

Abstract
Increased production of autoantibodies is a characteristic feature of systemic lupus erythematosus (SLE) and there is evidence that several of these autoantibodies may contribute to increased cardiovascular disease (CVD) in SLE. Autoantibodies against the apolipoprotein (apo) B-100 peptides p45 and p210 have been associated with a lower CVD risk in non-SLE cohorts. The aim of the present study was to investigate how SLE affects the occurrence of these potentially protective autoantibodies. The study cohort consisted of 434 SLE patients and 322 age- and sex-matched population controls. Antibodies against native and malondialdehyde (MDA)-modified p45 and p210 were measured by enzyme-linked immunosorbent assay (ELISA). SLE patients had significantly lower levels of p210 immunoglobulin (Ig)G and p45 IgM (both the native and malondialdehyde (MDA)-modified forms). SLE patients with manifest CVD (myocardial infarction, ischaemic cerebrovascular disease or peripheral vascular disease) had lower levels p210 IgG and p45 IgM than SLE patients without CVD. Decreased levels of these autoantibodies were also observed in SLE patients with permanent organ damage, as assessed by the Systemic Lupus International Collaborating Clinics/American College of Rheumatology (ACR) Damage Index (SDI). The present findings show that patients with SLE, a condition generally characterized by abundance of autoantibodies of multiple specificities, have reduced levels of antibodies against the apo B-100 antigens p45 and p210 and that the levels of these antibodies are reduced further in SLE patients with CVD. These observations suggest the possibility that an impaired antibody-mediated removal of damaged LDL particles may contribute to the development of vascular complications and organ damage in SLE.
Keywords: antibodies, human, systemic lupus erythematosus
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune systemic disease predominantly affecting females. It is associated with a high risk for myocardial infarction, overall the risk is 3 −10 fold enhanced compared with age- and sex-matched controls, with the highest relative risks among premenopausal women. This high cardiovascular risk remains after controlling for traditional Framingham cardiovascular risk factors [1–5]. SLE is in most studies characterized by a more aggressive development of atherosclerosis [6,7]. The mechanisms responsible for aggravated cardiovascular disease (CVD) in SLE remains to be fully elucidated but are likely to involve the immune cell activation and loss of tolerance for several self-antigens that characterize the disease [8–10]. This notion is well in line with accumulating evidence from experimental studies of atherosclerosis suggesting that loss of tolerance against oxidized LDL and other plaque antigens play an important role in driving vascular inflammation and plaque development [11–13]. An impaired capacity to clear apoptotic cells and necrotic debris has been implicated as an important factor for the loss of tolerance in both SLE and atherosclerosis [14,15]. Advanced atherosclerotic plaques contain not only significant accumulations of apoptotic cells but also large amounts of oxidized LDL which competes with the binding of apoptotic cell ligands to phagocyte scavenger receptors, further reducing the local efferocytosis capacity [16]. In addition to uptake by scavenger receptors, opsonins such as the complement factors C1q and C3b play a critical role by mediating binding of apoptotic cells to phagocyte endocytic receptors. There is evidence from both experimental and clinical studies that complement deficiency, a result of either genetic deficiency or consumption, is an important factor which contributes to impaired clearance of cellular debris in SLE [17].
One important consequence of the defective efferocytosis and loss of self-tolerance in SLE is the generation of autoantibodies. Antiphospholipid (aPL) antibodies, targeting either cardiolipin (aCL), β2-Glycoprotein I (anti-β2GPI) or a mixture of specificities responsible for positivity in the functional lupus anticoagulant (LA) test, are present in 30–40% of patients with SLE [18]. The pathophysiological role of aPL antibodies in SLE remains to be fully understood but both experimental and clinical studies have suggested that they contribute to the development of vascular events, both arterial and venous [19–23].
Autoantibodies against several types of antigens including phospholipids, -β2GPI, apolipoprotein (apo) B-100, apo A-1 and heat shock proteins, have been implicated in CVD [24].Many of these studies have focused on the role of autoantibodies against different LDL antigens [25]. Experimental and clinical studies of autoantibodies against specific molecular epitopes present in LDL such as phosphatidylcholine and different apo B-100 peptide sequences have suggested a possible protective role for these antibodies [26,27]. The apo B-100 peptides p45 (amino acids 661-680) and p210 (amino acids 3136-3155) have been identified as important targets for immune responses against LDL [28]. Autoantibodies against these LDL antigens are present in most individuals and have been associated with less atherosclerosis as well as with reduced risk for development of myocardial infarction [29–31]. In the present study we analyzed plasma levels of IgG and IgM against native and MDA-modified p45 and p210 in a cohort of SLE patients and matched controls. The result demonstrates that patients with SLE have decreased levels of p45 IgM and p210 IgG autoantibodies and that this is associated with an increased risk of CVD and other organ complications.
Methods
Patients and controls
Patients and controls were included between January 2004 and October 2013. All patients who fulfilled four or more of the 1982 revised American College of Rheumatology (ACR) classification criteria for SLE [32] and who received care for SLE at the Department of Rheumatology, Karolinska University Hospital Solna during this period were asked to participate. Patients were required to be older than 18 years, otherwise there were no exclusion criteria. Population controls, individually matched to the first 322 SLE patients were identified in the population registry. Matching was performed within one year of age, for sex, and region of living. Controls were contacted and asked to participate through a letter. The only exclusion criterion among controls was a diagnosis of SLE. The Local Ethics Committee of the Karolinska University Hospital approved the study protocol. All participants gave informed written consent to participate.
Data collection
All patients and controls were investigated in person by a rheumatologist. Traditional risk factors for CVD were tabulated. Hypertension was defined as a systolic BP> 140 mm Hg and/or a diastolic BP > 90 mm Hg or use of antihypertensive treatment. Diabetes was considered present if patients were previously diagnosed with diabetes. History of vascular events, defined as a history of objectively verified myocardial infarction, ischemic cerebrovascular disease or peripheral vascular disease was obtained though interview and review of medical files. In SLE patients, age at diagnosis, duration of disease, and lupus manifestations including autoantibodies were recorded. Lupus nephritis was defined according to the 1982 revised ACR classification criteria for nephritis [32]. Organ damage was assessed with Systemic Lupus International Collaborating Clinics/ACR Damage index (SDI) [33] All blood samples were taken after overnight fasting and laboratory examinations were performed blinded, either on fresh blood samples or after storage in −70°C.
Intima-media wall thickness measurements
Three hundred and two patients were investigated with carotid ultrasound using a duplex scanner (Acuson 128XP, Mountain View, California, USA) with a 7·0 MHz ART linear array transducer. Left and right carotids were examined. The IM thickness was defined as the distance between the leading edge of the luminal echo to the leading edge of the media/adventitia echo [34]. IM thickness was measured over one cm length just proximal to the bulb. The mean intima-media thickness (IMT) values for both sides were calculated for each subject. One technician recorded all measurements.
Determination of p45, p210 and β2GPI autoantibodies
Peptides corresponding to the amino acids from 661 to 680 (p45; IEIGLEGKGFEPTLEALFGK) and amino acids 3136-3155 (p210; KTTKQSFDLSVKAQYKKNKH) of human apoB-100 were synthesized (KJ Ross Petersen AS, Horsholm, Denmark) and used in ELISA. The peptides were modified by 0·5 M MDA for 3 h at 37°C and dialyzed against PBS containing 1 mM EDTA as described [28]. Native and MDA-modified peptides diluted in PBS pH 7·4 (20 µg/ml) were absorbed to microtiter plate wells (Nunc MaxiSorp, Nunc, Roskilde, Denmark) in an overnight incubation at 4°C. After washing with PBS containing 0·01% Tween-20 (PBS-T) the coated plates were blocked with SuperBlock in TBS (Pierce, Rockford, Illinois) for 30 min at RT followed by an incubation of test plasma, diluted 1/100 in TBS-0·01% Tween-20 (TBS-T) for 2 h at RT and overnight at 4°C. After rinsing, deposition of autoantibodies directed to the peptide was detected using biotinylated rabbit anti-human IgM (ICN, Biomedicals, Inc., Aurora, OH) or IgG antibodies (Abcam, ab 7159) appropriately diluted in TBS-T. After another incubation for 2 h at RT the plates were washed and the bound biotinylated antibodies detected by alkaline phosphatase conjugated streptavidin (BioLegend, 405211), incubated for 2 h at RT. The colour reaction was developed by using phosphatase substrate kit (Pierce) and the absorbance at 405 nm was measured after 1 h of incubation at RT. Values are presented as the ratio against a standard reference plasma. Data regarding the specificity and variability of the antibody ELISA have been published previously [28,30].
Anti-β2GPI antibodies IgM/IgG were determined with the multiplex immunoassays, BioPlex 2200 APLS (Bio-Rad Laboratories Inc., Hercules, CA, USA). Results were reported in the ranges between 1·9-160 U/ml for IgM and 1·9-160 U/ml for IgG. Results were handled as continuous variables. The multiplex assays are regarded as positive if ≥ 20 U/ml. This cut-off level corresponded to at least the 99th percentile of healthy blood donors.
Statistics
Clinical characteristics are presented as median (interquartile range, IQR) for continuous variables and as percentages for categorical variables. Continuous variables that were not normally distributed were log transformed. If not normally distributed after log transformation, non-parametric tests were used. Depending on data type, Students’ t-test, Mann Whitney or Chi square test were used to compare differences between groups. Correlations were investigated through calculating the Spearman rank correlation coefficients. Multivariable-adjusted logistic regression models were performed to evaluate the associations between autoantibodies and cardiovascular/organ damage outcomes. Partial correlations were calculated to determine the associations between autoantibodies and IMT controlling for age and sex.
Results
The clinical characteristics of the SLE patient and control groups are shown in Table 1. Around 90% of the study subjects were females and the median age was just below 50 years. The prevalence of clinically manifest CVD (myocardial infarction, stroke or peripheral artery disease) was 13-fold higher in the SLE group.
Table 1.
Clinical characteristic of systemic lupus erythematosus (SLE) patients and controls
| SLE patients (n = 434) median (IQR)* | Controls (n = 322) median (IQR)* | P-value | |
|---|---|---|---|
| Age (years) | 47·2 (34·2–57·8) | 48·2 (35 ·4–58·6) | n.s. |
| Female sex % | 86 | 92 | 0·01 |
| SLE characteristics | |||
| Number of SLE criteria | 6 (5–7) 17 missing | n.a. | |
| Disease duration year | 10·6 (2·8–20·9) | n.a. | |
| SLICC damage index (SDI) | 1 (IQR: 0–2, range 0–10) | n.a. | |
| Traditional risk factors and laboratory tests | |||
| Current smoking (%) | 18·8 | 14·6 | n.s. |
| Systolic blood pressure (mm Hg) | 120 (110–140) | 120 (110–135) | n.s. |
| Diastolic blood pressure (mm Hg) | 78 (70–80) | 75 (70–82) | n.s. |
| Hypertension treatment (%) | 37·2 | 13·7 | <0·0001 |
| Body mass index (kg/m2) | 24·0 (21·4–27·2) | 24·3 (22·0–27·6) | n.s. |
| Diabetes (%) | 1·4 | 0·9 | n.s. |
| Total cholesterol (mmol/l) | 4·9 (4·2–5·7) | 5·1 (4·4–5·9) | 0·009 |
| High-density lipoprotein (mmol/l) | 1·1 (1·1–1·6) | 1·5 (1·2–1·8) | 0·006 |
| Low-density lipoprotein (mmol/l) | 3·0 (2·5–3·6) | 3·2 (2·6–3·9) | 0·0002 |
| Triglycerides (mmol/l) | 1·0 (0·7–1·5) | 0·78 (0·55–1·10) | <0·0001 |
| Apolipoprotein A1 (mg/ml) | 1·5 (1·3–1·7) | 1·7 (1·4–1·9) | <0·0001 |
| Apolipoprotein B (mg/ml) | 0·81 (0·69–0·96) | 0·81 (0·66–0·97) | n.s. |
| Glucose | 4·8 (4·5–5·2) | 4·9 (4·6–5·2) | n.s. |
| High-sensitivity CRP | 1·7 (0·7–5·3) | 0·9 (0·4–0·9) | <0·0001 |
| Creatinine | 69 (58–84) | 66 (59–73) | 0·0005 |
| Cardiovascular disease | |||
| Cardiovascular event† (%) | 16·1 | 1·2 | <0·0001 |
| Ischaemic heart disease (%) | 6·5 | 0·3 | <0·0001 |
| Ischaemic cerebrovascular disease (%) | 8·7 | 1·6 | <0·0001 |
| Ischaemic peripheral vascular disease (%) | 2·8 | 0·6 | <0·0001 |
| IMT mm (mean of both sides) | 0·053 (0·048–0·063) | n.a. | |
| Treatment (ongoing) | |||
| Prednisolone % | 61·4 | ||
| Anti-malarials % | 37·1 | ||
| Azathioprine % | 17·4 | ||
| Mycophenolate mofetil % | 11·4 |
Distributions are given as median [interquartile range (IQR)] unless indicated otherwise.
Includes myocardial infarction, ischaemic cerebrovascular and peripheral artery disease.
IMT = intima-media thickness; CRP = C-reactive protein; SLIC = Systemic Lupus International Collaborating Clinics; n.a. = not applicable; n.s. = not significant.
SLE patients have lower levels of apo B p45 IgM and p210 IgG
We first studied if there were differences in the expression of autoantibodies against apo B between SLE patients and controls. This was determined by analyzing IgM and IgG antibodies against the native and malondialdehyde (MDA)-modified apo B sequences p45 and p210. Autoantibodies against β2GPI (also called apo H) were used to compare the pattern of apo B peptide autoantibodies with those against another antigen which binds to lipoproteins and to membrane phospholipids. SLE patients had significantly lower levels of p210 IgG and p45 IgM (both the native and MDA-modified forms), while IgM against native and MDA-p210 were increased (Table 2). Antibody levels against native peptides generally correlated strongly with the level of antibodies against the MDA-modified form of the same peptide but much more weakly with antibodies against the other apo B peptide. As an example, the Spearman correlation coefficient for p210 IgG against MDA-p210 IgG was 0·85, while it was only 0·13 and 0·14 for p210 IgG against p45 IgG and MDA-p45 IgG, respectively. Similar trends were also observed for p45 and p210 IgM (data not shown). As expected, SLE patients also had significantly elevated levels of anti-β2GPI IgG and IgM. The levels of both p210 and MDA-p210 IgM correlated with β2-GP-I IgG (r=0·19, P < 0·001 and r=0·18, P < 0·001; respectively) and β2-GP-I IgM levels (r=0·23, P = 0·001 and r=0·21, P < 0·01; respectively). The levels of p45 and MDA-p45 IgM both correlated with β2-GP-I IgM levels (r=0·13, P = 0·001 and r=0·14, P < 0·001; respectively), but otherwise there were no association between autoantibody levels against apo B peptides and anti-β2GPI. None of the common SLE medications in Table 1 were associated with autoantibodies against apo B, with the exception of antimalarials, which were negatively associated with MDA-p45 IgG (P = 0·03).
Table 2.
Apolipoprotein B and β2-glycoprotein-I (GPI) autoantibodies in systemic lupus erythematosus (SLE) patients and controls
| SLE patients (n = 434) median (IQR)* | Controls (n = 322) median (IQR)* | P | |
|---|---|---|---|
| Apolipoprotein B antibodies | |||
| p45 IgM | 0·64 (0·30–1·29) | 0·86 (0·47–1·72) | 0·001 |
| MDA-p45 IgM | 0·72 (0·37–1·43) | 0·92 (0·56–0·92) | 0·001 |
| p45 IgG | 0·49 (0·23–1·03) | 0·42 (0·20–0·90) | n.s. |
| MDA-p45 IgG | 0·52 (0·28–1·02) | 0·43 (0·23–0·95) | n.s. |
| p210 IgM | 0·77 (0·52–1·06) | 0·70 (0·53–0·89) | 0·007 |
| MDA-p210 IgM | 0·87 (0·63–1·03) | 0·79 (0·62–0·93) | 0·002 |
| p210 IgG | 0·48 (0·24–0·84) | 0·54 (0·35–0·89) | 0·02 |
| MDA-p210 IgG | 0·70 (0·51–0·98) | 0·82 (0·61–1·05) | 0·005 |
| β2GPI antibodies | |||
| β2GPI IgM | 1·9 (1·9–3·9) | 1·9 (1·9–2·5) | 0·002 |
| β2GPI IgG | 1·9 (1·9–10·2) | 1·9 (1·9–1·9) | 0·001 |
Distributions are given as median [interquartile range (IQR)].
Ig = immunoglobulin; MDA = malondialdehyde; n.s. = not significant.
Low levels of apo B p45 IgM and p210 IgG are associated with CVD in SLE
Next we compared autoantibody levels against apo B peptides and β2GPI in SLE patients with and without prevalent CVD. CVD patients had lower levels of native and MDAp45 IgM, native p210 IgM, native and MDA-p210 IgG, whereas β2GPI IgG levels were higher. MDA-p45 IgM, MDA-p210 IgG and β2GPI IgG levels remained significantly different when adjusting for age and sex (Table 3 and Fig. 1). To further determine the association between these autoantibodies and cardiovascular disease in patients with SLE we used measurements of carotid IMT as assessed by ultrasonography. Associations were in a negative direction between carotid IMT and all apo B autoantibodies. For p45, both native and MDA modified, IgM antibodies became significant after age and gender adjustments, but these association were generally so weak that the biological relevance is questionable. Crude associations for all p210 antibodies were significant, but these associations were not independent of age and sex (Table 4).
Table 3.
Apolipoprotein B and β2-glycoprotein-I (GPI) autoantibodies in systemic lupus erythematosus (SLE) patients with and without cardiovascular disease
| SLE |
||||
|---|---|---|---|---|
| CVD (n = 62) median (IQR)* | No CVD (n = 370) median (IQR)* | P | P adjusted | |
| Apo B antibodies | ||||
| p45 IgM | 0·47 (0·19–1·17) | 0·70 (0·33–1·29) | 0·01 | n.s. |
| MDA-p45 IgM | 0·55 (0·24–1·02) | 0·75 (0·41–1·48) | 0·003 | 0·04 |
| p45 IgG | 0·36 (0·17–0·90) | 0·54 (0·23–1·05) | n.s. | n.s. |
| MDA-p45 IgG | 0·46 (0·29–0·84) | 0·54 (0·28–1·04) | n.s. | n.s. |
| p210 IgM | 0·64 (0·42–0·99) | 0·78 (0·53–1·07) | 0·03 | n.s. |
| MDA-p210 IgM | 0·80 (0·51–1·02) | 0·54 (0·28–1·04) | n.s. | n.s. |
| p210 IgG | 0·32 (0·10–0·64) | 0·50 (0·25–0·88) | 0·004 | n.s. |
| MDA-p210 IgG | 0·60 (0·3–0·84) | 0·72 (0·53–1·00) | 0·001 | 0·05 |
| Apo H antibodies | ||||
| β2GPI IgM | 1·9 (1·9–3·5) | 1·9 (1·9–4·0) | n.s. | n.s. |
| β2GPI IgG | 4·0 (1·9–30·9) | 1·9 (1·9–8·7) | 0·02 | 0·01 |
Distributions are given as median [interquartile range (IQR)]. Ig = immunoglobulin; MDA = malondialdehyde; CVD = cardiovascular disease; n.s. = not significant.
Fig. 1.
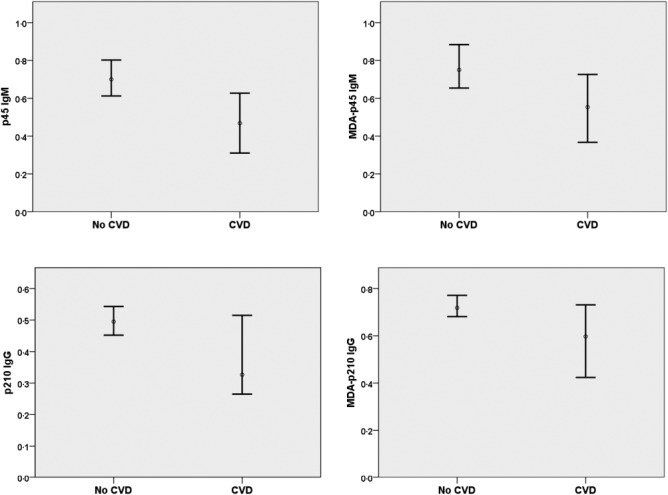
One-dimensional box-plots showing median and 95% confidence interval for p45 immunoglobulin (Ig)M, malondialdehyde (MDA)-p45 IgM, p210 IgG and MDA-p210 IgG in systemic lupus erythematosus (SLE) patients with and without cardiovascular disease (CVD).
Table 4.
Associations between apolipoprotein B and β2-glycoprotein-I (GPI) autoantibodies and carotid intima-media thickness (IMT) in systemic lupus erythematosus (SLE) patients
| SLE patients (n = 302) | P | P adjusted for age and gender | |
|---|---|---|---|
| Apolipoprotein B antibodies | |||
| p45 IgM | −0·05 | n.s. | 0·02 |
| MDA-p45 IgM | −0·08 | n.s. | 0·02 |
| p45 IgG | −0·10 | n.s. | n.s. |
| MDA-p45 IgG | −0·07 | n.s. | n.s. |
| p210 IgM | −0·23 | 0·001 | n.s. |
| MDA-p210 IgM | −0·16 | 0·005 | n.s. |
| p210 IgG | −0·20 | 0·001 | n.s. |
| MDA-p210 IgG | −0·19 | 0·001 | n.s. |
| Apolipoprotein H antibodies | |||
| β2GPI IgM | 0·04 | n.s. | n.s. |
| β2GPI IgG | −0·03 | n.s. | n.s. |
Partial correlations were calculated to determine independent associations between autoantibodies and IMT when controlling for age and sex. Ig = immunoglobulin; MDA = malondialdehyde; n.s. = not significant.
Low levels of apoB autoantibodies are associated with manifestation of organ damage in SLE
Finally we determined if plasma levels of apo B autoantibodies were associated with clinical signs of organ damage in SLE. SLE patients with permanent organ damage (SDI>1) had lower levels of p45 IgM (both native and MDA-modified), p210 IgM and p210 IgG (both native and MDA-modified) than SLE patients with a SDI≤1 (Table 5). When controlling for age and sex only the difference in MDA-p210 IgG remained significantly different between the groups (Table 5). In contrast, SLE patients with permanent organ damage had elevated levels of β2GPI IgG (Table 5).
Table 5.
Apolipoprotein B and β2-glycoprotein-I (GPI) autoantibodies in systemic lupus erythematosus (SLE) patients with and without organ damage (SDI > 1)
| SDI > 1 median (IQR)* | SDI ≤ 1 median (IQR)* | P | P adjusted for age and gender | |
|---|---|---|---|---|
| Apolipoprotein B antibodies | ||||
| p45 IgM | 0·53 (0·24–1·19) | 0·72 (0·36–1·38) | 0·02 | n.s. |
| MDA-p45 IgM | 0·60 (0·28–1·22) | 0·79 (0·44–1·55) | 0·003 | n.s. |
| p45 IgG | 0·48 (0·19–0·91) | 0·50 (0·24–1·04) | n.s. | n.s. |
| MDA-p45 IgG | 0·50 (0·30–0·97) | 0·56 (0·27–1·03) | n.s. | n.s. |
| p210 IgM | 0·68 (0·44–1·02) | 0·83 (0·57–1·07) | 0·006 | n.s. |
| MDA-p210 IgM | 0·80 (0·55–1·02) | 0·89 (0·68–1·05) | n.s. | n.s. |
| p210 IgG | 0·37 (0·16–0·66) | 0·54 (0·28–0·95) | 0·001 | n.s. |
| MDA-p210 IgG | 0·61 (0·41–0·84) | 0·76 (0·56–1·05) | 0·001 | 0·005 |
| Apolipoprotein H antibodies | ||||
| β2GPI IgM | 1·9 (1·9–5·9) | 1·9 (1·9–3·1) | n.s. | n.s. |
| β2GPI IgG | 2·1 (1·9–21·5) | 1·9 (1·9–7·6) | 0·02 | 0·006 |
Distributions are given as median [interquartile range (IQR)].
Ig = immunoglobulin; MDA = malondialdehyde; SDI = Systemic Lupus International Collaborating Clinics damage index; n.s. = not significant.
Since many of the associations between antibody levels and organ complications, including the cardiovascular, were found to be dependent on age we specifically analyzed the relationships between antibody levels and age. All apo B autoantibodies were found to decrease with age both in SLE patients and in controls, while no such trend was observed for β2GPI antibodies (Table 6).
Table 6.
Associations between apolipoprotein B and β2-glycoprotein-I (GPI) autoantibodies and age in systemic lupus erythematosus (SLE) patients and controls
| SLE patients (n = 434) | P | Controls (n = 322) | P | |
|---|---|---|---|---|
| Apolipoprotein B antibodies | ||||
| p45 IgM | −0·19 | 0·001 | −0·24 | 0·001 |
| MDA-p45 IgM | −0·22 | 0·001 | −0·24 | 0·001 |
| p45 IgG | −0·17 | 0·001 | −0·12 | 0·04 |
| MDA-p45 IgG | −0·16 | 0·001 | −0·07 | n.s. |
| p210 IgM | −0·26 | 0·001 | −0·26 | 0·001 |
| MDA-p210 IgM | −0·19 | 0·001 | −0·21 | 0·001 |
| p210 IgG | −0·29 | 0·001 | −0·28 | 0·001 |
| MDA-p210 IgG | −0·30 | 0·001 | −0·27 | 0·001 |
| Apolipoprotein H antibodies | ||||
| β2GPI IgM | 0·07 | n.s. | 0·12 | 0·04 |
| β2GPI IgG | 0·01 | n.s. | 0·09 | n.s. |
Ig = immunoglobulin; MDA = malondialdehyde; n.s. = not significant.
Discussion
Production of a multitude of autoantibodies is a characteristic feature of SLE and there is evidence that several of these autoantibodies, in particular aPL, contribute to increased CVD in SLE [5,20,35,36]. Autoantibodies against the apo B-100 peptides p45 and p210 are found in most individuals and have on the contrary been associated with a lower CVD risk in observational studies [37]. The present study investigated how SLE affects the occurrence of these potentially protective antibodies. Our findings demonstrate that subjects with SLE have reduced levels of p45 IgM and p210 IgG. Moreover, SLE patients with clinically manifest CVD had lower levels of p45 IgM and p210 IgG than those without CVD, but only the levels of MDA-p45 IgM and MDA-p210 IgG remained significantly after controlling for age and sex. One possible explanation for the stronger association with autoantibodies recognizing the MDA-peptides could be that these are more specific for epitopes present in oxidized LDL [38]. Also SLE patients with clinical manifestations of permanent organ damage as assessed by a SDI score >1 had lower levels of MDA-p210 IgG. In comparison, anti- β2GPI IgG levels were increased in SLE patients and those with prevalent CVD had higher levels than those without. Taken together these observations demonstrate that SLE is associated with suppression of a set of naturally occurring autoantibodies with potential protective effects and suggest that this may contribute to increased risk for development of organ damage and CVD in SLE. There is evidence that some medications used to treat SLE also have athero-protective effects [39]. However, we found no association between treatment with prednisolone, azathioprine or mycophenolate mofetil and apo B peptide autoantibodies in the present study while the use of antimalarias were associated with lower levels of MDA-p45 IgG.
There are several mechanisms through which autoantibodies to apo B antigens could protect against atherosclerosis and other types of organ damage in SLE. First, it is likely that such antigens are recognized by the immune system first when LDL is modified by oxidation. This oxidation is associated with degradation of the apo B protein into smaller peptide fragments as well as aldehyde-modifications. Aldehyde-modified apo B peptides are readily identified by the immune system but also non-modified apo B peptide sequences may be targeted by the immune system if normally embedded into phospholipid LDL membrane [38]. Oxidized LDL is cytotoxic for vascular cells and promotes the inflammation that leads to development and destabilization of atherosclerotic plaques [40]. Factors that facilitate an early removal of oxidized LDL are therefore likely to have an athero-protective effect. Both p45 and p210 IgG have been shown to promote the uptake of oxidized LDL in human monocyte/macrophages [41] and treatment of LDLr-/-/human apoB+/+ mice with MDA-p45 IgG lowers the plasma level of oxidized LDL [42]. Oxidized LDL/MDAp45 immune complexes have anti-inflammatory properties through activation of the inhibitory FcγRII receptor [43,44]. Treatment of hypercholesterolemic mice with recombinant malondialdehyde-p45 IgG has been shown to inhibit development of atherosclerosis and to promote plaque regression when combined with lowering of plasma cholesterol levels [41,43]. Low levels of autoantibodies against apo B peptides have been associated with more severe atherosclerosis and an increased risk for development of myocardial infarction [28,29,31,45,46]. Also IgM autoantibodies targeting phosphorylcholine (PC) in oxidized LDL have been attributed a protective role in cardiovascular disease [47]. Interestingly, several studies have shown that subclinical carotid disease in SLE patients is associated with lower levels of these autoantibodies [48,49] adding further support to the notion that autoantibodies against oxidized LDL antigens may protect against cardiovascular complications in SLE. In line with the notion that oxidized LDL contributes to vascular damage in autoimmune disease and that anti-PC antibodies may protect against this damage Ajeganova and coworkers reported that development of cardiovascular events in rheumatoid arthritis is associated with both elevated plasma levels of oxidized LDL and lower levels of anti-PC IgM [50].
β2GPI is an evolutionary conserved protein, which occurs abundantly in the human circulation. The function of β2GPI is still under investigation, but recent data indicate that β2GPI is mainly a scavenger molecule with capacity to bind and remove harmful bacterial products e.g. LPS. It is also involved in clearance of endogenous waste such as micro particles and cellular debris [51]. There is growing evidence that low affinity anti- β2GPI, in similarity to some anti-apoB antibodies, belong to the natural antibody repertoire, which defends us against well-conserved pathogenic structures e.g. bacterial antigens or products of oxidation. In most previous studies anti- β2GPI antibodies are regarded as present or absent according to cut-offs used in the APS criteria [18]. In this study, however, we have in similarity to anti-apoB antibodies investigated continuous titers and isotypes. Our results demonstrate that only anti-β2GPI antibodies of the IgG isotype occurred at higher titers in SLE patients as compared to controls, and high titers are especially common in the SLE subgroup with previous CVD. We also note that, unlike anti-apoB antibodies, anti- β2GPI antibodies do not decline with age, rather in the control group titers were higher among older subjects. Why a subgroup of SLE patients experience a transition from low to high affinity anti-β2GPI, a class switch from IgM to IgG and simultaneously rising levels that become persistent and predispose to thrombotic vascular events [52] is an urgent research question, but beyond the scope of this article. It is nevertheless interesting to note that pathogenic anti-β2GPI and protective anti-apoB antibodies occur with “opposite patterns of occurrence” both in SLE patients and in controls. A possible explanation may be that these antibodies are produced by different subsets of B-cells.
Loss of tolerance against abundant apoptotic cell antigens is an important pathogenic factor in SLE [14]. In atherosclerosis the loss of tolerance against apoptotic cell antigens appears to take place primarily within the environment of the atherosclerotic plaque where there is a similar loss of tolerance against antigens in oxidized LDL [15,53]. Taken together, these findings imply that the issue of tolerance control may be particularly critical in SLE atherosclerotic lesions. Oxidized LDL is enhanced in SLE [54] and may further aggravate pro-inflammatory responses to apoptotic cells in SLE atherosclerotic lesions by competing with the binding to phagocytic receptors, which mediate clearance of both apoptotic cells and oxidized LDL. It is likely that these mechanisms play a role in the accelerated atherosclerosis in SLE and that antibody-mediated removal of oxidized LDL could help to limit vascular and possibly also the general systemic inflammation in SLE.
There are some limitations of the present study that should be considered. Most importantly, the observational design of the study does not allow for conclusions regarding the functional role of p45 and p210 apo B-100 autoantibodies in SLE. Moreover, we only report retrospective data for the association between these antibodies and cardiovascular complications in CVD and the findings need to be further validated in prospective studies. The circumstance that both CVD and apo B-100 peptide autoantibodies depend on age also make the interpretation of the results more complex. However, although the statistical significance for the difference in apo B-100 peptide autoantibodies between SLE patients with and without CVD is weakened when adjusting for age and sex it still remained significant for MDA-p45 IgM and MDA-p210 IgG. Finally we did not analyze the biological target for apo B-100 peptide autoantibodies, e.g. oxidized LDL, in the present study. Previous studies have shown inverse associations between apo B peptide autoantibodies and the level of oxidized LDL in plasma suggesting that these antibodies may participate in the clearance of modified LDL particles [28]. It will be of considerable interest for future studies to determine if this is the case also in patients with SLE.
In conclusion, we report that patients with SLE, a condition generally characterized by a high production of autoantibodies, have reduced levels of athero-protective autoantibodies against the apo B-100 peptides p45 and p210. The level of these antibodies was further reduced in SLE patients that had developed CVD. We propose that an impaired antibody-mediated removal of oxidized LDL may contribute to loss of tolerance and increased inflammation in vascular tissues in SLE.
Acknowledgments
We are grateful to Eva Jemseby, Gull-Britt Almgren, Julia Boström and Gloria Rostvall for management of blood samples and to Anna-Britta Johansson for determination of anti-β2GPI antibodies. This work was supported by grants from the Swedish Research Council; the Swedish Heart-Lung foundation; the Swedish Foundation for Strategic Research and Skåne University Hospital Foundation, the Albert Påhlsson Foundation, the Swedish Rheumatism Association, the King Gustaf V 80th Birthday Fund,the Swedish Society of Medicine, the Åke Wiberg Foundation, the Clas Groschinsky Foundation, the Karolinska Institutet Foundations and ALF funding from Stockholm County Council and Karolinska Institutet.
Disclosures
Jan Nilsson is signed as co-inventor on patents describing immune-based therapies for atherosclerosis. The rights for these patents have been assigned to Forskarpatent, Lund, Sweden.
References
- 1.Esdaile JM, Abrahamowicz M, Grodzicky T, et al. Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis Rheum. 2001;44:2331–7. doi: 10.1002/1529-0131(200110)44:10<2331::aid-art395>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 2.Rahman P, Urowitz MB, Gladman DD, Bruce IN, Genest J., Jr Contribution of traditional risk factors to coronary artery disease in patients with systemic lupus erythematosus. J Rheumatol. 1999;26:2363–8. [PubMed] [Google Scholar]
- 3.Manzi S, Selzer F, Sutton-Tyrrell K, et al. Prevalence and risk factors of carotid plaque in women with systemic lupus erythematosus. Arthritis Rheum. 1999;42:51–60. doi: 10.1002/1529-0131(199901)42:1<51::AID-ANR7>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 4.Bengtsson C, Ohman ML, Nived O, Dahlqvist SR. Cardiovascular event in systemic lupus erythematosus in northern Sweden: incidence and predictors in a 7-year follow-up study. Lupus. 2012;21:452–9. doi: 10.1177/0961203311425524. [DOI] [PubMed] [Google Scholar]
- 5.Magder LS, Petri M. Incidence of and risk factors for adverse cardiovascular events among patients with systemic lupus erythematosus. Am J Epidemiol. 2012;176:708–19. doi: 10.1093/aje/kws130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Asanuma Y, Oeser A, Shintani AK, et al. Premature coronary-artery atherosclerosis in systemic lupus erythematosus. N Engl J Med. 2003;349:2407–15. doi: 10.1056/NEJMoa035611. [DOI] [PubMed] [Google Scholar]
- 7.Yiu KH, Wang S, Mok MY, et al. Pattern of arterial calcification in patients with systemic lupus erythematosus. J Rheumatol. 2009;36:2212–7. doi: 10.3899/jrheum.090312. [DOI] [PubMed] [Google Scholar]
- 8.Skaggs BJ, Hahn BH, McMahon M. Accelerated atherosclerosis in patients with SLE – mechanisms and management. Nat Rev Rheumatol. 2012;8:214–23. doi: 10.1038/nrrheum.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kahlenberg JM, Kaplan MJ. Mechanisms of premature atherosclerosis in rheumatoid arthritis and lupus. Annu Rev Med. 2013;64:249–63. doi: 10.1146/annurev-med-060911-090007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knight JS, Kaplan MJ. Cardiovascular disease in lupus: insights and updates. Curr Opin Rheumatol. 2013;25:597–605. doi: 10.1097/BOR.0b013e328363eba3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nilsson J, Hansson GK. Autoimmunity in atherosclerosis: a protective response losing control? J Intern Med. 2008;263:464–78. doi: 10.1111/j.1365-2796.2008.01945.x. [DOI] [PubMed] [Google Scholar]
- 12.Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12:204–12. doi: 10.1038/ni.2001. [DOI] [PubMed] [Google Scholar]
- 13.Lichtman AH, Binder CJ, Tsimikas S, Witztum JL. Adaptive immunity in atherogenesis: new insights and therapeutic approaches. J Clin Invest. 2013;123:27–36. doi: 10.1172/JCI63108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shao WH, Cohen PL. Disturbances of apoptotic cell clearance in systemic lupus erythematosus. Arthritis Res Ther. 2011;13:202. doi: 10.1186/ar3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Vre EA, Ait-Oufella H, Tedgui A, Mallat Z. Apoptotic cell death and efferocytosis in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:887–93. doi: 10.1161/ATVBAHA.111.224873. [DOI] [PubMed] [Google Scholar]
- 16.Schrijvers DM, De Meyer GR, Kockx MM, Herman AG, Martinet W. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler Thromb Vasc Biol. 2005;25:1256–61. doi: 10.1161/01.ATV.0000166517.18801.a7. [DOI] [PubMed] [Google Scholar]
- 17.Colonna L, Lood C, Elkon KB. Beyond apoptosis in lupus. Curr Opin Rheumatol. 2014;26:459–466. doi: 10.1097/BOR.0000000000000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS) J Thromb Haemost. 2006;4:295–306. doi: 10.1111/j.1538-7836.2006.01753.x. [DOI] [PubMed] [Google Scholar]
- 19.Svenungsson E, Jensen-Urstad K, Heimburger M, et al. Risk factors for cardiovascular disease in systemic lupus erythematosus. Circulation. 2001;104:1887–93. doi: 10.1161/hc4101.097518. [DOI] [PubMed] [Google Scholar]
- 20.Gustafsson J, Gunnarsson I, Borjesson O, Pettersson S, Moller S, Fei GZ, Elvin K, Simard JF, Hansson LO, Lundberg IE, Larsson A. Svenungsson E. Predictors of the first cardiovascular event in patients with systemic lupus erythematosus – a prospective cohort study. Arthritis Res Ther. 2009;11:R186. doi: 10.1186/ar2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simantov R, LaSala JM, Lo SK, et al. Activation of cultured vascular endothelial cells by antiphospholipid antibodies. J Clin Invest. 1995;96:2211–9. doi: 10.1172/JCI118276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gharavi AE, Pierangeli SS, Espinola RG, Liu X, Colden-Stanfield M, Harris EN. Antiphospholipid antibodies induced in mice by immunization with a cytomegalovirus-derived peptide cause thrombosis and activation of endothelial cells in vivo. Arthritis Rheum. 2002;46:545–52. doi: 10.1002/art.10130. [DOI] [PubMed] [Google Scholar]
- 23.Alarcon-Segovia D, Deleze M, Oria CV, et al. Antiphospholipid antibodies and the antiphospholipid syndrome in systemic lupus erythematosus. A prospective analysis of 500 consecutive patients. Medicine. 1989;68:353–65. doi: 10.1097/00005792-198911000-00003. [DOI] [PubMed] [Google Scholar]
- 24.Roux-Lombard P, Pagano S, Montecucco F, Satta N, Vuilleumier N. Auto-antibodies as emergent prognostic markers and possible mediators of ischemic cardiovascular diseases. Clin Rev Allergy Immunol. 2013;44:84–97. doi: 10.1007/s12016-010-8233-z. [DOI] [PubMed] [Google Scholar]
- 25.Hulthe J. Antibodies to oxidized LDL in atherosclerosis development–clinical and animal studies. Clin Chim Acta. 2004;348:1–8. doi: 10.1016/j.cccn.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 26.Binder CJ, Shaw PX, Chang MK, et al. The role of natural antibodies in atherogenesis. J Lipid Res. 2005;46:1353–63. doi: 10.1194/jlr.R500005-JLR200. [DOI] [PubMed] [Google Scholar]
- 27.Nilsson J, Nordin Fredrikson G, Schiopu A, Shah PK, Jansson B, Carlsson R. Oxidized LDL antibodies in treatment and risk assessment of atherosclerosis and associated cardiovascular disease. Curr Pharm Des. 2007;13:1021–30. doi: 10.2174/138161207780487557. [DOI] [PubMed] [Google Scholar]
- 28.Fredrikson GN, Hedblad B, Berglund G, et al. Identification of immune responses against aldehyde-modified peptide sequences in apo B-100 associated with cardiovascular disease. Arterioscler Thromb Vasc Biol. 2003;23:872–78. doi: 10.1161/01.ATV.0000067935.02679.B0. [DOI] [PubMed] [Google Scholar]
- 29.Fredrikson GN, Anand DV, Hopkins D, et al. Associations between autoantibodies against apolipoprotein B-100 peptides and vascular complications in patients with type 2 diabetes. Diabetologia. 2009;52:1426–33. doi: 10.1007/s00125-009-1377-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fredrikson GN, Schiopu A, Berglund G, Alm R, Shah PK, Nilsson J. Autoantibody against the amino acid sequence 661-680 in apo B-100 is associated with decreased carotid stenosis and cardiovascular events. Atherosclerosis. 2007;194:e188–92. doi: 10.1016/j.atherosclerosis.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 31.Sjogren P, Fredrikson GN, Samnegard A, et al. High plasma concentrations of autoantibodies against native peptide 210 of apoB-100 are related to less coronary atherosclerosis and lower risk of myocardial infarction. Eur Heart J. 2008;29:2218–26. doi: 10.1093/eurheartj/ehn336. [DOI] [PubMed] [Google Scholar]
- 32.Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271–7. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 33.Gladman D, Ginzler E, Goldsmith C, et al. The development and initial validation of the Systemic Lupus International Collaborating Clinics/American College of Rheumatology damage index for systemic lupus erythematosus. Arthritis Rheum. 1996;39:363–9. doi: 10.1002/art.1780390303. [DOI] [PubMed] [Google Scholar]
- 34.Wikstrand J, Wendelhag I. Methodological considerations of ultrasound investigation of intima-media thickness and lumen diameter. J Intern Med. 1994;236:555–9. doi: 10.1111/j.1365-2796.1994.tb00845.x. [DOI] [PubMed] [Google Scholar]
- 35.Narshi CB, Giles IP, Rahman A. The endothelium: an interface between autoimmunity and atherosclerosis in systemic lupus erythematosus? Lupus. 2011;20:5–13. doi: 10.1177/0961203310382429. [DOI] [PubMed] [Google Scholar]
- 36.Gustafsson JT, Simard JF, Gunnarsson I, et al. Risk factors for cardiovascular mortality in patients with systemic lupus erythematosus, a prospective cohort study. Arthritis Res Ther. 2012;14:R46. doi: 10.1186/ar3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bjorkbacka H, Fredrikson GN, Nilsson J. Emerging biomarkers and intervention targets for immune-modulation of atherosclerosis – a review of the experimental evidence. Atherosclerosis. 2013;227:9–17. doi: 10.1016/j.atherosclerosis.2012.10.074. [DOI] [PubMed] [Google Scholar]
- 38.Palinski W, Witztum JL. Immune responses to oxidative neoepitopes on LDL and phospholipids modulate the development of atherosclerosis. J Intern Med. 2000;247:371–80. doi: 10.1046/j.1365-2796.2000.00656.x. [DOI] [PubMed] [Google Scholar]
- 39.Iaccarino L, Bettio S, Zen M, et al. Premature coronary heart disease in SLE: can we prevent progression? Lupus. 2013;22:1232–42. doi: 10.1177/0961203313492871. [DOI] [PubMed] [Google Scholar]
- 40.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–95. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 41.Schiopu A, Bengtsson J, Soderberg I, et al. Recombinant human antibodies against aldehyde-modified apolipoprotein B-100 peptide sequences inhibit atherosclerosis. Circulation. 2004;110:2047–52. doi: 10.1161/01.CIR.0000143162.56057.B5. [DOI] [PubMed] [Google Scholar]
- 42.Strom A, Fredrikson GN, Schiopu A, et al. Inhibition of injury-induced arterial remodelling and carotid atherosclerosis by recombinant human antibodies against aldehyde-modified apoB-100. Atherosclerosis. 2006;190:298–305. doi: 10.1016/j.atherosclerosis.2006.03.032. [DOI] [PubMed] [Google Scholar]
- 43.Schiopu A, Frendeus B, Jansson B, et al. Recombinant antibodies to an oxidized low-density lipoprotein epitope induce rapid regression of atherosclerosis in apobec-1(-/-)/low-density lipoprotein receptor(-/-) mice. J Am Coll Cardiol. 2007;50:2313–8. doi: 10.1016/j.jacc.2007.07.081. [DOI] [PubMed] [Google Scholar]
- 44.Li S, Kievit P, Robertson AK, et al. Targeting oxidized LDL improves insulin sensitivity and immune cell function in obese Rhesus macaques. Mol Metab. 2013;2:256–69. doi: 10.1016/j.molmet.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fredrikson GN, Schiopu A, Berglund G, Alm R, Shah P, Nilsson J. Autoantibody against the amino acid sequence 661-680 in apoB-100 is associated with decreased carotid stenosis and cardiovascular events. Atherosclerosis. 2007;194:e188–92. doi: 10.1016/j.atherosclerosis.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 46.Engelbertsen D, Anand DV, Fredrikson GN, et al. High levels of IgM against methylglyoxal-modified apolipoprotein B100 are associated with less coronary artery calcification in patients with type 2 diabetes. J Intern Med. 2012;271:82–9. doi: 10.1111/j.1365-2796.2011.02411.x. [DOI] [PubMed] [Google Scholar]
- 47.Tsiantoulas D, Diehl CJ, Witztum JL, Binder CJ. B cells and humoral immunity in atherosclerosis. Circ Res. 2014;114:1743–56. doi: 10.1161/CIRCRESAHA.113.301145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gronwall C, Reynolds H, Kim JK, et al. Relation of carotid plaque with natural IgM antibodies in patients with systemic lupus erythematosus. Clin Immunol. 2014;153:1–7. doi: 10.1016/j.clim.2014.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anania C, Gustafsson T, Hua X, et al. Increased prevalence of vulnerable atherosclerotic plaques and low levels of natural IgM antibodies against phosphorylcholine in patients with systemic lupus erythematosus. Arthritis Res Ther. 2010;12:R214. doi: 10.1186/ar3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ajeganova S, de Faire U, Jogestrand T, Frostegard J, Hafstrom I. Carotid atherosclerosis, disease measures, oxidized low-density lipoproteins, and atheroprotective natural antibodies for cardiovascular disease in early rheumatoid arthritis – an inception cohort study. J Rheumatol. 2012;39:1146–54. doi: 10.3899/jrheum.111334. [DOI] [PubMed] [Google Scholar]
- 51.de Groot PG, Meijers JC. beta(2) -Glycoprotein I: evolution, structure and function. J Thromb Haemost. 2011;9:1275–84. doi: 10.1111/j.1538-7836.2011.04327.x. [DOI] [PubMed] [Google Scholar]
- 52.Vikerfors A, Johansson AB, Gustafsson JT, et al. Clinical manifestations and anti-phospholipid antibodies in 712 patients with systemic lupus erythematosus: evaluation of two diagnostic assays. Rheumatology (Oxf) 2013;52:501–9. doi: 10.1093/rheumatology/kes252. [DOI] [PubMed] [Google Scholar]
- 53.Stemme S, Faber B, Holm J, Wiklund O, Witztum JL, Hansson GK. T lymphocytes from human atherosclerotic plaques recognize oxidized low density lipoprotein. Proc Natl Acad Sci USA. 1995;92:3893–7. doi: 10.1073/pnas.92.9.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Frostegard J, Svenungsson E, Wu R, et al. Lipid peroxidation is enhanced in patients with systemic lupus erythematosus and is associated with arterial and renal disease manifestations. Arthritis Rheum. 2005;52:192–200. doi: 10.1002/art.20780. [DOI] [PubMed] [Google Scholar]