Downs, Jacob Crawford, Matteo Fumagalli, Thorfinn Korneliussen, Tyler Linderoth, Ed Louis, Athma Pai, George Perry, Jr., Christoph Schwitzer, Sylviane Volampeno and Karen Wong Miller, and as well as Dick Hudson, Ziyue Gao and other members of the PPS labs for helpful discussions, and we thank three anonymous reviewers for their valuable comments. Lemur data were collected under protocols approved by the Duke Institutional Animal Care and Use Committee and the Duke Lemur Center (approval nos. A053-09-02 and BS-8-11-1, respectively). This work was mostly completed while M. P. was a Howard Hughes Medical Institute Early Career Scientist at the University of Chicago. This is Duke Lemur Center publication #1292.
Data accessibility
The blue-eyed black lemur and black lemur reference assemblies have been deposited as Whole Genome Shotgun projects at DDBJ/EMBL/GenBank under the accessions LGHW00000000 (blue-eyed black lemur) and LGHX00000000 (black lemur). Sanger sequencing results, vcf files containing well-supported polymorphic and divergent sites within and between the blue-eyed black lemur and black lemur, fastq files used in PSMC, and annotated transcripts in regions within the 1% tail of FST for each species are available on Dryad (doi: 10.5061/dryad.rn745). Binary sequence alignment/map (BAM) files used in analyses are available on the NCBI Sequence Read Archive (SRA): Project PRJNA284191, Accession SRP058683.
Supporting information
Additional supporting information may be found in the online version of this article.
Appendix S1 Sample information and DNA extraction for whole-genome sequenced samples.
Appendix S2 Choice of sequencing libraries, library preparation, and sequencing for genome assemblies.
Appendix S3 Quality control on raw reads.
Appendix S4 Choice of pre-assembly correction method for PE reads.
Appendix S5 Choice of assembler.
Appendix S6 Evaluation of insert size distributions and resolution of ‘bimodal’ libraries.
Appendix S7 Command line parameters for assembly generation.
Appendix S8 Details of memory usage and run times.
Appendix S9 Aligning blue-eyed black lemur contigs to black lemur bacterial artificial chromosomes (BACs).
Appendix S10 Core Eukaryotic Gene Mapping Approach (CEGMA) analysis.
Appendix S11 SNP calling.
Appendix S12 Sanger-based resequencing of additional samples within the scaffold containing the OCA2 ortholog.
Appendix S13 Simulations of pairwise sequentially Markovian coalescent (PSMC) performance on scaffold data and with a population split.
Appendix S14 Estimating species split time.
Appendix S15 Choice of parameters for PSMC and scaling of PSMC output and species split time.
Appendix S16 Identification of candidate regions for recent positive selection in one species.
Appendix S17 Annotation of orthologs of OCA2 and additional human iris pigmentation candidate genes within the blue-eyed black lemur genome.
Appendix S18 Identification of candidate regulatory changes within the scaffold containing the OCA2 ortholog.
Appendix S19 Calculation of summary statistics from the combined sample using angsd and ngstools.
Appendix S20 Assessment of admixture in the combined sample.
Appendix S21 Annotation of candidate selected regions and gene ontology analysis.
Appendix S22 Genome size estimation.
Appendix S23 Identification of neighboring scaffolds to the scaffold containing the OCA2 ortholog.
Fig. S1 Overview of assembly and analysis pipeline.
Fig. S2 Peak memory consumption and time to completion for genome assembly steps.
Fig. S3 Simulations to assess impact of window size, scaffolds, generation time, mutation rate, adjustment for coverage, and population split on output of PSMC.
Fig. S4 Coverage distributions for mapped reads, q-mers, and k-mers.
Fig. S5 Principal components analysis (PCA) and estimation of admixture proportions indicate the absence of admixture in the combined sample.
Fig. S6 Bimodal distributions of estimated insert sizes indicate the presence of artifacts in some library preparations.
Fig. S7 Distributions of summary statistics from scans for selection in two-sample and full datasets.
Table S1 Read counts for each blue-eyed black lemur library.
Table S2 Statistics for Quake-corrected assembly (QCA) and SOAP-corrected assembly (SCA).
Table S3 Primers.
References
- Abecasis GR, Altshuler D, Auton A, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of Molecular Biology. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Amemiya CT, Alföldi J, Lee AP, et al. The African coelacanth genome provides insights into tetrapod evolution. Nature. 2013;496:311–316. doi: 10.1038/nature12027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrianjakarivelo V. 2004. Exploration de la zone en dehors de la peninsula Sahamalaza pour l’évaluation rapide de la population d'E. m. flavifrons, Report.
- Becquet C, Przeworski M. A new approach to estimate parameters of speciation models with application to apes. Genome Research. 2007;17:1505–1519. doi: 10.1101/gr.6409707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birky CW, Walsh JB. Effects of linkage on rates of molecular evolution. Proceedings of the National Academy of Sciences. 1988;85:6414–6418. doi: 10.1073/pnas.85.17.6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley BJ, Pedersen A, Mundy NI. Blue eyes in lemurs and humans: same phenotype, different genetic mechanism. American Journal of Physical Anthropology. 2009;139:269–273. doi: 10.1002/ajpa.21010. [DOI] [PubMed] [Google Scholar]
- Bultman SJ, Michaud EJ, Woychik RP. Molecular characterization of the mouse agouti locus. Cell. 1992;71:1195–1204. doi: 10.1016/s0092-8674(05)80067-4. [DOI] [PubMed] [Google Scholar]
- Chow CY, Zhang Y, Dowling JJ, et al. Mutation of FIG 4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature. 2007;448:68–72. doi: 10.1038/nature05876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett-Detig RB, Hartl DL, Sackton TB. Natural selection constrains neutral diversity across a wide range of species. PLOS Biology. 2015;13:e1002112. doi: 10.1371/journal.pbio.1002112. (NH Barton, ed.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genetics. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly MP, Paschou P, Grigorenko E, et al. A global view of the OCA2-HERC2 region and pigmentation. Human Genetics. 2012;131:683–696. doi: 10.1007/s00439-011-1110-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy DL, Montgomery GW, Chen W, et al. A three-single-nucleotide polymorphism haplotype in intron 1 of OCA2 explains most human eye-color variation. American Journal of Human Genetics. 2007;80:241–252. doi: 10.1086/510885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiberg H, Troelsen J, Nielsen M, et al. Blue eye color in humans may be caused by a perfectly associated founder mutation in a regulatory element located within the HERC2 gene inhibiting OCA2 expression. Human Genetics. 2008;123:177–187. doi: 10.1007/s00439-007-0460-x. [DOI] [PubMed] [Google Scholar]
- Frudakis T, Thomas M, Gaskin Z, et al. Sequences associated with human iris pigmentation. Genetics. 2003;165:2071–2083. doi: 10.1093/genetics/165.4.2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli M. Assessing the effect of sequencing depth and sample size in population genetics inferences. PLoS One. 2013;8:e79667. doi: 10.1371/journal.pone.0079667. (L Orlando, ed.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli M, Vieira FG, Korneliussen TS, et al. Quantifying population genetic differentiation from next-generation sequencing data. Genetics. 2013;195:979–992. doi: 10.1534/genetics.113.154740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesan AK, Ho H, Bodemann B, et al. Genome-wide siRNA-based functional genomics of pigmentation identifies novel genes and pathways that impact melanogenesis in human cells. PLoS Genetics. 2008;4:e1000298. doi: 10.1371/journal.pgen.1000298. (GS Barsh, ed.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnerre S, Maccallum I, Przybylski D, et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:1513–1518. doi: 10.1073/pnas.1017351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther CA, Tasic B, Luo L, Bedell MA, Kingsley DM. A molecular basis for classic blond hair color in Europeans. Nature Genetics. 2014;46:748–752. doi: 10.1038/ng.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl DL, Clark AG. Principles of Population Genetics. Sunderland, Massachusetts: Sinauer Associates, Incorporated; 2007. [Google Scholar]
- Haubold B, Pfaffelhuber P, Lynch M. mlRho – a program for estimating the population mutation and recombination rates from shotgun-sequenced diploid genomes. Molecular Ecology. 2010;19(Suppl 1):277–284. doi: 10.1111/j.1365-294X.2009.04482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauswirth R, Haase B, Blatter M, et al. Mutations in MITF and PAX3 cause “splashed white” and other white spotting phenotypes in horses. PLoS Genetics. 2012;8:e1002653. doi: 10.1371/journal.pgen.1002653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heliconius Genome Consortium. Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature. 2012;487:94–98. doi: 10.1038/nature11041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Camacho J, Cooper RW. The nonhuman primates of Colombia. In: Neotropical Primates: Field Studies and Conservation. In: Thorington RW, Heltne PG, editors. Natl. Acad. Sci. Washington, DC: 1976. pp. 35–69. [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols. 2008;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Research. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juraschko K, Meyer-Lindenberg A, Nolte I, Distl O. Analysis of systematic effects on congenital sensorineural deafness in German Dalmatian dogs. Veterinary Journal (London, England: 1997) 2003;166:164–169. doi: 10.1016/s1090-0233(02)00256-3. [DOI] [PubMed] [Google Scholar]
- Kelley DR, Schatz MC, Salzberg SL. Quake: quality-aware detection and correction of sequencing errors. Genome Biology. 2010;11:R116. doi: 10.1186/gb-2010-11-11-r116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. Preponderance of synonymous changes as evidence for the neutral theory of molecular evolution. Nature. 1977;267:275–276. doi: 10.1038/267275a0. [DOI] [PubMed] [Google Scholar]
- Kong A, Barnard J, Gudbjartsson DF, et al. Recombination rate and reproductive success in humans. Nature Genetics. 2004;36:1203–1206. doi: 10.1038/ng1445. [DOI] [PubMed] [Google Scholar]
- Korneliussen T, Albrechtsen A, Nielsen R. ANGSD: analysis of next generation sequencing data. BMC Bioinformatics. 2014;15:356. doi: 10.1186/s12859-014-0356-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leffler EM, Bullaughey K, Matute DR, et al. Revisiting an old riddle: what determines genetic diversity levels within species? PLoS Biology. 2012;10:e1001388. doi: 10.1371/journal.pbio.1001388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leman SC, Chen Y, Stajich JE, Noor MAF, Uyenoyama MK. Likelihoods from summary statistics: recent divergence between species. Genetics. 2005;171:1419–1436. doi: 10.1534/genetics.104.040402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy C, Khaled M, Fisher DE. MITF: master regulator of melanocyte development and melanoma oncogene. Trends in Molecular Medicine. 2006;12:406–414. doi: 10.1016/j.molmed.2006.07.008. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England) 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Inference of human population history from individual whole-genome sequences. Nature. 2011;475:493–496. doi: 10.1038/nature10231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics (Oxford, England) 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Zhu H, Ruan J, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Research. 2010;20:265–272. doi: 10.1101/gr.097261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindblad-Toh K, Garber M, Zuk O, et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature. 2011;478:476–482. doi: 10.1038/nature10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Wollstein A, Hysi PG, et al. Digital quantification of human eye color highlights genetic association of three new loci. PLoS Genetics. 2010;6:e1000934. doi: 10.1371/journal.pgen.1000934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Lande R. The critical effective size for a genetically secure population. Animal Conservation. 1998;1:70–72. [Google Scholar]
- MacLeod IM, Larkin DM, Lewin HA, Hayes BJ, Goddard ME. Inferring demography from runs of homozygosity in whole-genome sequence, with correction for sequence errors. Molecular Biology and Evolution. 2013;30:2209–2223. doi: 10.1093/molbev/mst125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- deMenocal PB. Plio-Pleistocene African climate. Science (New York, NY) 1995;270:53–59. doi: 10.1126/science.270.5233.53. [DOI] [PubMed] [Google Scholar]
- Meyer WK, Zhang S, Hayakawa S, Imai H, Przeworski M. The convergent evolution of blue iris pigmentation in primates took distinct molecular paths. American Journal of Physical Anthropology. 2013;151:398–407. doi: 10.1002/ajpa.22280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller W, Schuster SC, Welch AJ, et al. Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E2382–E2390. doi: 10.1073/pnas.1210506109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittermeier R, Konstant WR, Hawkins F, et al. Lemurs of Madagascar. Washington, DC: Conservation International; 2006. [Google Scholar]
- Mittermeier RA, Ganzhorn JU, Konstant WR, et al. Lemur diversity in Madagascar. International Journal of Primatology. 2008;29:1607–1656. [Google Scholar]
- Oetting WS, King RA. Molecular basis of albinism: mutations and polymorphisms of pigmentation genes associated with albinism. Human Mutation. 1999;13:99–115. doi: 10.1002/(SICI)1098-1004(1999)13:2<99::AID-HUMU2>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Parra G, Bradnam K, Ning Z, Keane T, Korf I. Assessing the gene space in draft genomes. Nucleic Acids Research. 2009;37:289–297. doi: 10.1093/nar/gkn916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perelman P, Johnson WE, Roos C, et al. A molecular phylogeny of living primates. PLoS Genetics. 2011;7:e1001342. doi: 10.1371/journal.pgen.1001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry GH, Melsted P, Marioni JC, et al. Comparative RNA sequencing reveals substantial genetic variation in endangered primates. Genome Research. 2012a;22:602–610. doi: 10.1101/gr.130468.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry GH, Reeves D, Melsted P, et al. A genome sequence resource for the aye-aye (Daubentonia madagascariensis), a nocturnal lemur from Madagascar. Genome Biology and Evolution. 2012b;4:126–135. doi: 10.1093/gbe/evr132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picardo M, Cardinali G. The genetic determination of skin pigmentation: KITLG and the KITLG/c-Kit pathway as key players in the onset of human familial pigmentary diseases. The Journal of Investigative Dermatology. 2011;131:1182–1185. doi: 10.1038/jid.2011.67. [DOI] [PubMed] [Google Scholar]
- Pingault V, Ente D, Dastot-Le Moal F, et al. Review and update of mutations causing Waardenburg syndrome. Human Mutation. 2010;31:391–406. doi: 10.1002/humu.21211. [DOI] [PubMed] [Google Scholar]
- Prado-Martinez J, Sudmant PH, Kidd JM, et al. Great ape genetic diversity and population history. Nature. 2013;499:471–475. doi: 10.1038/nature12228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przeworski M. The signature of positive selection at randomly chosen loci. Genetics. 2002;160:1179–1189. doi: 10.1093/genetics/160.3.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabarivola C, Meyers D, Rumpler Y. Distribution and morphological characters of intermediate forms between the black lemur (Eulemur macaco macaco) and the Sclater's lemur (E. m. flavifrons. Primates. 1991;32:269–273. [Google Scholar]
- Randriatahina GH, Rabarivola JC. Inventaire des lémuriens dans la partie nord-ouest de Madagascar et distribution d'Eulemur macaco flavifrons. Lemur News. 2004;9:7–9. [Google Scholar]
- Reich DE, Schaffner SF, Daly MJ, et al. Human genome sequence variation and the influence of gene history, mutation and recombination. Nature Genetics. 2002;32:135–142. doi: 10.1038/ng947. [DOI] [PubMed] [Google Scholar]
- Romiguier J, Gayral P, Ballenghien M, et al. Comparative population genomics in animals uncovers the determinants of genetic diversity. Nature. 2014;515:261–263. doi: 10.1038/nature13685. [DOI] [PubMed] [Google Scholar]
- Schwitzer C, Mittermeier RA, Davies N, Johnson SRJ, Razafindramanana J, Louis EERS., Jr . Lemurs of Madagascar: A Strategy for Their Conservation 2013–2016. Bristol, UK: IUCN SSC Primate Specialist Group, Bristol Conservation and Science Foundation, and Conservation International; 2013. [Google Scholar]
- Skotte L, Korneliussen TS, Albrechtsen A. Estimating individual admixture proportions from next generation sequencing data. Genetics. 2013;195:693–702. doi: 10.1534/genetics.113.154138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JM, Haigh J. The hitch-hiking effect of a favourable gene. Genetical Research. 1974;23:23–35. [PubMed] [Google Scholar]
- Smith SD, Kelley PM, Kenyon JB, Hoover D. Tietz syndrome (hypopigmentation/deafness) caused by mutation of MITF. Journal of Medical Genetics. 2000;37:446–448. doi: 10.1136/jmg.37.6.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Star B, Nederbragt AJ, Jentoft S, et al. The genome sequence of Atlantic cod reveals a unique immune system. Nature. 2011;477:207–210. doi: 10.1038/nature10342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern DL. Evolution, Development, & the Predictable Genome. Greenwood Village, Colo: Roberts and Co. Publishers; 2011. [Google Scholar]
- Sturm RA, Duffy DL, Zhao ZZ, et al. A single SNP in an evolutionary conserved region within intron 86 of the HERC2 gene determines human blue-brown eye color. American Journal of Human Genetics. 2008;82:424–431. doi: 10.1016/j.ajhg.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulem P, Gudbjartsson DF, Stacey SN, et al. Genetic determinants of hair, eye and skin pigmentation in Europeans. Nature Genetics. 2007;39:1443–1452. doi: 10.1038/ng.2007.13. [DOI] [PubMed] [Google Scholar]
- Visser M, Kayser M, Palstra R-J. HERC2 rs12913832 modulates human pigmentation by attenuating chromatin-loop formation between a long-range enhancer and the OCA2 promoter. Genome Research. 2012;22:446–455. doi: 10.1101/gr.128652.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volampeno MSN, Masters JC, Downs CT. A population estimate of blue-eyed black lemurs in Ankarafa Forest, Sahamalaza-Iles Radama National Park, Madagascar. Folia Primatologica; International Journal of Primatology. 2010;81:305–314. doi: 10.1159/000322231. [DOI] [PubMed] [Google Scholar]
- Wall JD. Estimating ancestral population sizes and divergence times. Genetics. 2003;163:395–404. doi: 10.1093/genetics/163.1.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagiwa J. Some external characters of the Japanese monkeys (Macaca fuscata. The Journal of Anthropological Society of Nippon. 1979;87:483–497. [Google Scholar]
- Yoder AD. The lemur revolution starts now: the genomic coming of age for a non-model organism. Molecular Phylogenetics and Evolution. 2013;66:442–452. doi: 10.1016/j.ympev.2012.08.024. [DOI] [PubMed] [Google Scholar]
- Zhan X, Pan S, Wang J, et al. Peregrine and saker falcon genome sequences provide insights into evolution of a predatory lifestyle. Nature Genetics. 2013;45:563–566. doi: 10.1038/ng.2588. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Sample information and DNA extraction for whole-genome sequenced samples.
Appendix S2 Choice of sequencing libraries, library preparation, and sequencing for genome assemblies.
Appendix S3 Quality control on raw reads.
Appendix S4 Choice of pre-assembly correction method for PE reads.
Appendix S5 Choice of assembler.
Appendix S6 Evaluation of insert size distributions and resolution of ‘bimodal’ libraries.
Appendix S7 Command line parameters for assembly generation.
Appendix S8 Details of memory usage and run times.
Appendix S9 Aligning blue-eyed black lemur contigs to black lemur bacterial artificial chromosomes (BACs).
Appendix S10 Core Eukaryotic Gene Mapping Approach (CEGMA) analysis.
Appendix S11 SNP calling.
Appendix S12 Sanger-based resequencing of additional samples within the scaffold containing the OCA2 ortholog.
Appendix S13 Simulations of pairwise sequentially Markovian coalescent (PSMC) performance on scaffold data and with a population split.
Appendix S14 Estimating species split time.
Appendix S15 Choice of parameters for PSMC and scaling of PSMC output and species split time.
Appendix S16 Identification of candidate regions for recent positive selection in one species.
Appendix S17 Annotation of orthologs of OCA2 and additional human iris pigmentation candidate genes within the blue-eyed black lemur genome.
Appendix S18 Identification of candidate regulatory changes within the scaffold containing the OCA2 ortholog.
Appendix S19 Calculation of summary statistics from the combined sample using angsd and ngstools.
Appendix S20 Assessment of admixture in the combined sample.
Appendix S21 Annotation of candidate selected regions and gene ontology analysis.
Appendix S22 Genome size estimation.
Appendix S23 Identification of neighboring scaffolds to the scaffold containing the OCA2 ortholog.
Fig. S1 Overview of assembly and analysis pipeline.
Fig. S2 Peak memory consumption and time to completion for genome assembly steps.
Fig. S3 Simulations to assess impact of window size, scaffolds, generation time, mutation rate, adjustment for coverage, and population split on output of PSMC.
Fig. S4 Coverage distributions for mapped reads, q-mers, and k-mers.
Fig. S5 Principal components analysis (PCA) and estimation of admixture proportions indicate the absence of admixture in the combined sample.
Fig. S6 Bimodal distributions of estimated insert sizes indicate the presence of artifacts in some library preparations.
Fig. S7 Distributions of summary statistics from scans for selection in two-sample and full datasets.
Table S1 Read counts for each blue-eyed black lemur library.
Table S2 Statistics for Quake-corrected assembly (QCA) and SOAP-corrected assembly (SCA).
Table S3 Primers.
Data Availability Statement
The blue-eyed black lemur and black lemur reference assemblies have been deposited as Whole Genome Shotgun projects at DDBJ/EMBL/GenBank under the accessions LGHW00000000 (blue-eyed black lemur) and LGHX00000000 (black lemur). Sanger sequencing results, vcf files containing well-supported polymorphic and divergent sites within and between the blue-eyed black lemur and black lemur, fastq files used in PSMC, and annotated transcripts in regions within the 1% tail of FST for each species are available on Dryad (doi: 10.5061/dryad.rn745). Binary sequence alignment/map (BAM) files used in analyses are available on the NCBI Sequence Read Archive (SRA): Project PRJNA284191, Accession SRP058683.