

Abstract
The interaction between neutrophils and activation of alternative complement pathway plays a pivotal role in the pathogenesis of anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV). ANCAs activate primed neutrophils to release neutrophil extracellular traps (NETs), which have recently gathered increasing attention in the development of AAV. The relationship between NETs and alternative complement pathway has not been elucidated. The current study aimed to investigate the relationship between NETs and alternative complement pathway. Detection of components of alternative complement pathway on NETs in vitro was assessed by immunostain and confocal microscopy. Complement deposition on NETs were detected after incubation with magnesium salt ethyleneglycol tetraacetic acid (Mg-EGTA)-treated human serum. After incubation of serum with supernatants enriched in ANCA-induced NETs, levels of complement components in supernatants were measured by enzyme-linked immunosorbent assay (ELISA). Complement factor B (Bb) and properdin deposited on NETs in vitro. The deposition of C3b and C5b-9 on NETs incubated with heat-inactivated normal human serum (Hi-NHS) or EGTA-treated Hi-NHS (Mg-EGTA-Hi-NHS) were significantly less than that on NETs incubated with NHS or EGTA-treated NHS (Mg-EGTA-NHS). NETs induced by ANCA could activate the alternative complement cascade in the serum. In the presence of EGTA, C3a, C5a and SC5b-9 concentration decreased from 800·42 ± 244·81 ng/ml, 7·68 ± 1·50 ng/ml, 382·15 ± 159·75 ng/ml in the supernatants enriched in ANCA induced NETs to 479·07 ± 156·2 ng/ml, 4·86 ± 1·26 ng/ml, 212·65 ± 44·40 ng/ml in the supernatants of DNase I-degraded NETs (P < 0·001, P = 0·008, P < 0·001, respectively). NETs could activate the alternative complement pathway, and might thus participate in the pathogenesis of AAV
Keywords: alternative complement pathway, ANCA, complement, neutrophil extracellular traps, vasculitis
Introduction
Anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) comprises granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA) and eosinophilic granulomatosis with polyangiitis (EGPA) [1]. AAV is a group of potentially life-threatening systemic autoimmune diseases characterized by necrotizing small-vessel vasculitis and frequently affects the kidneys [2]. As the serological hallmarks for AAV, ANCAs are predominantly IgG autoantibodies directed against neutrophil cytoplasmic constituents, in particular proteinase 3 (PR3) and myeloperoxidase (MPO) [1].
Neutrophils and their interaction with ANCAs play a critical role in the pathogenesis of AAV. In vitro, ANCAs activate primed neutrophils to undergo a respiratory burst and degranulation [3]. Recently, neutrophil extracellular traps (NETs) have gathered increasing attention in the pathogenesis of AAV. Kessenbrock et al. found that ANCA-stimulated neutrophils can induce the formation of NETs, which contain the ANCA target antigens, including MPO and PR3 [4]. In the renal tissue of AAV patients, NETs are located in close proximity to neutrophil infiltrates in affected glomeruli and interstitium [4]. It has also been reported that NETs can enhance the uptake of PR3 and MPO molecules by myeloid dendritic cells (mDCs) [5] and activate autoreactive B cells, leading to a significant induction of ANCAs [6]. Moreover, treatment with DNase I, which can selectively digest DNA threads of NETs, could prevent vessel inflammation, confirming the involvement of NETs in the development of AAV [6].
Conversely, increasing evidence from both animal models and human studies has demonstrated that activation of the complement system via the alternative pathway (AP) is another crucial component in the pathogenesis of AAV [7–11]. However, the interaction between NETs and complement has not yet been well elucidated. Leffler et al. discovered that NETs could activate the classical complement pathway due to the interaction with C1q [12]. It is well known that neutrophils contain various components of the alternative pathway but not the classical pathway [13]. In AAV, upon neutrophil activation mediated by ANCAs, activation of the alternative pathway dominates over the classical pathway [14]. However, the link between NETs and alternative complement pathway in the pathogenesis of AAV is far from clear. We hypothesized that ANCA-induced NETs can activate the alternative complement pathway, thereby initiating an inflammatory amplification cascade.
Materials and methods
Preparation of IgG
ANCA-positive immunoglobulin (Ig)G was prepared from plasma of patients with active MPO-ANCA- or PR3-ANCA-positive primary small vessel vasculitis. Plasma was filtered through a 0·22-mm syringe filter (Gelman Sciences, Ann Arbor, MI, USA) and applied to a High-Trap-protein G column on an AKTA-FPLC system (GE Biosciences, South San Francisco, CA, USA). Preparation of IgG was performed according to the methods described previously [15,16]. We obtained written informed consent from all participants. The research was in compliance with the Declaration of Helsinki and approved by the clinical research ethics committee of the Peking University First Hospital.
Neutrophil isolation
Peripheral blood polymorphonuclear cells (PMNs) were isolated from freshly drawn blood of healthy donors as described previously [17]. Briefly, whole blood was separated by spinning on a Histopaque 1119 cushion. The resulting neutrophil-rich fraction was washed and layered on a discontinuous Percoll gradient [85–65% in phosphate-buffered saline (PBS)] for centrifugation. Bands on the 80, 75 and 70% layers were collected, pooled and washed with PBS. The cells were counted and kept in RPMI-1640 medium with 0·5% heat-inactivated fetal bovine serum (FBS) for the induction experiment. Neutrophil purity was greater than 96% (Wright–Giemsa staining) with 98% cell viability (trypan blue).
NETs induction
PMNs were seeded on 13-mm glass cover slips in 24-well plates in 500 μl of RPMI supplemented with 0·5% heat-inactivated FBS at a density of 105 cells per well. The plates were incubated for 30 min at 37ºC to allow adhesion of the cells. Neutrophils were primed with 5 ng/ml tumour necrosis factor (TNF)-α (Sigma-Aldrich, Poole, UK), seeded on lysinated glass slides for 15 min and then treated with 250 μg/ml purified ANCA-positive IgG from AAV patients. Purified IgG from healthy subjects served as the negative control, and phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich) at a concentration of 50 nM and lipopolysaccharide (LPS; Sigma-Aldrich) at a concentration of 100 ng/ml served as positive controls. After 180 min, the cells were fixed with 4% paraformaldehyde (PFA) and the DNA was stained using 4',6-diamidino-2-phenylindole (DAPI) (Zhongshan Golden Bridge Biotechnology, Beijing, China). Using fluorescence microscopy, we determined the percentage of neutrophils releasing DNA fibres from blinded samples in at least five random microscopic fields using a ×20 objective.
NETs immunofluorescence
Coverslips with the fixed cells were removed from the plates and processed by floating on drops kept on parafilm sheet covering a test tube stand. After washing with PBS, the samples were permeabilized for 1 min with 0·5% Triton X-100 in PBS and washed again with PBS. Unspecific binding sites were blocked in PBS containing 5% donkey serum. The specimens were incubated with MPO-specific rabbit monoclonal antibody (1 : 100 dilution; Abcam, Cambridge, UK), rabbit anti-elastase antibody (1 : 100 dilution; Abcam) or anti-Cit-histone H3 antibody (1 : 50 dilution; Abcam) in blocking buffer combined with mouse anti-human complement factor B (Bb) (1 : 50 dilution; Quidel, San Diego, CA, USA) or with mouse anti-human properdin (1 : 50 dilution; LifeSpan BioScience, Seattle, WA, USA) for 1 h at 37ºC. After washing with PBS, AF488-labelled donkey anti-rabbit IgG or Cy3-labelled donkey anti-mouse IgG (both 1 : 500 dilution; Jackson ImmunoResearch Laboratories, Westgrove, PA, USA) were applied for 1 h. The samples were washed with PBS and distilled water, stained with 4′,6-diamidino-2-phenylindole (DAPI) and mounted with Mowiol. Negative controls were performed using normal rabbit IgG (1 mg/ml, GTX35035; GeneTex, Irvine, CA, USA) and mouse IgG2a (100 µg/ml, HI1004; Hycult Biotech, Uden, the Netherlands) diluted at concentrations equivalent to the primary antibodies. Confocal images were captured with a Zeiss LSM 780 confocal microscope (Zeiss, Jena, Germany). Images were taken using a ×63 PL Apo 1·4 oil objective, and were exported from the ZEN 2012 (blue edition) microscopy software.
Detection of complement deposition on NETs after incubation with magnesium salt ethyleneglycol tetraacetic acid (Mg-EGTA)-treated human serum
NETs were induced as described above. EGTA was added to inhibit the complement activation via the classical and mannose binding lectin (MBL) pathways [18]. Normal human serum (NHS) was added into an equal volume of 16·7 mM EGTA and 1·6 mM Mg2+ as Mg-EGTA-NHS, which can block the classical and MBL pathways of the complement activation [19]. The Mg-EGTA-NHS was then incubated with NETs for 40 min at 37°C with a final concentration of 10% for NHS. After incubation with serum, coverslips with NETs were washed, and complement components were detected using rabbit anti-human C3d polyclonal antibodies (1 : 2000 dilution; Dako A/S, Glostrup, Denmark) or mouse anti-human C5b-9 monoclonal antibodies (1 : 50 dilution; Abcam) as primary antibodies with secondary antibodies AF488-labelled donkey anti-rabbit IgG (1 : 500 dilution Jackson ImmunoResearch Laboratories) or AF488-labelled donkey anti-mouse IgG (1 : 500 dilution; Invitrogen, Carlsbad, CA, USA). NETs were visualized using propidium iodide (Invitrogen). Co-localization of NETs with C3d and C5b-9 were analysed as for complement deposition.
Incubation of serum with supernatants enriched in NETs
Isolated neutrophils were resuspended and stimulated to release NETs as described above. Cells were centrifuged and the supernatants were collected as described previously [20,21]. NETs were digested by DNase I (0·1 U/sample; Sigma Aldrich) for 20 min at 37°C. The use of DNase I, which selectively digests NET DNA threads, allowed discerning the contribution of NETs. Supernatants enriched in NETs or DNase I-degraded NETs were co-incubated with NHS or Mg-EGTA-NHS for 40 min at 37°C, with the final concentration of 30% for NHS. Thereafter, the complement activation was stopped by addition of 10 mM EDTA. Then samples were put on ice immediately, and stored in aliquots at −80°C until use for enzyme-linked immunosorbent assay (ELISA) analysis. When testing, after rapid thawing at 37°C, the frozen specimens were transferred immediately onto ice before use within 1 h. Repeated freeze/thaw cycles were avoided.
Quantification of complement components levels in supernatants
The terminal products of the complement cascade were tested by ELISA, including complement fragments C3a (Quidel, San Diego, CA, USA), C5a (Quidel) and soluble C5b-9 (SC5b-9, Quidel). All the complement components were assayed according to the manufacturer's instructions. The concentrations of C3a, C5a and SC5b-9 in the samples were then determined by comparing the optical density (OD) values of samples to the standard curve.
Statistical analysis
Data were analysed using GraphPad Prism (version 5; GraphPad Software, San Diego, CA, USA) and spss statistical software package version 11·0 (Chicago, IL, USA). The Shapiro–Wilk test was used to examine whether the data were normally distributed. Quantitative data were expressed as mean ± standard deviation (for data that were normally distributed) or median and range (for data that were not normally distributed). Differences of quantitative parameters between groups were assessed using the t-test (for data that were normally distributed) or Mann–Whitney U-test (for data that were not normally distributed), as appropriate. When the differences between more than two sets of data were analysed, we used the one-way analysis of variance. Differences were considered significant when the P-value < 0·05.
Results
Deposition of the components of alternative complement pathway on NETs in vitro
Neutrophils from healthy blood donors were stimulated as described above. TNF-α-primed neutrophils stimulated with ANCA-positive IgG could release robust NETs (Supporting information, Fig. S1). We also found that the isolated ANCA-positive IgG could bind to the NETs (Supporting information, Fig. S2). NETs are composed of decondensed extracellular DNA decorated with neutrophil-derived granules (Supporting information, Fig. S3). We then found that Bb and properdin, which are two important components in the alternative complement pathway, appeared to be present in some areas on NETs (Fig. 1). Factor B and properdin have been proved to be stored in neutrophils. After stimulation, activated neutrophils release NETs, which enables an assortment of neutrophil proteins to interact with these complement factors and could therefore lead to the deposition of Bb and properdin on NETs. We further found that NETs induced by PMA and LPS also have Bb and properdin deposition (Supporting information, Fig. S4). Immunoblotting of Bb and properdin further confirmed the results (Supporting information, Fig. S5).
Fig. 1.
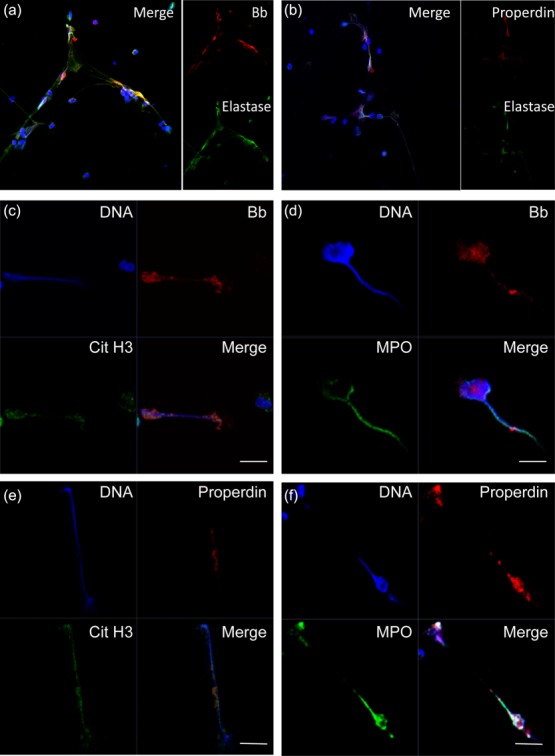
Detection of deposition of components of alternative complement on neutrophil extracellular traps (NETs) in vitro. Neutrophils isolated from healthy donors were primed with tumour necrosis factor (TNF)-α and incubated with 250 µg/ml anti-neutrophil cytoplasmic antibody (ANCA)-positive immunoglobulin (Ig)G. NETs induced by ANCA were identified by co-localization of DNA (blue), elastase, cit-histone H3 or myeloperoxidase (MPO) (green). Deposition of complement factor B (Bb) and properdin (red) on NETs was assessed by confocal microscopy. (a) Deposition of Bb (red) on NETs stained by DNA (blue) and elastase (green). Magnification ×400. (b) Deposition of properdin (red) on NETs stained by DNA (blue) and elastase (green). Magnification ×400. (c) Deposition of Bb (red) on NETs stained by DNA (blue) and cit-histone (green). (d) Deposition of Bb (red) on NETs stained by DNA (blue) and MPO (green). (e) Deposition of properdin (red) on NETs stained by DNA (blue) and cit-histone (green). (f) Deposition of properdin (red) on NETs stained by DNA (blue) and MPO (green). Representative images of six independent experiments are shown. (c–f) Scale bars = 10 μm.
Detection of alternative complement pathway activation on NETs by immunofluorescence
In order to confirm that NETs could activate the alternative complement pathway, we incubated NETs with NHS, Mg-EGTA-NHS or heat-inactivated normal human serum (Hi-NHS) and Mg-EGTA-Hi-NHS, respectively. EGTA has a higher affinity for calcium ions than magnesium ions. C1 and MBL require Ca2+ for activation and consequently do not function in the presence of EGTA, so only the alternative complement pathway remains functional in this circumstance [22]. Little, if any, deposition of C4d on NETs incubated with NHS or Hi-NHS in the presence of EGTA further confirmed the lack of classical and lectin pathway activation at the presence of EGTA (Supporting information, Fig. S6). Complement activation on NETs should result in the deposition of C3b and C5b-9. C3b, one of the products derived from C3 activation, could bind covalently via thioester bond to its acceptor molecule at the activated site [23]. The antibody against C3d, an epitope available on C3b, was used to detect bound C3b [12]. Therefore, the complement activation on NETs was detected by C3d and C5b-9 staining. As expected, regarding NETs induced by ANCA, we observed that C3b and C5b-9 deposited on NHS or Mg-EGTA-NHS-incubated NETs, while little or no deposition on Hi-NHS or Mg-EGTA-Hi-NHS incubated NETs (Fig. 2). We obtained similar results on NETs induced by phorbol myristate acetate (PMA) and lipopolysaccharide (LPS) (data not shown). We also quantified the mean fluorescence intensity of C3b staining on NETs after incubation with serum to further confirm our findings (Supporting information, Fig. S7). The deposition of C3b and C5b-9 on NETs incubated with Mg-EGTA-Hi-NHS was significantly less than that on NETs incubated with Mg-EGTA-NHS. These results suggested that at least some deposition of C3b and C5b-9 was through the alternative complement activation.
Fig. 2.
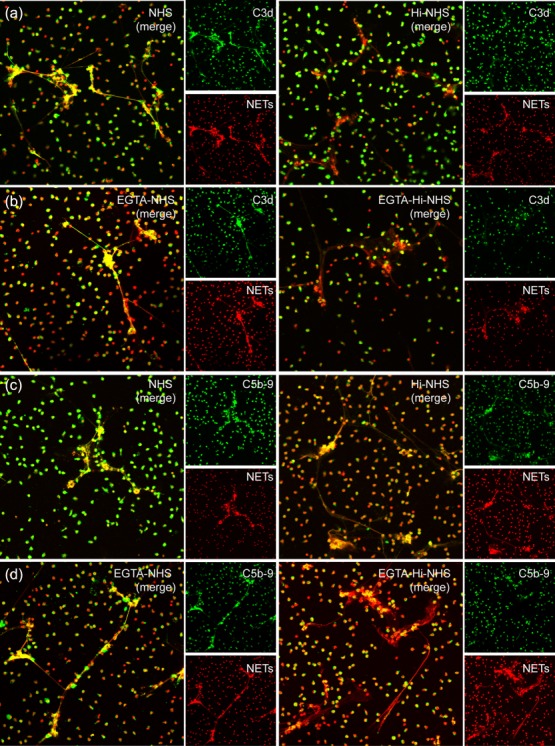
Immunofluorescence detection for alternative complement pathway activation on neutrophil extracellular traps (NETs). (a) C3d deposited on NETs during incubation with normal serum. Little or no deposition of C3d from heat-inactivated normal human serum (Hi-NHS) (lacking active complement) could be observed. (b) C3d deposited on NETs during incubation with magnesium salt (Mg)-ethyleneglycol tetraacetic acid (EGTA)-treated serum. Little or no deposition of C3d from Mg-EGTA-treated Hi-NHS (lacking active complement) could be observed. (c) C5b-9 deposited on NETs during incubation with normal serum. Little or no deposition of C5b-9 from Hi-NHS (lacking active complement) could be observed. (d) C5b-9 deposited on NETs during incubation with Mg-EGTA-treated serum. Little or no deposition of C5b-9 from Mg-EGTA-treated Hi-NHS (lacking active complement) could be observed. (a–d) Representative images of six independent experiments are shown. C3d and C5b-9 were stained in green and NETs were stained in red with propidium iodide (magnification ×200).
NETs could activate the complement cascade in the serum
By immunofluorescence assay, we demonstrated that the alternative complement pathway activation occurs on NETs, as described above. Then we further measured the terminal products of complement activation including C3a, C5a and SC5b-9 in liquid phase by specific ELISA. Neutrophils were primed with TNF-α and stimulated with human ANCA-positive IgG or normal human IgG, which was derived from healthy donors. We also stimulated neutrophils with ANCA-positive IgG alone without TNF-α priming. NETs were induced and supernatants were collected. The supernatants were incubated with NHS or Mg-EGTA-NHS for 40 min at 37°C. To confirm the activation of alternative complement pathway in this setting, C3a, C5a and SC5b-9 levels were detected with ELISA.
Figure 3 showed that NETs lead to C3a, C5a and SC5b-9 generation in NHS and EGTA-treated NHS. C3a concentration was 463·39 ± 73·88 ng/ml in the NHS buffer control, 580·64 ± 203·89 ng/ml in the supernatants of TNF-α-primed neutrophils stimulated with normal human IgG, 671·76 ± 92·48 ng/ml in the supernatants of neutrophils stimulated with ANCA-positive IgG without priming, 1420·66 ± 250·16 ng/ml in the supernatants enriched in ANCA-induced NETs (P < 0·001, P < 0·001, P < 0·001, respectively) and 735·51 ± 160·36 ng/ml in the supernatants of DNase I-degraded NETs (P < 0.001, compared with the ANCA-induced NETs group). In the presence of EGTA, C3a concentration was 395·25 ± 77·84 ng/ml in the Mg-EGTA-NHS buffer control, 414·18 ± 107·4 ng/ml in the supernatants of TNF-α-primed neutrophils stimulated with normal human IgG, 423·39 ± 71·3 ng/ml in the supernatants of neutrophils stimulated with ANCA-positive-IgG without priming, 800·42 ± 244·81 ng/ml in the supernatants enriched in ANCA-induced NETs (P < 0·001, P < 0·001, P < 0·001, respectively) and 479·07 ± 156·2 ng/ml in the supernatants of DNase I-degraded NETs (P < 0·001, compared with the ANCA-induced NETs group). C5a concentration was 9·31 ± 2·72 ng/ml in the NHS buffer control, 11·15 ± 3·76 ng/ml in the supernatants of TNF-α-primed neutrophils stimulated with normal human IgG, 12·33 ± 5·45 ng/ml in the supernatants of neutrophils stimulated with ANCA-positive IgG without priming, 21·76 ± 3·65 ng/ml in the supernatants enriched in ANCA-induced NETs (P < 0·001, P < 0·001, P = 0·003, respectively) and 14·64 ± 3·80 ng/ml in the supernatants of DNase I-degraded NETs (P = 0.004 compared with the ANCA-induced NETs group). In the presence of EGTA, C5a concentration was 2·36 ± 1·60 ng/ml in the Mg-EGTA-NHS buffer control, 3·67 ± 1·92 ng/ml in the supernatants of TNF-α-primed neutrophils stimulated with normal human IgG, 4·96 ± 2·39 ng/ml in the supernatants of neutrophils stimulated with ANCA-positive-IgG without priming, 7·68 ± 1·50 ng/ml in the supernatants enriched in ANCA-induced NETs (P < 0·001, P = 0·001, P = 0·032, respectively) and 4·86 ± 1·26 ng/ml in the supernatants of DNase I-degraded NETs (P = 0·008, compared with the ANCA-induced NETs group). SC5b-9 concentration was 358·36 ± 57·42 ng/ml in the NHS buffer control, 412·15 ± 136·25 ng/ml in the supernatants of TNF-α-primed neutrophils stimulated with normal human IgG, 525·47 ± 60·69 ng/ml in the supernatants of neutrophils stimulated with ANCA-positive IgG without priming, 875·70 ± 211·12 ng/ml in the supernatants enriched in ANCA-induced NETs (P < 0·001, P < 0·001, P < 0·001 respectively) and 621·67 ± 66·72 ng/ml in the supernatants of DNase I-degraded NETs (P < 0·001, compared with the ANCA-induced NETs group). In the presence of EGTA, SC5b-9 concentration was 176·23 ± 61·71 ng/ml in the Mg-EGTA-NHS buffer control, 188·41 ± 76·64 ng/ml in the supernatants of TNF-α-primed neutrophils stimulated with normal human IgG, 230·90 ± 65·60 ng/ml in the supernatants of neutrophils stimulated with ANCA-positive IgG without priming, 382·15 ± 159·75 ng/ml in the supernatants enriched in ANCA-induced NETs (P < 0·001, P < 0·001, P = 0·021, respectively) and 212·65 ± 44·40 ng/ml in the supernatants of DNase I-degraded NETs (P < 0.001, compared with the ANCA-induced NETs group). Collectively, in the presence of EGTA, the concentration of C3a, C5a and SC5b-9 in the supernatants enriched in the NETs group are much higher than that in the supernatants of DNase I-degraded NETs group. These results further supported that NETs, released from ANCA-activated neutrophils, could activate the alternative complement pathway.
Fig. 3.
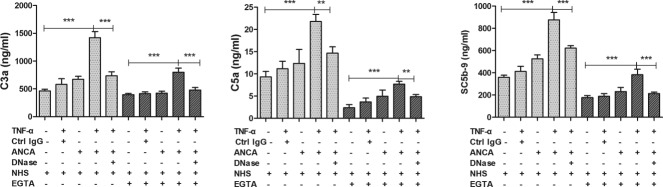
Neutrophil extracellular traps (NETs) induced by anti-neutrophil cytoplasmic antibody (ANCA) could activate the complement cascade in the serum. Neutrophils were stimulated under different conditions for 180 min. Bars represent mean ± standard deviation (s.d.). Each measured on neutrophils of 11 independent experiments and donors. Supernatants from the cell suspensions were incubated for 40 min with normal serum or magnesium salt (Mg)-ethyleneglycol tetraacetic acid (EGTA)-treated normal serum, and C3a, C5a, SC5b-9 generation was measured by specific enzyme-linked immunosorbent assay (ELISA). *P < 0·05, **P < 0·01, ***P < 0·001.
Regarding NETs induced by PMA and LPS, we obtained similar results indicating that the level of the terminal products of complement activation in the NETs group was significantly higher than that in the degraded NETs group in the presence of EGTA (Supporting information, Fig. S8). These results indicated that NETs could activate the complement through the alternative complement pathway.
Discussion
The interaction between neutrophils and activation of alternative complement pathway plays a pivotal role in the pathogenesis of AAV. Other than the well-known ANCA-mediated neutrophil respiratory burst and degranulation [3], Kessenbrock et al. [4] found a new pathogenic aspect of neutrophils in AAV that ANCA can lead TNF-α-primed neutrophils to produce NETs. Nakazawa et al. found that the NET induction ability of ANCA-positive IgG is correlated with disease activity [24], and rats immunized with abnormal NETs could produce MPO-ANCA [25], further suggesting the pathogenic role of NETs in AAV. Camous et al. discovered that neutrophil membranes and neutrophil-derived microparticles could activate the alternative complement pathway [26]. However, the relationship between NETs and alternative complement pathway activation has not been elucidated fully. The current study investigated the sequence of events of complement activation of ANCA-NETs in AAV, and whether NETs could activate the alternative complement pathway, which might further reveal the pathophysiological role of NETs in AAV.
In the present study, we found that the properdin and Bb were present on released NETs in vitro. It is well known that properdin stored in secondary granules of neutrophils is an essential positive regulatory component of the alternative complement pathway, which could recruit C3b and factor B and thus lead to the formation of the C3 convertase (C3bBb) of the alternative pathway [27]. The deposition of the components of the alternative pathway on NETs prompted us to explore whether NETs can activate the alternative complement pathway.
Schreiber et al. [28] found that ANCA-stimulated neutrophils release some factors which can activate the complement cascade in normal serum, resulting in the production of C5a. The neutrophil itself harbours proteases that are capable of activating the complement system [29]. Moreover, both clinical and experimental studies have proved that complement activation via the alternative pathway is involved in the pathogenesis of AAV [7–10,30,31]. In our study, neutrophils were primed with TNF-α, and then stimulated with ANCA-positive IgG to induce NETs formation. We found increased levels of C3a, C5a and SC5b-9 after incubation with normal serum or Mg-EGTA-treated normal serum. The concentration of these three terminal products of complement cascade in serum, i.e. C3a, C5a and SC5b-9, decreased after incubation with DNase I which can degrade NETs. These results suggested that although complement activation is reduced upon Mg-EGTA addition, there is still generation of complement activation products through the alternative pathway.
A recent study found that the neutrophils from AAV patients could release tissue factor (TF)-expressing NETs [32]. Given the abundant cross-talk between the coagulation and complement system [33,34], we further quantified the generation of C5a in serum after specific inhibition of TF or thrombin, respectively. We found that the inhibition of TF or thrombin has little effect on complement activation in PMA-induced NETs. However, the effect of ANCA-induced NETs on activating the complement is partly dependent upon TF and thrombin, indicating the novel step of TF and thrombin activation in the effect of ANCA-induced NETs on complement activation (Supporting information, Fig. S9).
Recently, another study also showed that MPO could bind properdin directly, which then served as a focus for alternative complement pathway activation [35]. Other than the special granules on NETs, the web-like structure of NETs could also provide the platform for the activation of the alternative complement pathway. A recent study by Camous et al. demonstrated that neutrophil membranes and neutrophil-derived microparticles could activate the alternative complement pathway [25], leading to the generation of C5a. C5a, through interaction with neutrophil C5aR, may attract and further prime neutrophils for full activation in response to ANCA, composing an amplification loop for ANCA-mediated neutrophil activation. NET induced by ANCA is another pathway that links the neutrophils and alternative complement activation, fulfilling the pathogenesis of AAV. It would be of great interest to carry out in-vivo studies in order to further confirm the findings in our in-vitro study.
However, there are some unspecific findings in our study that we need to determine. First, the ability of activating complement is not a specific trait of ANCA-induced NETs. NETs induced by PMA or LPS could also activate the complement. However, in the pathogenesis of AAV, ANCAs are the major stimulants of neutrophils to induce NETs formation. Considering the important roles of the interaction among neutrophils, ANCA and complement in the development AAV, NETs is an important link among these three factors, and thus comprises a vicious loop in the pathogenesis of AAV. Secondly, in our study we found that the level of the terminal products of complement activation was significantly higher in the NETs group alone than that in the NETs group with EGTA. We speculated that as well as the alternative complement pathway, NETs induced by ANCA could also activate complement via the classical and lectin pathways. Leffler et al. demonstrated that NETs in lupus could activate the classical complement pathway [12]. Our study further extended the finding by demonstrating that NETs could activate the alternative complement pathway. More importantly, however, it is the activation of the alternative pathway, rather than the classical and lectin pathways, that plays an important role in the development of AAV [8,9]. Collectively, in the context of pathogenesis of AAV, our finding that ANCA induced NETs to activate complement activation via multiple pathways, including the alternative pathway, is of interest. In conclusion, our study investigated the sequence of events of ANCA-NETs complement activation in AAV and discovered the novel relationship between NETs and the alternative complement pathway. NETs released from ANCA-activated neutrophils could activate the alternative complement pathway, and might thus participate in the pathogenesis of ANCA-associated vasculitis.
Acknowledgments
This study is supported by a grant from the Chinese 973 project (no. 2012CB517702), three grants from the National Natural Science Fund (no. 81425008, no. 81370829 and no. 81321064), and the ‘National Key Technology Research and Development (R&D) Program’ of the Ministry of Science and Technology of China (no. 2011BAI10B04).
Author contributions
H. W. recruited patients, collected samples and performed the experiment, analysed data and wrote the manuscript. C. W. recruited patients and analysed data. M. C. and M.-H. Z. designed and directed the study. M. C. is responsible for the interpretation of the data.
Disclosures
The authors confirm that there are no conflicts of interest.
Supporting information
Supporting information
Figure S1. Neutrophil extracellular trap (NET) induced by anti-neutrophil cytoplasmic antibody (ANCA). Neutrophils were primed with tumour necrosis factor (TNF)-α and incubated with 250 µg/ml ANCA-positive immunoglobulin (Ig)G or normal human IgG. We also stimulated neutrophils with ANCA-positive IgG alone without TNF-α priming. Phorbol myristate acetate (PMA)-activated neutrophils were used as the positive control. (a) After 180 min of incubation, the DNA release was visualized by fluorescence microscopy of 4',6-diamidino-2-phenylindole (DAPI)-stained specimens. A representative example of three independent experiments is shown. Magnification ×200. (b) Quantitative assessment of the percentage of NETs-forming cells revealed robust NET formation after ANCA activation. After ANCA activation, 24·26 ± 5·62% of neutrophils produced NETs, compared to 8·90 ± 2·78% of normal IgG-treated neutrophils, 13·51 ± 3·50% neutrophils treated with ANCA-positive IgG alone without TNF-α priming. Incubation with PMA, known as a strong inducer of NETs, triggered NETs production in 40·91 ± 6·50% of all neutrophils.
Figure S2. Isolated anti-neutrophil cytoplasmic antibody (ANCA)-positive immunoglobulin (Ig)G could bind to neutrophil extracellular trap (NETs). Neutrophils were stimulated with 50 nM PMA for 3 h at 37°C. After fixation with 4% paraformaldehyde (PFA), the samples were washed with phosphate-buffered saline (PBS) and then incubated with 250 µg/ml isolated ANCA-positive immunoglobulin (Ig)G (a) or normal human control IgG (b) for 1 h at 37°C. After washing with phosphate-buffered saline (PBS), AF488-conjugated anti-human IgG antibodies (1 : 500 dilution; Jackson Immuno Research Laboratories) were applied for 1 h at 37°C. Isolated ANCA-positive IgG bound to the NETs was seen. Representative figures are shown. Magnification ×200.
Figure S3. Immunofluorescence identifying neutrophil extracellular traps (NETs) induced by anti-neutrophil cytoplasmic antibodies (ANCA). Neutrophils were primed with tumour necrosis factor (TNF)-α and incubated with 250 µg/ml ANCA-positive-IgG. NETs induced by ANCA were identified by co-localization of DNA (blue), histone (red) and MPO (green). A representative example of three independent experiments is shown. Magnification ×400.
Figure S4. Detection of deposition of Bb and properdin on neutrophil extracellular trap (NETs) induced by phorbol myristate acetate (PMA) or lipopolysaccharide (LPS) in vitro. Neutrophils isolated from healthy donors were stimulated with 50 nM PMA or 100ng/ml LPS. NETs were identified by co-localization of DNA (blue) and elastase (green). Deposition of Bb and properdin (red) on NETs was assessed by confocal microscopy. Magnification ×400. (a) Deposition of Bb (red) on NETs induced by PMA stained by DNA (blue) and elastase (green). (b) Deposition of Bb (red) on NETs induced by LPS stained by DNA (blue) and elastase (green). (c) Isotype control for staining antibody was perfomed. Primary antibodies were replaced by normal rabbit immunoglobulin (Ig)G and mouse IgG2a. (d) Deposition of properdin (red) on NETs induced by PMA stained by DNA (blue) and elastase (green). (e) Deposition of properdin (red) on NETs induced by LPS stained by DNA (blue) and elastase (green). (f) Isotype control for staining antibody was performed. Primary antibodies were replaced by normal rabbit IgG and mouse IgG2a.
Figure S5. Immunoblotting of complements on neutrophil extracellular trap (NETs). Human neutrophils were left untreated, treated with anti-neutrophil cytoplasmic antibody (ANCA)-positive immunoglobulin (Ig)G after tumour necrosis factor (TNF)-α priming. Supernatants were collected, and Bb, properdin were examined by Western blotting. Supernatants enriched in NETs and degraded NETs (treated with commercial DNase I) were incubated by normal human serum in the presence of ethyleneglycol teraacetic acid (EGTA), C5b and C3d were examined by Western blotting. Primary antibodies were used as below: mouse anti-human Bb antibody (Quidel), rabbit anti-human properdin antibody (Abcam), rabbit anti-human C5b antibody (Abcam) and rabbit anti-human C3d anibody (Abcam). (a) Detection of Bb on NETs by immunoblotting. (b) Detection of properdin on NETs by immunoblotting. (c) Detection of C5b on NETs incubated with serum in the presence of EGTA by immunoblotting. (d) Detection of C3d on NETs incubated with serum in the presence of EGTA by immunoblotting.
Figure S6. Immunofluorescence detection for C4d on neutrophil extracellular trap (NETs). Neutrophils were primed with TNF-α and incubated with 250µg/ml anti-neutrophil cytoplasmic antibody (ANCA)-positive immunoglobulin (Ig)G. The induced NETs were then incubated with normal human serum (NHS) at the presence of ethyleneglycol teraacetic acid (EGTA) or not for 40 min at 37°C. After incubation with serum, coverslips with NETs were washed, and C4d was detected using rabbit anti-human C4d polyclonal antibodies (1 : 100 dilution; Abcam) as primary antibodies with secondary antibodies AF488-labelled donkey anti-rabbit IgG (1 : 500 dilution, Jackson ImmunoResearch Laboratories). NETs were visualized using 4',6-diamidino-2-phenylindole (DAPI) (blue). (a) C4d deposited on NETs during incubation with normal serum. (b) Little or no deposition of C4d on NETs during incubation with normal serum at the presence of EGTA could be observed. Representative images of three independent experiments are shown. Magnification ×200.
Figure S7. Quantification of mean fluorescence intensity of C3b staining on neutrophil extracellular trap (NETs) after incubation with serum. The mean fluorescence intensity (MFI) of C3b staining on NETs incubated with normal human serum (NHS), heat inactivated (Hi)-NHS (lack of active complements), magnesium salt-ethyleneglycol teraacetic acid (Mg-EGTA)-treated NHS, Mg-EGTA-treated Hi-NHS (lack of active complements). Data are presented as mean of three independent experiments ± standard deviation (s.d.). The MFI of C3b on NETs incubated with NHS was 49·74 ± 8·36, while the MFI of C3b on NETs incubated with Hi-NHS was 11·95 ± 2·85 (P<0·001). The MFI of C3b on NETs incubated with Mg-EGTA-treated NHS was 30·96 ± 5·37, while the MFI of C3b on NETs incubated with Mg-EGTA-treated Hi-NHS was 7·14 ± 1·40 (P<0·001).
Figure S8. Neutrophil extracellular trap (NETs) induced by phorbol myristate acetate (PMA) or lipopolysaccharide (LPS) could activate the alternative complement cascade in the serum. Neutrophils were stimulated by 50 nM PMA or 100 ng/ml LPS. Bars represent mean ± standard deviation (s.d.). Each measured on neutrophils of five independent experiments and donors. Supernatants from the cell suspensions were incubated for 40 min with normal serum or magnesium salt-ethyleneglycol teraacetic acid (Mg-EGTA)-treated normal serum, and C3a, C5a, SC5b-9 generation was measured by specific enzyme-linked immunosorbent assay (ELISA). *P < 0·05, **P < 0·01, ***P < 0·001.
Figure S9. Quantification of the generation of C5a in serum after specific inhibition of tissue factor or thrombin. Neutrophils were incubated with anti-TF antibody (10 μg/ml) for 30 min in 37°C or 1 μM D-Phe-Pro-Arg-choro-methylketone dihydrochloride (PPACK) for 40 min at room temperature, and were then stimulated by ANCA-positive immunoglobulin (Ig)G after tumour necrosis factor (TNF)-α-priming or 50 nM phorbol myristate acetate (PMA). Bars represent mean ± standard deviation (s.d.). Each measured on neutrophils of four independent experiments and donors. Supernatants from the cell suspensions were incubated for 40 min with normal serum or magnesium salt-ethyleneglycol teraacetic acid (Mg-EGTA)-treated normal serum, and C5a generation was measured by specific. D-Phe-Pro-Arg-choro-methylketone dihydrochloride (PPACK) was used to inactivate thrombin. Anti-TF antibody (murine monoclonal antibody against human tissue factor) was used to inhibit the pro-coagulant activity of tissue factor; n.s. no significant difference.
References
- 1.Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill consensus conference nomenclature of vasculitides. Arthritis Rheum. 2013;65:1–11. doi: 10.1002/art.37715. [DOI] [PubMed] [Google Scholar]
- 2.Chen M, Yu F, Zhang Y, Zhao MH. Antineutrophil cytoplasmic autoantibody-associated vasculitis in older patients. Medicine (Baltimore) 2008;87:203–9. doi: 10.1097/MD.0b013e31817c744b. [DOI] [PubMed] [Google Scholar]
- 3.Falk RJ, Terrell RS, Charles LA, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc Natl Acad Sci USA. 1990;87:4115–9. doi: 10.1073/pnas.87.11.4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kessenbrock K, Krumbholz M, Schonermarck U, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med. 2009;15:623–5. doi: 10.1038/nm.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lande R, Gregorio J, Facchinetti V, et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. 2007;449:564–9. doi: 10.1038/nature06116. [DOI] [PubMed] [Google Scholar]
- 6.Sangaletti S, Tripodo C, Chiodoni C, et al. Neutrophil extracellular traps mediate transfer of cytoplasmic neutrophil antigens to myeloid dendritic cells toward ANCA induction and associated autoimmunity. Blood. 2012;120:3007–18. doi: 10.1182/blood-2012-03-416156. [DOI] [PubMed] [Google Scholar]
- 7.Xing GQ, Chen M, Liu G, Zheng X, E J, Zhao MH. Differential deposition of C4d and MBL in glomeruli of patients with ANCA-negative pauci-immune crescentic glomerulonephritis. J Clin Immunol. 2010;30:144–56. doi: 10.1007/s10875-009-9344-2. [DOI] [PubMed] [Google Scholar]
- 8.Xiao H, Schreiber A, Heeringa P, Falk RJ, Jennette JC. Alternative complement pathway in the pathogenesis of disease mediated by anti-neutrophil cytoplasmic autoantibodies. Am J Pathol. 2007;170:52–64. doi: 10.2353/ajpath.2007.060573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gou SJ, Yuan J, Chen M, Yu F, Zhao MH. Circulating complement activation in patients with anti-neutrophil cytoplasmic antibody-associated vasculitis. Kidney Int. 2013;83:129–37. doi: 10.1038/ki.2012.313. [DOI] [PubMed] [Google Scholar]
- 10.Xing GQ, Chen M, Liu G, et al. Complement activation is involved in renal damage in human antineutrophil cytoplasmic autoantibody associated pauci-immune vasculitis. J Clin Immunol. 2009;29:282–91. doi: 10.1007/s10875-008-9268-2. [DOI] [PubMed] [Google Scholar]
- 11.Huugen D, van Esch A, Xiao H, et al. Inhibition of complement factor C5 protects against anti-myeloperoxidase antibody-mediated glomerulonephritis in mice. Kidney Int. 2007;71:646–54. doi: 10.1038/sj.ki.5002103. [DOI] [PubMed] [Google Scholar]
- 12.Leffler J, Martin M, Gullstrand B, et al. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J Immunol. 2012;188:3522–31. doi: 10.4049/jimmunol.1102404. [DOI] [PubMed] [Google Scholar]
- 13.Chen M, Daha MR, Kallenberg CG. The complement system in systemic autoimmune disease. J Autoimmun. 2010;34:J276–86. doi: 10.1016/j.jaut.2009.11.014. [DOI] [PubMed] [Google Scholar]
- 14.Van Timmeren MM, Chen M, Heeringa P. Review article: pathogenic role of complement activation in anti-neutrophil cytoplasmic auto-antibody-associated vasculitis. Nephrology (Carlton) 2009;14:16–25. doi: 10.1111/j.1440-1797.2009.01086.x. [DOI] [PubMed] [Google Scholar]
- 15.Schreiber A, Rolle S, Peripelittchenko L, et al. Phosphoinositol 3-kinase-gamma mediates antineutrophil cytoplasmic autoantibody-induced glomerulonephritis. Kidney Int. 2010;77:118–28. doi: 10.1038/ki.2009.420. [DOI] [PubMed] [Google Scholar]
- 16.Choi M, Rolle S, Rane M, Haller H, Luft FC, Kettritz R. Extracellular signal-regulated kinase inhibition by statins inhibits neutrophil activation by ANCA. Kidney Int. 2003;63:96–106. doi: 10.1046/j.1523-1755.2003.00718.x. [DOI] [PubMed] [Google Scholar]
- 17.Aga E, Katschinski DM, van Zandbergen G, et al. Inhibition of the spontaneous apoptosis of neutrophil granulocytes by the intracellular parasite Leishmania major. J Immunol. 2002;169:898–905. doi: 10.4049/jimmunol.169.2.898. [DOI] [PubMed] [Google Scholar]
- 18.Jiang H, Wang Z, Serra D, Frank MM, Amalfitano A. Recombinant adenovirus vectors activate the alternative complement pathway, leading to the binding of human complement protein C3 independent of anti-ad antibodies. Mol Ther. 2004;10:1140–2. doi: 10.1016/j.ymthe.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 19.Nakao M, Miura C, Itoh S, et al. A complement C3 fragment equivalent to mammalian C3d from the common carp (Cyprinus carpio): generation in serum after activation of the alternative pathway and detection of its receptor on the lymphocyte surface. Fish Shellfish Immunol. 2004;16:139–49. doi: 10.1016/S1050-4648(03)00057-3. [DOI] [PubMed] [Google Scholar]
- 20.Nakazawa D, Tomaru U, Suzuki A, et al. Abnormal conformation and impaired degradation of propylthiouracil-induced neutrophil extracellular traps: implications of disordered neutrophil extracellular traps in a rat model of myeloperoxidase antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum. 2012;64:3779–87. doi: 10.1002/art.34619. [DOI] [PubMed] [Google Scholar]
- 21.Saffarzadeh M, Juenemann C, Queisser MA, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLOS ONE. 2012;7:e32366. doi: 10.1371/journal.pone.0032366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mershon-Shier KL, Vasuthasawat A, Takahashi K, Morrison SL, Beenhouwer DO. In vitro C3 deposition on Cryptococcus capsule occurs via multiple complement activation pathways. Mol Immunol. 2011;48:2009–18. doi: 10.1016/j.molimm.2011.06.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sahu A, Lambris JD. Structure and biology of complement protein C3, a connecting link between innate and acquired immunity. Immunol Rev. 2001;180:35–48. doi: 10.1034/j.1600-065x.2001.1800103.x. [DOI] [PubMed] [Google Scholar]
- 24.Nakazawa D, Shida H, Tomaru U, et al. Enhanced formation and disordered regulation of NETs in myeloperoxidase-ANCA-associated microscopic polyangiitis. J Am Soc Nephrol. 2014;25:990–7. doi: 10.1681/ASN.2013060606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakazawa D, Tomaru U, Suzuki A, et al. Abnormal conformation and impaired degradation of propylthiouracil-induced neutrophil extracellular traps: implications of disordered neutrophil extracellular traps in a rat model of myeloperoxidase antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum. 2012;64:3779–87. doi: 10.1002/art.34619. [DOI] [PubMed] [Google Scholar]
- 26.Camous L, Roumenina L, Bigot S, et al. Complement alternative pathway acts as a positive feedback amplification of neutrophil activation. Blood. 2011;117:1340–9. doi: 10.1182/blood-2010-05-283564. [DOI] [PubMed] [Google Scholar]
- 27.Wirthmueller U, Dewald B, Thelen M, et al. Properdin, a positive regulator of complement activation, is released from secondary granules of stimulated peripheral blood neutrophils. J Immunol. 1997;158:4444–51. [PubMed] [Google Scholar]
- 28.Schreiber A, Xiao H, Jennette JC, Schneider W, Luft FC, Kettritz R. C5a receptor mediates neutrophil activation and ANCA-induced glomerulonephritis. J Am Soc Nephrol. 2009;20:289–98. doi: 10.1681/ASN.2008050497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huber-Lang M, Younkin EM, Sarma JV, et al. Generation of C5a by phagocytic cells. Am J Pathol. 2002;161:1849–59. doi: 10.1016/S0002-9440(10)64461-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen M, Xing GQ, Yu F, Liu G, Zhao MH. Complement deposition in renal histopathology of patients with ANCA-associated pauci-immune glomerulonephritis. Nephrol Dial Transplant. 2009;24:1247–52. doi: 10.1093/ndt/gfn586. [DOI] [PubMed] [Google Scholar]
- 31.Hao J, Meng LQ, Xu PC, Chen M, Zhao MH. p38MAPK, ERK and PI3K signaling pathways are involved in C5a-primed neutrophils for ANCA-mediated activation. PLOS ONE. 2012;7:e38317. doi: 10.1371/journal.pone.0038317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kambas K, Chrysanthopoulou A, Vassilopoulos D, et al. Tissue factor expression in neutrophil extracellular traps and neutrophil derived microparticles in antineutrophil cytoplasmic antibody associated vasculitis may promote thromboinflammation and the thrombophilic state associated with the disease. Ann Rheum Dis. 2014;73:1854–63. doi: 10.1136/annrheumdis-2013-203430. [DOI] [PubMed] [Google Scholar]
- 33.Huber-Lang M, Sarma JV, Zetoune FS, et al. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med. 2006;12:682–7. doi: 10.1038/nm1419. [DOI] [PubMed] [Google Scholar]
- 34.Krisinger MJ, Goebeler V, Lu Z, et al. Thrombin generates previously unidentified C5 products that support the terminal complement activation pathway. Blood. 2012;120:1717–25. doi: 10.1182/blood-2012-02-412080. [DOI] [PubMed] [Google Scholar]
- 35.O'Flynn J, Dixon KO, Faber Krol MC, Daha MR, van Kooten C. Myeloperoxidase directs properdin-mediated complement activation. J Innate Immun. 2014;6:417–25. doi: 10.1159/000356980. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Figure S1. Neutrophil extracellular trap (NET) induced by anti-neutrophil cytoplasmic antibody (ANCA). Neutrophils were primed with tumour necrosis factor (TNF)-α and incubated with 250 µg/ml ANCA-positive immunoglobulin (Ig)G or normal human IgG. We also stimulated neutrophils with ANCA-positive IgG alone without TNF-α priming. Phorbol myristate acetate (PMA)-activated neutrophils were used as the positive control. (a) After 180 min of incubation, the DNA release was visualized by fluorescence microscopy of 4',6-diamidino-2-phenylindole (DAPI)-stained specimens. A representative example of three independent experiments is shown. Magnification ×200. (b) Quantitative assessment of the percentage of NETs-forming cells revealed robust NET formation after ANCA activation. After ANCA activation, 24·26 ± 5·62% of neutrophils produced NETs, compared to 8·90 ± 2·78% of normal IgG-treated neutrophils, 13·51 ± 3·50% neutrophils treated with ANCA-positive IgG alone without TNF-α priming. Incubation with PMA, known as a strong inducer of NETs, triggered NETs production in 40·91 ± 6·50% of all neutrophils.
Figure S2. Isolated anti-neutrophil cytoplasmic antibody (ANCA)-positive immunoglobulin (Ig)G could bind to neutrophil extracellular trap (NETs). Neutrophils were stimulated with 50 nM PMA for 3 h at 37°C. After fixation with 4% paraformaldehyde (PFA), the samples were washed with phosphate-buffered saline (PBS) and then incubated with 250 µg/ml isolated ANCA-positive immunoglobulin (Ig)G (a) or normal human control IgG (b) for 1 h at 37°C. After washing with phosphate-buffered saline (PBS), AF488-conjugated anti-human IgG antibodies (1 : 500 dilution; Jackson Immuno Research Laboratories) were applied for 1 h at 37°C. Isolated ANCA-positive IgG bound to the NETs was seen. Representative figures are shown. Magnification ×200.
Figure S3. Immunofluorescence identifying neutrophil extracellular traps (NETs) induced by anti-neutrophil cytoplasmic antibodies (ANCA). Neutrophils were primed with tumour necrosis factor (TNF)-α and incubated with 250 µg/ml ANCA-positive-IgG. NETs induced by ANCA were identified by co-localization of DNA (blue), histone (red) and MPO (green). A representative example of three independent experiments is shown. Magnification ×400.
Figure S4. Detection of deposition of Bb and properdin on neutrophil extracellular trap (NETs) induced by phorbol myristate acetate (PMA) or lipopolysaccharide (LPS) in vitro. Neutrophils isolated from healthy donors were stimulated with 50 nM PMA or 100ng/ml LPS. NETs were identified by co-localization of DNA (blue) and elastase (green). Deposition of Bb and properdin (red) on NETs was assessed by confocal microscopy. Magnification ×400. (a) Deposition of Bb (red) on NETs induced by PMA stained by DNA (blue) and elastase (green). (b) Deposition of Bb (red) on NETs induced by LPS stained by DNA (blue) and elastase (green). (c) Isotype control for staining antibody was perfomed. Primary antibodies were replaced by normal rabbit immunoglobulin (Ig)G and mouse IgG2a. (d) Deposition of properdin (red) on NETs induced by PMA stained by DNA (blue) and elastase (green). (e) Deposition of properdin (red) on NETs induced by LPS stained by DNA (blue) and elastase (green). (f) Isotype control for staining antibody was performed. Primary antibodies were replaced by normal rabbit IgG and mouse IgG2a.
Figure S5. Immunoblotting of complements on neutrophil extracellular trap (NETs). Human neutrophils were left untreated, treated with anti-neutrophil cytoplasmic antibody (ANCA)-positive immunoglobulin (Ig)G after tumour necrosis factor (TNF)-α priming. Supernatants were collected, and Bb, properdin were examined by Western blotting. Supernatants enriched in NETs and degraded NETs (treated with commercial DNase I) were incubated by normal human serum in the presence of ethyleneglycol teraacetic acid (EGTA), C5b and C3d were examined by Western blotting. Primary antibodies were used as below: mouse anti-human Bb antibody (Quidel), rabbit anti-human properdin antibody (Abcam), rabbit anti-human C5b antibody (Abcam) and rabbit anti-human C3d anibody (Abcam). (a) Detection of Bb on NETs by immunoblotting. (b) Detection of properdin on NETs by immunoblotting. (c) Detection of C5b on NETs incubated with serum in the presence of EGTA by immunoblotting. (d) Detection of C3d on NETs incubated with serum in the presence of EGTA by immunoblotting.
Figure S6. Immunofluorescence detection for C4d on neutrophil extracellular trap (NETs). Neutrophils were primed with TNF-α and incubated with 250µg/ml anti-neutrophil cytoplasmic antibody (ANCA)-positive immunoglobulin (Ig)G. The induced NETs were then incubated with normal human serum (NHS) at the presence of ethyleneglycol teraacetic acid (EGTA) or not for 40 min at 37°C. After incubation with serum, coverslips with NETs were washed, and C4d was detected using rabbit anti-human C4d polyclonal antibodies (1 : 100 dilution; Abcam) as primary antibodies with secondary antibodies AF488-labelled donkey anti-rabbit IgG (1 : 500 dilution, Jackson ImmunoResearch Laboratories). NETs were visualized using 4',6-diamidino-2-phenylindole (DAPI) (blue). (a) C4d deposited on NETs during incubation with normal serum. (b) Little or no deposition of C4d on NETs during incubation with normal serum at the presence of EGTA could be observed. Representative images of three independent experiments are shown. Magnification ×200.
Figure S7. Quantification of mean fluorescence intensity of C3b staining on neutrophil extracellular trap (NETs) after incubation with serum. The mean fluorescence intensity (MFI) of C3b staining on NETs incubated with normal human serum (NHS), heat inactivated (Hi)-NHS (lack of active complements), magnesium salt-ethyleneglycol teraacetic acid (Mg-EGTA)-treated NHS, Mg-EGTA-treated Hi-NHS (lack of active complements). Data are presented as mean of three independent experiments ± standard deviation (s.d.). The MFI of C3b on NETs incubated with NHS was 49·74 ± 8·36, while the MFI of C3b on NETs incubated with Hi-NHS was 11·95 ± 2·85 (P<0·001). The MFI of C3b on NETs incubated with Mg-EGTA-treated NHS was 30·96 ± 5·37, while the MFI of C3b on NETs incubated with Mg-EGTA-treated Hi-NHS was 7·14 ± 1·40 (P<0·001).
Figure S8. Neutrophil extracellular trap (NETs) induced by phorbol myristate acetate (PMA) or lipopolysaccharide (LPS) could activate the alternative complement cascade in the serum. Neutrophils were stimulated by 50 nM PMA or 100 ng/ml LPS. Bars represent mean ± standard deviation (s.d.). Each measured on neutrophils of five independent experiments and donors. Supernatants from the cell suspensions were incubated for 40 min with normal serum or magnesium salt-ethyleneglycol teraacetic acid (Mg-EGTA)-treated normal serum, and C3a, C5a, SC5b-9 generation was measured by specific enzyme-linked immunosorbent assay (ELISA). *P < 0·05, **P < 0·01, ***P < 0·001.
Figure S9. Quantification of the generation of C5a in serum after specific inhibition of tissue factor or thrombin. Neutrophils were incubated with anti-TF antibody (10 μg/ml) for 30 min in 37°C or 1 μM D-Phe-Pro-Arg-choro-methylketone dihydrochloride (PPACK) for 40 min at room temperature, and were then stimulated by ANCA-positive immunoglobulin (Ig)G after tumour necrosis factor (TNF)-α-priming or 50 nM phorbol myristate acetate (PMA). Bars represent mean ± standard deviation (s.d.). Each measured on neutrophils of four independent experiments and donors. Supernatants from the cell suspensions were incubated for 40 min with normal serum or magnesium salt-ethyleneglycol teraacetic acid (Mg-EGTA)-treated normal serum, and C5a generation was measured by specific. D-Phe-Pro-Arg-choro-methylketone dihydrochloride (PPACK) was used to inactivate thrombin. Anti-TF antibody (murine monoclonal antibody against human tissue factor) was used to inhibit the pro-coagulant activity of tissue factor; n.s. no significant difference.