

Abstract
Biopharmaceuticals (BPs) represent a rapidly growing class of approved and investigational drug therapies that is contributing significantly to advancing treatment in multiple disease areas, including inflammatory and autoimmune diseases, genetic deficiencies and cancer. Unfortunately, unwanted immunogenic responses to BPs, in particular those affecting clinical safety or efficacy, remain among the most common negative effects associated with this important class of drugs. To manage and reduce risk of unwanted immunogenicity, diverse communities of clinicians, pharmaceutical industry and academic scientists are involved in: interpretation and management of clinical and biological outcomes of BP immunogenicity, improvement of methods for describing, predicting and mitigating immunogenicity risk and elucidation of underlying causes. Collaboration and alignment of efforts across these communities is made difficult due to lack of agreement on concepts, practices and standardized terms and definitions related to immunogenicity. The Innovative Medicines Initiative (IMI; http://www.imi-europe.org), ABIRISK consortium [Anti-Biopharmaceutical (BP) Immunization Prediction and Clinical Relevance to Reduce the Risk; http://www.abirisk.eu] was formed by leading clinicians, academic scientists and EFPIA (European Federation of Pharmaceutical Industries and Associations) members to elucidate underlying causes, improve methods for immunogenicity prediction and mitigation and establish common definitions around terms and concepts related to immunogenicity. These efforts are expected to facilitate broader collaborations and lead to new guidelines for managing immunogenicity. To support alignment, an overview of concepts behind the set of key terms and definitions adopted to date by ABIRISK is provided herein along with a link to access and download the ABIRISK terms and definitions and provide comments (http://www.abirisk.eu/index_t_and_d.asp).
Keywords: ABIRISK consortium, anti-drug antibodies, biopharmaceuticals, immunogenicity
Introduction
Biopharmaceuticals (BP) comprise a diverse and rapidly growing class of therapeutics that includes replacement factors for abnormal or deficient proteins, cytokines and growth factors that modulate biological functions, monoclonal antibodies targeting components of disease pathways, fusion proteins and protein–drug conjugates, both novel BP entities as well as copies of already licensed BPs, designated biosimilars [1,2]. An unwanted consequence of BP therapy is the development of immunogenic responses, which in some cases have little or no impact but in other cases affect safety significantly, with induction of infusion reactions, hypersensitivity reactions and autoimmune syndromes occurring due to cross-reactivity of anti-drug antibody (ADA) with endogenous counterparts of the BP [3–6], and/or decreased efficacy related to neutralization of the BP's biological activity or increasing its clearance [7–9]. For these reasons, immunogenic potential of a BP is determined during clinical trials by measurement and characterization of ADA that develop during treatment; in some cases, monitoring for biopharmaceutical and ADA levels may be conducted during routine use of approved BPs to guide patient care [10–12].
As the numbers of novel and biosimilar BPs continue to increase, there is considerable interest in standardizing methods for analysis and monitoring of immunogenicity and improving its management based on knowledge of factors that contribute to its development and consequences, including those related to the BP product itself, its mode of administration and the underlying disease or patient characteristics. These contributing factors are reviewed extensively elsewhere [13–18]. Key concepts and recommended definitions for standardizing the description and interpretation of immunogenicity are described further below in relation to practices for monitoring and mitigating immunogenicity during clinical use of BPs.
ABIRISK recommendations
Many terms and concepts pertaining to immunogenicity, in particular those pertaining to the reporting of ADA results, have been in common use throughout the medical, scientific and pharmaceutical communities involved in the use of BPs. However, various disease areas and communities may have used different terms or defined the same terms in inconsistent ways. Furthermore, these communities have focused largely on ADA, whereas data on cellular and pharmacogenomic markers of immunogenicity, results from predictive immunogenicity methods and additional measurements and outcomes of the anti-drug immune response (ADIR) that have not been commonly applied previously to the study of BP immunogenicity are now being generated. To bring this information together and advance the scientific understanding and clinical application of immunogenicity data, the ABIRISK [Anti-Biopharmaceutical (BP) Immunization Prediction and Clinical Relevance to Reduce the Risk; http://www.abirisk.eu] consortium has developed and agreed to use a set of common terms and definitions for describing immunogenicity of BPs.
As ABIRISK work proceeds, terms and definitions may be added or refined as necessary to enhance the interpretation of ADIR data. To reach broader consensus, ABIRISK will consult with other thought leaders, including the ABIRISK scientific advisory board and key associations, e.g. European Immunogenicity Platform (EIP), American Association of Pharmaceutical Scientists (AAPS), American and European Immunology Societies and regulatory agencies, some of which have already produced recommended practices for development and validation of ADA assays and assessment of immunogenicity risk profiles of BPs now used routinely during development of new BPs [19–24]. An AAPS focus group (AAPS Therapeutic Protein Immunogenicity Focus Group) also recognized the need for a harmonized approach for assessment and reporting of clinical immunogenicity and has recently published recommendations focused on terms and strategies related to reporting ADA results for analysed samples and patient populations [25], many of which have been adopted into the ABIRISK terms and definitions; where applicable, reporting of ABIRISK data will be consistent with those recommendations. As data generated by ABIRISK will be captured in a database, international efforts to create a suite of orthogonal interoperable reference ontologies in the biomedical domain will also be considered.
The comprehensive ABIRISK Terms and Definitions for Reporting Immunogenicity Results (http://www.abirisk.eu/index_t_and_d.asp) can be accessed at the ABIRISK website and comments may be provided at this site. Key concepts and definitions are described further below in relation to practices for monitoring and mitigating immunogenicity during clinical use of BPs.
Patient populations (evaluated subjects) and treatment history
The likelihood of development of immune responses and consequences of such responses can vary among different subject populations. Therefore, the meaningful interpretation and application of the data generated from clinical research of BPs will be highly dependent upon the knowledge of the disease and treatment history of the subjects that were evaluated. Evaluations may involve analysis of healthy subjects who have not been diagnosed with any disease, or patients who have been diagnosed with any of the specific disease(s) indicated for treatment with the BP. Differences in the immune status, underlying disease and prior or concomitant treatments (e.g. immunosuppressive drugs) have resulted in ADIR differences among these populations. For example, ADA differences reported in rituximab-treated low-grade or non-Hodgkins lymphoma patients (1·1%), rituximab plus methotrexate-treated rheumatoid arthritis (RA) patients (11%) and adult patients with granulomatosis with polyangiitis (Wegener's granulomatosis) and microscopic polyangiitis (MPA) treated in combination with glucocorticoids (23%) (rituximab prescribing information [26]) are attributed to the general immunosuppressive effects of prior treatments in the cancer population and methotrexate-reducing immune responses to the BP.
History of prior exposure as well as frequency, duration, consistency of treatment and total BP doses administered should be considered during immunogenicity risk assessment. Treatment regimens may be highly variable, for example with Factor VIII replacement therapy, where patients may be treated only to control bleeding episodes, sometimes receiving multiple treatments in a single day, whereas other patients may be treated on a regular prophylaxis schedule to prevent bleeds [27]. For other BPs, treatment regimens are more consistent. ‘Drug holidays’, interruptions in a chronic dosing schedule, e.g. to reduce adverse effects [28], have the potential to impact immunogenicity risk. Terms and definitions describing treatment history are shown in Table 1.
Table 1.
Terms and definitions describing patient biopharmaceuticals (BP) treatment history
| Term | Definition |
|---|---|
| Treatment-naive subject: | Subject not exposed previously to the active substance in the BP |
| Treatment days* | Days on which the subject received treatment with the BP |
| Exposure days* | Days over which the subject was exposed to the BP |
| Total doses | Total number of doses (administrations) of the BP received by the subject |
| Drug holiday | Interruption in regularly scheduled dose administrations of a BP that is intended for chronic administration on a regular schedule (e.g. weekly or monthly) |
| Cumulative treatment days | Total number of days on which the subject received treatment with the BP |
| Cumulative exposure days | Total number of days over which the subject was exposed to the BP |
| Time to end-point | The length of time from initial treatment to measured end-point |
The example in Table 2 shows how treatment days, cumulative treatment days and total doses are determined for individual subjects treated with a BP.
Table 2.
Determination of cumulative treatment days
| Treatment days and doses given |
|||||||
|---|---|---|---|---|---|---|---|
| Subject no. | Day 1 | Day 8 | Day 15 | Day 22 | Day 29 | Cumulative treatment days | Total (number of) doses |
| 1 | xx* | xx | xx | xx | xx | 5 | 10 |
| 2 | x | x | x | 3 | 3 | ||
x = dose given.
Because many BPs, in particular the monoclonal antibodies, have long half-lives, the number of days of exposure could be much greater than the number of treatment days. The extent to which immunogenicity is affected by total or cumulative doses, treatment or exposure days needs to be determined for each BP. While determination of doses and treatment days is relatively straightforward, exposure days are related to BP concentrations, usually measured in blood, and therefore dependent upon the analytical method sensitivity and timing of measurements. Exposure days may be determined based on BP measurements (e.g. number of days over which BP concentration in plasma or serum was above the assay limit of quantitation) or predicted by pharmacokinetic modelling (e.g. time divided by BP mean half-lives).
Assessments of clinical outcomes may be conducted at interim points throughout the duration of treatment or beyond its discontinuation, thus time to end-point is an additional variable to be taken into account in describing the impact of immunogenicity on outcomes.
Development of anti-drug immune response (ADIR) and characteristics of ADA
The ADIR is defined as the host immune system response to an administered BP. Typically, assessment of BP ADIRs has focused on adaptive ADA responses. However, it is important to understand that ADIR encompasses innate and adaptive immune systems. Either BP, co-administered impurities or disease-specific factors may elicit pro- or anti-inflammatory cytokines, chemokines and other signals that may impact upon the efficiency of antigen presentation and BP-specific T and B lymphocyte adaptive responses, ultimately determining whether the level of activation and maturation of B cells is sufficient to generate high-titre, high-affinity or isotype-switched ADA responses that could impact upon BP efficacy or mediate adverse events such as hypersensitivity. Components of the innate immune system and activated T lymphocytes also have the potential to mediate adverse events such as infusion reactions and delayed-type hypersensitivity. Thus, totality of the BP ADIR should be considered when defining its immunogenicity profile. ADA is measured routinely in most BP clinical studies and monitored for some BPs during clinical use, while measurements such as the innate and T cell responses described above have not been evaluated routinely.
Clinical consequences of BP immunogenicity are dependent upon characteristics of induced ADA which, by nature, is expected to be heterogeneous in composition, containing a mixture of different quantities of antibodies of different isotypes, specificities and affinities [31–33]. Low-affinity, transient IgM responses may be induced initially, but could be difficult to detect during routine ADA monitoring. However, immunoglobulin (Ig)M–BP complexes may activate complement, which may play a role in mediating infusion reactions [34]. If the BP activates T helper cells, switching to other classes or subclasses of ADA may occur along with random somatic mutation of the immunoglobulin complementarity determining region (CDR) genes, leading to production of ADA with higher affinity for the BP, a phenomenon known as ‘affinity maturation’. ADA isotype composition determines the potential for clinical effects such as complement activation, antibody-dependent cellular cytotoxicity (ADCC) and mast cell sensitization, and interactions with the specialized neonatal Fc receptor, which controls cross-epithelial and placental transport as well as the serum half-life of ADA [35]. IgG4 has been associated with chronic BP administration and high and persistent ADA responses to various BPs [36–40]. ADAs that bind epitope(s) close to the BP site of activity (e.g. target binding site) are likely to block or ‘neutralize’ the BP biological activity [31,39,41,42]. ADAs bound to the BP can affect the BP's pharmacokinetics (PK) by either increasing or decreasing clearance (referred to as clearing and sustaining ADA, respectively) [9,43] and can also interfere with quantification of BP in assays used to study PK [44]. If the immune response to the BP continues, further expansion and diversification of the BP-specific B cell population may occur, resulting in ADA becoming more diverse in its specificity for binding different structures on the BP, a phenomenon known as ‘epitope spreading’. Therefore, it is possible to observe low-affinity, non-neutralizing ADA at early stages of an ADA response and higher affinity, neutralizing ADA at later stages. Alternatively, ADA responses may decrease and even disappear over time, even when the BP treatment is being continued, due to the development of immune tolerance [45,46]. Consequently, at any given time point in an immune response, a unique mixture of ADA characteristics may be present, resulting in the varied ADA-associated clinical effects that can be observed during the course of BP treatment.
Terms for ADA characterization are defined in Table 3.
Table 3.
Terms and definitions for anti-drug antibody (ADA)
| Term | Definition |
|---|---|
| ADA | Host antibody specific for the biopharmaceutical (BP) molecule. Includes all antibodies that bind drug regardless of their functional activity. May include pre-existing and natural host antibodies cross-reactive with the BP (baseline ADA) as well as drug-induced or boosted ADA. Preferred term to ‘binding antibody’ (Bab), as all ADA bind the BP |
| Affinity | Refers to strength of a specific intermolecular interaction. Often expressed as an equilibrium dissociation or association constant (Kd/Ka) or ratio of dissociation/association rate constants (kd/ka); however, due to the multivalent binding and heterogeneity of affinities expected within a polyclonal ADA sample, other measurements (avidity, antigen binding capacity, neutralizing capacity) may be more appropriate for characterizing ADA–BP interaction |
| <Isotype/subclass-specific> ADA | ADA measured in assay designed to detect ADA of specific isotype(s), e.g. immunoglobulin (Ig)G ADA, BPIgG + IgM ADA, IgE ADA |
| Neutralizing ADA (neutralizing antibody, NAb) | ADA that inhibits or reduces the functional activity of the BP, as determined by an in-vitro test method, regardless of its in-vivo relevance (i.e. whether or not the NAb causes reduced efficacy) |
| Non-neutralizing ADA (non-neutralizing antibody, non-NAb) | ADA that binds to the BP but does not inhibit its functional activity in an in vitro test method, regardless of its in-vivo relevance (i.e. whether or not the non-NAb causes clinical impact) |
| Clearing ADA | ADA associated with increased clearance of the BP relative to its clearance rate in the absence of ADA |
| Sustaining ADA | ADA associated with apparent decreased clearance of the BP relative to its clearance rate in the absence of ADA; most frequently observed when the BP has a fast clearance rate relative to the rate of IgG clearance |
| Anti-<component/domain etc.> ADA | ADA against a particular component/domain of a BP, e.g. anti-Fc, anti-Fab, anti-receptor domain, anti-polyethylene glycol (PEG) moiety |
| Anti-idiotypic ADA | ADA specific for epitope(s) unique to a specific monoclonal antibody therapeutic; usually ADA specific for the unique antigen-binding/complementarity determining region (CDR) of monoclonal antibody (mAb) biopharmaceutical |
| Anti-allotypic ADA | Generally refers to ADA specific for allotypic (defined as a genetically inheritable determinant common to some but not all human immunoglobulin molecules) epitopes of a mAb or mAb fragment BP. Could also refer to ADA specific for allotypic determinants on non-immunoglobulin-based BPs |
ADA immune response assays
Considering the nature of ADA heterogeneity, it becomes obvious that the use of reliable analytical methods and thorough understanding of their limitations will be critical to describe the ADA response appropriately and determine potential relevance to clinical outcomes. A number of ADA assay formats are available, each with some bias in the type(s) of ADA measured and limitations in sensitivity and susceptibility to interferences. Recommended practices for developing and validating ADA assays for use in clinical development programmes have been developed through multiple collaborative efforts among the pharmaceutical and regulatory agencies and scientific communities under the sponsorship of organizations such as AAPS and EIP [19–24] and adopted into pharmaceutical regulatory agency guidelines [47–50].
While most BPs are immunogenic under certain conditions in some individuals, during the course of treatment only a fraction of patients’ samples will typically have measurable ADA levels, and therefore it is common practice to first screen samples for ADA and then characterize any positives using a tiered approach, as recommended by Koren et al. [23] (Fig. 1). Assay terms are defined in Table 4.
Fig. 1.
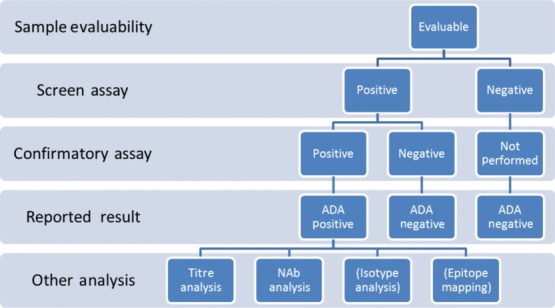
Typical tiered testing scheme for anti-drug antibody (ADA) testing and characterization. In the first tier, all evaluable samples are run in the screen assay. Samples that score positive in the screen assay are then analysed in a confirmatory assay (tier 2). Samples that score positive in the screen and confirmatory assay are reported as positive, while samples that score negative in either the screen or confirmatory assay are reported as negative. Further tiered testing of positive samples frequently includes analysis of titres and neutralizing activity. In some cases, isotype analysis or epitope mapping may also be performed.
Table 4.
Terms and definitions for anti-drug antibody (ADA) assays
| Term | Definition |
|---|---|
| ADA assay | Bioanalytical method used to determine if a sample is qualitatively positive or negative for ADA and that can provide quasi-quantitative information about the amount of ADA (typically reported as an antibody titre). Often considered synonymous with binding ADA assay (assay designed to detect antibodies that bind the biopharmaceutical (BP) regardless of the functional activity of the ADA) and total ADA assay (assay that measures all ADA in the sample, although usually not capable of binding immunoglobulin (Ig)E due to its low levels). |
| Free ADA assay | ADA assay that measures ADA not bound to the BP. Generally a binding ADA assay can be considered a free ADA assay unless specific ADA–BP dissociating conditions are incorporated into the assay procedure. |
| <Isotype/subclass x> ADA assay | ADA assay designed to detect ADA of specific isotype(s) or group of isotypes; e.g. IgE ADA assay |
| Neutralizing antibody assay | Assay used to determine whether ADA in a sample can neutralize some aspect of drug activity. Encompasses bioassay (cell-based or enzymatic) or competitive ligand-binding assay |
| <Epitope/domain> ADA assay | Assay designed to detect ADA specific for a particular epitope or domain of the BP, e.g. Fab assay |
| Screening assay | In a tiered testing strategy, the assay used to distinguish potentially positive samples (based on screening cut-point) versus negative samples |
| Confirmatory assay | An assay conducted on samples found to be potentially positive in the screening assay in a tiered testing strategy to identify false and true positives (based on confirmatory cut-point) |
| ADA characterization assay | Investigational assay that is designed to obtain additional information on the specificity or type of antibodies present in a sample. Information obtained from these assays may include, but is not limited to, the following: titre, neutralizing antibody assay, isotyping assay (see definitions below and above) |
| Qualitative assay | Assay that reports test results as positive/negative |
| Quasi-quantitative Assay | Assay that reports a relative magnitude of ADA present in a sample (e.g. ADA titre) |
| Titre assay | A quasi-quantitative assay providing titre as the unit of the amount of antibody in a sample. The titre is often defined as the reciprocal of the lowest dilution of a sample generating a signal that is above the assay cut-point. Alternatively, the titre is defined as the reciprocal of the dilution of a sample generating a signal that is equivalent to the assay cut-point, calculated by an interpolation formula provided in an assay-specific bioanalytical method |
| Relative concentration assay | A quasi-quantitative assay providing sample results reported in relative mass units, determined by comparing the assay signal generated by the sample relative to a signal generated by a diluted positive control sample. Because the positive control generally contains a different mixture of antibodies than the sample, concentrations reported by this result are generally not accurate and should be reported as ‘relative concentrations’ or defined units |
| Cut-point | An assay signal threshold that distinguishes positive samples from negative samples, as defined in an assay-specific analytical procedure. The cut-point is usually set based on statistical analysis with treatment-naive samples representative of the study population (bioanalytical cut-point) but could be based on a biological (e.g. change in pharmacodynamics marker; biological cut-point) or clinical end-point (e.g. loss of efficacy; clinical cut-point). Cut-points are typically set for each assay in the tiered analysis strategy (screening assay, confirmatory assay, neutralizing assay cut-points) |
Using current technology and reagents, it is possible to develop highly sensitive ADA screening assays capable of detecting the most prevalent classes of ADA (IgM, IgG and IgA); however, due to the low circulating levels of IgE, BP-specific IgE is unlikely to be detectable in these assays, therefore a separate IgE ADA assay may be required when IgE detection is needed.
Cut-points for distinguishing positive and negative sample results should be established initially based on a statistical analysis of samples from the BP treatment-naive patient population (if available) or from healthy donors. In BP clinical studies, it is common practice to establish a cut-point that allows sufficient sensitivity in order to ensure the detection of most true positive samples, while limiting the frequency of false negative results, i.e. a cut-point set at the 95th percentile of the distribution of the values obtained with BP treatment-naive donors, corresponding to a 5% false positive rate. Samples scoring below the screening assay cut-point are defined as ADA-negative, whereas samples scoring equal to or above the cut-point are then tested in the second tier of the immunogenicity testing strategy, the confirmatory assay, in order to further distinguish true positive from false positive samples.
The confirmatory assay usually consists of a competitive inhibition assay, in which samples are tested spiked and unspiked with an excess of the BP. The excess BP binds the specific ADA, preventing it from binding to the assay capture reagent, thus causing an inhibition in signal in the assay relative to the signal generated by the unspiked sample. Confirmatory assay cut-points are usually set in order to report a low percentage (typically 1–0·1%) of false positive samples using BP treatment-naive patient samples (if available) or samples from healthy donors and determining the percentage of signal inhibition corresponding to the 99th or 99.9th percentile of the distribution. Samples spiked with BP that exhibit reduced signal (% inhibition) in the assay relative to their respective unspiked signal at or above the confirmatory assay cut-point (% inhibition) are considered positive for BP-specific ADA, while those that score below the cut-point are considered negative for BP-specific ADA (false positive).
Qualitative ADA results (positive/negative) may be supplemented with quasi-quantitative measurements of the relative magnitude of ADA responses (e.g. titres or units) to provide more useful information for interpretation of ADA data and determining relationships to clinical outcomes. However, it is neither accurate nor meaningful to report ADA data in absolute quantitative readouts (i.e. units of mass concentration) calculated from a positive control antibody curve. Quantitative measurements of mass concentration require standards with the same composition as the unknown samples, while the spectra of ADA affinities, isotypes and epitope specificities are expected to differ in composition across patients and across time-points during the treatment of an individual patient. Therefore, it is not possible to establish standards or assay calibrators that are representative of the complex heterogeneity of ADA in all samples. Hence, ADA data should typically be reported using quasi-quantitative units, such as titres, which are usually determined by running positive samples in a dilution series and reporting the titre as the reciprocal of the last dilution at which the sample scores above the cut-point or the dilution that scores at the cut-point, calculated based on interpolation. Data from relative concentration assays should be designated clearly as ‘units of relative concentration’, or preferably reported in other quasi-quantitative units defined relative to a known amount of an ADA-positive control sample. Other more quantitative measurements such as binding affinity and in-vitro antigen binding or neutralizing capacity have also been reported [51,52]; however, these are cumbersome to perform on large numbers of samples and results may also be affected by ADA heterogeneity and the assay system used.
It is important to note that the statistically established cut-points used to identify positive samples are bioanalytical cut-points that reflect the analytical variability of the assay and biological variability of the treatment-naive study population. Therefore, they are not ‘clinically relevant’, in the sense that identifying a sample as positive for ADA is not an indicator that the level of ADA present in the sample is sufficient to cause a biologically or clinically meaningful effect (e.g. alter PK, efficacy or cause adverse effects). However, sensitive assays capable of detecting all ADA present help establish definitively whether there are any relationships between clinical effects and the measured ADA when initially studying the immunogenicity profile in clinical studies. Where sufficient clinical data demonstrating a clear relationship between ADA positivity or titres, and clinical effect are available, it may be possible to establish a ‘biological’ or ‘clinical’ for subsequent use to distinguish clinically relevant from irrelevant ADA responses. Sensitive ADA assays are also needed to correlate ADA development with underlying causes of immunogenicity and identify markers of immune response; e.g. markers of early events in triggering an immune response to the BP or indications of incomplete activation of the immune system that could lead to immune tolerance by the BP.
Other assays may be developed to characterize ADA-positive samples further and provide additional data to understand and interpret more extensively the clinical relevance of the ADA response to a BP. For example, an assay that specifically measures ADA that neutralize the biological effect of the BP may be useful in determining the impact of ADA on pharmacodynamic effect or efficacy while an assay that measures BP-specific IgE or IgM may help to identify the underlying cause of a hypersensitivity reaction.
A variety of different formats may be used for screening, confirmatory and characterization assays, and limitations are associated with each. Some tend to be more sensitive for the detection of high-affinity ADA while others tend to be more sensitive for detection of low-affinity ADA [53,54]; some are biased against detection of certain classes of ADA and some more susceptible to interferences by BP, BP target or other serum components that may be present in samples [19,22,55–60]. Table 5 summarizes analytical issues encountered with these assays. BP or drug interference is considered one of the most common and significant issues, as steady state trough levels of many BPs are frequently above the levels that could potentially interfere with reliable ADA detection. ADA–BP immune complexes present in samples may interfere with detection of either ADA or BP levels and therefore may contribute to under-reporting of ADA prevalence and incidence in the study population and erroneous BP PK characterization profiles [61]. The concentration of BP that interferes with ADA detection will be highly dependent upon the ADA levels in the sample (i.e. the drug tolerance level will be higher for samples with higher levels of ADA and lower for samples with lower levels of ADA). Whenever possible, drug interference should be minimized using various strategies, including sample acidification pretreatment, more drug-tolerant assay formats and platforms, use of competing antibodies or monovalent versions of multivalent BP and longer incubation times [39,59,62–65]. However, there are caveats to each of these strategies, as they may dissociate incompletely the immune complexes, may denature and reduce the binding activity of some ADA, may reduce detection of ADA against some epitopes or may increase the level of target interference. While drug interference could result in under-reporting of ADA data, target interference, due to the presence of multi-valent BP target molecules in samples, have been reported to cause false positive ADA assay results in the bridge assay format [55,56]. Because of the potential for BP, BP target or serum component interferences, ABIRISK includes the definition of an ‘inconclusive’ sample: ‘for which the assay result cannot be reported as incontrovertibly negative or positive for ADA’, and recommended that any positive or negative results that could be questionable due to assay limitations should be noted in reporting the results and considered in interpretation of the clinical or analytical outcomes. Because of these various known assay limitations, described above and in Table 5, whenever possible ADA data should be interpreted in combination with pharmacokinetic (or BP concentration), pharmacodynamic and efficacy and safety data to define the immunogenicity profile of the BP and determine the clinical relevance.
Table 5.
Analytical issues encountered in anti-drug antibody (ADA) measurement
| Issue | Definition and explanation | Mitigation or investigational strategies |
|---|---|---|
| Drug interference | Alteration in ADA detection (usually impaired detection) in an assay due to the presence of biopharmaceuticals (BP) in the sample | Adjust sample collection time-points to achieve no/lowest BP concentrations (before administration, after washout, during drug holiday); utilize assay formats with a higher degree of drug tolerance (bridge assay, high-density surface, long incubation times); incorporate dissociation step prior to analysis |
| Target interference | Alteration in ADA detection. In bridge assay formats, multivalent target may cause false positive results (note target levels can increase after BP administration) | Evaluate potential target interference during validation; remove or denature target; add anti-target monoclonal antibodies (mAbs) to block bridging |
| Pre-existing antibodies | Antibodies reactive with the BP before initiation of treatment | Affinity removal of antibodies or select pool of negative samples to establish negative control. Statistical analysis to identify true negative population for establishing negative control cut-point. Establish individual cut-points using baseline samples. Evaluate increases and decreases from baseline in final population analysis to determine level of BP-induced ADA |
| Rheumatoid factor (RF) interference | Rheumatoid factor present in sample may elicit positive result | Evaluate potential RF interference during validation; use assay formats that minimize RF interference. Evaluate increases and decreases from baseline in final population analysis to determine level of BP-induced ADA |
Prior to use, ADA assays should be validated analytically to assure reliable performance over time and establish which changes in ADA measurements are likely to be meaningful [20,22,47,49,50]. Analytical validation typically involves analysis of positive and negative quality control (QC) samples to characterize inter- and intra-assay variability (precision), sensitivity to small changes in conditions (robustness) and interlaboratory variability (ruggedness). As noted previously, it is not possible to identify a positive control that contains a representative mixture of the ADAs present in all samples, therefore caution should be used when translating information on performance of positive controls to performance of actual study samples. In particular, parameters such as sensitivity and drug tolerance are unique to the properties of the positive control and will differ for individual patient samples. While assays should be validated analytically for use in BP clinical research, a higher standard would be needed to demonstrate that the assay is clinically validated, in particular that it correlates with clinical outcomes [i.e. correlates with loss of efficacy (LoE) or pharmacodynamic response], such that it may be used for clinical treatment decision-making.
Study design, patient monitoring and analysis of ADIR results
In order to establish a clear causal relationship between presence or type of ADA and safety signals or LoE in patients, time-points at which ADA are measured should be scheduled as closely as possible to time-points for safety or efficacy measurements. For example, the chance of identifying clearing ADA or neutralizing antibody (Nab) as the cause of LoE would be enhanced if a sample(s) collected from the patient near time of observed LoE is determined to be positive for these parameters. To comprehend relevance to patient safety and efficacious response and to define the immunogenicity risk profile of the BP in the patient population, ADIR results should be evaluated at the sample, patient and population levels.
As described previously, ADA sample results are reported typically as positive or negative based on a tiered analysis approach (Fig. 1). These results can then be used to determine the ADA status of patients as well as whether they have had treatment-induced or treatment-boosted responses (Fig. 2, Table 7). Typically, subjects are expected to be negative prior to BP exposure; if a subject becomes positive after treatment, the subject is designated as having a treatment-induced ADA response. It is well recognized that some BP treatment-naive subjects may have pre-existing antibodies to the BP [66,67], which may originate from the natural population of antibodies or from exposure to cross-reactive environmental antigens or other BPs containing the same or homologous ingredients. Cases in which exposure to the BP results in post-treatment increases in ADA titres relative to a positive baseline titre are referred to as treatment-boosted ADA responses; in other cases, exposure results in no post-treatment increases in ADA titres relative to the baseline titres (treatment unaffected). Over time, ADA titres may persist at the same level, increase (persistent response) or decrease or become negative (transient response). When sample collection is infrequent or sporadic, it may be difficult to establish persistence or transience of the response. None the less, in many cases, merely designating whether a subject has developed ADA or was positive at some point during the study may be insufficient to demonstrate causal relationships between ADA and clinical outcomes.
Fig. 2.
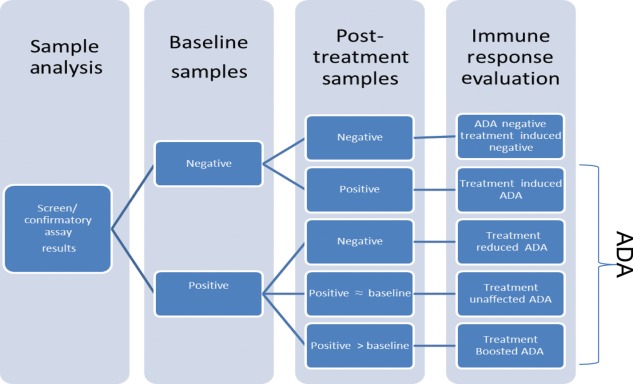
Interpretation of anti-drug immune response. After sample results have been determined, it will then be possible to categorize the anti-drug antibody (ADA) status of the subjects, and determine whether the positive subjects’ ADA originated from treatment induced, boosted or unaffected ADA responses.
Table 7.
Interpretation of anti-drug antibody (ADA) status, prevalence and incidence
| Time-point (month) |
||||||||
|---|---|---|---|---|---|---|---|---|
| Baseline M0 | M1 | M3 | Last M9 | Follow-up M18 | ||||
| Sample result neg/pos |
||||||||
| Subject no. | Sample result titre (ns = no sample) | ADA status | Induced ADA | Boosted ADA | ||||
| 1 | Neg | Neg | Neg | Neg | Neg | Neg | Neg | |
| 2 | Neg | Neg | Pos | Pos | Neg | Pos | Pos | |
| 225 | 1350 | |||||||
| 3 | Neg | Pos | Pos | Neg | Neg | Pos | Pos | |
| 75 | 225 | |||||||
| 4 | Pos | Pos | Pos | NS | Pos | Pos | Neg | |
| 225 | 225 | 225 | 225 | |||||
| 5 | Pos | Pos | Pos | Pos | Pos | Pos | Pos | |
| 75 | 1350 | 16200 | 8100 | 16200 | ||||
| 6 | NS | Pos | Pos | Pos | Pos | Pos | ||
| 75 | 225 | 75 | 225 | |||||
| Prevalence | 40% | 67% | 83% | 60% | 50% | In study: 83% (5/6) | ||
| n = 2/5 | n = 4/6 | n = 5/6 | n = 3/5 | n = 3/6 | ||||
| Incidence | 60% n = 3/5 | |||||||
In this example, subject 1 remained negative throughout the study while subjects 2–6 had one or more positive samples and are therefore designated ADA-positive. Because subjects 2 and 3 were negative at baseline and were positive at one or more post-treatment time-points, both are considered positive for treatment-induced ADA. Subjects 4 and 5 were positive at baseline but only subject 5 had a post-treatment increase in titre, therefore subject 4 is negative for boosted ADA response while subject 5 is positive for boosted ADA response. At the time of the follow-up, subjects 2 and 3 had become negative and were therefore considered transient immune responders. The prevalence at each time-point was calculated based on the no. of positive subjects at each time-point as % of/no. of evaluable subjects at that time-point and the incidence was calculated as the no. of subjects positive for induced or boosted ADA as % of no. of evaluable subjects in the study. Because subject 4 had no sample at M9 and subject 6 had no sample at M0 (baseline), they were not evaluable for calculation of prevalence at M9 or incidence across the study.
ADA frequency in a population should be described with the terms of incidence and prevalence (Table 6). Incidence is a cumulative measurement of the number of subjects who, within a defined period of time, have developed an immune response to the BP (includes treatment-induced and treatment-boosted responses), and is useful to predict the probability that a particular BP will induce an ADA response in that population. Prevalence is a measure of the number of subjects who are positive at a particular time or during a particular time-frame and is most useful to assess relationships between ADA and clinical outcomes, particularly when the outcomes are immediately apparent (e.g. type I or immediate-type hypersensitivity response due to IgE ADA or LoE due to NAb development in haemophilia). When clinical outcomes are delayed (e.g. LoE in multiple sclerosis), prevalence within the study or interim measurements of incidence may be more useful to assess the clinical relationship.
Table 6.
Terms for anti-drug antibody (ADA) response origin and frequency
| Term | Definition |
|---|---|
| Treatment-induced ADA-positive subject | Subject with ADA developed de novo (seroconverted) following biopharmaceuticals (BP) administration (i.e. formation of ADA any time after the initial drug administration in a subject without pre-existing antibodies) |
| Treatment-boosted ADA-positive subject | Subject with pre-existing antibodies that develops an increased level of ADA following BP administration (i.e. any time after the initial drug administration the ADA titre is disproportionately greater by a biologically relevant margin, such as two- or threefold relative to the baseline titre) |
| Treatment-unaffected ADA-positive subject | Subject with pre-existing ADA level that does not change following the BP administration |
| Incidence of ADA (rate of ADA development) | Measure of the rate of BP-specific ADA immune responses during a defined observation period, usually equal to the sum total of treatment-induced and treatment-boosted ADA-positive subjects (but not treatment-unaffected) as a percentage of the evaluable subject population. Incidence is a cumulative measurement. Incidence rates for treatment-induced ADA treatment-boosted ADA, or specific types of ADA [e.g. non-neutralizing antibodies (Nabs) or immunoglobulin (Ig)E] may also be considered separately. Incidence includes transient and persistent positive patients and may also be referred to as cumulative incidence |
| Prevalence of ADA (frequency of ADA-positive subjects) | The percentage of subjects positive for ADA in a defined population at a particular time-point or within a particular defined time-frame. For example, antibody prevalence at baseline (pretreatment) is determined by dividing the number of evaluable subjects with antibody-positive pre-treatment samples by the number of subjects with a pretreatment sample result, expressed as a percentage. May also be useful when subjects with diverse treatment histories are sampled randomly |
Table 7 shows the proposed interpretation of ADA status, induced and boosted ADA and prevalence and incidence based on different patterns of ADA results. Although vast differences in study design and immunogenicity data-reporting strategies can be observed across published studies, common concepts and terms can be applied retrospectively in describing these study designs and results for comparative purposes as indicated in Table 8.
Table 8.
Descriptions of immunogenicity reported in studies of approved biopharmaceuticals using ABIRISK recommended terms and definitions (definitions in [] are study-specific definitions defined in the publications)
| BP and study designation, study population | Assays: types of assays (and reported results) used) | Samples: times of collection | Samples: ADA/NAb status definitions | ADA status of subject population | NAb status of subject population |
|---|---|---|---|---|---|
| Natalizumab AFFIRM study in: treatment naive MS patients [68] | ADA: ELISA bridge assay: screening (qualitative) and titre (quasi-quantitative) NAb: cell-based blocking bioassay;screening and titre assays | Baseline, then every 12 weeks over 2 years | ADA:cut-point: OD of 50 μg/ml calibrator control; positive sample: OD ≥ cut-point OD; Titre: sample dilution with OD = cut-point NAb: reduced fluorescence intensity from bound phycoerythrin-conjugated natalizumab below predetermined limit | ADA-positive subjects: 9% (57/625) Persistent positive [ADA positive at 2 or more time-points ≥6 weeks apart] subjects:6% (37/57); Transient-positive [ADA-positive at a single time point] subjects:3% (20/57)Incidence of ADA: 9% | NAb-positive subjects: 100% of ADA-positive subjects |
| Interferon-β PRISMS study in: treatment-naive MS patients treated with 44 μg three times weekly (TIW) or 22 μg TIW [69] | ADA: ELISA direct binding assay. Screening (qualitative), confirmatory, and titre (quasi-quantitative)NAb: cell-based bioassay: VSV-induced cytopathic effect (CPE) assay (quasi-quantitative) | Baseline, then every 6 months over 4 yrs | ADA:cut-point: 2 s.d. above mean OD for pooled normal human serum. Positive sample: OD > cut-point OD and positive in confirmatory assay NAb: Cut-point: titre ≥ 20 NU/ml where NU = (f x N)/10 and f = 1/sample dilution at ED50, and N = IFN concentration (LU/ml) | ADA-positive subjects:44% (82/186) at 22 μg TIW; 37% (67/182) at 44 μg TIW;41% overall | NAb-positive subjects: 30% (55/186) at 22 μg TIW; 19% (35/182) at 44 μg TIW; 24% overall;Persistent positive [NAb positive at final assessment] subjects: 24% (44/186) at 22 μg TIW;14% (26/182) at 44 μg TIW;19% overall |
| Factor VIII Turoctocog alfa study in: treatment-experienced subjects with severe hemophilia A with no history of Nabs [70] | NAb: non-cell-based bioassay: Nijmegan-modified Bethesda inhibitor assay clotting assay (quasi-quantitative) | Visits 1, 2, 4, 5, 6, 7, 8, 9 | NAb:cut-point: Bethesda units (BU) > 0·6 | NAb-positive subjects: [patient tested positive >0·6 BU in 2 consecutive test samples]; 0% (0/150) of patients developed NAbs | |
| Infliximab re-initiation study in: treatment-experienced patients who restarted infliximab after drug holiday [71] | ADA: acidification pretreatment homogenous mobility shift assay (quasi-quantitative) [72] | T–1, T0, T +1, T +2* | ADA:cut-point: 7.95 U/ml [1 unit corresponding to signal generated by positive control serum approx. 0.18 µg] | ADA-positive subjects:13.3% at T–1*;0% at T0 (0/124);40% at T+1 (31/77);29% at T+2 (19/65) | |
| Adalimumab Long-term follow-up study in: treatment-naive RA patients [73] | ADA: radioimmunoassay (qualitative and quasi-quantitative) | Baseline, 4, 16, 28, 40, 52, 78, 104, 130, 156 weeks | ADA:cut-point:12 AU (arbitrary units)/ml [1 AU defined based on signal generated by ∼ 12 ng of a positive control serum] | ADA-positive subjects: [titres> cut-point on at least 1 occasion in combination with serum adalimumab levels < 5.0 mg/l]: 28% (76/272) | |
| Adalimumab assay comparison study [65](from study above [73]) | ADA assays:(a–d) radioimmunoassay with (a) no modification (b) acidification (c) acidification + anti-adalimumab Fab idiotype (d) overnight incubation at 37C(e) bridge format electrochemiluminescent with acidification (qualitative and quasi-quantitative) | Baseline, 16, 28, 52 weeks | ADA:cut-points for assays (a–e):(a) 12 AU (arbitrary units)/mL(b) 30 AU/ml (c) 48 AU/ml (d) 33 AU/ml (e) 3·9 AU/ml | ADA positive subjects [titres> assay cut-point on at least 1 time point] in assays (a–e):a) 14·9%b) 66·0%c) 51·1%d) 57·4%e) 57·4% | |
T–1 = last sample during previous course (prior to drug holiday), T0 = day of restart before first reinfusion, T+1 early time-point after re-exposure (i.e. just before second or third infusion), T +2 = later sample (i.e. before third or fourth reinfusion). ADA = anti-drug antibody; MS = multiple sclerosis; RA = rheumatoid arthritis; ELISA = enzyme-linked immunosorbent assay; OD = optical density; VSV = vesicular stomatitis virus; ED50 = effective median dose; Nab = neutralizing antibody; s.d. = standard deviation.
Establishing clinical relevance of an ADA response
Once ADA status and response of individual subjects and incidence and prevalence for the study population have been determined, their association and potential relationship to PK/PD profiles, efficacy and safety outcomes should be evaluated. Patients could be stratified into various groups (for example: positive/negative for ADA, NAb or pre-existing antibodies; for boosted or induced ADA; based on time of ADA onset; transience, persistence and duration of response; or relative magnitude of ADA or NAb) for comparison of clinical outcomes and immune response measurements. Rationale and reliability of stratification approaches should be determined for each patient population being studied.
In some cases, causal relationships can be identified from a limited amount of clinical experience. However, due to the different types of ADA responses that may develop over time, it is expected that some associations between treatment and risk of ADA development and between ADA development and clinical consequences of ADA may be subtle, requiring extensive accumulation of clinical data and multivariate analysis to identify or establish causal relationships. Relationships can be especially challenging to discern when the underlying disease and/or drug pharmacology affect the immune system.
For most BP, it is common practice to conduct immunogenicity monitoring in pre- and post-marketing clinical trials. For products with higher immunogenicity risk, longer-term monitoring during chronic treatment may be necessary. However, there are no common practices to generate and analyse these data to understand fully the immunogenicity profiles of BPs used in various treatment scenarios. Several recommendations have been made for common approaches to presenting and analysing ADA data, including receiver operating characteristic (ROC) curves, to estimate clinically relevant thresholds of variables such as titre, onset and duration of ADA, and the Classification and Regression Tree (CART) ‘partition’ analysis to predict categorical or continuous clinical outcomes [25]. Several recent publications have proposed mathematical modelling approaches to assess the impacts of ADA on PK and of BP exposure on ADA development [9,43,74–76]. Depending on the risk level and clinical impact, ADA or NAb testing for approved BPs may be performed either routinely to monitor treatment or only if needed, based on clinical observations such as altered PK, loss of efficacy or AEs. For example, testing for neutralizing antibodies is routine in haemophilia treatment [12,27], while ADA testing may be performed only in tumour necrosis factor (TNF)-α antagonist non-responders who have low or undetectable drug concentrations after treatment [10,11]. ABIRISK is evaluating data analysis strategies actively. Appropriate strategies to utilize ADIR data in treatment decisions may need to be developed for each BP.
Contributions of ABIRISK
Data from ABIRISK pertaining to immunogenicity development, prediction and safety will be collected in a single database, requiring standardization of information originating from multiple types of investigational designs (e.g. prospective and retrospective investigations), multiple BPs (including coagulation factor VIII, β-interferons, TNF-α antagonists, natalizumab and rituximab), different diseases (e.g. haemophilia A, multiple sclerosis, rheumatoid arthritis, other autoimmune rheumatic conditions, inflammatory bowel diseases), different cohorts for a given disease and various preclinical, clinical and immune monitoring factors. ABIRISK will collect and publish data in accordance with the recommended terms and definitions and promote their use in the broader communities.
Conclusions
Production of ADA represents one outcome of a complex ADIR process that follows initial exposure to a BP and involves innate and adaptive immune responses. To minimize the risk of ADA induction and sustain BP efficacy, insight into the complex mechanisms that drive ADIR is needed, including characteristics intrinsic to the molecular structures of BPs and the roles of inflammation and underlying patient characteristics. A first step that will facilitate the scientific process of elucidating these mechanisms is the adoption of clear definitions for terms and concepts related to immunogenicity, its prediction and associated clinical events across all communities involved in the immunogenicity area. Thus, ABIRISK is providing their recommendations as a resource to these communities for their use and to obtain their feedback on the relevance to long-term immunogenicity monitoring practices.
Acknowledgments
This work has received support from the Innovative Medicines Initiative Joint Undertaking (IMI JU) under grant agreement no. 115303, the resources of which are composed of financial contributions from the European Union's Seventh Framework Programme (FP7/2007-2013) and European Federation of Pharmaceutical Industries and Associations (EFPIA) companies' in-kind contributions. The following members of ABIRISK are acknowledged for contributions to reviewing this document: Michael Tovey, Cornelia Sabine Viebahn, Zoe Waibler and Arsalan Zharazmi. We also thank Sophie Turbot for her assistance in coordinating the review.
Disclosure
Authors B. R., C. C., J. D., D. F., A. H., C. H., J. L., H. K., D. K., A. L., V. M., C. R.-P., D. S. and S. S. belong to EFPIA (European Federation of Pharmaceutical Industries and Association) member companies in the IMI JU and costs related to their part in the research were carried by the respective companies as in kind contributions under the IMI JU scheme. The following authors have received funding from ABIRISK: T. A., P. B., P. C., N. de V., A. F.-H., E. H., E. J., S. L.-D., B. M., X. M., D. M., J. O., G. P., M. P., R. S. and F. D. In addition, all authors have completed the ICMJE uniform disclosure form at http://www.icmje.org/coi_disclosure.pdf. Outside the submitted work, the following authors report receiving fees or grants or other support from the following companies: T. A. (Baxter, Bayer, Biogen-Idec, Biotest, CSL-Behring, Grifols, Novo Nordisk, Octapharma, Pfizer), M. A. (MSD, Abbvie, Ferring, Janseen-Cilag, Novo Nordisk, Genentech, Pfizer, UCB), P. C. (Merz Pharmaceuticals GmbH), N. de V. (Pfizer and Roche), A. F.-H. (Biogen Idec, Pfizer), G. P. (Genzyme, Novartis, Roche Pharma), X. M., (BMS, Pfizer, Roche, UCB) and J. O. (Baxter, Bayer, Biogen Idec, Biotest, CSL-Behring, Grifols, Inspiration, NovoNordisk, Octapharma, Swedish Orphan Biovitrum and Pfizer/Wyeth). No other financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years (except as their employers or as listed above) and no other relationships or activities that could appear to have influenced the submitted work were reported. F. D. has participated in meetings sponsored by or received honoraria for acting as an adviser/speaker for Bayer Healthcare, Biogen Idec, Genzyme-Sanofi, Merck, Novartis Pharma and Teva-Ratiopharm. His institution has received financial support for participation in randomized controlled trials of INFb-1b (Betaferon, Bayer Schering Pharma), IFN-β1a (Avonex, Biogen Idec; Rebif, Merck Serono), glatiramer acetate (Copaxone, Teva Pharmaceuticals) and Natalizumab (Tysabri, Biogen Idec), in multiple sclerosis. He is section editor of the MSARD Journal (Multiple Sclerosis and Related Disorders). N. R. received honoraria for consultancy of speaker's bureau from the following pharmaceutical companies: Abbott, AbbVie, Amgen, Biogenidec, Astellas, Alter, AstraZeneca, Boehringer, BMS, CDPharma, Celgene, CrescendoBio, EMD Serono,Hoffman-La Roche, Italfarmaco, Janssen, MedImmune, Medac, Novartis, Novo Nordisk, Pfizer, Sanofi Aventis, Servier and Vertex. The Gaslini Hospital, which is the public Hospital where N. R. works as full-time public employee, has received contributions from the following industries: Abbott, BMS, ‘Francesco Angelini’, GlaxoSmithKline (GSK), Hoffman-La Roche, Italfarmaco, Janssen, Novartis, Pfizer, Sanofi Aventis, Schwarz Biosciences, Sobi, Xoma and Wyeth. This money has been reinvested for the research activities of the hospital in a fully independent manner besides any commitment with third parties. S. S. is a full-time employee of and has stocks and stock options in Novartis.
Author's contributions
First author/corresponding author B. R. and last author, F. D., were the primary writers for the manuscript. In addition, all authors contributed equally in proposing, refining and endorsing terms and definitions adopted by the ABIRISK consortium and in providing concepts, supporting literature and regulatory guidance references communicated in this manuscript. All authors reviewed and approved the final version.
References
- 1.Dimitrov DS. Therapeutic proteins. Methods Mol Biol. 2012;899:1–26. doi: 10.1007/978-1-61779-921-1_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leader B, Baca QJ, Golan DE. Protein therapeutics: a summary and pharmacological classification. Nat Rev Drug Discov. 2008;7:21–39. doi: 10.1038/nrd2399. [DOI] [PubMed] [Google Scholar]
- 3.Li J, Yang C, Xia Y, et al. Thrombocytopenia caused by the development of antibodies to thrombopoietin. Blood. 2001;98:3241–8. doi: 10.1182/blood.v98.12.3241. [DOI] [PubMed] [Google Scholar]
- 4.McKoy JM, Stonecash RE, Cournoyer D, et al. Epoetin-associated pure red cell aplasia: past, present, and future considerations. Transfusion. 2008;48:1754–62. doi: 10.1111/j.1537-2995.2008.01749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hansel TT, Kropshofer H, Singer T, Mitchell JA, George AJ. The safety and side effects of monoclonal antibodies. Nat Rev Drug Discov. 2010;9:325–38. doi: 10.1038/nrd3003. [DOI] [PubMed] [Google Scholar]
- 6.Warrier I, Ewenstein BM, Koerper MA, et al. Factor IX inhibitors and anaphylaxis in hemophilia B. J Pediatr Hematol Oncol. 1997;19:23–7. doi: 10.1097/00043426-199701000-00003. [DOI] [PubMed] [Google Scholar]
- 7.Wolbink GJ, Vis M, Lems W, et al. Development of antiinfliximab antibodies and relationship to clinical response in patients with rheumatoid arthritis. Arthritis Rheum. 2006;54:711–5. doi: 10.1002/art.21671. [DOI] [PubMed] [Google Scholar]
- 8.Bertolotto A, Sala A, Malucchi S, et al. Biological activity of interferon betas in patients with multiple sclerosis is affected by treatment regimen and neutralising antibodies. J Neurol Neurosurg Psychiatry. 2004;75:1294–9. doi: 10.1136/jnnp.2004.037259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chirmule N, Jawa V, Meibohm B. Immunogenicity to therapeutic proteins: impact on PK/PD and efficacy. Am Assoc Pharm Sci J. 2012;14:296–302. doi: 10.1208/s12248-012-9340-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garces S, Antunes M, Benito-Garcia E, da Silva JC, Aarden L, Demengeot J. A preliminary algorithm introducing immunogenicity assessment in the management of patients with RA receiving tumour necrosis factor inhibitor therapies. Ann Rheum Dis. 2014;73:1138–43. doi: 10.1136/annrheumdis-2013-203296. [DOI] [PubMed] [Google Scholar]
- 11.Vincent FB, Morand EF, Murphy K, Mackay F, Mariette X, Marcelli C. Antidrug antibodies (ADAb) to tumour necrosis factor (TNF)-specific neutralising agents in chronic inflammatory diseases: a real issue, a clinical perspective. Ann Rheum Dis. 2013;72:165–78. doi: 10.1136/annrheumdis-2012-202545. [DOI] [PubMed] [Google Scholar]
- 12.Collins PW, Chalmers E, Hart DP, et al. Diagnosis and treatment of factor VIII and IX inhibitors in congenital haemophilia (4th edition). UK Haemophilia Centre Doctors Organization. Br J Haematol. 2013;160:153–70. doi: 10.1111/bjh.12091. [DOI] [PubMed] [Google Scholar]
- 13.Rosenberg AS, Worobec AS. A risk-based approach to immunogenicity concerns of therapeutic protein products. Part 1. Considering consequences of the immune response to a protein. Biopharm Int. 2004;17:22–6. [Google Scholar]
- 14.Rosenberg AS, Worobec AS. A risk-based approach to immunogenicity concerns of therapeutic protein products. Part 2. Considering host-specific and product-specific factors impacting immunogenicity. Biopharm Int. 2004;17 [Google Scholar]
- 15.Rosenberg AS, Worobec AS. A risk-based approach to immunogenicity concerns of therapeutic protein products. Part 3. Effects of manufacturing changes in immunogenicity and the utility of animal immunogenicity studies. Biopharm Int. 2005;18 [Google Scholar]
- 16.Ebbers HC, Crow SA, Vulto AG, Schellekens H. Interchangeability, immunogenicity and biosimilars. Nat Biotechnol. 2012;30:1186–90. doi: 10.1038/nbt.2438. [DOI] [PubMed] [Google Scholar]
- 17.Singh SK. Impact of product-related factors on immunogenicity of biotherapeutics. J Pharm Sci. 2011;100:354–87. doi: 10.1002/jps.22276. [DOI] [PubMed] [Google Scholar]
- 18.Ghaderi D, Taylor RE, Padler-Karavani V, Diaz S, Varki A. Implications of the presence of N-glycolylneuraminic acid in recombinant therapeutic glycoproteins. Nat Biotechnol. 2010;28:863–7. doi: 10.1038/nbt.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mire-Sluis AR, Barrett YC, Devanarayan V, et al. Recommendations for the design and optimization of immunoassays used in the detection of host antibodies against biotechnology products. J Immunol Methods. 2004;289:1–16. doi: 10.1016/j.jim.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 20.Shankar G, Devanarayan V, Amaravadi L, et al. Recommendations for the validation of immunoassays used for detection of host antibodies against biotechnology products. J Pharm Biomed Anal. 2008;48:1267–81. doi: 10.1016/j.jpba.2008.09.020. [DOI] [PubMed] [Google Scholar]
- 21.Gupta S, Indelicato SR, Jethwa V, et al. Recommendations for the design, optimization, and qualification of cell-based assays used for the detection of neutralizing antibody responses elicited to biological therapeutics. J Immunol Methods. 2007;321:1–18. doi: 10.1016/j.jim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 22.Gupta S, Devanarayan V, Finco D, et al. Recommendations for the validation of cell-based assays used for the detection of neutralizing antibody immune responses elicited against biological therapeutics. J Pharm Biomed Anal. 2011;55:878–88. doi: 10.1016/j.jpba.2011.03.038. [DOI] [PubMed] [Google Scholar]
- 23.Koren E, Smith HW, Shores E, et al. Recommendations on risk-based strategies for detection and characterization of antibodies against biotechnology products. J Immunol Methods. 2008;333:1–9. doi: 10.1016/j.jim.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 24.Buttel IC, Chamberlain P, Chowers Y, et al. Taking immunogenicity assessment of therapeutic proteins to the next level. Biol J Int Assoc Biol Standard. 2011;39:100–9. doi: 10.1016/j.biologicals.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 25.Shankar G, Arkin S, Cocea L, et al. Assessment and reporting of the clinical immunogenicity of therapeutic proteins and peptides-harmonized terminology and tactical recommendations. Am Assoc Pharm Sci J. 2014;16:658–73. doi: 10.1208/s12248-014-9599-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.26. 2014. Rituxan. US Prescribing information.
- 27.O'Mahony B, Noone D, Giangrande PL, Prihodova L. Haemophilia care in Europe – a survey of 35 countries. Haemophilia. 2013;19:e239–47. doi: 10.1111/hae.12125. [DOI] [PubMed] [Google Scholar]
- 28.West TW, Cree BA. Natalizumab dosage suspension: are we helping or hurting? Ann Neurol. 2010;68:395–9. doi: 10.1002/ana.22163. [DOI] [PubMed] [Google Scholar]
- 29.European Medicines Agency (EMA) 2012. Guideline on the clinical investigation of recombinant and human plasma-derived factor IX products. Doc Ref. EMA/CHMP/BPWP/144552/2009. London, UK: EMA.
- 30.European Medicines Agency (EMA) 2012. Guideline on the clinical investigation of recombinant and plasma-derived FVIII products. Doc Ref. EMA/CHMP/BPWP/144533/2009. London, UK: EMA.
- 31.van Schie KA, Hart MH, de Groot ER, et al. The antibody response against human and chimeric anti-TNF therapeutic antibodies primarily targets the TNF binding region. Ann Rheum Dis. 2015;74:311–4. doi: 10.1136/annrheumdis-2014-206237. [DOI] [PubMed] [Google Scholar]
- 32.Gneiss C, Tripp P, Ehling R, et al. Interferon-beta antibodies have a higher affinity in patients with neutralizing antibodies compared to patients with non-neutralizing antibodies. J Neuroimmunol. 2006;174:174–9. doi: 10.1016/j.jneuroim.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 33.Deisenhammer F, Reindl M, Berger T. Immunoglobulin subclasses in patients with neutralizing and nonneutralizing antibodies against IFN-beta1b. J Interferon Cytokine Res. 2001;21:167–71. doi: 10.1089/107999001750133195. [DOI] [PubMed] [Google Scholar]
- 34.van der Kolk LE, Grillo-Lopez AJ, Baars JW, Hack CE, van Oers MH. Complement activation plays a key role in the side-effects of rituximab treatment. Br J Haematol. 2001;115:807–11. doi: 10.1046/j.1365-2141.2001.03166.x. [DOI] [PubMed] [Google Scholar]
- 35.Ward ES, Ghetie V. The effector functions of immunoglobulins: implications for therapy. Ther Immunol. 1995;2:77–94. [PubMed] [Google Scholar]
- 36.Tatarewicz SM, Mytych DT, Manning MS, Swanson SJ, Moxness MS, Chirmule N. Strategic characterization of anti-drug antibody responses for the assessment of clinical relevance and impact. Bioanalysis. 2014;6:1509–23. doi: 10.4155/bio.14.114. [DOI] [PubMed] [Google Scholar]
- 37.Moorehead PC, Thibeault L, Tuttle A, et al. Rapid acquisition of immunologic tolerance to factor VIII and disappearance of anti-factor VIII IgG4 after prophylactic therapy in a hemophilia A patient with high-titer factor VIII inhibitor. J Pediatr Hematol Oncol. 2015;37:e220–2. doi: 10.1097/MPH.0000000000000287. [DOI] [PubMed] [Google Scholar]
- 38.Lundkvist M, Engdahl E, Holmen C, et al. Characterization of anti-natalizumab antibodies in multiple sclerosis patients. Mult Scler. 2013;19:757–64. doi: 10.1177/1352458512462920. [DOI] [PubMed] [Google Scholar]
- 39.Svenson M, Geborek P, Saxne T, Bendtzen K. Monitoring patients treated with anti-TNF-alpha biopharmaceuticals: assessing serum infliximab and anti-infliximab antibodies. Rheumatology. 2007;46:1828–34. doi: 10.1093/rheumatology/kem261. [DOI] [PubMed] [Google Scholar]
- 40.van Schouwenburg PA, Krieckaert CL, Nurmohamed M, et al. IgG4 production against adalimumab during long term treatment of RA patients. J Clin Immunol. 2012;32:1000–6. doi: 10.1007/s10875-012-9705-0. [DOI] [PubMed] [Google Scholar]
- 41.van Schouwenburg PA, Kruithof S, Votsmeier C, et al. Functional analysis of the anti-adalimumab response using patient-derived monoclonal antibodies. J Biol Chem. 2014;289:34482–8. doi: 10.1074/jbc.M114.615500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gharagozlou S, Sharifian RA, Khoshnoodi J, et al. Epitope specificity of anti-factor VIII antibodies from inhibitor positive acquired and congenital haemophilia A patients using synthetic peptides spanning A and C domains. Thromb Haemost. 2009;101:834–9. [PubMed] [Google Scholar]
- 43.Chen X, Hickling T, Kraynov E, Kuang B, Parng C, Vicini P. A mathematical model of the effect of immunogenicity on therapeutic protein pharmacokinetics. Am Assoc Pharm Sci J. 2013;15:1141–54. doi: 10.1208/s12248-013-9517-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kelley M, Ahene AB, Gorovits B, et al. Theoretical considerations and practical approaches to address the effect of anti-drug antibody (ADA) on quantification of biotherapeutics in circulation. Am Assoc Pharm Sci J. 2013;15:646–58. doi: 10.1208/s12248-013-9468-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vande Casteele N, Gils A, et al. Antibody response to infliximab and its impact on pharmacokinetics can be transient. Am J Gastroenterol. 2013;108:962–71. doi: 10.1038/ajg.2013.12. [DOI] [PubMed] [Google Scholar]
- 46.Wight J, Paisley S. The epidemiology of inhibitors in haemophilia A: a systematic review. Haemophilia. 2003;9:418–35. doi: 10.1046/j.1365-2516.2003.00780.x. [DOI] [PubMed] [Google Scholar]
- 47.Food and Drug Administration (FDA) 2009. FDA guidance for industry: assay development for immunogenicity testing of therapeutic proteins (draft guidance). Silver Spring, MD: FDA.
- 48.Food and Drug Administration (FDA) 2014. Guidance for industry: immunogenicity assessment for therapeutic protein products. Silver Spring, MD: FDA.
- 49.European Medicines Agency (EMA) 2008. Guideline on immunogenicity assessment of biotechnology-derived therapeutic proteins. Doc Ref. EMEA/CHMP/BMWP/14327/2006. London, UK: EMA.
- 50.European Medicines Agency (EMA) 2012. Guideline on immunogenicity assessment of monoclonal antibodies intended for in vivo clinical use. Doc Ref. EMA/CHMP/BMWP/86289/2010. London, UK: EMA.
- 51.Casadevall N, Nataf J, Viron B, et al. Pure red-cell aplasia and antierythropoietin antibodies in patients treated with recombinant erythropoietin. N Engl J Med. 2002;346:469–75. doi: 10.1056/NEJMoa011931. [DOI] [PubMed] [Google Scholar]
- 52.Rougeot C, Marchand P, Dray F, et al. Comparative study of biosynthetic human growth hormone immunogenicity in growth hormone deficient children. Hormone Res. 1991;35:76–81. doi: 10.1159/000181877. [DOI] [PubMed] [Google Scholar]
- 53.Lofgren JA, Dhandapani S, Pennucci JJ, et al. Comparing ELISA and surface plasmon resonance for assessing clinical immunogenicity of panitumumab. J Immunol. 2007;178:7467–72. doi: 10.4049/jimmunol.178.11.7467. [DOI] [PubMed] [Google Scholar]
- 54.Li J, Schantz A, Schwegler M, Shankar G. Detection of low-affinity anti-drug antibodies and improved drug tolerance in immunogenicity testing by Octet((R)) biolayer interferometry. J Pharm Biomed Anal. 2011;54:286–94. doi: 10.1016/j.jpba.2010.08.022. [DOI] [PubMed] [Google Scholar]
- 55.Zhong ZD, Dinnogen S, Hokom M, et al. Identification and inhibition of drug target interference in immunogenicity assays. J Immunol Methods. 2010;355:21–8. doi: 10.1016/j.jim.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 56.Chen K, Page JG, Schwartz AM, et al. False-positive immunogenicity responses are caused by CD20+ B cell membrane fragments in an anti-ofatumumab antibody bridging assay. J Immunol Methods. 2013;394:22–31. doi: 10.1016/j.jim.2013.04.011. [DOI] [PubMed] [Google Scholar]
- 57.Rispens T, de Vrieze H, de Groot E, et al. Antibodies to constant domains of therapeutic monoclonal antibodies: anti-hinge antibodies in immunogenicity testing. J Immunol Methods. 2012;375:93–9. doi: 10.1016/j.jim.2011.09.011. [DOI] [PubMed] [Google Scholar]
- 58.Tatarewicz S, Miller JM, Swanson SJ, Moxness MS. Rheumatoid factor interference in immunogenicity assays for human monoclonal antibody therapeutics. J Immunol Methods. 2010;357:10–6. doi: 10.1016/j.jim.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 59.Bendtzen K. Personalized medicine: theranostics (therapeutics diagnostics) essential for rational use of tumor necrosis factor-alpha antagonists. Discov Med. 2013;15:201–11. [PubMed] [Google Scholar]
- 60.Gorovits B, McNally J, Fiorotti C, Leung S. Protein-based matrix interferences in ligand-binding assays. Bioanalysis. 2014;6:1131–40. doi: 10.4155/bio.14.56. [DOI] [PubMed] [Google Scholar]
- 61.Wang Y-MC, Fang L, Zhou L, Wang J, Ahn H-Y. A survey of applications of biological products for drug interference of immunogenicity assays. Pharm Res. 2012;29:3384–92. doi: 10.1007/s11095-012-0833-2. [DOI] [PubMed] [Google Scholar]
- 62.Llinares-Tello F, Rosas-Gomez de Salazar J, Senabre-Gallego JM, et al. Practical application of acid dissociation in monitoring patients treated with adalimumab. Rheumatol Int. 2014;34:1701–8. doi: 10.1007/s00296-014-3032-0. [DOI] [PubMed] [Google Scholar]
- 63.Patton A, Mullenix MC, Swanson SJ, Koren E. An acid dissociation bridging ELISA for detection of antibodies directed against therapeutic proteins in the presence of antigen. J Immunol Methods. 2005;304:189–95. doi: 10.1016/j.jim.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 64.van Schouwenburg PA, Bartelds GM, Hart MH, Aarden L, Wolbink GJ, Wouters D. A novel method for the detection of antibodies to adalimumab in the presence of drug reveals ‘hidden’ immunogenicity in rheumatoid arthritis patients. J Immunol Methods. 2010;362:82–8. doi: 10.1016/j.jim.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 65.Bloem K, van Leeuwen A, Verbeek G, et al. Systematic comparison of drug-tolerant assays for anti-drug antibodies in a cohort of adalimumab-treated rheumatoid arthritis patients. J Immunol Methods. 2015;418:29–8. doi: 10.1016/j.jim.2015.01.007. [DOI] [PubMed] [Google Scholar]
- 66.Xue L, Fiscella M, Rajadhyaksha M, et al. Pre-existing biotherapeutic-reactive antibodies: survey results within the American Association of Pharmaceutical Scientists. Am Assoc Pharm Sci J. 2013;15:852–5. doi: 10.1208/s12248-013-9492-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xue L, Rup B. Evaluation of pre-existing antibody presence as a risk factor for posttreatment anti-drug antibody induction: analysis of human clinical study data for multiple biotherapeutics. Am Assoc Pharm Sci J. 2013;15:893–6. doi: 10.1208/s12248-013-9497-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Calabresi PA, Giovannoni G, Confavreux C, et al. The incidence and significance of anti-natalizumab antibodies: results from AFFIRM and SENTINEL. Neurology. 2007;69:1391–403. doi: 10.1212/01.wnl.0000277457.17420.b5. [DOI] [PubMed] [Google Scholar]
- 69.Francis GS, Rice GP, Alsop JC, Group PS. Interferon beta-1a in MS: results following development of neutralizing antibodies in PRISMS. Neurology. 2005;65:48–55. doi: 10.1212/01.wnl.0000171748.48188.5b. [DOI] [PubMed] [Google Scholar]
- 70.Lentz SR, Misgav M, Ozelo M, et al. Results from a large multinational clinical trial (guardian1) using prophylactic treatment with turoctocog alfa in adolescent and adult patients with severe haemophilia A: safety and efficacy. Haemophilia. 2013;19:691–7. doi: 10.1111/hae.12159. [DOI] [PubMed] [Google Scholar]
- 71.Baert F, Drobne D, Gils A, et al. Early trough levels and antibodies to infliximab predict safety and success of reinitiation of infliximab therapy. Clin Gastroenterol Hepatol. 2014;12:1474–81 e2. doi: 10.1016/j.cgh.2014.01.033. quiz e91. [DOI] [PubMed] [Google Scholar]
- 72.Wang SL, Ohrmund L, Hauenstein S, et al. Development and validation of a homogeneous mobility shift assay for the measurement of infliximab and antibodies-to-infliximab levels in patient serum. J Immunol Methods. 2012;382:177–88. doi: 10.1016/j.jim.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 73.Bartelds GM, Krieckaert CL, Nurmohamed MT, et al. Development of antidrug antibodies against adalimumab and association with disease activity and treatment failure during long-term follow-up. JAMA. 2011;305:1460–8. doi: 10.1001/jama.2011.406. [DOI] [PubMed] [Google Scholar]
- 74.Chen X, Hickling TP, Vicini P. A mechanistic, multiscale mathematical model of immunogenicity for therapeutic proteins: part 1-theoretical model. Pharmacometrics Syst Pharmacol. 2014;3:e133. doi: 10.1038/psp.2014.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen X, Hickling TP, Vicini P. A mechanistic, multiscale mathematical model of immunogenicity for therapeutic proteins: part 2-model applications. Pharmacometrics Syst Pharmacol. 2014;3:e134. doi: 10.1038/psp.2014.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Perez Ruixo JJ, Ma P, Chow AT. The utility of modeling and simulation approaches to evaluate immunogenicity effect on the therapeutic protein pharmacokinetics. Am Assoc Pharm Sci J. 2013;15:172–82. doi: 10.1208/s12248-012-9424-8. [DOI] [PMC free article] [PubMed] [Google Scholar]