

Abstract
In this work, effects of bortezomib on apoptosis, clonal progenitor growth, cytokine production, and NF-κB expression in patients with MDS with cytopenias requiring transfusion support are examined. Bortezomib increased apoptosis in marrow mononuclear cells but had no effects on CFU-GM, BFU-E, or CFU-L content. No consistent effects on NF-κB activation in vivo were noted. To further define the role of bortezomib in AML and MDS, we examined it in combination with several targeted agents and chemotherapeutic agents in vitro. Combinations with arsenic trioxide, sorafenib, and cytarabine demonstrated synergistic in vitro effects in AML cell lines.
Keywords: Leukemias, Signal transduction, Treatment
INTRODUCTION
In addition to activity in plasma cell dyscrasias and various subtypes of lymphoma, proteasome inhibition is postulated to have therapeutic benefit for other diseases such as acute myelogenous leukemia (AML) and myelodysplastic syndromes (MDS). The proteasome is present in all eukaryotic cells and is the primary component of the protein degradation pathway of the cell known as the ubiquitin-proteasome pathway. The proteasome consists of two functional components, a 26S complex responsible for the proteolytic activity and a 19S regulatory subunit (1). Proteasome inhibition by compounds such as bortezomib occurs because of rapid but reversible binding to a single threonine in the active site of the 26S proteolytic core that leads to increased apoptosis and affects p53, NF-κB, and Bcl-xL expression. Such inhibition also results in downregulation of the cascade triggered by Interleukin-6 (IL-6) (2).
Proteasome inhibitors such as bortezomib also block degradation of the NF-κB regulator IκB, and NF-κB appears to be a therapeutic target in leukemias, given its constitutive activation in primitive AML cells and in acute lymphoblastic leukemia cells (3, 4). NF-κB regulates apoptosis, cell proliferation, and differentiation as well as inflammation, angiogenesis, and tumor migration (5). Members of the NF-κB family form heterodimers (p50 and p65), which are retained in the cytoplasm through association with IκB (inhibitor of NF-κB). With activation, IκB is phosphorylated and degraded by the ubiquitin-proteasome pathway. This frees NF-κB to translocate to the nucleus where it activates target genes (5).
For reasons not completely understood, proteasome inhibitors induce apoptosis in tumor as opposed to normal cells (6). Bortezomib may also increase the generation of reactive oxygen species (ROS) (7), and it interferes with folded protein degradation (8). Unlike normal CD34+ cells that do not express activated NF-κB, AML CD34+ cells have NF-κB activation as do populations enriched for AML stem cells (CD34+/CD38−/CD123+ cells) (3). This has suggested that AML cells might be selectively targeted by proteasome inhibitor therapy. NF-κB is also activated at baseline in myelodysplasia (MDS) marrows (9) and in the P39 MDS/AML cell line, where bortezomib was able to down-regulate antiapoptotic target genes of NF-κB. Marrow cells from highrisk MDS patients also showed constitutive NF-κB activation, and p65 NF-κB was translocated to the nucleus and was associated with suppression of apoptosis, suggesting it might be involved in transformation from MDS to AML (9). In this work, we examine the role of proteasome inhibition in AML and MDS and present some correlative data from patients with MDS treated with bortezomib on a phase II single agent protocol.
The effect of combining bortezomib with other small molecule inhibitors on leukemia cell proliferation and apoptosis was also examined in vitro. Based on the mechanism of NF-κB inhibition in isolation, bortezomib would not be anticipated to have effect in lowrisk MDS where NF-κB is not activated. However, because bortezomib as a proteasome inhibitor targets many other pathways known to be aberrant in MDS, it was hypothesized that this agent might have therapeutic activity in these disorders irrespective of MDS subclassification or risk stratification based on the International Prognostic Scoring System (10). In myelodysplasia, hematopoietic progenitors do not respond normally to cytokine signals, and premature apoptotic death ensues. Local production of cytokines such as tumor necrosis factor –α (TNF-α), IL-1β, and increased susceptibility to Fas ligand are thought to play a role in the enhanced apoptosis (11). Abnormalities in the marrow microenvironment and other angiogenic mediators may also play a role in MDS pathogenesis (11). Since bortezomib is a potent and reversible inhibitor of NF-κB, it indirectly inhibits many NF-κB dependent genes, which are important in MDS and AML, including cell adhesion molecules (E-selectin, ICAM-1, and VCAM-1), apoptosis mediators such as Bcl-2 and p53, cytokines such as IL-6 and TNF-α, and other angiogenesis mediators (12, 13). A previous study of bortezomib in MDS patients published as an abstract showed a decrease in TNF-α levels in about one-third of cases (14).
CLINICAL AND LABORATORY MATERIALS AND METHODS
Description of clinical trial
A Phase II pilot study of bortezomib in patients with MDS was conducted at the University of Rochester. All patients gave written informed consent as approved by the Research Subjects Review Board of the University of Rochester before any procedures related to the study were begun. The primary objective of this trial was to determine the safety and efficacy of bortezomib to improve cytopenias in patients with MDS who required red cell transfusions or other treatment interventions. Patients could have had previous therapies, and they had to have been diagnosed at least 8 weeks prior to enrolment to determine transfusion dependence. IPSS scores of 1.5 or less were required for enrolment, and IPSS 2 (>20% blasts) and CMML cases were excluded. Adequacy of renal, hepatic, and other organ function was required, and those patients with greater than or equal to grade 2 peripheral neuropathy were excluded. All patients were treated as outpatients at a dose of 1.3 mg/m2 on days 1, 4, 8, and 11 on a 21 day cycle; the FDA approved regimen in multiple myeloma. Up to 12 cycles were allowed, and response assessments were made after the 3rd, 6th, and 12th cycles of therapy. The International Working Group criteria for response determination in MDS were utilized (15). Marrow morphology analysis and flow cytometry to determine percentage of blasts were conducted by the hematopathology service at times of response analysis. Bortezomib was provided by Millennium™ from commercial drug sources. Samples of blood and marrow were obtained at designated time points to determine effects of bortezomib on apoptosis, progenitor cell outgrowth, and stromal cell cytokine milieu. Effects on NF-κB expression were also conducted on those samples where cell numbers allowed.
Patients were allowed to continue erythropoietin at a stable dose and to receive needed red cell and/or platelet transfusion support. Colony stimulating factor was utilized in the cases of fever with infection. To assess safety, incidence of cytopenias, neuropathy, gastrointestinal symptoms, and other effects were noted and graded. This trial was registered at ClinicalTrials.gov before patient accrual (Registration number NCT00262873). Patients enrolled on this trial between 2005 and 2008.
Reagent sources
All cell lines were obtained from the ATCC (Rockville, MD). For in vitro studies, cells were cultured in RPMI medium with 10% fetal bovine serum (Hyclone, Logan, UT or Atlanta Biologicals, Lawrenceville, GA). Sorafenib (Bay 43–9006) was obtained from Bayer Pharmaceuticals Corporation, West Haven, CT and was dissolved in DMSO. Arsenic trioxide was from Cell Therapeutics, Inc., Seattle, WA and was dissolved in tissue culture medium, and tipifarnib (R115777) was from Johnson and Johnson (Beerse, Belgium) and was dissolved in DMSO. L-744832 was obtained from Merck. ELISA kits for TNF-α, IL-6, and VEGF were obtained from R&D Systems, Minneapolis, MN. Cytarabine, 5-azacytidine, and bortezomib were purchased from pharmaceutical sources.
Cell isolation
Marrow was obtained from consenting patients during screening prior to therapy and for those subsequently enrolled on the protocol at specified time points. Samples were subjected to ficoll-hypaque density gradient centrifugation. In some cases, cells were selected for CD34 expression using antibody coated magnetic beads and a magnetic column (Miltenyi Biotec). Cell viability was determined by trypan blue exclusion, and cell counts were determined by hemacytometer or by MTT assay (R&D Systems). For specific determination of apoptosis, staining for Annexin V (R&D Systems) was performed. Simultaneous determination of propidium iodide (PI) staining allowed for determination of apoptosis versus necrosis.
Determination of bortezomib effects on apoptosis
The CD34+ fraction of light density marrow obtained from patients at baseline was exposed in vitro to bortezomib at 0–6 nM, concentrations found inhibitory in leukemia cell lines and which correspond to clinically achievable concentrations. At 24, 48, and 72 hr, the effects of bortezomib exposure on apoptosis were assessed through measurement of Annexin V (assay obtained form R&D Systems) and by flow cytometry analysis. Samples obtained from marrow or blood from patients receiving bortezomib were also analyzed by Annexin V staining of mononuclear cells to determine effects on in vivo apoptosis. Cell counts, viabilities, and cell cycle analysis (PI staining) were quantified at indicated time points.
Determination of the effects of bortezomib on MDS precursors
Colony forming unit-granulocyte-macrophage (CFU-GM) progenitors, erythroid burst forming units (BFU-E), and leukemia colony forming units (CFU-L) were measured at day 0 and day 14 of cycle 1. Five × 10(4) light density cell for granulocyte-macrophage colony forming unit (CFU-GM) or erythroid burst forming unit (BFU-E) assays were plated in 0.9% methylcellulose, 30% FCS, 2 mmol/L L-glutamine, 10−4 mol/L β-mercaptoethanol, and 1% BSA with 3U/ml human erythropoietin, 10 ng/ml GM-CSF, 10 ng/ml IL-3, and 50 ng/ml stem cell factor (SCF) (c-kit ligand). For leukemia colony forming units (CFU-Ls), the plating mixture was comparable with the exception that the cytokines utilized were 4 U/ml erythropoietin, 10 ng/ml GM-CSF, 10 ng/ml IL-3, 100 ng/ml c-kit ligand, and 100 ng/ml Flt3 ligand. The methylcellulose mixture and associated reagents were purchased from Stem Cell Technologies (Vancouver, BC). Colonies were scored at Day 14 and were defined as > 20 grouped cells.
Effects of bortezomib on circulating cytokine levels
In multiple myeloma, bortezomib has been found to inhibit stromal cell cytokine release, to alter cell adhesion interactions, and to inhibit microenvironment determined drug resistance and angiogenic mediators (12, 13). Therefore, TNF-α, IL-6, and VEGF levels were measured by ELISA (R&D Systems) in serum from MDS patients exposed to bortezomib. Levels were measured at Day 0 and Day 14 of cycle 1 of the clinical trial. Each of these cytokines is altered in MDS (11) and all are potentially related to NF-κB activation.
Measurement of NF-κB activation
NF-κB activity was quantified by ELISA using the Trans-AM NF-κB p65 Transcription Factor Assay Kit (Active Motif North America, Carlsbad, CA) according to the manufacturer’s instructions. Nuclear extracts were prepared from mononuclear cells and these were incubated in 96 well plates coated with immobilized oligonucleotides with a binding site for the p65 subunit of NF-κB. Binding was quantified by incubation with primary antibody specific for the activated form of p65, which was visualized by anti-IgG horseradish peroxidase and measured on an ELISA reader.
Measurement of effects on the Ras/MEK/ERK and PI3-K/AKT Pathways
In some MDS and AML samples, baseline expression of phospho-AKT and phospho-ERK and changes in expression, which occur after bortezomib were determined by immune blotting with actin controls. To assess AKT or ERK phosphorylation, leukemic blasts isolated as described above were harvested and lysed after Ficoll-Hypaque density gradient centrifugation, lysed with buffer containing 50 nM Tris, pH 8.0, 120 mM NaCl, 0.5% Nonidet P40, and protease inhibitor cocktail (Roche). The protein concentration of the lysate was determined by using the Bradford method (Biorad, Hercules, CA). Equal amounts of protein were separated on a 4–20% Tris-Glycine gel (Invitrogen) and transferred to a BioTrace PVDF membrane. Blots were probed with primary antibody to AKT or phospho-AKT (Ser 473) or to ERK or phospho-ERK (Cell Signaling). After washing and incubating with secondary antibody, immunoreactive proteins were visualized by using the ECL Plus detection system (Amersham Biosciences).
Statistics
For analysis of laboratory effects, values were represented as means ±SD. The significance of differences between experimental variables was determined by the Student t-test or two-tailed paired t-test. A p value of ≤.05 was considered significant. Combination indices were determined using Calcusyn software (Biosoft™).
RESULTS
Clinical responses of MDS patients to bortezomib
Nine patients were enrolled on the trial to examine the ability of bortezomib to improve cytopenias in MDS. Three patients were not able to start study drug despite completion of screening, one because of transformation to AML on screening marrow; one because of a diagnosis of primary myelofibrosis versus MDS on screening marrow, and one because of active fungal infection, which developed during the screening period. In addition to the patients who enrolled on the trial, 14 other patients were screened after signing informed consent. They did not meet study eligibility criteria and therefore did not complete the screening process or correlative laboratory testing. Three were not eligible because of a concurrent malignancy, two because of transformation to AML, five because of other co-morbidities such as > grade 3 neuropathy or renal insufficiency, two because of lack of transfusion or erythropoietin dependency, and two because of eventual patient refusal. Out of the six patients who received bortezomib, two patients completed only first or second cycles of therapy. In one of these cases, bortezomib was discontinued because of development of a pneumonia related to underlying neutropenia with subsequent respiratory failure and in the other because of painful grade 3 neuropathy, which improved after medication discontinuation. This patient had type II diabetes mellitus. One patient withdrew after four cycles because of progressive disease. He had also developed significant constipation and ileus on bortezomib. The remaining three patients completed > 4 cycles and were evaluated for responses. None of the six patients receiving the drug had clonal chromosomal abnormalities, so cytogenetic responses could not be assessed. No patient had clonal evolution while on trial. In three patients, decreased blast percentages were noted on marrow aspirates during the course of therapy (22–6%, 9–2%, and 8–1%) as assessed by aspirate differential. No patient had improvement in transfusion requirements during bortezomib therapy. Beyond the toxicities listed above, there were no other medication related grade 3 or 4 adverse events.
Bortezomib effects on apoptosis in MDS
Our laboratory and others have previously shown that bortezomib does neither inhibit normal CD34+ cell proliferation nor induce apoptosis in normal CD34+ cells (16). In the patients with MDS screened for the trial of bortezomib where marrow samples were available, as assessed by Annexin V staining, the baseline marrow apoptosis rate in mononuclear cells was 9.5 ± 4.1% (n = 0 11; range 1–50%). In patients who received bortezomib, effects on annexin V expression were determined after 14 days of in vivo exposure or in vitro after 48 or 72 hr of exposure. Bortezomib increased apoptosis in marrow mononuclear cells by day 14 in the five patients in whom pre and postbortezomib marrow samples were available (Figure 1); n = 0 5 p = .05. Similar effects were noted with in vitro exposure (not shown). The sixth patient enrolled on the trial did not have an aspirable marrow.
Figure 1.

Effect of in vivo bortezomib on apoptosis. Marrow was sampled from patients enrolled on the clinical trial before bortezomib exposure and at day 14 after four doses of bortezomib. Shown is the percentage of light density cells in apoptosis as assessed by Annexin V staining. Data are shown as Mean ± SEM for the five pre and postbortezomib samples. N = 5; p = .05 by paired t-test.
Effects of bortezomib on progenitor proliferation in MDS
In those patients where both day 0 and day 14 marrow samples were available for clonogenic assays, bortezomib treatment resulted in no consistent effects on CFU-GM, BFU-E, or CFU-L. As was the case with baseline apoptosis assessment, there was a high degree of variation in baseline clonogenic precursors in these myelodysplastic marrows (Figure 2).
Figure 2.

Effect of in vivo bortezomib on progenitor cells. In vivo exposure to bortezomib did not significantly affect the outgrowth of clonogenic progenitors (CFU-GM (A), BFU-E (B), or CFU-L(C)) between screening marrow (day 0) and 14 days after the start of bortezomib. N = 5. Data shown are individual values and means ± SD.
Effects of bortezomib on circulating cytokine levels
Tumor necrosis factor levels were negligible or undetectable in serum of patients pre and postbortezomib exposure. The serum levels of Interleukin-6 did not change appreciably (6.8 ± 1.7 pg/ml prebortezomib and 8.6 ± 3.3 pg/ml postbortezomib), whereas levels of vascular endothelial growth factor (VEGF) decreased after bortezomib exposure (402 ± 105 prebortezomib and 254 ± 69 postbortezomib) (Figure 3).
Figure 3.

Effects of bortezomib on IL-6 and VEGF levels. Fourteen days after the start of bortezomib, the serum levels of IL-6 had not changed significantly, whereas the levels of VEGF were significantly decreased (p < .05). n = 5. Bars shown are Mean ± SD.
Effects of bortezomib on levels of active NF-κB
In blood, some degree of active NF-κB could be detected in all MDS patients tested and usually to a greater extent than in marrow. Figure 4 demonstrates the degree of variability in levels of active NF-κB in blood. Out of the six patients who received bortezomib, four were assessable for pre and day 14 NF-κB expression. In two cases, there was a minimal decrease in levels of active NFκB at day 14, and the other two demonstrated an increase at day 14 (Figure 5). In one patient with an initial increase in blood active NF-κB expression at day 14, a later decline was noted after cycles 4 and 7 (Figure 6).
Figure 4.

Expression of active NF-κB in MDS patients. Shown is the degree of active NF-κB expression at baseline in the blood of six MDS patients. p65 NF-κB activity was assayed using the Trans-AM assay kit and quantified by ELISA. The Y-axis represents OD units.
Figure 5.

Effect on expression of active NF-κB after 14 days of bortezomib therapy. Shown is the pre and postlevels of expression of active NF-κB after 14 days of bortezomib. P65 NF-κB activity was assayed using the Trans-AM assay kit and quantified by ELISA. The Y-axis represents OD units. Data from patients 1 through 3 is from paired marrow samples, and data from patient 4 is from blood due to inaspirable marrow.
Figure 6.

Time course of expression of active NF-κB in blood mononuclear cells after exposure to bortezomib. In this patient, an initial increase in activation was followed by a decline after additional cycles of bortezomib were administered.
Effects of bortezomib in combination with arsenic trioxide, farnesyltransferase inhibitors, and sorafenib in vitro
Because of minimal activity noted in the initial group of patients on the clinical trial with single agent bortezomib, we have begun to explore whether combination of bortezomib with other inhibitory substances would increase activity against AML and MDS cells and progenitors. Just as proteasome inhibitors block degradation of the NF-κB regulator, IκB, resulting in inhibition of NF-κB nuclear localization, arsenic trioxide (AsO3) has been shown to inhibit NF-κB activation in leukemia and MDS (17). When arsenic trioxide and bortezomib were combined, proliferation of AML cell lines was inhibited to a greater extent than with either alone (shown in Figure 7 for HL60). Combination index (CI) analysis for HL60 exposed to 2 nM bortezomib and 2 uM arsenic trioxide indicated synergistic activity (CI 0.535). The combination of bortezomib and AsO3 resulted in decreased phospho-ERK but not of phospho-AKT expression (Figure 8). Phospho-IKB was also increased with the combination of arsenic and bortezomib (not shown). The MV411 cell line, which is FLT3 ITD positive, is very sensitive to arsenic trioxide (2 uM IC50). In this cell line, the combination of bortezomib (PS341) and arsenic trioxide resulted in increased rates of apoptosis as measured by Annexin V as compared with each agent alone (not shown).
Figure 7.
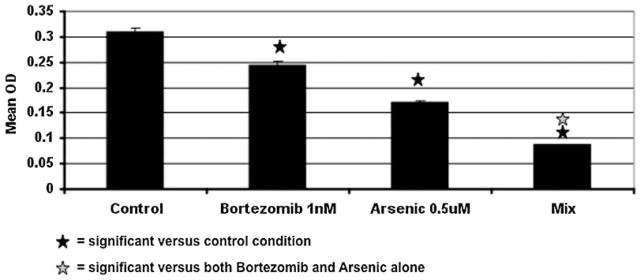
Effect of concurrent bortezomib and arsenic trioxide on proliferation of HL60 cells as measured by MTT. Bortezomib (1 nM) and arsenic (0.5 μM) were added to HL60 cells, and proliferation was determined by MTT assay at 48 hr. Data shown is Mean ± SEM with n = 3.
Figure 8.

Effect of bortezomib and arsenic trioxide on phospho-ERK and phospho-AKT expression in the HL60 cell line. Proteins were extracted, electropheresed as described, and probed with the appropriate antibody. This experiment was completed twice in the HL60 cell line with comparable results and normalization to an actin control (not shown given equidensity ERK bands).
The farnesyltransferase inhibitors (FTIs) also inhibit AML proliferation (18), and their effects in combination with bortezomib have also been explored. Because FTIs may inhibit signal transduction pathways independent of those affected by NF-κB inhibition, and because FTIs may indirectly inhibit NF-κB function via Akt inhibition, the effects of combining these agents with bortezomib on AML/MDS cells in vitro have also been explored. The nonpeptidomimetic FTI, R115777 (Tipifarnib), did not inhibit NF-κB at concentrations up to 100 nM, concentrations which effectively inhibit farnesylation of target proteins, whereas the FTI, L-744832 (Merck), was able to inhibit NF-κB expression at 1 μM up to 72 hr of exposure (Figure 9). In the HL60 line, inhibited by FTI and bortezomib independently, the combination did not appear to have additive or synergistic effects (not shown). Furthermore, the effects of combined exposure to FTI and bortezomib on expression of activated caspase-3 or activated PARP cleavage were no greater than with bortezomib alone. Likewise, combination effects on expression of phosphorylated AKT or ERK were not observed (not shown).
Figure 9.

Effect of the farnesyltransferase inhibitor, L-744832 on levels of active NF-κB expression as measured by ELISA for the p65 component in the HL60 cell line.
The combination of bortezomib with sorafenib, which inhibits Raf, Flt3, and other tyrosine kinases inhibited proliferation beyond that seen with each agent alone (Figure 10) (n = 3). This appeared to be mediated in part through apoptosis induction (Figure 10). Each of these agents was effective in both tissue culture plastic culture conditions as well as with culture over fibronectin coated surfaces, suggesting that adhesion molecule related resistance was not seen with these agents.
Figure 10.

The combination of sorafenib and bortezomib inhibited proliferation in AML cells lines beyond that seen with either agent alone (A). Apoptosis was also increased with the combination as compared with the control (B). Shown is the Mean ± SD of cell proliferation as measured in the MTT assay (OD) or percentage of cells in apoptosis as measured by Annexin V at 48 hr in the MV411 cell line. (n = 3 independent experiments).
Effects of combining bortezomib with cytarabine and 5-azacytidine
When cytarabine, 100 nM, was combined with bortezomib at 1–4 nM, greater inhibition of proliferation was seen with this combination than with individual agents as demonstrated in MTT assays with CI calculations suggesting synergism. Figure 11 shows such enhanced effect in a primary AML sample. Apoptosis was increased with the combination as evidenced by an increase in cleaved PARP and active caspase-3 expression, again shown in a primary AML sample (Figure 12).
Figure 11.

Effect of concurrent cytarabine and bortezomib exposure on primary AML blasts. Concurrent exposure decreased proliferation as compared with either agent alone. Shown is a representative experiment of 3. Similar effects were noted with leukemia cell lines. B = bortezomib.
Figure 12.

Effect of cytarabine (ARA-C) and bortezomib on expression of cleaved PARP and activated caspase-3 in a primary AML sample. Beta-actin served as a control to assure equal protein loading.
The concurrent exposure of KG1a cells to the hypomethylating agent, 5-azacytidine and bortezomib did not result in synergistic suppression of proliferation or apoptosis induction (Figure 13). This combination remained equally effective in inhibition of KG1a growth in both plastic and stromal cell coculture conditions. With the MV-4-11 cell line, no additive or synergistic effect of concurrent 5-azacytidine and bortezomib addition was noted (Figure 14), but apoptosis in this cell line was less with stromal coculture than in plastic tissue culture conditions (Not shown).
Figure 13.

Effect of 5-azacytidine(Vidaza™) (V) and bortezomib (B) on viability in the KG1a leukemia cell line. Cells were cultured in plastic flasks for 24–72 hr. No significant effects of the combination were noted. N = 3.
Figure 14.

Effect of 5-azacytidine (V) and bortezomib (V) on viability in the MV-411 cell line. The combination did not result in increased apoptosis (viability measured as Annexin V expression) as compared with the control conditions. N = 3.
DISCUSSION
Based on previous reports of NF-κB activation in both AML and MDS, we conducted a trial of single agent bortezomib in MDS. Accrual to this trial which opened in 2005 was lower than anticipated due to FDA approval of 5-azacytidine, decitabine, and lenalidomide (19–21). While bortezomib significantly increased the rate of apoptosis noted in MDS marrow, it had neither consistent effects on outgrowth of hematopoietic progenitors nor on NF-κB expression, and furthermore, no patient studied had improvement in cytopenias while on bortezomib. Some patients had blast reduction in marrow, but most patients completed only four cycles before discontinuation, usually due to lack of benefit or other toxicity, usually unrelated to bortezomib which was overall well-tolerated in this patient group. Based on this experience, we have begun to examine combinations of bortezomib with other agents in vitro and to re-examine the role that proteasome inhibition might play in the therapy of AML or MDS.
There are many in vitro studies to suggest that AML blasts are sensitive to bortezomib. Both bortezomib, a reversible proteasome inhibitor and depoxomicin, an irreversible inhibitor, have been found to inhibit proliferation of AML blasts at nanomolar concentrations, and both drugs enhanced apoptosis (22). In one series, 18 out of 30 AML cases were sensitive to proapoptotic effects of bortezomib, and monocytic (FAB M4 and M5) subtypes and Flt3 positive cases were the most sensitive (23). In the series reported here of MDS patients exposed in vivo to bortezomib, apoptosis was also increased. The role of apoptosis induction in successful MDS therapy as compared with AML therapy is often uncertain since many MDS marrows express increased apoptosis at baseline (11). It has also been shown that the pattern of proteosomal subunit activity in primary AML cells influences sensitivity (24), and the level of the 26S proteasome may be positively correlated with responsiveness to bortezomib (25). Whether this is also the case in MDS has not been examined. MDR proteins and p53 levels in blasts have not been found to affect bortezomib cytotoxicity or in vitro interactions with anthracyclines or cytarabine (26).
Other clinical trials examining bortezomib use in AML and MDS have been published (27, 28). In one trial, bortezomib 1.3 mg/m2 was added to induction chemotherapy with standard cytarabine and daunorubicin on days 1, 4, 8, and 11 with a complete remission rate of 67% observed with acceptable toxicities (27). Reports of combining bortezomib with anthracyclines in AML and high risk MDS have also appeared in preliminary form (29).
The in vitro studies described in this work suggest that future studies combining proteasome inhibitors with standard chemotherapeutic agents or with other inhibitors of NF-κB may have greater effects in MDS or AML by inducing apoptosis in a synergistic fashion. These in vitro studies utilized cell lines and primary AML samples and provide the basis for rational combination of bortezomib with other agents. In addition, since DNA methyltransferase (DNMT) inhibitors such as 5-aza-2′-deoxycytidine and histone deacetylase (HDAC) inhibitors have been shown to induce apoptosis correlating with an inhibition of NFκB through reduced phosphorylation of IKKα/β, it is possible that synergy could be seen with these agents combined with proteasome inhibitors (30). In myeloma, the histone deacetylase inhibitor SAHA enhances cell death stimulated by bortezomib and by disrupting bortezomib-induced aggresome formation (30). Whether this also occurs in AML and MDS is uncertain, but similar findings have been reported in CML cells (31). The in vitro data shown here suggest that bortezomib has synergistic activity with cytarabine, but this was not demonstrated with 5-azacytidine. This may reflect the difficulty in assessing hypomethylation effects with short in vitro exposures as most clinical responses to these agents are not observed until after several cycles of therapy.
There is evidence that proteasome inhibition can increase sensitivity to idarubicin. THP-1 cells exposed to either bortezomib or the proteasome inhibitor, MG132 along with idarubicin underwent apoptosis with synergy demonstrated. This was correlated with accumulation of Bax and Bim. This effect was also demonstrated in primary AML cells (32). Synergistic effects have also been noted between bortezomib and cytarabine (33), as also demonstrated here.
It has also been noted that TRAIL, a member of the TNF family which induces apoptosis in tumor cells while sparing normal cell mediated killing can be modulated by low dose bortezomib (34). Bortezomib primed TRAIL resistant AML cells for enhanced TRAIL-mediated killing. Bortezomib–induced cell death upregulated TRAIL-R1, TRAIL-R2, and activated executioner caspases (34). TRAIL has also been reported to potentiate bortezomib via activated caspase-8 and caspase-3 and decreased cellular FLICE (Fas-associated death domain-like interleukin-1 beta converting enzyme) (35)
BAY11-7082, an IKK inhibitor, has been found to demonstrate synergistic killing with NF-κB inhibitors in conjunction with nutrient starving. This is characterized by early loss of mitochondrial transmembrane potential, release of cytochrome c, activation of caspase-3, and phosphatidylserine exposure on the plasma membrane surface and nuclear chromatin condensation suggesting autophagic stress followed by apoptotic cell death (36). Arsenic trioxide down-regulates constitutive IκB kinase (IKK) as well as NF-κB activity and induced apoptosis, which was caspase-8 and caspase-3 dependent. Arsenic also down-regulated NF-κB target genes including TRAF-1, IAP2, and CCR7 (37). In this work, we show that arsenic in combination with bortezomib demonstrated synergistic inhibitory activity with bortezomib in some leukemia cell lines. In the in vitro studies here with AML cell lines and primary samples, sorafenib (BAY43-9006) was also found to have synergistic inhibitory effects with bortezomib, indicating that multi-kinase inhibitors can also synergize with bortezomib in AML/MDS.
Tipifarnib has been proposed to act synergistically with bortezomib in AML (38), and here we have shown that some farnesyltransferase inhibitors do inhibit NF-κB. Others have shown that tipifarnib and bortezomib are synergistic and overcome cell adhesion-mediated drug resistance in both myeloma and AML (39). Activation of the endoplasmic reticulum stress response was enhanced and correlated with apoptosis and reversal of the adhesion mediated resistance.
In myelodysplasia, hematopoietic progenitors do not respond to cytokine signals, and premature apoptotic death ensues. Local production of cytokines such as tumor necrosis factor-α, IL-1β, and increased susceptibility to Fas ligand are thought to play a role in the enhanced apoptosis noted (11). Which progenitors undergo apoptosis in MDS remains controversial, and how the balance of clonal proliferation and apoptosis/ineffective erythropoiesis affects MDS clinical course and evolution remains unclear. The frequency of cells with activated NF-κB nuclear p65 has been noted to correlate with blast counts and disease progression (9). This might suggest that bortezomib would be most effective in patients with highrisk MDS where NF-κB might play a role in apoptosis suppression and the transformation from AML to MDS. This could involve Bcl-xL or cIAP2 modulation (9). On the other hand, bortezomib has effects on numerous other cytokines and pathways such as inhibition of angiogenesis, disruption of survival signal cascades, aggresome disruption, and ROS generation (7). This would suggest that even in MDS cases where NF-κB activation is not prominent, proteasome inhibition may have a beneficial effect.
Abnormalities in the marrow microenvironment and other angiogenesis mediators may also play a role in MDS pathogenesis. Molecules inhibited by NF-κB, which are altered in MDS include cell adhesion molecules (E-selectin, ICAM-1, and VCAM-1) as well as Bcl-1 and IL-6 (12, 13). Since TNFα expression is usually elevated in MDS, it is hypothesized that inhibition of NF-κB would result in abrogation of TNF effects on the marrow microenvironment and on MDS progenitors themselves. In the serum assays performed on patients receiving bortezomib, we did not detect any inhibition of TNF-α nor of IL-6. The levels of VEGF were decreased, however. It has been shown (40) that VEGFR-1 and VEGFR-2 initiate a PI-3 kinase dependent clonogenic effect in AML, so if bortezomib inhibits VEGF, this could have a beneficial impact in inhibiting AML proliferation. AML cells may also produce angiopoietin-1, a ligand for Tie-2, and this can also be inhibited by bortezomib (41).
Based on the findings that bortezomib has ability to inhibit AML and MDS cells in vitro, and based on early results of its use in clinical trials for AML and MDS, it would appear that proteasome inhibition may have an anti proliferative and apoptosis—inducing role in these disorders, especially in combination with other antiproliferative agents and agents which might synergize to inhibit activated NF-κB. Many factors such as optimal doses, optimal dose schedules, and optimal means of utilizing proteasome inhibitors in combination with other agents remain to be determined.
Acknowledgments
This work was supported by NIH grant R21CA112835 (JLL). Bortezomib for the clinical trial was provided by Millennium. We thank Nicole Proia for clinical trial and regulatory coordination, and Elva Mikk for aid in manuscript preparation.
Footnotes
The clinical trial described here is registered at http://.clincaltrials.gov (#NCT00262873).
DECLARATION OF INTEREST
The authors report no conflicts of interest.
References
- 1.Wu WK, Cho CH, Lee CW, Wu K, Fan D, Yu J, Sung JJ. Proteasome inhibition: a new therapeutic strategy to cancer treatment. Cancer Lett. 2010;293:15–22. doi: 10.1016/j.canlet.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 2.Navon A, Ciechanover A. The 26 S proteasome: from basic mechanisms to drug targeting. J Biol Chem. 2009;284:33713–33718. doi: 10.1074/jbc.R109.018481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guzman ML, Neering SJ, Upchurch D, Grimes B, Howard DS, Rizzieri DA, Luger SM, Jordan CT. Nuclear factor-kB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood. 2001;98:2301–2307. doi: 10.1182/blood.v98.8.2301. [DOI] [PubMed] [Google Scholar]
- 4.Cilloni D, Martinelli G, Messa F, Baccarani M, Saglio G. Nuclear factor kB as a target for new drug development in myeloid malignancies. Haematologica. 2007;92:1224–1229. doi: 10.3324/haematol.11199. [DOI] [PubMed] [Google Scholar]
- 5.Karin M, Greten FR. NF-κB: linking inflammation and immunity to cancer development and progression. Nat Rev. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 6.Servida F, Soligo D, Delia D, Henderson C, Bracolini C, Lombardi L, Delliers GL. Sensitivity of human multiple myelomas and myeloid leukemias to the proteasome inhibitor I. Leukemia. 2005;19:2324–2331. doi: 10.1038/sj.leu.2403987. [DOI] [PubMed] [Google Scholar]
- 7.Dasmahapatra G, Rahmani M, Dent P, Grant S. The tryphostin adaphostin interacts synergistically with proteasome inhibitors to induce apoptosis in human leukemia cells through a reactive oxygen species (ROS)-dependent mechanism. Blood. 2006;107:323–340. doi: 10.1182/blood-2005-06-2302. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 8.Moore HE, Davenport EL, Smith EM, Muralikrishnan S, Dunlop AS, Waler BA, Drige D, Drummond AH, Hooftman L, Morgan GJ, Davies FE. Aminopeptidase inhibition as a targeted treatment strategy in myeloma. Mol Cancer Ther. 2009;8:762–770. doi: 10.1158/1535-7163.MCT-08-0735. [DOI] [PubMed] [Google Scholar]
- 9.Braun T, Carvalho G, Coquelle A, Vozenin M-C, Lepelley P, Hirsch F, Kiladjian J-J, Ribrag V, Fenaux P, Kroemer G. NF-KB constitutes a potential therapeutic target in high-risk myelodysplastic syndrome. Blood. 2006;107:1156–1165. doi: 10.1182/blood-2005-05-1989. [DOI] [PubMed] [Google Scholar]
- 10.Sanz GF, Sanz MA, Greenberg PA. Prognostic factors and scoring systems in myelodysplastic syndromes. Haematologica. 1998;83:358–368. [PubMed] [Google Scholar]
- 11.Liesveld JL, Jordan CT, Phillips GL., II The hematopoietic stem cell in myelodysplasia. Stem Cells. 2004;22:590–599. doi: 10.1634/stemcells.22-4-590. [DOI] [PubMed] [Google Scholar]
- 12.Anderson KC. Targeted therapy of multiple myeloma based upon tumor-microenvironmental interaction. Exp Hematol. 2007;35:156–162. doi: 10.1016/j.exphem.2007.01.024. [DOI] [PubMed] [Google Scholar]
- 13.Hideshima T, Chauhan D, Hayashi T, Akiyama M, Mitsiades N, Mitsiades C, Podar K, Munshi NC, Richardson PG, Anderson KC. Proteasome inhibitor PS-341 abrogates IL-6 triggered signaling cascades via caspase-dependent downregulation of gp130 in multiple myeloma. Oncogene. 2003;20:8386–8393. doi: 10.1038/sj.onc.1207170. [DOI] [PubMed] [Google Scholar]
- 14.Shetty V, Verspoor F, Nguyen H, Gallegos A, Mundle S, Chaudhry N, Imran M, Mahmud G, Mumtaz M, Candoni A, Yousiff M, Alvi S, Reddy PL, Galili N, Gezer S, Venugopal P, Raza A. Effect of proteasome inhibition by bortezomib on tumor necrosis factor alpha (TNF-alpha) and apoptosis in patients with myelodysplastic syndromes (MDS) Blood. 2003;102:422a. [Google Scholar]
- 15.Cheson BD, Bennett JM, Kantarjian H, Pinto A, Schiffer CA, Nimer SD, Lowenberg B, Beran M, deWitte TM, Stone RM, Mittleman M, Sanz GF, Oijermans PW, Gore S, Greenberg PL. Report of an international working group to standardize response criteria for myelodysplastic synderomes. Blood. 2000;96:3671–3674. [PubMed] [Google Scholar]
- 16.Liesveld JL, Rosell KE, Lu C, Bechelli J, Phillips G, Lancet JE, Abboud CN. Acute myelogenous leukemia—microenvironment interactions: role of endothelial cells and proteasome inhibition. Hematology. 2005;10:463–494. doi: 10.1080/10245330500233452. [DOI] [PubMed] [Google Scholar]
- 17.Mathas S, Lietz A, Janz M, Hinz M, Jundt F, Scheidereit C, Bommert K, Dorken B. Inhibition of NF-kappaB essentially contributes to arsenic-induced apoptosis. Blood. 2003;102:1028–1034. doi: 10.1182/blood-2002-04-1154. [DOI] [PubMed] [Google Scholar]
- 18.Karp JE, Lancet JE. Tipifarnib in the treatment of newly diagnosed acute myelogenous leukemia. Biologics. 2008;2:491–500. doi: 10.2147/btt.s3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Silverman LR. DNA methyltransferase inhibitors in myelodysplastic syndrome. Best Pract Res Clin Haematol. 2004;17:585–594. doi: 10.1016/j.beha.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 20.Kantarjian H, Oki Y, Garcia-Manero G, Huang X, O’Brien S, Cortes J, Faderl S, Bueso-Ramos C, Ravandi F, Estrov Z, Ferrojoli A, Wierda W, Shan J, Davis J, Giles F, Saba HI, Issa J-PJ. Results of a randomized study of 3 schedules of low-dose decitabine in higher-risk myelodysplastic syndrome and chronic myelomonocytic leukemia. Blood. 2007;109:52–57. doi: 10.1182/blood-2006-05-021162. [DOI] [PubMed] [Google Scholar]
- 21.List A, Kurtin S, Roe DJ, Buresh A, Mahadevan D, Fuchs D, Rimsza L, Heaton R, Knight R, Zeldis JB. Efficacy of lenalidomide in myelodysplastic syndromes. N Engl J Med. 2005;352:549–557. doi: 10.1056/NEJMoa041668. [DOI] [PubMed] [Google Scholar]
- 22.Stapnes C, Doskeland AP, Hatfield K, Ersvaer E, Ryningen A, Lorens JB, Gjertsen BT, Bruserud O. The proteasome inhibitors bortezomib and PR-171 have antiproliferative and proapoptotic effects on primary human acute myeloid leukaemia cells. Br J Haematol. 2007;136:814–828. doi: 10.1111/j.1365-2141.2007.06504.x. [DOI] [PubMed] [Google Scholar]
- 23.Riccioni R, Senese M, Diverio D, Riti V, Buffolino S, Mariani G, Boe A, Cedrone M, Coco F, Foa R, Peschle C, Testa U. M4 and M5 acute myeloid leukaemias display a high sensitivity to bortezomib-mediated apoptosis. Br J Haematol. 2007;139:194–205. doi: 10.1111/j.1365-2141.2007.06757.x. [DOI] [PubMed] [Google Scholar]
- 24.Kraus M, Ruckrich T, Reich M, Gogel J, Beck A, Kammer W, Berkers CR, Burg D, Overkleeft H, Ovaa H, Driessen C. Activity patterns of proteasome subunits reflect bortezomib sensitivity of hematologoic malignancies and are variable in primary human leukemia cells. Leukemia. 2007;21:84–92. doi: 10.1038/sj.leu.2404414. [DOI] [PubMed] [Google Scholar]
- 25.Matondo M, Bousquet-Dubouch MP, Gallay N, Utternweiler-Joseph S, Recher C, Payrastre B, Manenti S, Monsarrat B, Burlet-Schiltz O. Proteasome inhibitor-induced apoptosis in acute myeloid leukemia: a correlation with the proteasome status. Leuk Res. 2010;34:498–506. doi: 10.1016/j.leukres.2009.09.020. [DOI] [PubMed] [Google Scholar]
- 26.Minderman H, Zhou Y, O’Loughlin KL, Baer MR. Bortezomib activity and in vitro interactions with anthracyclines and cytarabine in acute myeloid leukemia cells are independent of multidrug resistance mechanisms and p53 status. Cancer Chemother Pharmacol. 2007;60:245–55. doi: 10.1007/s00280-006-0367-6. [DOI] [PubMed] [Google Scholar]
- 27.Attar EC, Donohue KA, Amrein PC, Wadleigh M, DeAngelo DJ, Kolitz JE, Powell BL, Voorhess PM, Wang ES, Blum W, Booth A, Stone RM, Moser BK, Larson R. Phase II study of bortezomib added to standard daunorubicin and cytarabine induction and dose escalation of bortezomib with intermediate-dose cytarabine consolidation therapy for patients with previously untreated acute myeloid leukemia age 60–75 years: cancer and leukemia group B. (CALGB) study 10502. Blood. 2010;116:150a. doi: 10.1200/JCO.2012.45.2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gil L, Styczynski J, Dytfeld D, Debski R, Kazmierczak M, Kolodziej B, Rafinska B, Kubicka M, Nowicki A, Komarnicki M, Wysocki M. Activity of bortezomib in adult de novo and relapsed acute myeloid leukemia. Anticancer Res. 2007;27:4021–4025. [PubMed] [Google Scholar]
- 29.Howard D, Liesveld J, Phillips GL, Guzman ML, Jordan CT. A phase I study using Bortezomib with weekly idarubicin for the treatment of elderly (>60 years) and relapsed patients with acute myeloid leukemia (AML) Blood. 2008;112:1014a. doi: 10.1016/j.leukres.2013.09.003. (abstract) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nawrocki ST, Carew JS, Maclean KH, Courage JF, Huang P, Houghton JA, Cleveland JL, Giles FJ, McConkey DJ. Myc regulates aggresome formation, the induction of Noxa, and apoptosis in response to the combination of bortezomib and SAHA. Blood. 2008;112:2917–2926. doi: 10.1182/blood-2007-12-130823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu C, Rahmani M, Conrad D, Subler M, Dent P, Grant S. The proteasome inhibitor bortezomib interacts synergistically with histone deacetylase inhibitors to induce apoptosis in Bcr/Abl + cells sensitive and resistant to STI571. Blood. 2003;102:2765–3774. doi: 10.1182/blood-2003-03-0737. [DOI] [PubMed] [Google Scholar]
- 32.Pigneux A, Mahon FX, Moreau-Gaudry F, Uhalde M, de Vermeuil H, Lacombe F, Reiffers J, Milipied N, Praloran V, Belloc F. Proteasome inhibition specifically sensitizes leukemic cells to anthracycline-induced apoptosis through the accumulation of Bim and Bax pro-apoptotic proteins. Cancer Biol Ther. 2007;6:603–11. doi: 10.4161/cbt.6.4.4226. [DOI] [PubMed] [Google Scholar]
- 33.Hamada A, Kawaguchi T, Nakano M. Clinical pharmacokinetics of cytarabine formulations. Clin Pharmacokinet. 2002;4:705–718. doi: 10.2165/00003088-200241100-00002. [DOI] [PubMed] [Google Scholar]
- 34.Conticello C, Adamo L, Vicari L, Giuffrida R, Iannolo G, Anastasi G, Canuso L, Moschetti G, Cupri A, Palumbo GA, Gulisano M, De Maria R, Giustolisi R, DiRaimondo F. Antitumor activity of bortezomib alone and in combination with TRAIL in human acute myeloid leukemia. Acta Haematol. 2008;120:19–30. doi: 10.1159/000151511. [DOI] [PubMed] [Google Scholar]
- 35.Collison A, Foster PS, Mattes J. Emerging role of tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) as a key regulator of inflammatory responses. Clin Exp Pharacol Physiol. 2009;36:1049–1053. doi: 10.1111/j.1440-1681.2009.05258.x. [DOI] [PubMed] [Google Scholar]
- 36.Fabre C, Carvalho G, Tasdemir E, Braun T, Ades L, Grosjean J, Boehrer S, Metivier D, Souquere S, Pierron G, Fenaux P, Kroemer G. NF-kappaB inhibition sensitizes to starvation-induced cell death in high-risk myelodysplastic syndrome and acute myeloid leukemia. Oncogene. 2007;26:4071–4083. doi: 10.1038/sj.onc.1210187. [DOI] [PubMed] [Google Scholar]
- 37.Hu Y, Jin X, Snow ET. Effect of arsenic on transcription factor AP-1 and NF-kappaB DNA binding activity and related gene expression. Toxicol Lett. 2002;133:33–45. doi: 10.1016/s0378-4274(02)00083-8. [DOI] [PubMed] [Google Scholar]
- 38.Armand J-P, Burnett AK, Drach J, Harousseau J-L, Lowenberg B, San Miguel J. The emerging role of targeted therapy for hematologic malignancies: update on bortezomib and tipifarnib. The Oncologist. 2007;12:281–290. doi: 10.1634/theoncologist.12-3-281. [DOI] [PubMed] [Google Scholar]
- 39.Yanamandra N, Colaco NM, Parquet NA, Buzzeo RW, Boulware D, Wright G, Perez LE, Dalton WS, Beaupre DM. Tipifarnib and bortezomib are synergistic and overcome cell adhesion—mediated drug resistance in multiple myeloma and acute myeloid leukemia. Clin Cancer Res. 2006;12:591–599. doi: 10.1158/1078-0432.CCR-05-1792. [DOI] [PubMed] [Google Scholar]
- 40.Richardson PG, Mitsiades CS, Hideshima T, Anderson KC. Novel biological therapies for the treatment of multiple myeloma. Best Pract Res Haematol. 2005;18:619–634. doi: 10.1016/j.beha.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 41.Reikvam H, Hatfield KJ, Lassalle P, Kittang AO, Ersvaer E, Bruserud O. Targeting the angiopoietin (Ang)/Tie-2 pathways in the crosstalk between acute myeloid leukaemia and endothelial cells: studies of Tie-2 blocking antibodies, exogenous Ang-2 and inhibition of constitutive agonistic Ang-2 release. Expert Opin Investig Drugs. 2010;19:169–183. doi: 10.1517/13543780903485659. [DOI] [PubMed] [Google Scholar]