

Abstract
The Fc receptor I for IgA (FcαRI) down-regulates humoral immune responses and modulates the risk of autoimmunity. This study aimed to investigate whether FcαRI targeting can affect progression of pristine-induced lupus nephritis. In the first experiment (early intervention), four groups of animals were evaluated: untreated FcαRI/FcRγ transgenic (Tg) mice and Tg mice administered control antibody (Ctr Fab), saline and anti-FcαRI Fab [macrophage inflammatory protein (MIP)-8a], respectively, three times a week for 29 weeks, after being injected once intraperitoneally with 0·5 ml pristane. In the second experiment, antibody injection started after the onset of nephritis and was carried out for 2 months, with similar groups as described above. MIP-8a improved proteinuria, decreased the amounts of glomerular injury markers, serum interleukin (IL)-6, IL-1 and monocyte chemoattractant protein (MCP)-1, and F4/80 macrophages in the interstitium and glomeruli, in both experiments. When MIP-8a was used as early intervention, a decrease in mouse serum anti-nuclear antibody (ANA) titres and reduced deposition of immunoglobulins in glomeruli were observed. This effect was associated with reduced serum titres of immunoglobulin (Ig)G2a but not IgG1, IgG2b and IgG3. Furthermore, pathological analysis showed lower glomerular activity index and less fibronectin in MIP-8a treated mice. This study suggests that FcαRI targeting could halt disease progression and lupus activation by selective inhibition of cytokine production, leucocyte recruitment and renal inflammation. Our findings provide a basis for the use of FcαRI as a molecular target for the treatment of lupus.
Keywords: IgA Fc receptor I, MIP-8 alpha, molecular targeting, pristane, systemic lupus erythematosus
Introduction
Inflammation is a feature of lupus nephritis (LN), and it is well accepted that the inflammatory process in LN is initiated by the interaction between immunoglobulin (Ig)G-containing immune-complexes and myeloid-derived effect or cells, particularly monocytes/macrophages [1]. This interaction might be mediated by the cellular receptor for the Fc portion of IgG (FcγR). Indeed, FcγRI and III activate inflammatory cells through the immunoreceptor tyrosine-based activation motif (ITAM); FcγRIIB, which contains an immunoreceptor tyrosine-based inhibitory motif (ITIM), acts as an inhibitor of the inflammatory process [2,3].
FcαRI (CD89) is the only Fc receptor specific for IgA expressed on blood myeloid cells including macrophages, monocytes, dendritic cells, Kupffer cells, neutrophils and eosinophils [4]. FcαRI is expressed with or without physical association to the FcγR adaptor, which contains an ITAM. As a unique member of FcR, FcαRI displays a dual role in immune cell inhibition and activation [4,5]. The inhibitory signal is generated when monomeric IgA binds to two FcαRI molecules or when the receptor binds to an anti FcαRI antibody. Partial phosphorylation of FcγR initiates the recruitment of Src-homology 1 (SHP-1), which has inhibitory potential [6]. This leads to deactivation of the inflammatory reaction, therefore preventing the autoimmune process. Previous studies have shown the involvement of FcγR signal activation through the ITAM-containing FcγR adaptor in diseases [7,8]. Indeed, mice deficient in cell surface FcγR develop lupus nephritis and anti-glomerular basement membrane (GBM) antibody-mediated glomerulonephritis [9–11]. Interestingly, monovalent targeting of FcαRI has been reported to effectively induce the development of several immune and non-immune mediated diseases in experimental models, such as anti-GBM-induced glomerulonephritis, unilateral ureteral obstruction (UUO) nephritis and horse spleen apoferritin (HAF)-induced aggressive glomerulonephritis [5,7,12].
The pristane model is a well-established mouse model of systemic lupus erythematosus, in which the symptoms in mice resemble human lupus erythematosus with anti-nuclear antibodies, haemolytic anaemia, proteinuria and glomerulonephritis [13–15]. Pristane is a natural hydrocarbon oil, also called TMPD (2,6,10,14-tetramethylpentadecane). A single injection of pristane leads to typical systemic lupus erythematosus (SLE) autoantibodies and immune complex-mediated glomerulonephritis in non-autoimmune mice, inducing the development of glomerular IgGs and complement deposits, cell proliferation and proteinuria [14–16]. C57BL/6 mice have been reported to develop mesangical glomerulonephritis (class II) after pristane injection, and this model has been used to assess the pathogenic role of unbalanced cytokines such as interferon (IFN)-γ, interleukin (IL)-6 and IL-12 [17–24].
The FcαRI, with ITAM in the cytoplasmic domain, could down-regulate humoral immune responses and modulate the risk for autoimmunity in animal models. In this study, pristine-induced progressive LN autoimmune mice were used to assess whether early and/or late FcαRI targeting during the disease course can affect the progression of renal disease.
We demonstrated that FcαRI targeting could prevent lupus activation by selective inhibition of cytokine production, leucocyte recruitment and renal inflammation.
Materials and methods
Animals
Transgenic (Tg) mice expressing the human FcαRIR209L/FcRγ [12] chimeric protein were generated in mouse facilities of the Research Institute for Diseases of Old Age (Juntendo University School of Medicine, Tokyo, Japan), in a C57BL/6J background with more than eight consecutive crosses. Genotyping was performed by polymerase chain reaction (PCR) analysis of DNA extracted from tail biopsies. As reported previously, there was no difference in phenotype between Tg and wild-type C57BL/6 mice [12]. A total of 32 8-week-old female C57BL/6J Tg mice [body weight (BW): 17·1 ± 0·5 g] were used in this study. The animals were maintained in a specific pathogen-free (SPF) mouse centre at Juntendo University. All animal procedures were conducted in accordance with national guidelines.
Induction of lupus nephritis
According to a previously reported method [14], 8-week-old Tg mice were injected once intraperitoneally (i.p.) with 0·5 ml pristane (2,6,10,14-tetramethylpentadecane; Sigma Chemical Co., St Louis, MO, USA). Proteinuria was monitored by dipstick analysis and urinary albumin was measured by immunoassays (DCA 2000 system; Bayer Diagnostics, Elkhart, IN, USA). Serum was collected before injection and at 1-month intervals after administration.
Early intervention study
Immediately following pristane administration, Tg mice were given control antibody [(AER-37) Fab, ebioscience, San Diego, CA, USA], anti-FcαRI fragment antigen binding (Fab)-macrophage inflammatory protein (MIP-8a) (affinity-purified monoclonal mouse anti human CD89 antibody; AbDSerotec, Oxford, UK) at 20 µg/20 g BW three times/week or normal saline (200 µl, three times/week) for the G4P, G3P or G2P groups, respectively. Another control group included untreated Tg mice (G1P). Mice were killed 7 months after pristane injection and kidneys were processed for light and immunofluorescent microscopy, immunohistochemistry and electron microscopy.
Treatment of established disease
Experiments were designed to treat mice with established lupus nephritis as defined by proteinuria. Proteinuria and body weight were measured twice a week after 0·5 ml pristane i.p. injection. As reported previously, LN in C57BL/6 Tg mice was class II LN; the disease beginning was defined as the appearance of proteinuria [13,14]. In this study, these symptoms were obtained at approximately 6 months. Six months after pristane administration, treatment was performed three times per week for 8 weeks with anti FcαRI mAb (MIP-8a) Fab (G3T), control antibody (G4T) or normal saline (G2T), as described above. Another control group included untreated Tg mice (G1T). Complete remission was defined as UACR 30 mg/g.Cr or less, maintained for 2 weeks. Mice were observed for 2 additional weeks after treatment for proteinuria to determine complete remission before euthanasia.
Biochemical assays
Morning urine samples were collected monthly in every group. Urinary albumin was measured by immunoassay using the DCA 2000 system (Bayer Diagnostics, Elkhart, IN, USA). Blood samples were collected from retro-orbital venous plexus and serum blood urea nitrogen (BUN) and creatinine were determined enzymatically on an autoanalyser (Fuji Dry-Chem 5500; Fujifilm, Tokyo, Japan).
Enzyme-linked immunosorbent assay (ELISA)
Total levels of various immunoglobulin isotypes were determined by ELISA. Plates were coated at 20°C for 2 h with 100 µl of 0·05% Tween and blocked with 1% bovine serum albumin (BSA) in phosphate-buffered saline (PBS). Mouse serum samples were diluted at 1 : 200 000 with 1% BSA in PBS containing 0·05% Tween and added to wells for 2 h at 20°C. Immunoglobulin isotype standards [IgA, total IgG, IgG1, IgG2a IgG2b and IgG3 (Ethyle, Japan)] were used for standard curve. After washing, labelled goat anti-mouse antibodies specific for IgA, total IgG, IgG1, IgG2a, IgG2b and IgG3 were added for 1 h at room temperature. Finally, 3,3′,5,5′-tetramethylbenzidine (TMB) substrate was used for detection at 405 nm.
Then, serum monocyte chemoattractant protein (MCP)-1, IL-6, IL-1beta and tumour necrosis factor (TNF)-alpha levels were determined according to protocols provided by the BD OptEIA™ ELISA kits (mouse MCP-1, cat. 555260; mouse IL-6 cat. 550950; mouse IL-1β, cat. 559603, mouse TNF-alpha cat. 555268).
Finally, anti-nuclear antibody (ANA) titres were determined by a standard protocol using the mouse anti-dsDNA ELISA kit (AKRDD-061; Shibayagi, Gunma, Japan).
Histological and immunohistochemical staining
Kidney tissues were processed immediately for light, immunofluorescence and immunohistochemistry following standard techniques. For light microscopy, kidneys were fixed in 20% formalin, dehydrated and embedded in paraffin, and stained with haematoxylin and eosin (H&E), Masson stain and periodic acid-Schiff. We assessed glomerular activity index by scoring glomeruli modified from Austin et al. and Pirani et al. [25,26] (Table 1).
Table 1.
Activity index of glomeruli
| Activity index score |
|||
|---|---|---|---|
| Variables | 1 | 2 | 3 |
| Cell number | 41–50 | 51–60 | >60 |
| Leucocyte infiltration | 2 | 2–5 | >5 |
| Karyorrhexis (%) | <25 | 25–50 | >50 |
| Fibrinoid (%) | <25 | 25–50 | >50 |
| Wire-loop (%) | <25 | 25–50 | >50 |
| Hyaline thrombi (%) | <25 | 25–50 | >50 |
| Cellular crescents (%) | <25 | 25–50 | >50 |
For direct immunofluorescence, 3-μm cryostat sections were cut and stained with rhodamine-conjugated rabbit anti-mouse IgG, fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgM, FITC-conjugated rabbit anti-mouse IgA, FITC-conjugated rabbit anti-mouse complement 3 (C3) and FITC-conjugated anti-C1q antibodies (ICN, Aurora, OH, USA), respectively, to assess the localization and strength of immunoglobulin and complement deposition. Sections were examined by confocal microscopy (Leica TCS SP8, Berlin, Germany).
Immunohistochemistry detection of fibronectin and F4/80 in renal tissue sections was performed as follows. Cryostat sections were dried and fixed in cold acetone for 10 min. Then, endogenous peroxidase activity was inhibited by addition of methanol containing 0·3% H2O2 for 10 min. Afterwards, non-specific binding was blocked by incubation with the blocking solution [PBS, pH 7·4, containing 2% BSA, 2% fetal calf serum (FCS) and 0·2% fish gelatin] for 30 min. The following primary antibodies were tested: rabbit anti-mouse fibronectin polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and rat anti-mouse F4/80 antigen (MCA497G, 1 : 250; AbD Serotec). We determined the number of positive cells in 10 randomly selected high-power fields (HPF).
Electron microscopy
Electron microscopy (EM) examination was conducted on kidney tissues fixed in 2·5% glutaraldehyde, post-fixed in osmium tetroxide and embedded in Epok 812 (Ouken-syouji Co., Tokyo, Japan). One-μm thick slides were prepared and stained with toluidine blue and selected tissues with glomeruli were sectioned at 70–80 nm, double-stained with uranyl acetate and lead citrate, for observation under a Hitachi 7100 Transmission Electron Microscope (Mito, Honshu, Japan).
Statistical analysis
All statistical analyses were conducted using SPSS version 13·0 (SPSS Inc., Chicago, IL, USA) and SAS version 9·3 (SAS Institute, Cary, NC, USA). Normally distributed data are expressed as means ± standard deviation (s.d.). Statistical significance among groups was evaluated by spss using one-way analysis of variance (anova) with least significant difference (LSD) test for post-hoc analysis. Non-normally distributed data are presented as median and interquartile range (Q25, Q75), and were analysed using the Kruskal–Wallis test with the Bonferroni method for adjusting the alpha threshold [P < 0·017 (0·05/3)]. Statistical significance among time-points was evaluated by sas using repeated-measures anova with pairwise testing using the Tukey's method. If not specified otherwise, statistical significance was indicated by two sided P-values < 0·05.
Results
After treatment, six mice were excluded due to death or infections. The remaining 26 were used in the experiments: G1P and G1T groups contained four mice, respectively; G2P, G2T, G3P, G3T, G4P and G4T contained three mice, respectively.
Effects of early intervention with anti FcαRI monoclonal antibody on markers of lupus associated glomerulonephritis induced by pristane
As shown in Fig. 1a, urinary albumin/creatinine ratio (ACR) was lower in the MIP-8a treatment (G3P) group (22·78 ± 3·54 mg/g creatinine) compared with normal saline treatment (G2P) (28·90 ± 2·99 mg/g creatinine) and control antibody (G4P) (34·00 ± 5·89 mg/g creatinine) groups (both P < 0·05). The same trend was observed with serum BUN levels with 15·5 ± 3·9, 18·8 ± 2·2 and 22·6 ± 3·0 mg/dl observed in the G3P, G2P and G4P groups (Fig. 1b). However, G3P had lower ACR than G2P (P = 0·038). No significant increase was noticed in serum creatinine levels (Fig. 1c). Of note, there was no statistical difference in body weight among groups (data not shown).
Fig. 1.
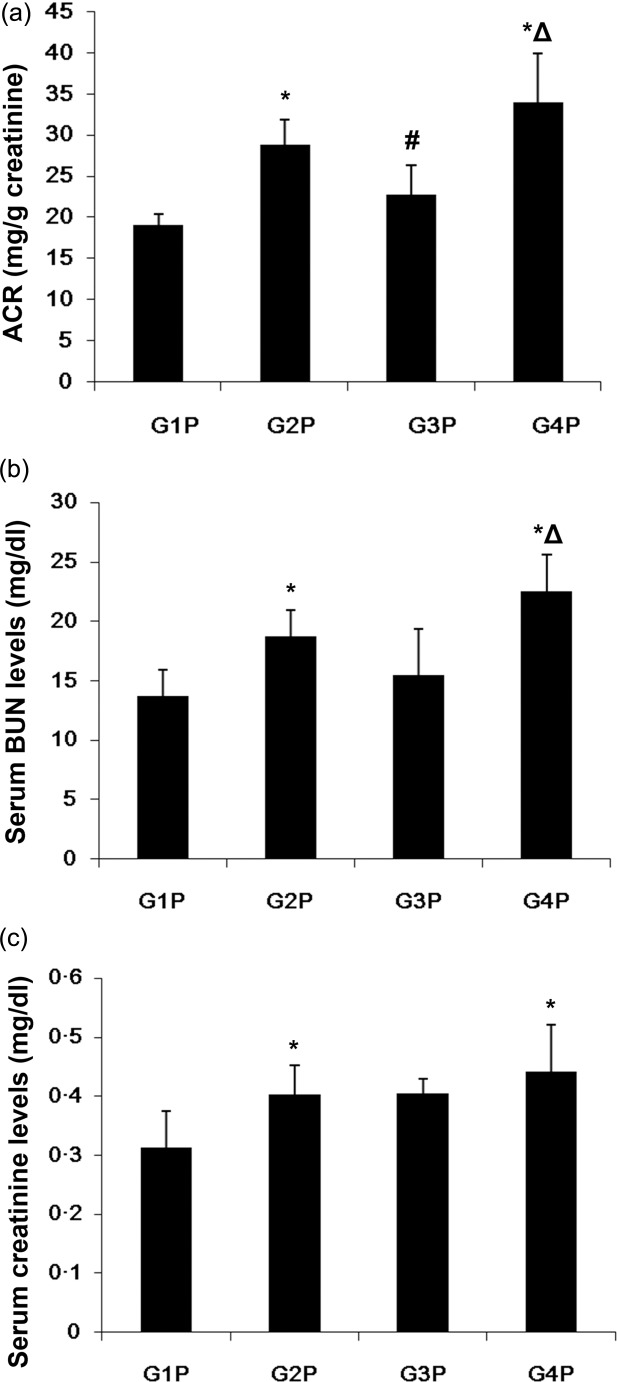
Early monovalent targeting of Fc receptor I for immunoglobulin (Ig)A (FcαRI) decreased the albuminuria and serum blood urea nitrogen (BUN) but not serum creatinine levels in lupus nephritis. (a) Urinary albumin/creatinine ratio (ACR); (b) serum urea nitrogen (BUN); and (c) serum creatinine levels. Data are presented as means ± standard deviation (s.d.) (n = 3 for G2P, G3P, G4P; n = 4 for G1P). *P < 0·05 versus G1P; #P < 0·05 versus G2P; ΔP < 0·05 versus G3P. G1P = transgenic (Tg) mice; G2P = pristane-induced lupus Tg mice treated with normal saline (early intervention); G3P = pristane-induced lupus Tg mice administered macrophage inflammatory protein (MIP)-8a (early intervention); G4P = pristane-induced lupus Tg mice treated with control Fab (early intervention).
Early intervention with anti FcαRI monoclonal antibody inhibits aggressive humoral response in lupus-associated glomerulonephritis induced by pristane
To determine the mechanism underlying the effect of FcαRI targeting, the humoral response was assessed in Tg mice. Serum immunoglobulin production was monitored, with major immunoglobulin isotypes analysed by ELISA. Figure 2a–f displays the time-course of total IgG, IgG1, IgG2a, IgG2b, IgG3 and IgA, respectively, in Tg mouse serum samples after treatment with control Fab (G4P), anti-FcαRI Fab (MIP-8a) (G3P) and normal saline (G2P), or non-pristine-treated Tg mice (G1P). For the control groups treated with normal saline or control antibody, immunoglobulins generally increased with time after pristane injection. Compared with these control groups, MIP-8a treatment resulted in significantly lower serum titres of immunoglobulin IgG2a from the fourth month of pristane injection and IgG3 from the fifth month, but not IgG1, IgG2b and IgA (Fig. 2). Interestingly, ANA titres were lower in the MIP-8a treatment group (16·5 ± 6·2) compared with the saline control (40·5 ± 4·4) and control Fab (40·0 ± 7·5) groups (both P < 0·01) (Fig. 2g).
Fig. 2.
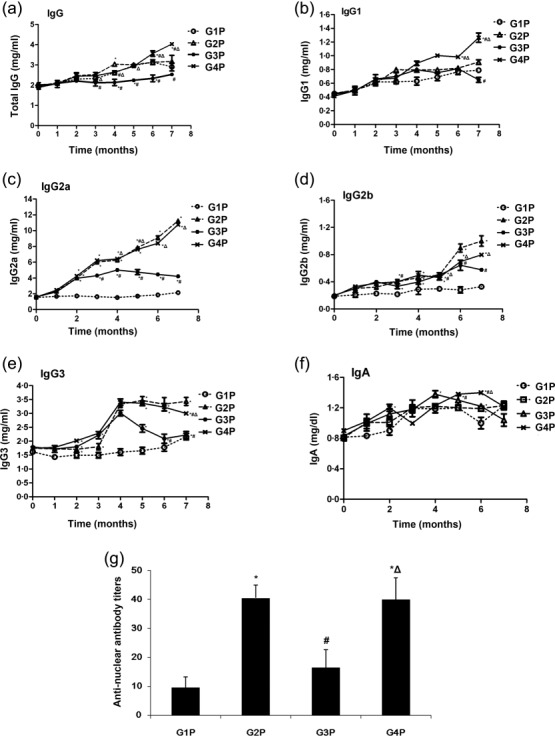
Early intervention with anti-Fc receptor I for immunoglobulin (Ig)A (FcαRI) monoclonal antibody inhibits aggressive humoral response in lupus-associated glomerulonephritis induced by pristane. Serum levels and time–course of total IgG (a), IgG1 (b), IgG2a (c), IgG2b (d), IgG3 (e) and IgA (f), respectively, from the G1P, G2P, G3P and G4P groups. (g) Anti-nuclear antibody titres. Detection was carried out by enzyme-linked inmmunosorbent assay (ELISA). Data represent means ± standard deviation (s.d.) (n = 3 for G2P, G3P, G4P; n = 4 for G1P). *P < 0·05 versus G1P; #P < 0·05 versus G2P; ΔP < 0·05 versus G3P.
Lupus-related serum cytokines are down-regulated after early intervention with anti-FcαRI in lupus-associated glomerulonephritis induced by pristane
Given that cytokines such as IL-6, IL-1β, TNF-α and MCP-1 might play a role in the pathogenesis of autoantibodies in pristine-induced lupus, their levels in peripheral blood of pristine-treated Tg mice were assessed by ELISA. Compared with saline and AER-37 Fab controls that showed significantly increased IL-6 production at 7 months after injection, the MIP-8a group showed a threefold decrease in IL-6 levels (both P < 0·05) (Fig. 3a). Similarly, IL-1β and MCP-1 levels were significantly lower in MIP-8a-treated animals (all P < 0.05) (Fig. 3b,c). Of note, no significant difference in serum levels of TNF-α was observed among the G2P, G3P and G4P groups (Fig. 3d).
Fig. 3.
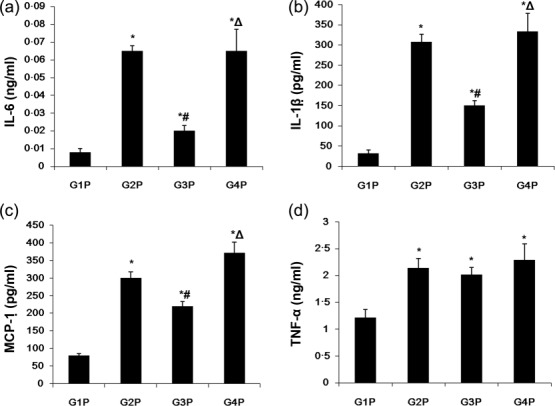
Changes of lupus-related serum cytokines after early intervention with anti-Fc receptor I for immunoglobulin (Ig)A (FcαRI) in lupus-associated glomerulonephritis induced by pristane. Serum levels of interleukin (IL)-6 (a), monocyte chemoattractant protein (MCP)-1 (b), IL-1β (c) and tumour necrosis factor (TNF)-α (d) obtained from transgenic (Tg) mice were determined by enzyme-linked inmmunosorbent assay (ELISA). Data represent means ± standard deviation (s.d.) (n = 3 for G2P, G3P, G4P; n = 4 for G1P). *P < 0·05 versus G1P; #P < 0·05 versus G2P; ΔP < 0·05 versus G3P.
Early intervention with anti-FcαRI monoclonal antibody inhibits renal inflammation and macrophage infiltration in lupus-associated glomerulonephritis induced by pristane
Deposition of circulating autoimmune complexes and complement factors in glomerular tissues may lead to the development of glomerulonephritis in pristine-induced experimental lupus. We therefore investigated whether anti FcαRI Fab therapy plays a role in glomerular immunoglobulin deposition and glomerulonephritis in Tg mice with pristine-induced lupus. As shown in Fig. 4, MIP-8a-treated mice showed reduced glomerular cellular proliferation (P < 0·017; Fig. 4a) and weaker IgG, IgM, IgA, C1q and C3 deposits in glomeruli (P < 0·017; Fig. 4b), compared with controls (G2P and G4P). In addition, fibronectin was hardly observed in Tg mice (G1P), while high accumulations were observed in the glomeruli of pristine-challenged mice treated with saline (G2P) and control Fab (G4P). Interestingly, fibronectin accumulation was decreased in anti FcαRI antibody-treated animals (G3P) compared with the latter control groups (Fig. 4a). A similar trend was observed for renal F4/80-positive cells (P < 0·017; Fig. 4a,d) (both P < 0·017 versus controls). These findings were in agreement with the lower glomerular activity index observed in MIP-8a-treated animals [median (Q25, Q75): 2·00 (1·00, 3·00)] compared with saline [5·00 (1·75, 6·25)] and control Fab [4·00 (2·00, 6·25)] groups, but there was no significant difference (both P > 0·017), as shown in Fig. 4a,c.
Fig. 4.
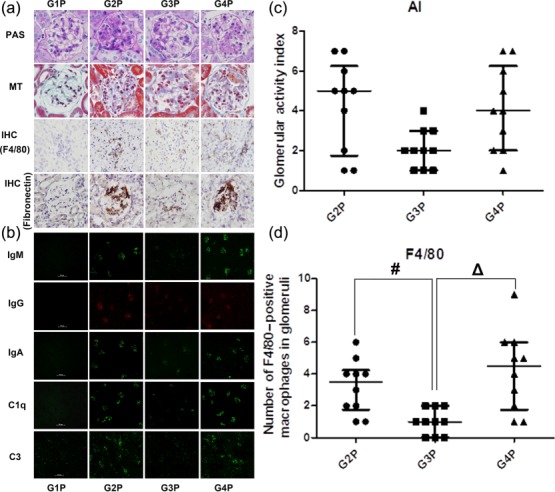
Early intervention with anti-Fc receptor I for immunoglobulin (Ig)A (FcαRI) monoclonal antibody inhibits renal inflammation and macrophage infiltration in lupus-associated glomerulonephritis induced by pristane. (a) Histological analysis [periodic acid-Schiff (PAS) and Masson's trichrome (MT) stain] and immunohistological (IHC) analysis (F4/80 and fibronectin) of kidney sections from transgenic (Tg) mice (magnification: ×400). (b) Mesangial immunoglobulin and complement deposition were observed in the control groups treated with normal saline (G2P) or control antibody (G4P) by fluorescent microscopy (magnification: ×200). Macrophage inflammatory protein (MIP)-8a-treated group (G3P) showed less deposition. Green = fluorescein isothiocyanate (FITC); red = rhodamine. The MIP-8a-treated group (G3P) had a lower glomerular activity index (c) and fewer F4/80-positive macrophages in glomeruli (d). Of note, the glomerular activity index and number of F4/80-positive macrophages in glomeruli obtained for G1P were zero. Non-normally distributed data are presented as the median (Q25, Q75) (n = 3 for G2P, G3P, G4P; n = 4 for G1P), and were analysed using the Kruskal–Wallis test with Bonferroni's method for adjusting the alpha (P < 0·017 is considered statistically significant). #P < 0·017 G3P versus G2P; ΔP < 0·017 G4P versus G3P.
Effects of established lupus treated with anti-FcαRI monoclonal antibody in lupus-associated glomerulonephritis induced by pristane
No significant differences between proteinuria, serum creatinine, ANA titres and IgG levels between MIP-8a and control antibody/saline groups were detected (data not shown). As shown in Fig. 5a, urinary ACR was not different between the MIP-8a and control antibody/saline groups.
Fig. 5.
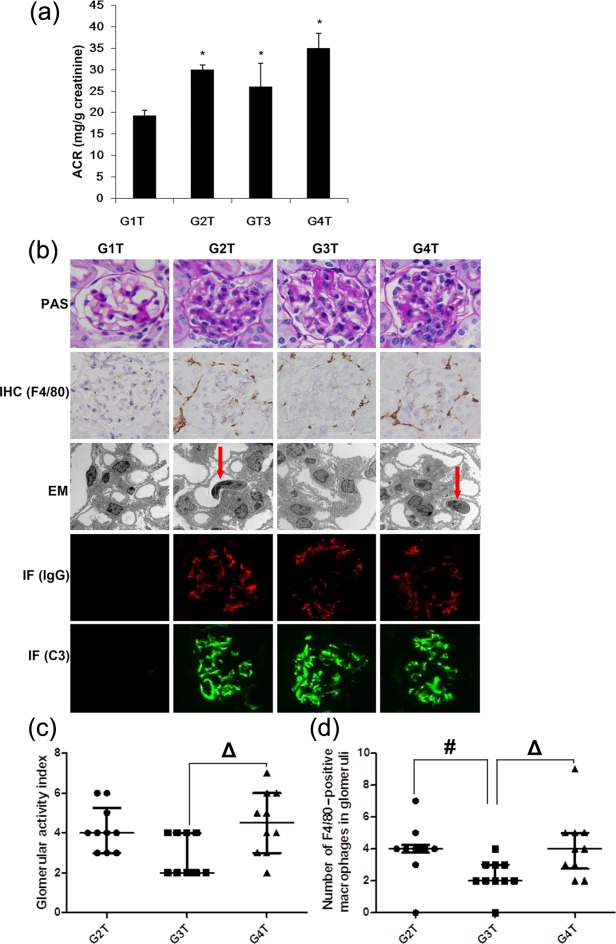
Effects of established lupus treated with anti-Fc receptor I for immunoglobulin (Ig)A (FcαRI) monoclonal antibody in lupus associated glomerulonephritis induced by pristane. (a) Urinary albumin/creatinine ratio (ACR) was determined 34 weeks after pristane administration to determine complete remission. Data are presented as means ± standard deviation (s.d.) (n = 3 for G2T, G3T, G4T; n = 4 for G1T). *P < 0·05 versus G1T. (b) Histological analysis [periodic acid-Schiff (PAS) stain] and immunohistological (IHC) (F4/80) analysis (magnification: ×400) of kidney sections from transgenic mice (Tg) mice. The macrophage inflammatory protein (MIP)-8a-treated group (G3T) had less leucocyte infiltration (arrows) in glomeruli as observed by electron microscopy (EM) examination (magnification: ×3000). Mesangial IgG and C3 deposition were observed by immunofluorescence (IF) staining (magnification: ×400). Green = fluorescein idothiocyanate (FITC); red = phycoerythrin (PE). The MIP-8a-treated group (G3T) had a lower activity index (c) and fewer F4/80-positive macrophages in glomeruli (d). Of note, the glomerular activity index and number of F4/80-positive macrophages in glomeruli obtained for G1P were zero. Non-normally distributed data are presented as the median (Q25, Q75) (n = 3 for G2T, G3T, G4T; n = 4 for G1T), and were analysed using the Kruskal–Wallis test with Bonferroni's method for adjusting the alpha (P < 0·017 is considered statistically significant). #P < 0·017 G3P versus G2P; ΔP < 0·017 G4P versus G3P. G1T = transgenic (Tg) mice; G2T = pristane-induced lupus Tg mice treated with normal saline (treatment of established disease); G3T = pristane-induced lupus Tg mice administered MIP-8a (treatment of established disease); G4T = pristane-induced lupus Tg mice treated with control Fab (treatment of established disease).
However, histological and immunohistochemical analyses showed that MIP-8a-treated animals (G3T) displayed lower glomerular activity index (P < 0·017; Fig. 5b,c), fewer F4/80-positive cells (P < 0·017; Fig. 5b,d) and less leucocyte infiltration in glomeruli, as observed by EM (P < 0·017; Fig. 5b), compared with established lupus treated with normal saline (G2T) and control Fab (G4T). However, no differences were observed in IgG- and C3-positive areas detected by immunofluorescence microscopy (P < 0·017; Fig 5b).
With regard to serum cytokine levels, the MIP-8a-treated animals (G3T) showed lower MCP-1 (Fig. 6a) and IL-6 levels (Fig. 6b) compared with established lupus treated with normal saline (G2T) and control Fab (G4T) (all P < 0·05). Of note, no difference in serum levels of TNF-α was observed among the G2T, G3T and G4T groups (Fig. 6c).
Fig. 6.
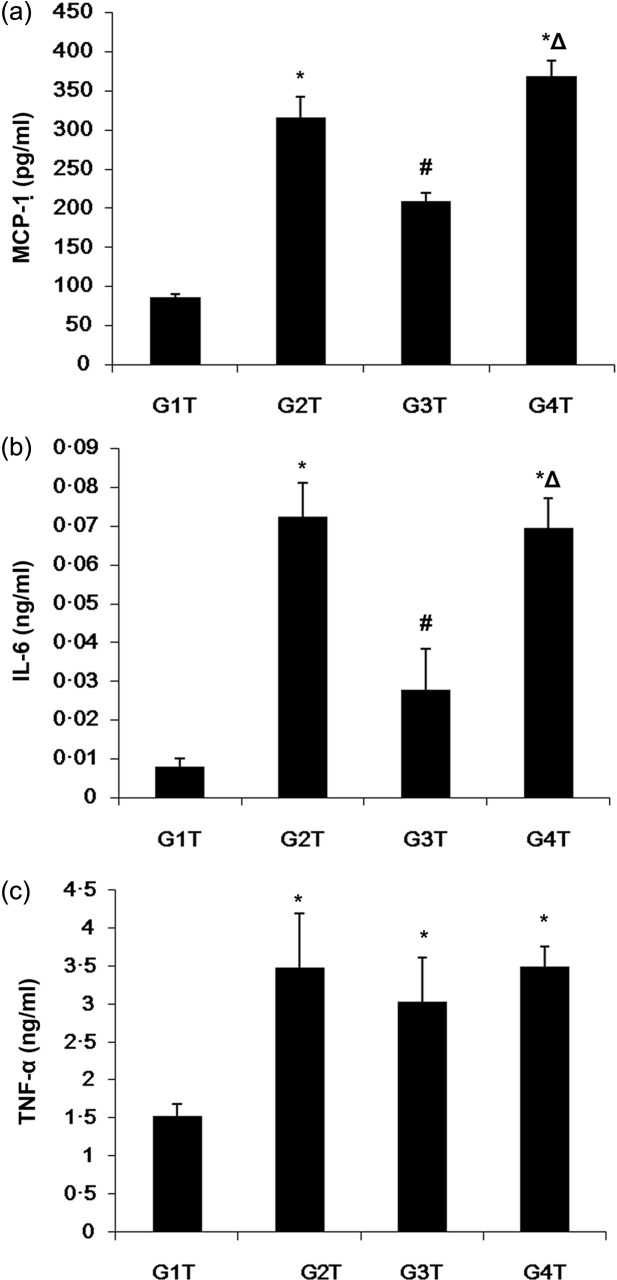
Changes of lupus-related serum cytokines after established lupus treated with anti Fc receptor I for immunoglobulin (Ig)A (FcαRI) monoclonal antibody in lupus-associated glomerulonephritis induced by pristane. Serum levels of monocyte chemoattractant protein (MCP)-1 (a), interleukin (IL)-6 (b) and tumour necrosis factor (TNF)-α (c) obtained from transgenic mice (Tg) as measured by enzyme-linked inmmunosorbent assay (ELISA). Data are presented as means ± standard deviation (s.d.) (n = 3 for G2T, G3T, G4T; n = 4 for G1T). *P < 0·05 versus G1T; #P < 0·05 versus G2T; ΔP < 0·05 versus G3T.
Discussion
A recent study reporting anti FcαRI Fab treatment of Toll-like receptor-9 (TLR) signalling-induced glomerulonephritis has demonstrated that renal inflammation could induce FcαRI inhibitory effects [12]. It is known that TLR-9 plays a key role in the pristane LN model by promoting cytokine production and activating B cells [27]. Therefore, we hypothesized that the inhibitory signal induced by FcαRI could be used to control LN. To test this hypothesis, we used a lupus model in which FcαRI209L/FcRγTg mice were induced by intraperitoneal injection of pristane. We found that early intervention using anti-FcαRI monoclonal antibody inhibited the over-activation of humoral immunity and reduced cytokine production, therefore improving renal lesions. Intervention at late LN stages decreased the influx of inflammatory cells.
The FcαRI209L/FcRγ Tg mouse model expressed the human FcαRI209L/FcRγ chimerical receptor. However, the kidney in chimerical Tg mice are free from natural inflammation, and no soluble FcαRI could be detected in their serum [12]. According to previous reports describing the pristane model, type I interferons, IL-6, IFN-γ, IL-12 and IL-1β promote autoantibody production in TMPD lupus; indeed, a cross-talk was suggested between the IFN-α, -β, -γ and IL-6 signalling pathways [19–23]. For instance, IL-6 is involved in hypergammaglobulinaemia and is critical for the production of anti-DNA and anti-chromatin autoantibodies by pristane-treated mice, mediating inflammation with effects on plasma cell differentiation in pristane models [19,21]. In addition, the increase of serum IL-6 occurs much earlier than the development of autoantibodies [23]. IL-6 deficiency abrogates the induction of IgG anti-DNA and chromatin, and IL-6-deficient mice are highly resistant to renal disease induction [20]. Cytokine transgenic mice that over-express IL-6 produce anti-nuclear antibodies spontaneously [28]. Therefore, the inhibitory effect of anti-FcαRI monoclonal antibody on hypergammaglobulinaemia described in this study may result from the inhibition of IL-6 production. Recent data suggest that IgA may inhibit IL-12 secretion by activated monocytes and dendritic cells and that FcαRI is involved in the process [29]. However, in the present study, IgA levels were unchanged, and further study is necessary to address this point. Our data also supported the hypothesis that inhibition of particular cytokine or shifting cytokine balance may be enough to verify certain type of autoimmunity.
Previous reports assessing the effect of anti FcαRI monoclonal antibody treatment on renal diseases in FcαRI Tg mice have shown its effectiveness in controlling inflammation and fibrosis in the kidney [5,30]. In addition, a recent report showed that anti FcαRI monoclonal antibody treatment of induced acute glomerular nephritis in FcαRI209L/FcRγTg mice displayed not only an anti-inflammatory effect, but also affected immunoglobulin deposition in the glomeruli and complement activation; this inhibitory effect was proposed to result from decreased expression of FcγRIIb and DC-SIGN (dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin) [12]. The data presented herein demonstrated that early intervention could inhibit the over-activated humoral immunity, for instance, by alleviating hypergammaglobulinaemia or decreasing multi-deposition of immunoglobulins and reducing autoantibody titres. These results are consistent with the previously reported inhibitory effect in the HAF-induced acute glomerulonephritis model [12]. Although the effects were not significant for late stage treatment, we speculate that the observed alterations might result from the monotarget therapy, which affects over-activated immune cells through modulation of FcγRIIb expression and cytokine production, as demonstrated by ELISA.
Importantly, MIP-8a treatment resulted in significantly lower serum titres of IgG2a and IgG3 subtypes, but not IgG1, IgG2b and IgA. When we compared our results with the HAF model, in which other phenotypes of immunoglobulin were also decreased in FcαRI targeting, it appeared to be an interesting phenomenon. Indeed, in the horse apoferritin cytosine–guanine–dinucleotide (HAF-CpG)-induced glomerulonephritis, MIP-8a fragment antigen binding (Fab)-treated Tg mice have showed decreased infiltration of CD11b+/F4/80+ macrophages in glomeruli and interstitial tissue, indicating a stark efficacy for MIP-8a Fab against glomerulonephritis [12]. However, we found a different effect of MIP-8a on immunoglobulins: deposition of IgM, IgG2a, IgG1 and C3 disappeared completely after MIP-8a Fab treatment, indicating that different mechanisms exist between both models.
In the present study, activating the IgA/FcαRI pathway had protective effects in a mouse model of LN. However, IgA are involved directly in IgA nephropathy. Indeed, patients with IgA nephropathy often have elevated serum IgA levels and the IgA molecules are often aberrant; these aberrant IgA activate mesangial cells, inducing proliferation and secretion of extracellular matrix and cytokines that result in renal injury [31]. However, a recent study has shown that there is no genetic susceptibility overlapping between lupus and IgA nephropathy [32], suggesting that the two diseases are caused by different mechanisms. However, it is unknown from our results if activating the IgA/FcαRI pathway could have detrimental long-term effects. Further study is necessary on these mechanisms.
The mechanisms leading to the inhibition of pristine-induced nephritis through Fc-alpha-RI is mediated, at least in part, by the extracellular-regulated kinase (ERK) pathway. In the present study, after preincubation with 10% of fetal calf serum for 1, 3, 6 or 12 h, FcαRIR209L/FcRγ transfectant (I3D) from a mouse macrophage cell line (RAW264-7) [12] were treated with serum from pristane models for 5 min; then, p38 and ERK protein expression were determined by Western blot; the results showed that levels of p38 and ERK were increased in the presence of serum from pristane models (data not shown). The inhibition of mitogen-activated protein kinase (MAPK) by FcαRI target has been shown using Western blot, as reported by Kanamaru et al. [7] and Watanabe et al. [12]. In Kanamaru et al.'s study, BALB/c-derived (IgG1) mouse monoclonal antibodies specific for FcαRI (clone A77) and a control antibody (clone 320) were used, while Watanabe et al.'s study used MIP-8a. The inhibition of MAPK by FcαRI target was independent from the stimuli activating the MAPK pathway (especially the ERK pathway), and the inhibition was dose- and time-dependent. Key events in MCP-1 mediated chemotaxis such as p38 and p42–44 ERK MAPK phosphorylation were strongly inhibited. SHP-1 is involved in the mechanism of this inhibition, as the knock-down of SHP-1 significantly reversed FcαRI target inhibition of MCP-1-induced and TNF-α-mediated MAPK activation [7]. Further study is necessary to address the exact mechanisms involved.
A limitation of this study is that treatment effect on the established disease was not assessed over 2 months, to detect potential relapse. Therefore, it is not clear whether prolonged treatment would be needed for the inhibition of hypergammaglobulinaemia and immune deposition in the kidney. The effect of treatment in the late disease stage, which was limited to controlling inflammatory cell infiltration, might be explained by the short treatment time. IgA was assessed as a whole, and no data are available about the different IgA isoforms. In addition, no cross-linking experiment was performed. Finally, we did not inject MIP-8a Fab to wild-type animals, but a previous study has shown that there was no difference between Tg and wild-type animals [12]. Additional studies will need to examine the mechanisms of action of MIP-8a Fab in pristine-induced lupus nephritis.
Overall, these findings indicate that FcαRI targeting could halt disease progression and lupus activation, and provide a basis for the use of FcαRI as a molecular target for the treatment of lupus. However, further experimental and clinical studies are needed to unveil the underlying mechanisms and safety.
Acknowledgments
We thank Ms Shibata for her excellent technical support.
Disclosure
The authors declare that they have no conflicts of interest.
References
- 1.Koffler D, Agnello V, Thoburn R, Kunkel HG. Systemic lupus erythematosus: prototype of immune complex nephritis in man. J Exp Med. 1971;134:169–79. [PMC free article] [PubMed] [Google Scholar]
- 2.Clynes R, Dumitru C, Ravetch JV. Uncoupling of immune complex formation and kidney damage in autoimmune glomerulonephritis. Science. 1998;279:1052–4. doi: 10.1126/science.279.5353.1052. [DOI] [PubMed] [Google Scholar]
- 3.Ravetch JV, Bolland S. IgG Fc receptors. Annu Rev Immunol. 2001;19:275–90. doi: 10.1146/annurev.immunol.19.1.275. [DOI] [PubMed] [Google Scholar]
- 4.Pasquier B, Launay P, Kanamaru Y, et al. Identification of FcalphaRI as an inhibitory receptor that controls inflammation: dual role of FcRgamma ITAM. Immunity. 2005;22:31–42. doi: 10.1016/j.immuni.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 5.Kanamaru Y, Pfirsch S, Aloulou M, et al. Inhibitory ITAM signaling by Fc alpha RI-FcR gamma chain controls multiple activating responses and prevents renal inflammation. J Immunol. 2008;180:2669–78. doi: 10.4049/jimmunol.180.4.2669. [DOI] [PubMed] [Google Scholar]
- 6.Bartoe JL, Nathanson NM. Differential regulation of leukemia inhibitory factor-stimulated neuronal gene expression by protein phosphatases SHP-1 and SHP-2 through mitogen-activated protein kinase-dependent and -independent pathways. J Neurochem. 2000;74:2021–32. doi: 10.1046/j.1471-4159.2000.0742021.x. [DOI] [PubMed] [Google Scholar]
- 7.Kanamaru Y, Arcos-Fajardo M, Moura IC, et al. Fc alpha receptor I activation induces leukocyte recruitment and promotes aggravation of glomerulonephritis through the FcR gamma adaptor. Eur J Immunol. 2007;37:1116–28. doi: 10.1002/eji.200636826. [DOI] [PubMed] [Google Scholar]
- 8.Kanamaru Y, Tamouza H, Pfirsch S, et al. IgA Fc receptor I signals apoptosis through the FcRgamma ITAM and affects tumor growth. Blood. 2007;109:203–11. doi: 10.1182/blood-2006-06-025882. [DOI] [PubMed] [Google Scholar]
- 9.Nakamura A, Yuasa T, Ujike A, et al. Fcgamma receptor IIB-deficient mice develop Goodpasture's syndrome upon immunization with type IV collagen: a novel murine model for autoimmune glomerular basement membrane disease. J Exp Med. 2000;191:899–906. doi: 10.1084/jem.191.5.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bolland S, Ravetch JV. Spontaneous autoimmune disease in Fc(gamma)RIIB-deficient mice results from strain-specific epistasis. Immunity. 2000;13:277–85. doi: 10.1016/s1074-7613(00)00027-3. [DOI] [PubMed] [Google Scholar]
- 11.Bolland S, Yim YS, Tus K, Wakeland EK, Ravetch JV. Genetic modifiers of systemic lupus erythematosus in FcgammaRIIB(-/-) mice. J Exp Med. 2002;195:1167–74. doi: 10.1084/jem.20020165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watanabe T, Kanamaru Y, Liu C, et al. Negative regulation of inflammatory responses by immunoglobulin A receptor (FcalphaRI) inhibits the development of Toll-like receptor-9 signalling-accelerated glomerulonephritis. Clin Exp Immunol. 2011;166:235–50. doi: 10.1111/j.1365-2249.2011.04452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Satoh M, Reeves WH. Induction of lupus-associated autoantibodies in BALB/c mice by intraperitoneal injection of pristane. J Exp Med. 1994;180:2341–6. doi: 10.1084/jem.180.6.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Satoh M, Kumar A, Kanwar YS, Reeves WH. Anti-nuclear antibody production and immune-complex glomerulonephritis in BALB/c mice treated with pristane. Proc Natl Acad Sci USA. 1995;92:10934–8. doi: 10.1073/pnas.92.24.10934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reeves WH, Lee PY, Weinstein JS, Satoh M, Lu L. Induction of autoimmunity by pristane and other naturally occurring hydrocarbons. Trends Immunol. 2009;30:455–64. doi: 10.1016/j.it.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koffler D, Agnello V, Kimkel HG. Polynucleotide immune complexes in serum and glomeruli of patients with systemic lupus erythematosus. Am J Pathol. 1974;74:109–24. [PMC free article] [PubMed] [Google Scholar]
- 17.Kelley VR, Wüthrich RP. Cytokines in the pathogenesis of systemic lupus erythematosus. Semin Nephrol. 1999;19:57–66. [PubMed] [Google Scholar]
- 18.Patten C, Bush K, Rioja I, et al. Characterization of pristane-induced arthritis, a murine model of chronic disease: response to antirheumatic agents, expression of joint cytokines, and immunopathology. Arthritis Rheum. 2004;50:3334–45. doi: 10.1002/art.20507. [DOI] [PubMed] [Google Scholar]
- 19.Finck BK, Chan B, Wofsy D. Interleukin 6 promotes murine lupus in NZB/NZW F1 mice. J Clin Invest. 1994;94:585–91. doi: 10.1172/JCI117373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lattanzio G, Libert C, Aquilina M, et al. Defective development of pristane-oil-induced plasmacytomas in interleukin-6-deficient BALB/c mice. Am J Pathol. 1997;151:689–96. [PMC free article] [PubMed] [Google Scholar]
- 21.Richards HB, Satoh M, Shaw M, Libert C, Poli V, Reeves WH. Interleukin 6 dependence of anti-DNA antibody production: evidence for two pathways of autoantibody formation in pristane-induced lupus. J Exp Med. 1998;188:985–90. doi: 10.1084/jem.188.5.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nacionales DC, Kelly-Scumpia KM, Lee PY, et al. Deficiency of the type I interferon receptor protects mice from experimental lupus. Arthritis Rheum. 2007;56:3770–83. doi: 10.1002/art.23023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Satoh M, Kuroda Y, Yoshida H, et al. Induction of lupus autoantibodies by adjuvants. J Autoimmun. 2003;21:1–9. doi: 10.1016/s0896-8411(03)00083-0. [DOI] [PubMed] [Google Scholar]
- 24.Mitani Y, Takaoka A, Kim SH, et al. Cross talk of the interferon-alpha/beta signalling complex with gp130 for effective interleukin-6 signalling. Genes Cells. 2001;6:631–40. doi: 10.1046/j.1365-2443.2001.00448.x. [DOI] [PubMed] [Google Scholar]
- 25.Austin HA, III, Muenz LR, Joyce KM, et al. Prognostic factors in lupus nephritis. Contribution of renal histologic data. Am J Med. 1983;75:382–91. doi: 10.1016/0002-9343(83)90338-8. [DOI] [PubMed] [Google Scholar]
- 26.Pirani CL, Pollak VE, Schwartz FD. The reproducibility of semiquantitative analyses of renal histology. Nephron. 1964;1:230–7. doi: 10.1159/000179336. [DOI] [PubMed] [Google Scholar]
- 27.Summers SA, Hoi A, Steinmetz OM, et al. TLR9 and TLR4 are required for the development of autoimmunity and lupus nephritis in pristane nephropathy. J Autoimmun. 2010;35:291–8. doi: 10.1016/j.jaut.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 28.Seery JP, Carroll JM, Cattell V, Watt FM. Antinuclear autoantibodies and lupus nephritis in transgenic mice expressing interferon gamma in the epidermis. J Exp Med. 1997;186:1451–9. doi: 10.1084/jem.186.9.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lecocq M, Detry B, Guisset A, Pilette C. FcalphaRI-mediated inhibition of IL-12 production and priming by IFN-gamma of human monocytes and dendritic cells. J Immunol. 2013;190:2362–71. doi: 10.4049/jimmunol.1201128. [DOI] [PubMed] [Google Scholar]
- 30.van der Steen LP, Bakema JE, Sesarman A, et al. Blocking Fcalpha receptor I on granulocytes prevents tissue damage induced by IgA autoantibodies. J Immunol. 2012;189:1594–601. doi: 10.4049/jimmunol.1101763. [DOI] [PubMed] [Google Scholar]
- 31.Suzuki H, Kiryluk K, Novak J, et al. The pathophysiology of IgA nephropathy. J Am Soc Nephrol. 2011;22:1795–803. doi: 10.1681/ASN.2011050464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vuong MT, Gunnarsson I, Lundberg S, et al. Genetic risk factors in lupus nephritis and IgA nephropathy – no support of an overlap. PLOS ONE. 2010;5:e10559. doi: 10.1371/journal.pone.0010559. [DOI] [PMC free article] [PubMed] [Google Scholar]