

Abstract
Background:
With few exceptions, members of the Leishmania donovani complex such as L. donovani, L. infantum and L. chagashi are the etiological agents of visceral leishmaniasis or kala-azar. Promastigotes of Leishmania spp. lose their Pathogenicity; the ability to establish infection in a susceptible host, after prolonged culture. The molecular basis of this evolution of pathogenic to nonpathogenic culture has not been very well understood. It has been proposed that the loss of pathogenicity is associated with the gradual disappearance of selective parasite proteins. An alternative hypothesis is that during prolonged culture, the pathogenic clonal population of the parasite is deleted from the mixed population due to their selection pressure. This clonal deletion is proposed to be responsible for the emergence of the nonpathogenic population.
Study Methodology and Results:
We have a done a series of two-dimensional polyacrylamide gel electrophoresis followed by western blot experiments to study the antigenic profile of few L. donovani isolates of Indian origin. We observed a gradual and significant downregulation of expression of a group of low molecular weight proteins (LMW, molecular weight 20–30 kDa) which are associated with loss of pathogenicity. These proteins are recognized only by antiserum raised against the whole cell extract of one of the pathogenic Indian L. donovani isolates, Ag83, and remained undetected by antiserum raised against the nonpathogenic AG83 isolates. These LMW proteins were also present in the nonpathogenic extract in very low levels and remained undetected by the virulent serum, indicating a phenomenon of simultaneous downregulation of the expression and altered immunogenicity. LMW proteins were universally expressed in all early passage Indian isolate we tested and also detected in two clones obtained from pathogenic parasite culture. The antigenic patterns of none of the eight clones obtained from nonpathogenic culture were not exactly similar with the pathogenic clones.
Conclusion:
Therefore, our data strongly support the hypothesis that the loss of pathogenicity of L. donovani is associated with a change in antigenic profile, but not due the selective deletion of pathogenic clones.
KEY WORDS: Two-dimensional polyacrylamide gel electrophoresis, Leishmania donovani, pathogenic promastigotes
INTRODUCTION
Trypanosomatid protozoan parasite Leishmania is responsible for the disease leishmaniasis. The parasites infect several million people worldwide, particularly in the sub-Saharan Africa, Brazil, and Indian sub-continents and causes a spectrum of disease ranging from simple, self-healing innocuous oriental shore to fatal visceral leishmaniasis or commonly known as kala-azar. The unicellular, flagellated, motile form of the parasite, the promastigotes, infect the human hosts during the bite of the vector (sandfly). After entering blood circulation, the parasite invades the macrophages and transforms into nonmotile, aflagellated amastigote form, and multiplies within the hostile phagolysosomal environment. L. donovani, L. infantum, and L. chagasi are the members of L. donovani complex and considered as etiological agents for visceral leishmaniasis. L. donovani is the sole causative agent for visceral leishmaniasis particularly in the Indian sub-continent, and it is fatal if it remains untreated.
It is not very well documented how Leishmania establishes infection in a mammalian host. Several factors, either membrane-associated, or secretory have been proposed to be the contributors, if not exclusively, at least collectively, to determine the pathogenicity of this organism. Lipophosphoglycan (LPG), glycoprotein 63 (GP63) and glycoinositolphospholipids belong to the membrane-associated group, whereas cysteine proteases and secreted acid proteases have been shown to be released through the flagellar pocket as virulence co-factors.[1] Major importance was given in last two decades to establish the role of LPG and GP63 as virulence factors, but cumulative evidences are in the favor of the fact that none of them is indispensable to determine the virulence of this organism. For example, LPG plays major roles from the attachment and development of the parasite within the mid-gut of the sandfly vector to the infection and survival within the macrophages.[2,3,4,5,6,7] However, LPG mutant Leishmania maxicana has been reported to be as virulent as wild type strain to the BALB/c mice.[8] Zinc-dependent metalloprotease GP63 is also a multifunctional protein and participates in several important events such as modulating infectivity of Leishmania, protecting parasites by complement-mediated lysis,[9] intra macrophage survival[10] and alternation of host macrophage signaling.[11] Targeted deletion of GP63 genes in L. major showed no effect in parasite survival within macrophages or lesion formation in BALB/c mice.[12] Similarly, no major difference was observed in infectivity of mutant Leishmania deficient in the synthesis of glycosylphosphotidylinositol anchors,[13] cysteine protease B[14] and secreted acid phosphatases.[15] Therefore, it is clear that genetic ablation of any of the major known virulence factors is insufficient to determine the pathogenicity of this organism leaving ample scope to develop new models to identify factors or co-factors contributing to the pathogenesis.
It has been known for a long time that promastigotes loose pathogenicity during prolonged in vitro culture. Handman et al. proposed that axenic culture of promastigotes is a mixture of pathogenic and nonpathogenic clones and during in vitro culture, noninfective parasite clones take the advantage of vegetative growth to out-grow the infective clones.[16,17] However, it has not been well studied whether; in vitro adaptation has any effect on continuous change in the gene expression pattern of promastigotes.
To understand the effect of in vitro adaptation on the expression of parasite proteins as well as pathogenicity, in this study, we have analyzed the protein profiles of promastigotes, maintained in in vitro culture, by two-dimensional polyacrylamide gel electrophoresis (2D-PAGE) followed by immunoblot experiment using antiserum raised against one of the pathogenic and nonpathogenic L. donovani strains of Indian origin (AG83). A group of 3–4 low molecular weight (LMW) immunoreactive proteins were identified which are present ubiquitously in all pathogenic Indian isolates, and their expressions are downregulated during prolonged culture. Interestingly, those proteins were not detected by the antiserum raised against nonpathogenic promastigotes. On the other hand, nonpathogenic AG83 expressed those proteins in very low level and were detectable only by antiserum raised against nonpathogenic promastigotes. In order to check clonal deletion hypothesis on Indian isolates, pathogenic and nonpathogenic AG83 promastigotes were put through serial dilution to generate clonal populations. The immunoreactive protein profiles of all pathogenic clones are very similar and also identical to the mixed parent population. Among the eight clones obtained from nonpathogenic culture, immunoreactive protein patterns of all are significantly different from the pathogenic population and very similar to the parent nonpathogenic population.
MATERIALS AND METHODS
Parasite
L. donovani promastigotes were subcultured once in a week at 22°C in M-199 (GIBCO-BRL, USA) supplemented with 10% FBS (GIBCO-BRL), penicillin (100 U/ml) and streptomycin (100 μg/ml) and L-Glutamin (BIGCO-BRL, USA). To obtain pathogenic promastigotes, spleens from infected BALB/c mice were dissected into small pieces and dispersed in a medium M-199 supplemented with 20% FBS for 4–5 days. Transformed promastigotes were subcultured at a density of 2 × 106 cells/ml up to 2 months. After that, parasites were maintained in culture up to 2 years or more at a density of 2 × 105 cells/ml.
To maintain the pathogenicity, 1 × 107 promastigotes were suspended in 0.1 ml phosphate-buffered saline (PBS) and injected to BALB/c mice intracardially. After 2 months, mice were sacrificed, and spleen cells were cultured in Medium-199 supplemented with 20% FBS to get the pathogenic promastigotes. L. domnovani isolates, RS and SL were isolated from two different visceral leishmaniasis patients (R. Shaw and S. Lal). Approximately, 0.150 ml of bone marrow aspirate was diluted to 1.0 ml with M-199 containing 20% FBS. Promastigotes were visible after 3–5 days incubation at 22°C.
Biosynthetic labeling
Actively growing promastigotes were harvested at 5000 ×g for 10 min, washed 2 times with PBS and resuspended (2.0 × 107 cells/ml) in Hank's Balanced Salt Solution (HBSS, GIBCO-BRL, USA) supplemented with 45 μCi/ml of 35S-methionin for 4 h at 22°C. Cells were washed twice to remove unincorporated radioactive methionine.
Surface iodination
Promastigotes were harvested and resuspended in PBS (5.0 × 107 cells/ml) for radio-iodination with the addition of 100 μCi 125I-NaI for 10 min at 4°C in a total volume of 1 ml using Iodogen as a catalyst described by Howard et al.[9] Reaction was stopped by removing the parasite from the reaction tube and washed thoroughly with PBS to remove excess radioactivity. More than 90% cells were observed to be viable after radio-iodination.
Two-dimensional polyacrylamide gel electrophoresis
Approximately, 2 × 108 promastigotes were harvested and the cell pellet was resuspended in a 0.2 ml of lysis buffer containing 9.5M Urea (BioRad, USA), 4.0% NP-40 (Sigma, USA), 5% β-mercaptoethanol (Sigma, USA) and 2% ampholine, pH 3.5–10 (Pharmacia, Upsala, Sweden) and incubated at 37°C for 1 h with occasional mixing. Extracts were centrifuged at 10,000 ×g for 10 min at 4°C to collect the supernatant. Promastigote proteins were analyzed by 2D-PAGE according to the method described by O’Farrell et al.[10] with some modifications. Briefly, parasite proteins (approximately 100 μg for each run) were loaded in a 4.0% polyacrylamide gel made in glass tube (12.0 × 0.3 cm) containing 9.5M urea, 4.0% NP–40, 2.0% ampholine pH 3.5–10 and the electrophoresis was carried out in three steps; 200V for 15 min, 300V for 30 min and 400V for 2 h using 10 mm phosphoric acid and 20 mM NaOH as anode and cathode solution, respectively. Gels were ejected from the tube and incubated 1.5 h in a buffer containing 63 mM Tris-HCl pH 6.8, 10.0% glycerol, 2.3% sodium dodecyl sulfate (SDS), and 5.0% β-marcaptoethanol for equilibration. The tube gels were placed on 12.5% SDS-PAGE to analyze protein in the second dimension.
Isolation of amastigotes and in vitro transformation
Spleens from infected BALB/c mice were collected aseptically, dissected into small pieces, homogenized and centrifuged at 800 ×g for 5 min at 4°C to remove cell debris. The supernatant was then collected and re-centrifuged at 5000 ×g for 5 min at 4°C. The pellet containing mainly amastigotes was suspended in M-199 supplemented with 20% FBS and incubated at 22°C for transformation for 3–4 days.
Protein estimation
To minimize the interference of detergents and ampholines, promastigote protein extracts were measured by DOC-TCA method as described by Patterson, GL.[11] Briefly, 10 μl extract was diluted to 1 ml by adding water. To this solution, 100 μl each of 0.15% DOC and 72.0% TCA were added for precipitation. The suspension was centrifuged at 10,000 ×g for 10 min and the pellet was resuspended in 1.0 ml of water. Protein estimation was performed after addition of reagent A (10.0% Na2CO3, 0.1% CuSO4, 5H2O, and 0.2% sodium potassium tartrate) and ×0.5 diluted Folin's reagent after 30 min incubation at room temperature with BSA as a standard at OD750 nm.
Antisera
Promastigotes (5.0 × 109 cells) were harvested and resuspended in PBS containing 0.25% SDS, 0.25% DOC, 0.5% NP-40, 1 mm each of ethylene glycol tetraacetic acid, phenylmethanesulfonyl fluoride and iodoacetic acid, 2 mm ethylenediaminetetraacetic acid and 5 mm benzamidine HCl. The suspension was incubated at 4°C for 1 h with constant stirring and centrifuged at 100,000 ×g for 1 h. Solubilized promastigote proteins (2.0 mg protein/dose/rabbit) were mixed with complete Freund's adjuvants and injected subcutaneously in rabbits. Two booster dosages were given with the same amount of protein mixed with incomplete adjuvants.
Immunoblot
After SDS-PAGE analysis, proteins were transferred to nitrocellulose membrane and incubated with 1:20 dilution of antiserum for1 h at room temperature followed by overnight incubation at 4°C. After washing 1 h in PBS supplemented with 0.05% tween-20, the membranes were treated with protein A-alkaline phosphatase conjugate (2 μg/ml in 150 mm NaCl, 50 mm Tris-HCl, pH 7.5 and 1.0% gelatin, Santa Cruz Biotech, USA) for 1 h. The blots were developed using a diluted mixture of (nitroblue tetrazolium, 50 μg/ml) and 5-bromo-4-chololoro-3-indolyl phosphate, 30 μg/ml in 100 mm Tris-HCl pH 8.8.
Cloning by dilution
Culture supernatant from exponentially growing AG83 promastigotes in M-199 containing 20.0% FBS was filter sterilized and used as conditioned medium for all cloning work. Exponentially growing pathogenic or nonpathogenic AG83 promastigotes were harvested, washed and serially diluted in M-199 culture medium supplemented with 20.0% FBS, 50 μg/ml gentamycin, 100 U/ml penicillin and 100 μg/ml streptomycin. Parasite suspension at the concentration of approximately 1.0 cell/ml was dispensed in a 96 well plate (0.1 ml/well and 0.1 ml conditioned medium) and incubated at 22–25°C. Plates were monitored routinely up to 3 weeks for parasite growth.
RESULT
Identification of a group of low molecular weight immunoreactive proteins highly expressed in pathogenic promastigotes
Initially, we attempted to analyze the total parasite proteins isolated from both pathogenic and nonpathogenic population by SDS-PAGE followed by silver staining. To compare between two protein profiles by this way was not successful because of the very “congested” protein pattern generated by the sensitive silver staining method. In order to get more distinguishable protein pattern, whole parasite proteins were analyzed by two-dimensional gel electrophoresis (2D-PAGE). Unlike combined isoelectrophoresis followed by SDS-PAGE,[12] we used nonequilibrium pH gradient electrophoresis (NEPHGE) because it is less time consuming and separates proteins based on their isoelectric point.[10] The optimum resolution was obtained when proteins were run at 1000 VH in a mixture of ampholines with pH 3.5–10.0 and 4.0–6.0 at 1:2 ratio [Supplement Figure 1a and b]. To obtain total parasite protein profile, promastigotes were incubated with 35S-methionine for 4 h and extracts were analyzed by NEPHGE followed by SDS-PAGE. We observed around 50 major and several 100 minor clearly distinguishable protein spots in each gel [Figure 1a and b]. Though the background of these radioactive gels was very high, but we could still find few differences in the protein profile of pathogenic and nonpathogenic promastigotes. For example, if we consider the relative position of the peptide “a” and “b,” in Figure 1a and b, then we observed those peptides were present only in pathogenic extract. Similarly, few minor spots present in Figure 1b (indicated by arrow), appeared to be missing in pathogenic extract. Therefore, Figure 1a and b indicated for the 1st time that expression of proteins in two forms of promastigotes is not exactly identical. Next, we wanted to compare immunoreactive protein profile of both promastigotes because those protein may contribute directly or indirectly to the pathogenesis. Promastigote proteins were analyzed by 2D-PAGE followed by western blotting using antiserum raised against virulent (virulent serum) and avirulent AG83 promastigote extracts. At first, we western blotted pathogenic promastigote extract with virulent and avirulent serum and observed a significant difference between two protein profiles [Figure 2a and b]. Few proteins (marked by arrow) were expressed in pathogenic promastigotes were recognized by virulent serum. However, the most significant difference was observed in the case of a group of 3–4 LMW protein located inside the square. Two proteins (indicated by arrow inside the square) in this group were absolutely recognized by virulent serum but not by the avirulent serum. Other two minor proteins located at the edge of the square were also not recognized by avirulent serum. It, therefore, appeared that those LMW proteins are either not present in avirulent extract, or they expressed with altered antigenicity. To explore that possibility, we analyzed the nonpathogenic promastigote extract by 2D-PAGE-immunoblot experiments using the virulent and avirulent serum. We observed that avirulent serum recognized few high molecular weight proteins more strongly and few extra protein spots than virulent serum recognized in the same extract. Interestingly, the two proteins (indicated by arrow inside the box) very weakly detected by the serum and one of the two proteins remained undetected [Figure 2c]. These LMW proteins in nonpathogenic parasite extracts were not detected by the virulent serum [Figure 2d]. Above result, therefore, indicated that a group of LMW proteins were highly expressed in pathogenic promastigotes and their expression is downregulated in nonpathogenic counterpart perhaps with altered anitigenicity. All proteins detected in Western blot experiments by antiserum were of parasite origin and did not cross-react with proteins either from spleen tissue of mice or from serum [Supplement Figure 2a and b].
Supplement Figure 1.
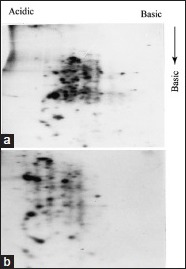
Combination of ampholines is required for better resolution. 35S-methionine labeled AG83 promastigotes were analyzed in first dimension 1000 vhr with two different ampholine compositions. One with pH 3.5–10.0 only (a) and other with 1:2 mixture of ampholine pH 3.5–10.0 and pH 4.0–6.0
Figure 1.
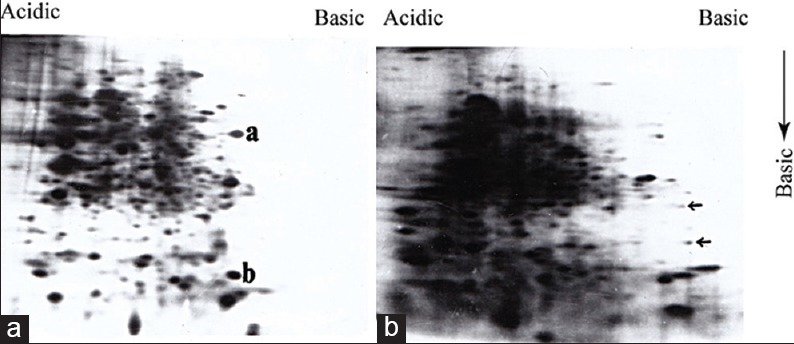
Analysis of metabolically labeled promastigote proteins by two-dimensional polyacrylamide gel electrophoresis identified different protein expression in pathogenic and nonpathogenic promastigotes: 35s-methionine labeled proteins (~2 × 106 cpm) of pathogenic (a) and nonpathogenic (b) AG83 promastigotes were analyzed by two-dimensional polyacrylamide gel electrophoresis. Proteins marked as ‘a’ and ‘b’ in (a) and arrows (..) in (b) indicated proteins which are expressed distinctly in two different promastigotes
Figure 2.
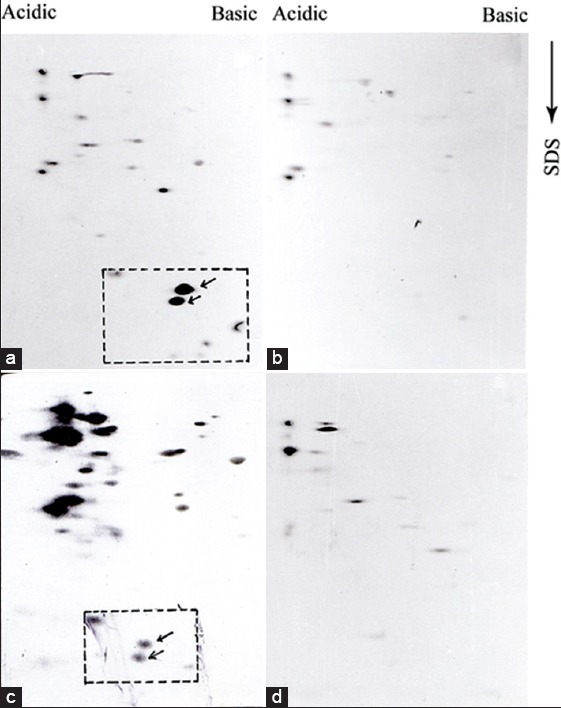
A distinct difference is observed in immunoreactive protein pattern of pathogenic and nonpathogenic promastigote proteins: Protein extracts (~100 μg) of pathogenic (a and b) and nonpathogenic (c and d) promastigotes were analyzed by two-dimensional polyacrylamide gel electrophoresis and western blotted with virulent antisera (a and d) and avirulent sera (b and c) at 1:20 dilution. The low molecular weight proteins are enclosed with rectangle with arrows
Supplement Figure 2.
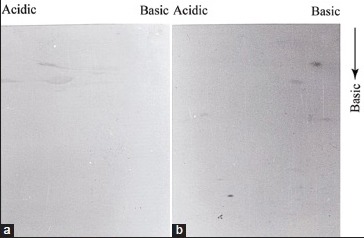
Virulent serum recognized specifically parasite proteins. Two-dimensional polyacrylamide gel electrophoresis followed by immunoblot analysis was carried out with virulent serum using precipitated FBS protein (a) and spleen cell protein extract form uninfected BALB/c mice (b) about 100 μg protein was loaded in each gel
Loss of pathogenicity is associated with the downregulation of low molecular weight proteins
From Figure 2, it is clear that we identified the high-level expression of a group of LMW protein which is differentially expressed in pathogenic promastigotes. We were more interested to know whether this high expression has any association with the ‘age’ of the parasite culture. AG83 promastigotes were maintained in a culture for more than 9 months, and parasite cultures were harvested with an interval of few months and extracts were analyzed as 2D-PAGE followed by immunoblotting experiments with virulent antiserum. From Figure 3a-d, it is clear that besides other minor changes in the protein profiles, the most significant difference was observed in the group of 3–4 LMW proteins. The level of those proteins remained nearly same in promastigotes maintained in the culture up to 4 months. After 7 months, the expression of all but one was significantly reduces, and only two of them could be detected in the extracts, when parasites were maintained in continuous culture for 9 months. It was observed that for AG83 promastigotes, pathogenicity started to decline when parasites were in continuous culture around 5–6 months (tested by BALB/c mice infection followed by measuring the spleen size after few months interval). After 9 months, we found that the infectivity of the parasite was 5–6-fold reduced (measured by the spleen size) in comparison to the very early passage parasite. Therefore, we conclude that both differential expression and altered composition of immunoreactive proteins are associated with the loss of pathogenicity of this L. donovani isolate. The LMW proteins were not detected in the amastigote extracts (data not shown), indicating they are specifically expressed in promastigotes. To check the promastigote specific expression, amastigotes were isolated from the spleen of the infected BALB/c mice and incubated in vitro at 22°C for transformation. We observed under our conditions, the transformation took 48 h. Therefore, parasite extracts were isolated and analyzed SDS-PAGE followed by immunoblotting by pathogenic and nonpathogenic serum. Four protein bands in between ~20 and 30 kDa, detected by pathogenic serum [Figure 4, left panel] but not by the nonpathogenic serum [Figure 4 right panel], corresponded well with our previous 2D-PAGE-immunoblot data showing the high-level expression of LMW proteins in pathogenic extracts. However, we are intrigued by the fact that amastigote extracts or “0” h extract had a very poor recognition by those two antiserums.
Figure 3.
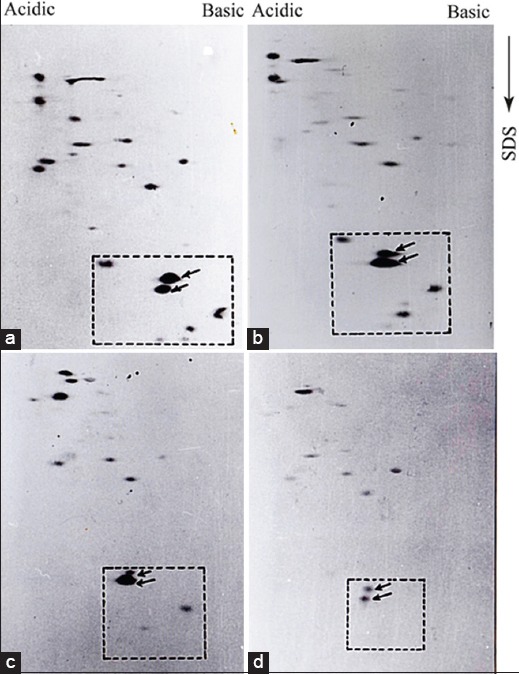
The expression of low molecular weight promastigote proteins are downregulated during prolong culture. AG83 promastigotes were harvested after one (a), four (b), seven (c), and nine (d) months in culture after transformation from amastigotes. Proteins (100 μg) from harvested parasite stocks were analyzed by two-dimensional polyacrylamide gel electrophoresis followed by immunoblotting with virulent aniserum. Low molecular weight proteins are marked by arrows inside the box
Figure 4.
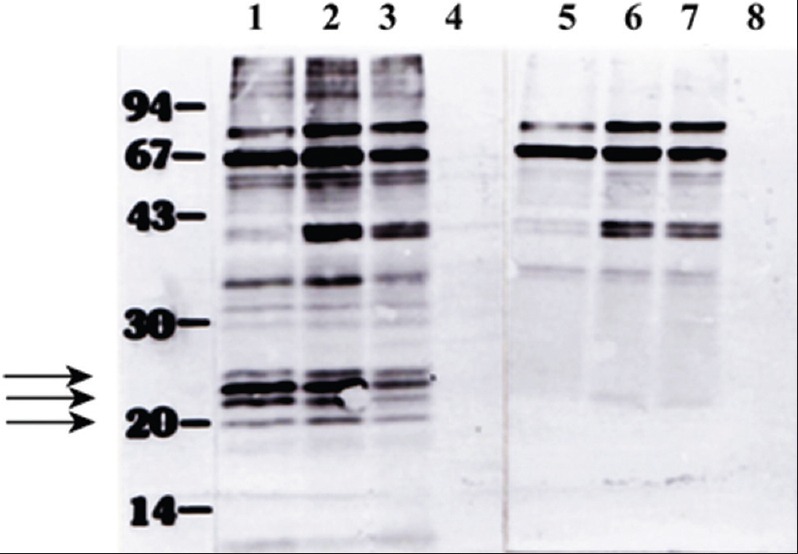
Low molecular weight antigens were appeared in promastigotes during the transformation from amastigotes in vitro. Parasite proteins during in vitro transformation from promastigotes and amastigotes were collected at regular intervals such as 0 h (lanes 4 and 8), 48 h (lanes 3 and 7), 72 h (lanes 2 and 6), and 96 h (lanes 1 and 5) and extracts (~25 μg) were analyzed in 12.5% SDS-PAGE and immunoblotted using virulent (lanes 1-4) and avirulent (lanes 5-8) serum. The low molecular weight proteins are indicated by arrows
The low molecular weight proteins are present in all Leishmania donovani isolates available
Antigenic diversity in parasites such as Leishmania is a very well established phenomenon. In one of the recent demonstrations, Forgber et al. showed a distinct diversity among the immunoreactive L. donovani proteins isolated from several endemic zones of a particular region of India.[13] The GE1 and GE2 isolates were obtained in one of the endemic zones in India (Bihar), and RS and SL were isolated from patients from another distinct endemic zone in India (Kolkata). We were also interested to check the expression of this LMW immunoreactive parasite proteins among the isolates collected from several endemic areas. Therefore, besides AG83, we analyzed immunoreactive protein profiles of pathogenic promastigote isolates such as GE1, GE2, RS, and SL using the virulent and avirulent serum. An overall similarity in immunoreactive protein profile were observed among the L. donovani isolates generated by the virulent serum as depicted in Figure 5a-e. Few extra cross reactive proteins are also detected in GE2 [Figure 5c] and SL [Figure 5e] isolates (marked by arrow) indicating the antigenic diversity among the isolates. However, besides this antigenic diversity, the LMW proteins (enclosed by the box) are expressed by all L. donovani isolates tested. In order to confirm their pathogenic promastigote specific expression, GE1, GE2, and SL isolate extracts were analyzed by 2D-PAGE-immunoblot experiments using avirulent antiserum. As it has been observed previously for AG83 isolates, these LMW proteins were not detected by this antiserum. It is also important to note that majority of the immunoreactive proteins of pathogenic isolates show a very weak recognition by avirulent serum [Figure 6]. Therefore, we conclude that: (i) Antigenic polymorphism exists among the Indian L. donovani isolates; (ii) a significant change is observed in immunoreactivity of pathogenic and nonpathogenic proteins; and (iii) among the immunoreactive proteins a group of LMW proteins with conserved antigenicity is expressed ubiquitously in all L. donovani isolates of Indian origin.
Figure 5.
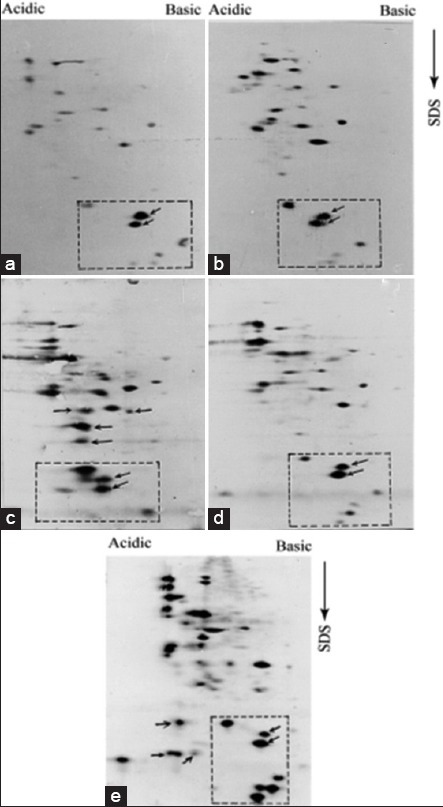
All Leishmania donovani Indian isolates express low molecular weight proteins besides maintaining noticeable antigenic diversity. All Indian isolates of Leishmania donovani promastigotes extracts were analyzed by two-dimensional polyacrylamide gel electrophoresis followed by immunoblot experiment with virulent serum. AG83 (a), GE1 (b), GE2 (c), RS (d) and SL (5e) express low molecular weight enclosed by square. Isolate specific immunoreactive proteins, such as in GE1 and SL, are also marked by arrows outside the square
Figure 6.
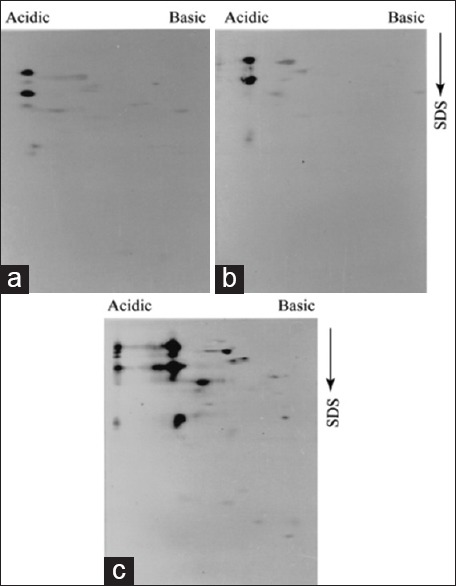
Low molecular weight and antigenically diverse immunoreactive proteins are not recognized by avirulent serum. Promastigote proteins (100 μg) were analyzed by 2-D PAGE followed by immunoblot experiments with 1:20 dilution of avirulent serum. GE1 (a), GE2 (b), and SL (c) display very similar antigen pattern though few additional spots were detected in SL extract
Cell surface proteins of promastigotes are more immunogenic in nature than proteins that functionally participate in parasites metabolic pathways mostly because they are modified by other macromolecules such as polysaccharides, long chain fatty acids, etc. To determine the expression of LMW on the surface of the parasite, live AG83 promastigotes were radio-iodinated, and the radiolabeled parasite extract was analyzed by 2D-PAGE followed by western blotting by virulent serum. The same blot was exposed to detect the radiolabeled proteins. Though few proteins were detected in the autoradiogram, LMW proteins were not detected by radio-iodination [Figure 7].
Figure 7.
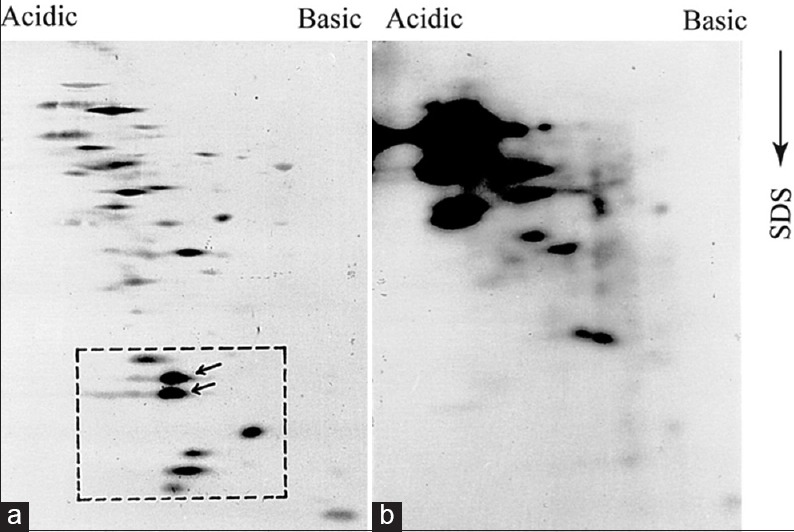
Low molecular weight promastigote proteins are not exposed to the parasite surface. Approximately, 4 × 106 AG83 pathogenic promastigotes were harvested and subjected to radio-iodination. Extract of radio labelled parasite protein (100μg) were analysed by 2D-PAGE-immunoblotting experiment using virulent serum.
Loss of pathogenicity in Leishmania donovani is associated with the change in antigenic profile not clonal deletion
It appears from our data that adaptation of promastigotes in culture is strongly associated with a gradual loss of a group of LMW proteins. However, in case of L. tropica, it has been demonstrated that loss of pathogenicity during adaptation is a result of the overwhelming growth of noninfective parasite clone over the infective clones.[16] In order to explore that possibility in L. donovani, we used serial dilution method to seed single promastigote in a well of a 96 well culture plate supplemented with conditioned medium. This serial dilution method was used for both pathogenic and nonpathogenic culture to isolate the virulent and avirulent clones, if exist, in both populations. Just after transformation from amastigotes, we observed a very slow growth rate of promastigotes until up to 6–8 weeks. Several attempts to isolate clones within that period of time were unsuccessful. However, two clones – C6 and D6 (named according to the well number of a 96-well plate) – were obtained from a promastigote stock that was kept in culture for more than 2 months. To confirm pathogenicity of C6 and D6, we used both clones in BALB/c mice infection and observed that infectivity that was very much comparable to the pathogenic promastigote culture estimated by the amount of swelling of the spleen with respect to the noninfected mice (data not shown). Extracts of promastigote clones C6 and D6 were analyzed by SDS-PAGE and 2D-PAGE followed by western blotting by virulent and avirulent serum. Extract of C6 [Figure 8 (Upper panel), lanes 2 and 5], D6 [Figure 8 (Upper panel), lanes 3 and 6] and pathogenic promastigotes [Figure 8 (Upper panel), lane 1 and 4] were analyzed SDS-PAGE followed by immunoblotting with virulent (lanes 1–3) and avirulent (lanes 4–6) serum. As it was observed in Figure 8, virulent serum recognized four LMW proteins in between 15 and 25 kDa which corresponds well with the proteins (marked by a square) in 2D-PAGE western blot data. Though both antiserum detected few proteins between 40 and 70 kDa, however, those LMW proteins were not detected by avirulent serum. A 2D-PAGE followed by western blot analysis also supported this same observation. C6 and D6 clones and virulent AG83 promastigote generated very similar immunoblot profiles with high-level expression of LMW proteins [Figure 8a-c (Lower panel)]. When we used nonpathogenic AG83 promastigotes in serial dilution to isolate clonal parasite population, we were successful to generate eight clones. In order to determine the nature of those clonal populations, promastigotes extracts of all clones were analysed by SDS-PAGE followed by immunoblotting using avirulent serum [Figure 9a] and virulent serum [Figure 9b]. It was observed that three major immunoreactive proteins (65, 60, and 38 kDa) were present in all clones as well as the uncloned population of AG83. Similarly, a number of proteins in the range of 18–20 kDa was also present in each clone but not in virulent AG83 population and a 52 kDa protein is present in variable amounts in all clones along with avirulent population – but not in the virulent counterpart [Figure 9a]. An additional 25 kDa protein is observed in one of the clones F10 (lane 7) and to some lesser extent in F11 (lane 8) and H2 (lane 9) but it is not in the uncloned populations (lanes 1 and 2). Therefore, apart from few minor differences, immunoreactive protein profiles of all clones are nearly identical, and the pattern is closer to the uncloned avirulent population. This conclusion was further confirmed by immunoblot analysis of all clones with virulent serum [Figure 9b]. An overall similarity among the immunoreactive protein profiles of all clones are observed though the relative amount of 25, 14, and 13 kDa proteins were varied among the clones. In comparison to the noninfective clones, the immunoreactive protein profile of AG83 clone D6 was found to be significantly different. Protein profile of D6 is characterized by the presence of two major immunoreactive proteins (23 and 24 kDa, Figure 9b, lane 1) instead of three (21, 22, and 23 kDa, lanes 3–10). It was also observed, that those three proteins are not present equally in all clones. For example, they are expressed in equal amount in clone H2 (lane 10) but one of them is missing in clone F10 (lane 8) and in clone D8 (lane 6). All those were significantly less expressed in nonvirulent AG83 uncloned population (lane 2). More significantly, the 24 kDa protein is only expressed in clone D6. Taken together, this result clearly indicated the change antigenic profile not the clonal deletion is associated with the loss of pathogenicity in Indian L. donovani isolates.
Figure 8.
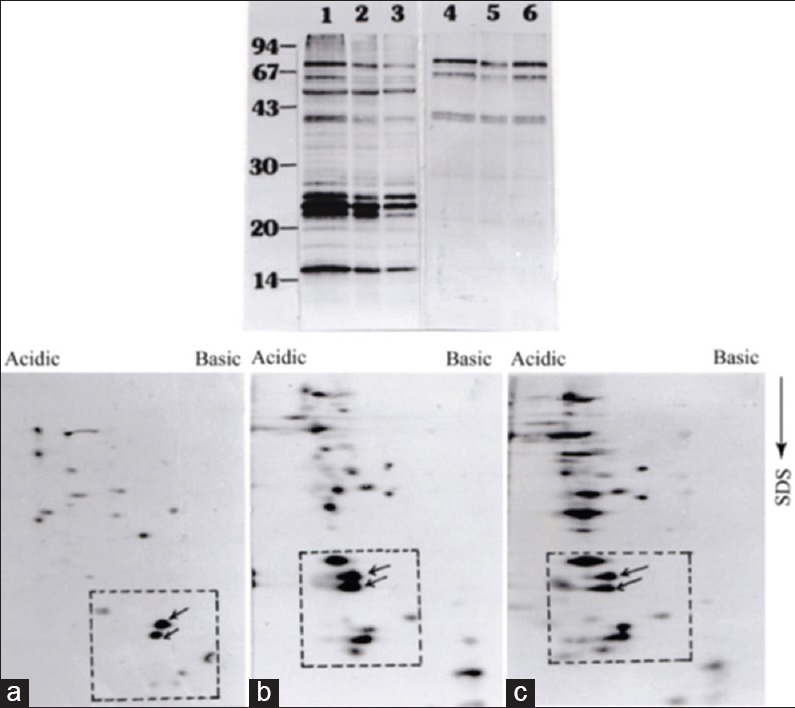
Pathogenic clones and an axenic mixed pathogenic population of Leishmania donovani promastigotes show identical immunoreactive protein pattern. Upper panel: Extracts of axenic culture of virulent AG83 promastigotes (lanes, 1 and 4), virulent clone C6 (lanes, 2 and 5), and D6 (lanes 3 and 6) were analyzed by immunoblotting with 1:20 dilution of virulent antiserum (lanes, 1-3) and avirulent antiserum (lanes, 4-6). Lower panel: Extracts of virulent AG83 (a), clones C6 (b), and D6 (c) were analyzed by two-dimensional polyacrylamide gel electrophoresis followed by immunoblotting with virulent serum. Low molecular weight proteins with in the box were marked
Figure 9.
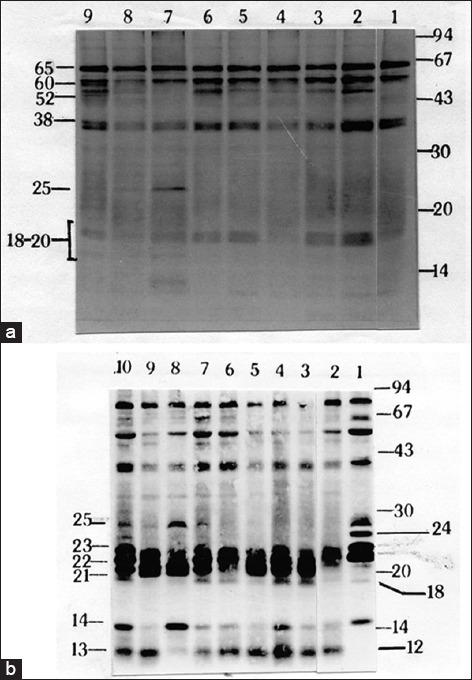
An antigenic pattern of non-pathogenic clones is very similar but significantly different from the pathogenic clone. Immunoblot analysis was carried out using extracts (50 μg) of nonpathogenic clones (lanes 1-9; wild type avirulent AG83, clones, A5, C2, C4, D8, F7, F10, F11, and H2 respectively) using avirulent (a) and (lanes 1-10; wild-type virulent clone D6, wild type avirulent AG83, clones, A5, C2, C4, D8, F7, F10, F11, and H2 respectively) using virulent (b) serum. The uniquely expressed 24 kDa protein by virulent promastigotes is mentioned
DISCUSSION
Summary
Loss of pathogenicity during prolonged in vitro culture provides a unique opportunity to understand the sequential change in gene expression of L. donovani promastigotes from pathogenic to nonpathogenic transition. Interestingly, not many investigations were done at the molecular level to understand this transformation. We wanted to utilize this phenomenon as a model system to establish a link between coherent protein expression pattern and loss of pathogenicity and thus tried to address the hypotheses to explain the loss of pathogenicity phenomenon of this parasite. In the first section of our results, the 2D-PAGE followed by western blot experiments identified a change in the protein profile associated with the adaptation. Our data, clearly established that all pathogenic L. donovani isolates of Indian origin highly express a group of LMW proteins which are either not expressed in a nonpathogenic parasites or expressed with alter immunoreactivity. During prolonged culture, the expression of these LMWs was significantly downregulated which correlated well with the loss of pathogenicity. In the second section, we wanted to establish the association of the high expression of LMAs proteins in parasite clones originated from a pathogenic and nonpromastigote stock by serial dilution method. Both pathogenic (C6 and D6) were virulent which was observed by the animal infection study and their antigenic expression profiles analyzed in both SDS-PAGE and 2D-PAGE are very similar to the virulent AG83 populations with the expression of significant high-levels of those LMW's. On the other hand none of the clones (A2, C2, C4, D8, F7, F10, F11 and H2) originated from nonpathogenic populations have antigenic profile exactly similar to the pathogenic clone though we observed some antigenic variations among these clones. Our results indicated that a group of LMWs are expressed consistently in all L. donovani isolates of Indian origin we have tested along with the pathogenic clones and strongly support the change in antigenic profile rather than ablation of clones associated with the loss of pathogenicity during adaptation.
LMAs are not appeared to be exposed on the parasite surface
It appears those LMWs are not exposed to the surface of the parasite because surface iodination of intact parasite failed to detect those proteins [Figure 7]. Parasites surface proteins are more immunodominant than proteins participating in metabolic activities mostly because they are very often conjugated with carbohydrate moieties[14] or long chain fatty acids. The strong immonoreactivity of those LMWs and gradual disappearance during adaptation clearly indicated that they are probably associated with the cell surface to take part in the antigenic variability or to determine the pathogenicity of this parasite. Alternatively, it is possible that these small molecule antigens are not exposed to the surface of the parasite– a phenomenon that can be called “antigen masking” due to the presence of widely prevalent surface macro molecules such as LPG.[15]
Amastigote antigens failed to cross-react with promastigote specific antisera
We are intrigued by the fact that amastigote proteins were poorly detected by the antisera raised agaist L. donovani promastigotes in this case. One of the reasons for this poor detection is due to the very little similarity between antigens of these two forms of the parasite. A significant difference was reported in antigenic pattern between those two forms of the parasite. In case of L. donovani, absolutely no cross-reactivity is observed between antigens when immune serum was raised against membrane part of both forms of parasites.[18] In case of Leishmania mexicana pifanoi, it has been reported that antigenic pattern of amastigotes isolated from culture, from the footpads of infected hamsters or from a J774 macrophage cell line show similar antigenic profile but differ from the promastigotes.[19] Therefore, it can be at least partly conceivable that the poor recognition by those antisera is possibly due to the wide diversity of immunoreactive proteins between those two forms of the parasite.
Antigenic diversity at the micro level
An overall similarity in the immunoreactive protein profile of all L. donovani isolates AG83, GE1, GE2, RS, and SL has been observed though few differences can be noticed very clearly. In case of RS few extra spots were detected on top of the box (marked by arrow) and in case of SL few additional spots were detected within as well as outside the box. All Indian isolates were obtained from the bone marrow aspirates of patients which excludes the possibility of host tissue-induced modifications. AG83, GE1, and GE2 were maintained more than 2 years through animal passaging, and the RS and SL both were recent isolates and maintained such as other three isolates. Therefore, differences in immunoreactive protein pattern between isolates may be due the antigenic diversity among the isolates. Using 2D-PAGE followed by MALDI-TOF analysis Forgber et al.[13] showed a wide heterogeneity in the antigenic profiles among the L. donovani isolated from several endemic zones of India. Our analysis indicated even minor differences in the immunoreactive proteins among the nonpathogenic clones originated from the AG83 population which were not observed among the pathogenic clones. Therefore, it is possible that parasites are very well equipped to change their gene expression pattern. Genes, associated with the pathogenicity, are less likely to be diversified than genes which are not. The LMW identified here, which are highly expressed in all pathogenic isolates and disappear during adaptation, may belong to this conserve group of genes whose expression are required to maintain the pathogenicity.
Clonal deletion versus change in antigenic profiles
Loss of pathogenicity in Leishmania spp. during prolonged in vitro culture is a very well established phenomenon. Two major hypotheses though proposed several decades ago, could not gather enough supportive evidence to establish them. Schneider and Hertig[20] proposed that this decreased infectivity is a due loss of antigens associated with invasiveness. In case of L. tropica (LCR-L137), Handman et al. (1993) showed, that loss of infectivity is a result of deletion of pathogenic clones due to prolong culture which was supported by the fact that one out of seven clones obtained from L. tropica was found to be pathogenic. We obtained two clones from pathogenic promastigote culture whereas eight clones from the nonpathogenic promastigote culture of AG83 promastigotes. Those difference in numbers indicated that nonpathogenic parasites certainly have a growth advantage over their pathogenic counterpart. Therefore, if the pathogenic culture is a mixed population, then it is conceivable that we would have got few nonpathogenic clones along with the pathogenic clones. Interestingly, the immunoreactive protein profiles of two pathogenic clones are very similar. In case of nonpathogenic clones, our western blot data indicated that their immunoreactive protein profiles are different than the pathogenic clones and some minor differences are also observed between clones. Therefore, keeping promastigotes in continuous culture, which is less hostile environment than human body, is another way of introducing a selective pressure on parasites to change the gene expression which eventually leads to shut down or downregulation of virulence associated gene; whereas, invasiveness on the other hand, is necessary for the continual survival of the parasite inside the host. This selection pressure can help to introduce minor changes in the gene expression of the survived clones. It was beyond the scope of this study, to test the animal infectivity of each individual clones we obtained from nonpathogenic culture. Our data, therefore, is supportive to the loss of invasive antigen hypothesis, at least in case of L. donovani.
What are those proteins?
It was beyond our scope to further characterize the strongly immunoreactive LMW antigens. However, their consistent expression in the virulent stage of the promastigotes indicated that they have yet to be discovered role to determine the pathogenicity of this organism. Several reports are in the favor of the fact that LMW proteins of Leishmania can be a potentially protective antigen against this parasite. It has been shown that T-cell clone that provides immunity against Leishmania recognized antigens around 8–35 kDa.[21] Coelho et al., have recently identified several new proteins between 16 and 35 kDa range by 2D-PAGE followed by immunoblot experiment using pooled symptomatic and asymptomatic visceral leishmaniasis dog serum. More interestingly, the pooled serum recognized several hypothetical proteins within 18–36 kDa which are expressed in the active infection.[22] Recently, thiol-specific antioxidant protein, 22.1 kDa, which elicits a Th1 response in mouse and provides significant protection during L. major infection, has been shown as a major vaccine candidate.[23,24,25]
CONCLUSION
Identification of universal immunoreactive proteins in the case of protozoan parasite Leishmania is very challenging, because of the presence of a high degree of antigenic diversity exists within the isolates from the single endemic zone. Our studies have identified a group of LMW proteins present in all L. donovani isolates collected over a period of several years. These LMW proteins are highly expressed in pathogenic parasites, and their downregulated expression is associated with the loss of pathogenicity. Therefore, our model was capable to identify specific antigens which are differentially expressed on pathogenic parasites and can be considered as a marker for virulence. Further studies to characterize and forced expression or knocking down the expression of those proteins in parasites can possibly open new windows to understand the role of those proteins in pathogenesis in this parasite.
Acknowledgments
The whole research work was carried out in the laboratory of Dr. Swadesh Duttagupta, of Indian Institute of Chemical Biology, Jadavpur, West Bengal, and India.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Olivier M, Atayde VD, Isnard A, Hassani K, Shio MT. Leishmania virulence factors: Focus on the metalloprotease GP63. Microbes Infect. 2012;14:1377–89. doi: 10.1016/j.micinf.2012.05.014. [DOI] [PubMed] [Google Scholar]
- 2.Desjardins M, Descoteaux A. Inhibition of phagolysosomal biogenesis by the Leishmania lipophosphoglycan. J Exp Med. 1997;185:2061–8. doi: 10.1084/jem.185.12.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beverley SM, Turco SJ. Lipophosphoglycan (LPG) and the identification of virulence genes in the protozoan parasite Leishmania. Trends Microbiol. 1998;6:35–40. doi: 10.1016/S0966-842X(97)01180-3. [DOI] [PubMed] [Google Scholar]
- 4.Descoteaux A, Turco SJ. Glycoconjugates in Leishmania infectivity. Biochim Biophys Acta. 1999;1455:341–52. doi: 10.1016/s0925-4439(99)00065-4. [DOI] [PubMed] [Google Scholar]
- 5.Pimenta PF, Saraiva EM, Rowton E, Modi GB, Garraway LA, Beverley SM, et al. Evidence that the vectorial competence of phlebotomine sand flies for different species of Leishmania is controlled by structural polymorphisms in the surface lipophosphoglycan. Proc Natl Acad Sci U S A. 1994;91:9155–9. doi: 10.1073/pnas.91.19.9155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sacks DL, Pimenta PF, McConville MJ, Schneider P, Turco SJ. Stage-specific binding of Leishmania donovani to the sand fly vector midgut is regulated by conformational changes in the abundant surface lipophosphoglycan. J Exp Med. 1995;181:685–97. doi: 10.1084/jem.181.2.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sacks DL, Modi G, Rowton E, Späth G, Epstein L, Turco SJ, et al. The role of phosphoglycans in Leishmania-sand fly interactions. Proc Natl Acad Sci U S A. 2000;97:406–11. doi: 10.1073/pnas.97.1.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ilg T. Lipophosphoglycan is not required for infection of macrophages or mice by Leishmania mexicana. EMBO J. 2000;19:1953–62. doi: 10.1093/emboj/19.9.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Howard RJ, Kaushal DC, Carter R. Radioiodination of parasite antigens with 1,3,4,6-tetrachloro-3 alpha, 6 alpha-diphenylglycoluril (IODOGEN): Studies with zygotes of Plasmodium gallinaceum. J Protozool. 1982;29:114–7. doi: 10.1111/j.1550-7408.1982.tb02891.x. [DOI] [PubMed] [Google Scholar]
- 10.O’Farrell PZ, Goodman HM, O’Farrell PH. High resolution two-dimensional electrophoresis of basic as well as acidic proteins. Cell. 1977;12:1133–41. doi: 10.1016/0092-8674(77)90176-3. [DOI] [PubMed] [Google Scholar]
- 11.Peterson GL. A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal Biochem. 1977;83:346–56. doi: 10.1016/0003-2697(77)90043-4. [DOI] [PubMed] [Google Scholar]
- 12.O’Farrell PH. High resolution two-dimensional electrophoresis of proteins. J Biol Chem. 1975;250:4007–21. [PMC free article] [PubMed] [Google Scholar]
- 13.Forgber M, Basu R, Roychoudhury K, Theinert S, Roy S, Sundar S, et al. Mapping the antigenicity of the parasites in Leishmania donovani infection by proteome serology. PLoS One. 2006;1:e40. doi: 10.1371/journal.pone.0000040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olafson R, Dwek R, Rademacher T, Chang KP, Ferguson MJ. Structure function studies on Leishmania surface membrane proteins. In: Hart DT, editor. Leishmaniasis. US: Springer; 1989. pp. 619–25. [Google Scholar]
- 15.Karp CL, Turco SJ, Sacks DL. Lipophosphoglycan masks recognition of the Leishmania donovani promastigote surface by human kala-azar serum. J Immunol. 1991;147:680–4. [PubMed] [Google Scholar]
- 16.Handman E, Hocking RE, Mitchell GF, Spithill TW. Isolation and characterization of infective and non-infective clones of Leishmania tropica. Mol Biochem Parasitol. 1983;7:111–26. doi: 10.1016/0166-6851(83)90039-7. [DOI] [PubMed] [Google Scholar]
- 17.Segovia M, Artero JM, Mellado E, Chance ML. Effects of long-term in vitro cultivation on the virulence of cloned lines of Leishmania major promastigotes. Ann Trop Med Parasitol. 1992;86:347–54. doi: 10.1080/00034983.1992.11812677. [DOI] [PubMed] [Google Scholar]
- 18.Chattopadhayay R, Kaur S, Ganguly NK, Mahajan RC. Antigenic differences between axenic amastigotes and promastigotes of Leishmania donovani. Indian J Med Res. 1996;104:349–54. [PubMed] [Google Scholar]
- 19.Pan AA. Leishmania mexicana pifanoi: Analysis of the antigenic relationships between promastigotes and amastigotes by gel diffusion, immunoelectrophoresis, and immunoprecipitation. J Protozool. 1986;33:192–7. doi: 10.1111/j.1550-7408.1986.tb05588.x. [DOI] [PubMed] [Google Scholar]
- 20.Schneider CR, Hertig M. Immunodiffusion reactions of Panamanian Leishmania. Exp Parasitol. 1966;18:25–34. doi: 10.1016/0014-4894(66)90004-x. [DOI] [PubMed] [Google Scholar]
- 21.Scott P, Caspar P, Sher A. Protection against Leishmania major in BALB/c mice by adoptive transfer of a T cell clone recognizing a low molecular weight antigen released by promastigotes. J Immunol. 1990;144:1075–9. [PubMed] [Google Scholar]
- 22.Coelho VT, Oliveira JS, Valadares DG, Chávez-Fumagalli MA, Duarte MC, Lage PS, et al. Identification of proteins in promastigote and amastigote-like Leishmania using an immunoproteomic approach. PLoS Negl Trop Dis. 2012;6:e1430. doi: 10.1371/journal.pntd.0001430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Webb JR, Campos-Neto A, Ovendale PJ, Martin TI, Stromberg EJ, Badaro R, et al. Human and murine immune responses to a novel Leishmania major recombinant protein encoded by members of a multicopy gene family. Infect Immun. 1998;66:3279–89. doi: 10.1128/iai.66.7.3279-3289.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Méndez S, Belkaid Y, Seder RA, Sacks D. Optimization of DNA vaccination against cutaneous leishmaniasis. Vaccine. 2002;20:3702–8. doi: 10.1016/s0264-410x(02)00376-6. [DOI] [PubMed] [Google Scholar]
- 25.Campos-Neto A, Webb JR, Greeson K, Coler RN, Skeiky YA, Reed SG. Vaccination with plasmid DNA encoding TSA/LmSTI1 leishmanial fusion proteins confers protection against Leishmania major infection in susceptible BALB/c mice. Infect Immun. 2002;70:2828–36. doi: 10.1128/IAI.70.6.2828-2836.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]