

Abstract
Background
Blood platelet numbers are correlated with growth and aggressiveness of several tumor types, including hepatocellular carcinoma (HCC). We previously found that platelet lysates (hPLs) both stimulated HCC cell growth and migration, and antagonized the growth-inhibitory and apoptotic effects of Regorafenib, multikinase growth inhibitor, on HCC cell lines. We evaluated the effects of human insulin-like growth factor-1 (IGF1), a mitogen contained in platelets, on the Regorafenib-mediated growth inhibition.
Methods
An Elisa kit was used to evaluate hPL IGF1 concentrations. The effects of IGF1 on cell proliferation were assessed with MTT assay and analysis of cell cycle progression. Apoptosis assays, scratch assay and Transwell assay were performed to measure apoptosis, cell migration and invasion respectively. Western blots were performed by standard protocols.
Results
IGF1 antagonized growth inhibition exerted by Regorafenib on HCC cell lines. Moreover the mitogen blocked Regorafenib-induced apoptosis and decreased the rate of cell migration and invasion. The IGF1 effects were in turn antagonized by actions of a potent IGF1 receptor inhibitor, GSK1838705A, showing that the IGF1 receptor was involved in the mechanisms of IGF1-mediated blocking of Regorafenib action. GSK1838705A also partially blocked the effects of hPLs in antagonizing Regorafenib-mediated growth inhibition, showing that IGF1 was an important component of hPL actions.
Conclusions
These results show that IGF1 antagonized Regorafenib-mediated growth, migration and invasion inhibition, as well as the drug-mediated induction of apoptosis in HCC cells and reinforce the idea that microenvironmental factors can influence cancer drug actions.
Keywords: Platelets, Insulin growth factor, Regorafenib, HCC cells
Introduction
Hepatocellular carcinoma (HCC) occurs most frequently in livers that have been chronically damaged by hepatitis B or C, chronic alcohol ingestion, or a wide range of chronic metabolic disturbances. Most cases develop in association with liver fibrosis or cirrhosis, especially in the presence of hepatitis C, although chronic infection with hepatitis B can lead to HCC without cirrhosis and HCC [1, 2]. A consequence of the liver fibrosis is portal hypertension, with associated splenomegaly, that can cause thrombocytopenia. The latter has been considered to be a warning sign of impending HCC development in patients with chronic virus hepatitis or cirrhosis [3–5]. Thrombocytopenia-associated HCC has recently been shown to be associated with smaller-size tumors [6, 7]. In contrast, several reports have shown that large size HCCs often have normal or elevated (thrombocytosis) platelet counts [8–13], likely due to less portal hypertension. There are multiple reports of thrombosis and thrombocytosis in various cancer types [14–20].
Platelets have been shown to be involved in tumor metastasis, as well as in HCC growth, in addition to their well-recognized role in blood coagulation [21, 22]. Human platelet lysates (hPL) are a source of many growth factors and have been recently introduced into clinical practice as an adjunct to wound healing [23–27]. We previously found that hPL have anti-apoptotic effects in HCC cells and can antagonize the apoptotic and cell growth inhibitory actions of the multikinase inhibitors, Sorafenib and Regorafenib [28, 29]. The current work extends previous findings, by showing that growth inhibitory actions mediated by Regorafenib [30] in HCC cells in vitro can be antagonized by insulin like growth factor 1 (IGF1), one of the well-described mitogens contained in platelets [25–27]. Furthermore, an IGF1 receptor inhibitor can partially block the drug resistance actions of hPL, supporting the idea that platelet-associated IGF1 may modulate HCC resistance to multikinase inhibitor effects.
Materials and methods
Cells and drugs
Regorafenib was gifts from the Bayer Corp (West Haven, CT, USA), recombinant human IGF1 was purchased from Pepro-Tech (Rocky Hill, NJ, USA), GSK1838705A was purchased from Selleckchem (Houston, TX, USA).
Hep3B, HepG2 and PLC/PRF/5 human HCC cells were purchased from the American Type Culture Collection (ATCC, Rockville, MD, USA). The culture medium was Dulbecco’s Modified Eagle’s Medium (DMEM). All cell culture components were purchased from Sigma- Aldrich (Milan, Italy).
Cell culture
Cells were cultured in DMEM in monolayer culture, and supplemented with 10 % fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin, and incubated at 37 °C in a humidified atmosphere containing 5 % CO2 in air.
Platelet lysates
The hPL were blood bank time-expired bags, from healthy volunteers. The study protocol was approved by the institutional review boards of the University of Bari and “Saverio de Bellis” Institute of Castellana G. (BA), Italy. Additionally, written informed consent was obtained from participants for the use of their blood in this study. The platelet-rich plasma was obtained using an automated hemapheresis procedure in a local blood transfusion centre. The platelets obtained from different volunteers were pooled and then divided into aliquots. Each aliquot was subjected to three freeze-thaw cycles to disrupt their membranes and release the growth factors stored in the granules, producing hPLs.
IGF1 concentrations in platelet lysates
The Human IGF1 ELISA kit (Wuhan Boster Biological Technology LTD, Wuhan, China) was used for the in vitro quantitative determination of human IGF1 in FBS (control) and serial dilution of hPL, according to the user’s guide.
Growth assay
The cells were cultured in 1 % FBS medium containing IGF1 40 ng/ml, the concentration was derived from the IGF1 ELISA dosage in hPL, or hPL corresponding to 3.75 × 107 platelets/ml or equivalent percentage of FBS in presence of 1 μM (HepG2 cells) or 5 μM (Hep3B and PLC/RFP/5) of Regorafenib. In the same growth condition, HCC cells were cultured in absence or presence of IGFR inhibitor, GSK1838705A 1 μM. After defined incubation times, the proliferative response was estimated by colorimetric 3-(4,5 di-methylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) test. The trypan blue exclusion assay was used to evaluate cell viability. Each experiment was performed in triplicate and repeated three times.
Cell cycle analysis
PLC/PRF/5 were synchronized by using thymidine 0.2 M added to the medium. After 18 h of incubation, the medium containing thymidine was replaced with fresh medium for 9 h, and then cells were treated with thymidine for an additional 17 h. Cells were separated into two groups: one group was collected for cell cycle analysis and the other one continued culturing; Regorafenib 5 μM, IGF1 40 ng/ml and GSK1838705A 1 μM were added, and after 6 h of treatment cells were collected to be processed, according to the user’s guide, with the Muse Cell Cycle Kit (Millipore, Darmstadt, Germany) which determines the percentage of cells in the G0/G1, S and G2/M phases of cell cycle with the Muse Cell Analyzer.
Migration assay
A scratch assay was performed as previously described [31, 32]. Briefly, a wound was generated with a pipette tip, after rinsing, medium containing IGF1 40 ng/ml or 1 % FBS (control) alone or in combination with Regorafenib 1 μM and/or GSK1838705A 1 μM. Photographs were taken of each well immediately (T0) and after 24 h (T1), 48 h (T2) and 72 h (T3). The values were expressed as percentage of migration, with 100 % being when the wound was completely closed. The results were representative of three independent experiments.
Invasion assay
Cell invasion assays were performed using Matrigel (BD Transduction, San Jose, CA, USA)-coated Transwells (8 μm pore PET membrane, Millipore, Billerica, MA, USA) as previously described [31]. Briefly, Regorafenib 5 μM and/or GSK1838705A 1 μM treated cells were suspended in low serum medium. Medium containing IGF 40 ng/ml or FBS was added to the bottom wells. After incubation of 24 h, the invading cells were fixed and stained. The images were acquired and analyzed counting the cells with Image J Software (National Institute of Health, USA). Values obtained were expressed as percentage of invading cells, setting the cell counts of control cells as 100 %. Results were representative of three independent experiments.
Apoptosis assays - Annexin V
The Muse Annexin V/Dead Cell Assay Kit (Millipore, Darmstadt, Germany) for quantitative analysis of live, early/ late apoptotic and dead cells was used with a Muse Cell Analyzer (Millipore). Briefly, the assay utilizes Annexin V to detect PS on the external membrane of apoptotic cells. A dead cell marker (7-AAD) is also used. PLC/PRF/5 cell line, including positive and negative controls, were cultured in 1 % FBS medium in presence of Regorafenib 5 μM alone (control cells) or in combination with IGF1 40 ng/ml or IGF1 40 ng/ml and GSK1838705A 1 μM for 48 h. The cells were then processed as described in the user’s guide.
Western blots
MAPK signaling and anti-apoptosis markers in cells treated with Regorafenib 5 μM and IGF1 40 ng/mL were analyzed by Western blot, as previously described [31]. Briefly, cells were washed twice with cold PBS and then lysed in RIPA buffer (Sigma-Aldrich, Milan; Italy). After quantization of protein concentration, equal amount of protein (50 μg) were resolved on SDS–PAGE and transferred to polyvinyldifluoride (PVDF) filters. The blots were blocked with 5 % (w/v) nonfat dry milk for 2 h at room temperature and then probed with primary antibody overnight at 4 °C.
The primary antibodies were directed against the following proteins: IGFR-1 and phospho-IGFR-1 (p-IGFR-1), ERK and phospho-ERK (p-ERK), BRAF and phospho-BRAF (p-BRAF), c-Myc and phospho-c-Myc (p-c-Myc), JNK and phospho-JNK (p-JNK), STAT3 and phospho-STAT3 (Tyr705, Ser727) (p-STAT3), p38 and phospho-p38 (p-p38), Bim, Bid, Bad, phospho-survivin (p-survivin), survivin, Bcl-xL, Bcl-2 and β-actin (Cell Signaling, Beverly, MA, USA). After three washes, incubation was followed by the reaction with horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature. The immunoreactive bands were visualized and analyzed using enhanced chemiluminescence detection reagents, according to the manufacturer’s instructions, and chemiluminescence detection system (ChemiDoc XRS apparatus and software, Bio-Rad, Milan, Italy).
Statistical analysis
GraphPad Prism 5.0 software (La Jolla, CA, USA) was used for all statistical analysis. Mann–Whitney nonparametric test was employed to assess the statistical significance of differences between two groups. For multiple comparisons was used one-way Anova test followed by appropriate post-test. p < 0.05 was considered statistically significant. All experiments were done in triplicate and data are presented as mean ± standard deviation (SD).
Results
Antagonism by IGF1 of Regorafenib-mediated inhibition of HCC cell growth
hPL were previously examined for the ability to antagonize Regorafenib-mediated inhibition of human HCC cell line growth [29]. To further investigate the role of IGF1, one of the known platelet growth factors, in antagonizing Regorafenib actions on HCC cells, we initially measured IGF1 levels in hPL as described in Materials and Methods. The results showed concentrations of 40 ng/ml of IGF1 were present in hPL, corresponding to 3.75 × 107 platelets/ml. This IGF1 concentration range was used in the subsequent experiments.
Hep3B, PLC/PRF/5 and HepG2 human HCC cell lines were treated in log phase growth in culture dishes with Regorafenib 1–5 μM or IGF1 40 ng/ml alone or in combination with appropriate controls and proliferation was evaluated by MTT assay.
We found that IGF1 significantly antagonized the growth inhibitory actions of Regorafenib. We found that in all HCC cell lines that were examined, IGF1, added in combination with Regorafenib, significantly increased the proliferation rate of about 60 %, compared to the same cells treated only with Regorafenib. This effect was only partial because the proliferation rate of IGF1/R treated cells was still lower than both IGF1 (42 %) and control (15 %) cells and was blocked by GSK1838705A, a potent inhibitor of IGF1 receptor, used at a non- toxic concentration (1 μM) that did not by itself affect proliferation (Fig. 1a).
Fig. 1.
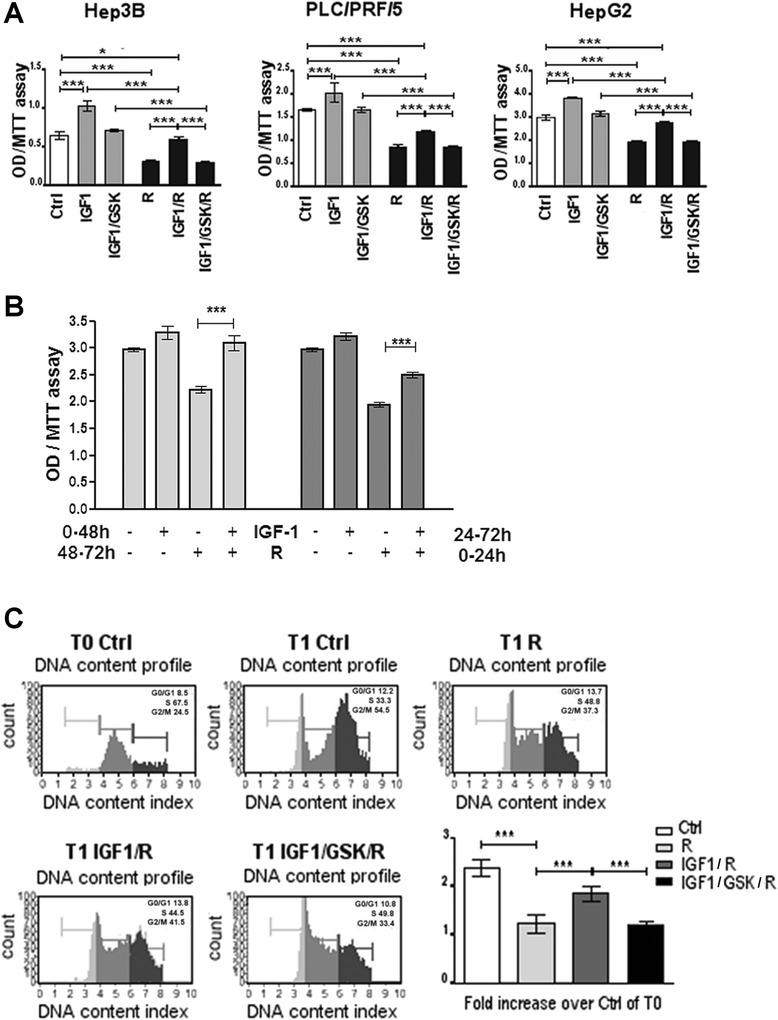
Antagonism by IGF1 of Regorafenib-mediated growth inhibition of HCC cell Lines. a. Hep3B, PLC/PRF/5 and HepG2 cells were cultured in presence of Regorafenib 1–5 μM, IGF1 40 ng/ml, GSK1838705A 1 μM. MTT assay was assessed after 48 h (48 h). b. PLC/PRF/5 cells were cultured in presence of Regorafenib 5 μM and IGF1 40 ng/ml in different time conditions. In the first group (light gray) the cells previously treated with IGF1 for 48 h received Regorafenib for the following 24 h. In the second group (dark gray) the cells, previously treated with Regorafenib for 24 h, received IGF for the following 48 h. MTT assay was assessed after 72 h. c. PLC/PRF/5 cells were synchronized in the S phase of the cell cycle using thymidine 0.2 M (T0), after 6 h from block release (T1), the cells were processed with the Cell Cycle Kit and analyzed with Muse Cell Analyzer to evaluate the percentage of cells in G0/G1, S and G2/M phases. The panels represent an example of DNA content profile in different treatment conditions. The mean of three independent experiments was plotted in the relative graph, where the values are calculated as fold increase of the cells in G2/M at T1 respect to control cells at T0. The results of three independent experiments are expressed as means ± SD. ***p < 0.0001
We next investigated if the timing of the IGF1 addition might affect Regorafenib-mediated growth inhibition. For this purpose, two different culture conditions were used. In the first condition, cells that had been previously cultured for 48 h with IGF1 40 ng/ml or equivalent percentage of FBS (control) were then treated with Regorafenib 5 μM for the next 24 h. In the second condition, cells that had been pre-treated for 24 h with Regorafenib were subsequently cultured for the next 48 h in the presence of IGF1 or FBS. In Regorafenib-treated cells, IGF1 pretreatment led to an increase in the proliferation rate (40 %) compared to the proliferation rate observed in cells treated only with drug (Fig. 1b). Moreover, IGF1 partially reversed (28 %) the Regorafenib-mediated growth inhibition if added after drug administration. These results confirmed our previous findings in which the reversible effect of Regorafenib was shown [33].
The antagonism exerted by IGF1 on Regorafenib-mediated growth inhibitory actions was also observed on cell cycle progression. Regorafenib caused an inhibition in the progression from S phase of the cell cycle to G2/M phase. After 6 h (T1) from block release (T0), Regorafenib treated cells in G2/M phase were only 0.2 times more than the control cells at T0, while the number of control cells in G2/M phase at T1 were 1.5 times more with respect the number of control cells at T0 (Fig. 1c). IGF1 counteracted the Regorafenib-mediated block in cell cycle progression (0.85 times more than the control cells at T0), and the IGFR antagonist, GSK1838705A, abrogated this effect.
Antagonism by IGF1 of Regorafenib-mediated induction of apoptosis
The effects of IGF1 on Regorafenib–mediated apoptosis, a major aspect of its growth-inhibitory effects, were then examined. Regorafenib induced an increase in cellular Annexin V. When IGF1 was also added to the cell medium together with Regorafenib, a pronounced and significant antagonism of apoptosis induction was found and this antagonism was abrogated by the IGFR inhibitor GSK1838705A 1 μM (Fig. 2a). Major apoptosis markers in cells treated with Regorafenib alone or in combination with IGF1 were then examined. We found that pro-apoptotic marker (Bim, tBid and Bad) levels increased in the presence of Regorafenib alone and anti-apoptotic markers (p-survivin, Bcl-xL and Bcl-2) decreased under the same conditions. However, in cells treated with Regorafenib in combination with IGF1 (Fig. 2b), we found that IGF1 antagonized these Regorafenib effects on induction of apoptosis.
Fig. 2.
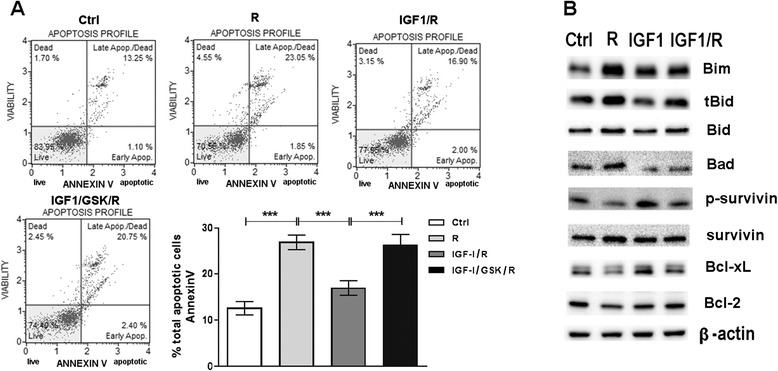
Antagonism by IGF1 of Regorafenib-mediated induction of apoptosis. a. PLC/PRF/5 cell line cultured in 1 % FBS medium was treated with IGF1 40 ng/ml in combination with Regorafenib 5 μM and GSK 1 μM. The Muse Annexin V kit was used to evaluate the percentage of apoptotic cells. The means ± SD of three independent experiments is plotted in the relative graph. ***p < 0.0001. b. Representative Western blot that shows anti-apoptotic (phospho-survivin, Bcl-xL and Bcl-2) and pro-apoptotic (Bim, truncated-Bid and Bad) proteins in PLC/PRF/5 cells treated as described above
Antagonism by IGF-1 of Regorafenib-mediated inhibition of cell migration and invasion
Regorafenib inhibits both HCC cell migration, as well as cell invasion through Matrigel membranes [31]. IGF1 40 ng/ml was then added to cells in the presence of Regorafenib 5 μM, a concentration that can inhibit both migration and invasion in HCC cells. We found that IGF1 antagonized the inhibition by Regorafenib of both migration and invasion and these effects were also abrogated by GSK1838705A 1 μM (Fig. 3a/b).
Fig. 3.
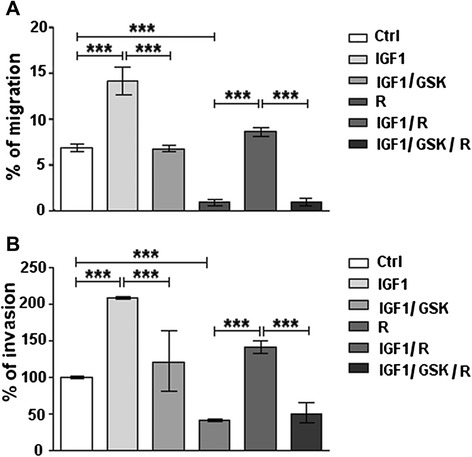
Antagonism by IGF1 of regorafenib-mediated inhibition of migration and invasion. PLC/PRF/5 cell line cultured in 1 % FBS medium was treated with IGF1 40 ng/ml alone or in combination with Regorafenib 5 μM and GSK 1 μM. a. Migration assay was performed as described and the microscopic analysis was assessed at the time of the scratch (T0) and after 48 h (T2). The values were expressed as percentage of migration, where 100 % represents the scratch completely closed. b. The percentage of invasion was calculated comparing the invading drug-treated cells to drug-untreated control cells (100 %). The results of three independent experiments are expressed as means ± SD. ***p < 0.0001
IGF-1 as a major contributor to hPL actions
The above results using GSK1838705A, showed that IGF1 receptors were involved in the mechanisms of IGF1-mediated antagonism of Regorafenib effects. We then examined the possibility that the blocking effects of hPL on Regorafenib that had been previously noted, might be attributable mostly to IGF1 presence in platelet lysates [28]. We found that the platelet lysates increased the proliferative rate of Regorafenib-treated cells by 84 %. This effect was attributed to IGF1 action for 41 %, as demonstrated adding GSK1838705A 1 μM to the platelet lysates, and the remaining 43 % was due to the combined effects exerted by all the other growth factors included in platelets (Fig.4).
Fig. 4.
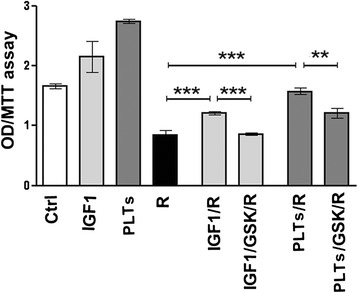
Effects of IGF1 receptor (IGFR1) inhibitor (GSK1838705A) on Regorafenib-mediated growth inhibition in PLC/PRF/5 cells. PLC/PRF/5 cells were cultured in 1 % FBS medium in presence of IGF1 40 ng/ml or platelet lysate corresponding to 3.75 × 107 platelets/ml, Regorafenib 5 μM and GSK1838705A 1 μM, using the conditions indicated in the graph. MTT assay was assessed after 48 h. The results of three independent experiments are expressed as mean ± SD. **p < 0.001; ***p < 0.0001
Effects of IGF1 on cell signaling
Regorafenib has previously been shown [28] to cause a decrease in p-ERK levels, consequent on Raf inhibition. We next examined the effects of Regorafenib on cellular MAPK signaling pathway marker levels in cells that were treated in the absence or presence of IGF1. We found that IGF1 antagonized the Regorafenib-mediated decrease in p-BRAF and p-ERK levels, and also antagonized the decreases in the levels of p-c-Myc, p-p38 and p-STAT3 (Tyr705, Ser727). By contrast, the Regorafenib-mediated increases in p-JNK levels were decreased by the addition of IGF1 to the cultures (Fig. 5a). These actions of IGF1 were in turn blocked by the presence in the cultures of GSK1838705A, indicating the involvement of the IGF1 receptors. IGF1 thus counteracted Regorafenib actions on signaling intracellular markers (Fig. 5b/c).
Fig. 5.
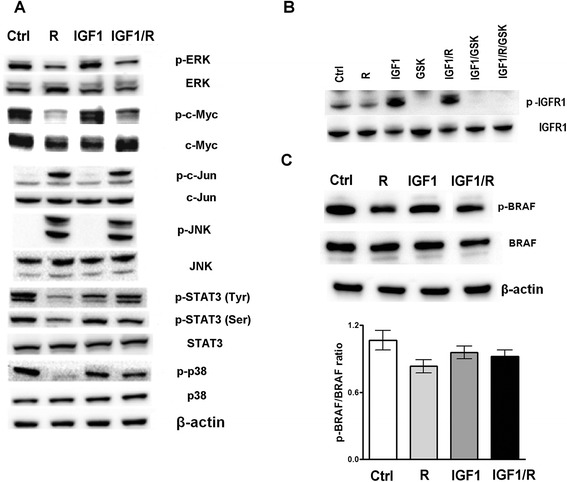
MAPK signaling and IGFR1 expression in PLC/PRF/5 cells. a. Western blot of MAPK proteins that shows IGF1 antagonism effect on Regorafenib action in PLC/PRF/5 cells. b. Activated IGF1 receptor (IGFR1) after treatment with Regorafenib (R), IGF1, IGF1 + R, IGF1 + GSK and IGF1 + R + GSK, respectively. c. IGF1 effect on the BRAF protein that is direct Regorafenib target
Discussion
Platelets and their contained growth factors have become of great interest in tumor biology, due to their contribution to the tumor microenvironment, their content of many growth and inflammatory factors, and to their identification as important contributors to tumor growth and metastasis, including HCC, as well as to normal liver regeneration [21, 22, 34–36]. Amongst the many factors in platelet granules, IGF1 has received considerable attention, due to its involvement in HCC growth and prognosis [37–39] and as a possible target of anti-cancer and anti-HCC therapies.
Regorafenib is a multikinase inhibitor and its action is similar of Sorafenib [40]. Sorafenib is a small multi-target inhibitor that has broad-spectrum activity against several tyrosine kinases. Although initially described as a b-Raf inhibitor, Sorafenib inhibits the activity of several cell surface receptor tyrosine kinases [40]. A constitutive activation of receptor tyrosine kinases is a common feature of many types of cancers, particularly those of soft tissue origin where their concerted expression has been shown to promote tumor growth and survival [41]. Regorafenib blocks ERK action, an intracellular signaling involved in HCC cell proliferation [32].
Also Regorafenib has an indirect effect of p38 kinase, JNK and Stat3 involved in tumorigenesis and HCC proliferation. In this study we explored the action of IGF1 in HCC cells in vitro. We found that IGF1 antagonized the growth inhibitory effects of Regorafenib. The insulin-like growth factor (IGF) pathway has highly conserved functions in mammals and plays a critical role in energy metabolism and cell renewal in response to nutrients. IGF pathway is not only involved in cell growth in tissue culture, but it also promotes cell proliferation, migration and transformation into malignancy [42]. The anti-apoptotic property of IGF-1R was shown in its response to p53, the tumor suppressor gene that promotes apoptosis [42]. Wild type p53 expression inhibited the gene expression of IGF-1R, while mutant p53 increased the gene expression of IGF-1R. Oncogenes such as Src kinase and Akt kinase both stimulated the gene expression of IGF-1R, providing more evidence that IGF-1R is vital in carcinogenesis [43]. In addition, IGF-1R also stimulates cell mobility, as demonstrated by its activity in melanoma cell lines. Another important role of IGF-1R in carcinogenesis is its ability to transform and maintain the transformed phenotype [42]. IGF1 and its binding to its receptor induce activations of two major intracellular cascades, the phosphatidyl inositol 3-kinase (PI3K) and the mitogen-activated protein kinase (MAPK), both of which result in cell differentiation, proliferation and anti-apoptosis [43]. Particularly in HCC, IGF-1R is overexpressed and can induce carcinogenesis. In a study where 10 HCC cell lines (including PLC/PRF/5 cell line) were tested, all of them showed elevated IGF-1R mRNA. Furthermore, the addition of IGF-1 to the PLC/PRF/5 cell line induced increased cell proliferation in a dose dependent manner, showing that the major tumor promoting effects of IGF ligands on HCC are exerted through IGF-1R [42–44].
The IGF1 signaling pathway provides an important regulatory mechanism for tumorigenesis and drug resistance in HCC [41].
We found that IGF1, used in the same concentration as was measured in hPL, significantly antagonized the growth inhibitory actions of Regorafenib. This effect was blocked by GSK1838705A, a potent inhibitor of IGF1 receptors, used at non-toxic concentrations that do not affect proliferation. We next found that IGF pre-treatment protected the cells from subsequent addition of Regorafenib to the cultures and antagonized Regorafenib-mediated growth inhibition, which was reduced by 40 % when the cells were pre-treated with IGF1. This suggested that IGF1 signalling is implicated in the observed Regorafenib resistance or that a major mechanism of Regorafenib-mediated growth control is exerted through interference in the IGFR pathway, which is then counteracted by addition of IGF1 [42]. Moreover, the Regorafenib pre-treatment modified the stimulatory action ensuing to the addition of the IGF1. Regorafenib-mediated inhibition of cell growth was only partially rescued by subsequent IGF1 treatment (28 %), showing that Regorafenib treatment, even if reversible, modified the cells [43].
A corollary is that IGFR inhibitors might enhance the growth inhibitory actions of Regorafenib, since IGF1 can block the drug effects on cell growth. Our results on cell cycle progression support the idea of antagonism exerted by IGF on Regorafenib-mediated growth inhibition.
An important mechanism of resistance to IGFR inhibitors is the compensatory activation of related signalling pathways [44]. We observed that IGF1 changed intracellular signalling in HCC cells. We found that levels of the proliferation markers p-BRAF, p-ERK, p-p38 and p-Stat3 (Tyr705, Ser727) were decreased by Regorafenib action. By contrast, IGF1 addition to the cultures antagonized Regorafenib action and levels of the proliferation markers increased. We, also, found that levels of apoptosis markers were influenced by IGF1 actions, since pro-apoptotic markers (p-JNK, p-c-Jun, Bim, Bad and Bid) decreased with IGF1, while anti-apoptosis markers (survivin, BCL-xL and Bcl-2) increased. Survivin is an apoptosis-inhibitory protein that is over-expressed in multiple cancer types, including HCC, plays critical roles in regulating apoptosis, cell proliferation and survival and has been shown to be a direct downstream target of IGF1 pathway [44].
Regorafenib decreased p-survivin levels and this was also antagonized by addition of IGF1. GSK1838705A reversed the IGF1 actions, showing that the antagonist effects of IGF are exerted through its receptor. The antagonism exerted by IGF on Regorafenib effects was also shown for drug mediated apoptosis, as a decrease in Annexin V levels and in the Caspase 3/7 activation. Pre-clinical studies have shown that the efficacy in anti-cancer therapy for HCC can be improved by inhibiting the IGF signalling pathway in HCC cells [45].
Several new inhibitors of IGF1 or its receptor are in current clinical trials for HCC, including Octreotide (Novartis), a Somatostatin analog and MEDI 573 (Astrazeneca), targeting either IGF1 and or IGFII, as well as Linsitinib (OSI Pharmaceuticals), Cixutumumab (ImClone), AVE1642 (Sanofi-Aventis), which are IGF receptor antagonists. However, the relationship of IGF1 levels in serum and tumor tissue and of its receptor levels with HCC growth and prognosis are very complex [37]. It has recently been shown that one of the many mechanisms of Sorafenib (Regorafenib is Fluoro-Sorafenib) action on HCC cell growth is via an inhibition of IGF1 [39]. Furthermore, IGF1 has been shown to be involved in resistance to cytotoxic chemotherapy by several drugs and on a variety of tumor types, mainly through an anti-apoptotic effect [33, 46–51], as we have found in the current work.
Conclusion
These experiments highlight the importance of the microenvironment, including IGF1, in modulating the growth inhibitory effects of anti-HCC therapeutic drugs. By contrast, anti-IGF or anti-IGF receptor agents might be predicted to be therapeutically useful in enhancing the activity of anti-HCC therapies. Finally, we re-enforce the concept of drug resistance mediated by factors in the tumor microenvironment, and also summarize potential drug targets based on the current knowledge of the tumor microenvironment [52].
Acknowledgments
This research was supported by Italian Ministry of Public Health.
Abbreviations
- HCC
Hepatocellular carcinoma
- hPL
Human platelet lysates
- ERK
Extracellular signal-regulated kinase
- JNK
c-Jun NH2-terminal kinase
- STAT
Signal transducer and activator of transcription-3
- WB
Western blot
- MTT
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- BRdU
BrdU 5-bromo-2’-deoxy-uridine
- IGF1
Insulin like growth factor 1
Footnotes
Competing interest
The authors declare that they have no competing interests.
Authors’ contribution
BIC and CL are fully responsible for study designing, drafting and finalizing the manuscript; MGR and RD’A executed in vitro experiments; NC and CL carried out protein measurement by Western blot; AC and CM conducted statistical analysis and participated in coordination manuscript providing important suggestions. All authors read and approved the final manuscript.
Contributor Information
Catia Lippolis, Email: catia.lippolis20@libero.it.
Maria Grazia Refolo, Email: mgrefolo@libero.it.
Rosalba D’Alessandro, Email: dalessandro.rosalba@libero.it.
Nicola Carella, Email: nicolacarella@gmail.com.
Caterina Messa, Email: caterina.messa@irccsdebellis.it.
Aldo Cavallini, Email: cavaldo@libero.it.
Brian Irving Carr, Phone: +1 412 980 4518, Email: brianicarr@hotmail.com.
References
- 1.Nordenstedt H, White DL, El-Serag HB. The changing pattern of epidemiology in hepatocellular carcinoma. Dig Liver Dis. 2010;42:S206–14. doi: 10.1016/S1590-8658(10)60507-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carr BI. Understanding Liver Cancer: a tale of two diseases. New York: Springer Press; 2014. [Google Scholar]
- 3.Lu SN, Wang JH, Liu SL, Hung CH, Chen CH, Tung HD, et al. Thrombocytopenia as a surrogate for cirrhosis and a marker for the identification of patients at high-risk for hepatocellular carcinoma. Cancer. 2006;107:2212–22. doi: 10.1002/cncr.22242. [DOI] [PubMed] [Google Scholar]
- 4.Kumada T, Toyoda H, Kiriyama S, Sone Y, Tanikawa M, Hisanaga Y, et al. Incidence of hepatocellular carcinoma in patients with chronic hepatitis B virus infection who have normal alanine aminotransferase values. J Med Virol. 2010;82:539–45. doi: 10.1002/jmv.21686. [DOI] [PubMed] [Google Scholar]
- 5.Lok AS, Seeff LB, Morgan TR, di Bisceglie AM, Sterling RK, Curto TM, et al. HALT-C Trial Group. Incidence of hepatocellular carcinoma and associated risk factors in hepatitis C-related advanced liver disease. Gastroenterology. 2009;136:138–48. doi: 10.1053/j.gastro.2008.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carr BI, Guerra V, Pancoska P. Thrombocytopenia in relation to tumor size in patients with hepatocellular carcinoma. Oncology. 2012;83:339–45. doi: 10.1159/000342431. [DOI] [PubMed] [Google Scholar]
- 7.Carr BI, Guerra V, De Giorgio M, Fagiuoli S, Pancoska P. Small hepatocellular carcinomas and thrombocytopenia. Oncology. 2012;83:331–8. doi: 10.1159/000341533. [DOI] [PubMed] [Google Scholar]
- 8.Trevisani F, D’Intino PE, Caraceni P, Pizzo M, Stefanini GF, Mazziotti A, et al. Etiologic factors and clinical presentation of hepatocellular carcinoma. Differences between cirrhotic and noncirrhotic Italian patients. Cancer. 1995;75:2220–32. doi: 10.1002/1097-0142(19950501)75:9<2220::AID-CNCR2820750906>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 9.Truant S, Boleslawsky E, Duhamel A, Bouras AF, Louvet A, Febvay C, et al. Tumor size of hepatocellular carcinoma in noncirrhotic liver: A controversial predictive factor for outcome after resection. Eur J Surg Oncol. 2012;38:1189–96. doi: 10.1016/j.ejso.2012.07.112. [DOI] [PubMed] [Google Scholar]
- 10.Carr BI, Guerra V, Giannini EG, Farinati F, Ciccarese F, Rapaccini GL, et al. Significance of platelet and AFP levels and liver function parameters for HCC size and survival. Int J Biol Markers. 2014;29:e215–23. doi: 10.5301/jbm.5000092. [DOI] [PubMed] [Google Scholar]
- 11.Carr BI, Guerra V. Features of massive hepatocellular carcinomas. Eur J Gastroenterol Hepatol. 2014;26:101–8. doi: 10.1097/MEG.0b013e3283644c49. [DOI] [PubMed] [Google Scholar]
- 12.Carr BI, Guerra V. Thrombocytosis and hepatocellular carcinoma. Dig Dis Sci. 2013;58:1790–6. doi: 10.1007/s10620-012-2527-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hwang SJ, Luo JC, Li CP, Chu CW, Wu JC, Lai CR, et al. Thrombocytosis: a paraneoplastic syndrome in patients with hepatocellular carcinoma. World J Gastroenterol. 2004;10:2472–7. doi: 10.3748/wjg.v10.i17.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levin J, Conley CL. Thrombocytosis associated with malignant disease. Arch Int Med. 1964;114:497–500. doi: 10.1001/archinte.1964.03860100079008. [DOI] [PubMed] [Google Scholar]
- 15.Stone RL, Nick AM, McNeish IA, Balkwill F, Han HD, Bottsford-Miller J, et al. Paraneoplastic thrombocytosis in ovarian cancer. N Engl J Med. 2012;366:610–8. doi: 10.1056/NEJMoa1110352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Voutsadakis IA. Thrombocytosis as a prognostic marker in gastrointestinal cancers. World J Gastrointest Oncol. 2014;6:34–40. doi: 10.4251/wjgo.v6.i2.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li FX, Wei LJ, Zhang H, Li SX, Liu JT. Significance of thrombocytosis in clinicopathologic characteristics and prognosis of gastric cancer. Asian Pac J Cancer Prev. 2014;15:6511–7. doi: 10.7314/APJCP.2014.15.16.6511. [DOI] [PubMed] [Google Scholar]
- 18.Kim M, Chang H, Yang HC, Kim YJ, Lee CT, Lee JH, et al. Preoperative thrombocytosis is a significant unfavorable prognostic factor for patients with resectable non-small cell lung cancer. World J Surg Oncol. 2014;12:37. doi: 10.1186/1477-7819-12-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Njolstad TS, Engerud H, Werner HM, Salvesen HB, Trovik J. Preoperative anemia, leukocytosis and thrombocytosis identify aggressive endometrial carcinomas. Gynecol Oncol. 2013;131:410–5. doi: 10.1016/j.ygyno.2013.08.032. [DOI] [PubMed] [Google Scholar]
- 20.Goubran HA, Stakiw J, Radosevic M, Burnouf T. Platelets effects on tumor growth. Semin Oncol. 2014;41:359–69. doi: 10.1053/j.seminoncol.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 21.Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell. 2011;20:576–90. doi: 10.1016/j.ccr.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Everts PA, Brown Mahoney C, Hoffmann JJ, Schönberger JP, Box HA, van Zundert A, et al. Platelet-rich plasma preparation using three devices: implications for platelet activation and platelet growth factor release. Growth Factors. 2006;24:165–71. doi: 10.1080/08977190600821327. [DOI] [PubMed] [Google Scholar]
- 23.Christgau M, Moder D, Hiller KA, Dada A, Schmitz G, Schmalz G. Growth factors and cytokines in autologous platelet concentrate and their correlation to periodontal regeneration outcomes. J Clin Periodontol. 2006;33:837–45. doi: 10.1111/j.1600-051X.2006.00991.x. [DOI] [PubMed] [Google Scholar]
- 24.Eppley BL, Woodell JE, Higgins J. Platelet quantification and growth factor analysis from platelet-rich plasma: implications for wound healing. Plast Reconstr Surg. 2004;114:1502–8. doi: 10.1097/01.PRS.0000138251.07040.51. [DOI] [PubMed] [Google Scholar]
- 25.Weibric G, Buch RS, Kleis WK, Hafner G, Hitzler WE, Wagner W. Quantification of thrombocyte growth factors in platelet concentrates produced by discontinuous cell separation. Growth Factors. 2002;20:93–7. doi: 10.1080/08977190290031950. [DOI] [PubMed] [Google Scholar]
- 26.Karey KP, Marquardt H, Sirbasku DA. Human platelet-derived mitogens. Identification of insulin like growth factors I and II by purification and N alpha amino acid sequence analysis. Blood. 1989;74:1084–92. [PubMed] [Google Scholar]
- 27.Carr BI, Cavallini A, D'Alessandro R, Refolo MG, Lippolis C, Mazzocca A, et al. Platelet extracts induce growth, migration and invasion in human hepatocellular carcinoma in vitro. BMC Cancer. 2014;14:43. doi: 10.1186/1471-2407-14-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.D'Alessandro R, Refolo MG, Lippolis C, Giannuzzi G, Carella N, Messa C, et al. Antagonism of Sorafenib and Regorafenib actions by platelet factors in hepatocellular carcinoma cell lines. BMC Cancer. 2014;14:351. doi: 10.1186/1471-2407-14-351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ettrich TJ1, Seufferlein T. Regorafenib. Recent Results. Cancer Res. 2014;201:185–96. doi: 10.1007/978-3-642-54490-3_10. [DOI] [PubMed] [Google Scholar]
- 30.Sartore-Bianchi A1, Zeppellini A, Amatu A, Ricotta R, Bencardino K, Siena S. Regorafenib in metastatic colorectal cancer. Expert Rev Anticancer Ther. 2014;14:255–65. doi: 10.1586/14737140.2014.894887. [DOI] [PubMed] [Google Scholar]
- 31.Carr BI, D’Alessandro R, Refolo MG, Iacovazzi PA, Lippolis C, Messa C, et al. Effects of low concentrations of Regorafenib and Sorafenib on human HCC cell AFP, migration, invasion, and growth In Vitro. J Cell Physiol. 2013;228:1344–50. doi: 10.1002/jcp.24291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carr BI, Cavallini A, Lippolis C, D'Alessandro R, Messa C, Refolo MG, et al. Fluoro Sorafenib (Regorafenib) effects on hepatoma cells: growth inhibition, quiescence, and recovery. J Cell Physiol. 2013;228:292–7. doi: 10.1002/jcp.24148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.D'Alessandro R, Refolo MG, Lippolis C, Messa C, Cavallini A, Rossi R, et al. Reversibility of Regorafenib effects in hepatocellular carcinoma cells. Cancer Chemother Pharmacol. 2013;72:869–77. doi: 10.1007/s00280-013-2269-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morimoto Y, Nouso K, Wada N, Takeuchi Y, Kinugasa H, Miyahara K, et al. Involvement of platelets in extrahepatic metastasis of hepatocellular carcinoma. Hepatol Res. 2014;44:e353–9. doi: 10.1111/hepr.12315. [DOI] [PubMed] [Google Scholar]
- 35.Hoshi R, Murata S, Matsuo R, Myronovych A, Hashimoto I, Ikeda H, et al. Freeze-dried platelets promote hepatocyte proliferation in mice. Cryobiology. 2007;55:255–60. doi: 10.1016/j.cryobiol.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 36.Enguita-Germán M, Fortes P. Targeting the insulin-like growth factor pathway in hepatocellular carcinoma. World J Hepatol. 2014;6:716–37. doi: 10.4254/wjh.v6.i10.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaseb AO, Morris JS, Hassan MM, Siddiqui AM, Lin E, Xiao L, et al. Clinical and prognostic implications of plasma insulin-like growth factor-1 and vascular endothelial growth factor in patients with hepatocellular carcinoma. J Clin Oncol. 2011;29:3892–9. doi: 10.1200/JCO.2011.36.0636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shao YY, Huang CC, Lin SD, Hsu CH, Cheng AL. Serum insulin- like growth factor-1 levels predict outcomes of patients with advanced hepatocellular carcinoma receiving antiangiogenic therapy. Clin Cancer Res. 2012;18:3992–7. doi: 10.1158/1078-0432.CCR-11-2853. [DOI] [PubMed] [Google Scholar]
- 39.Sprinzl MF, Puschnik A, Schlitter AM, Schad A, Ackermann K, Esposito I, et al. Sorafenib inhibits macrophage-induced growth of HCC cells by interference with insulin-like growth factor-1 secretion. J Hepatol. 2014;62:863–70. doi: 10.1016/j.jhep.2014.11.011. [DOI] [PubMed] [Google Scholar]
- 40.Maruwge W, D’Arcy P, Folin A, Brnjic S, Wejde J, Davis A, et al. Sorafenib inhibits tumor growth and vascularization of rhabdomyosarcoma cells by blocking IGF-1R-mediated signaling. Onco Targets Ther. 2008;1:67–78. doi: 10.2147/OTT.S3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Caro JF, Poulos J, Ittoop O, Pories WJ, Flickinger EG, Sinha MK. Insulin-like growth factor I binding in hepatocytes from human liver, human hepatoma, and normal, regenerating, and fetal rat liver. J Clin Invest. 1988;81:976–81. doi: 10.1172/JCI113451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tovar V, Alsinet C, Villanueva A, Hoshida Y, Chiang DY, Sole M, et al. IGF activation in a molecular subclass of hepatocellular carcinoma and pre-clinical efficacy of IGF-1R blockage. J Hepatol. 2010;52:550–9. doi: 10.1016/j.jhep.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ou DL, Lee BS, Lin LI, Liou JY, Liao SC, Hsu C, et al. Vertical blockade of the IGFR- PI3K/Akt/mTOR pathway for the treatment of hepatocellular carcinoma: the role of survivin. Mol Cancer. 2014;13:2. doi: 10.1186/1476-4598-13-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gao J, Chang YS, Jallal B, Viner J. Targeting the insulin-like growth factor axis for the development of novel therapeutics in oncology. Cancer Res. 2012;72:3–12. doi: 10.1158/0008-5472.CAN-11-0550. [DOI] [PubMed] [Google Scholar]
- 45.Scharf JG, Schmidt-Sandte W, Pahernik SA, Ramadori G, Braulke T, Hartmann H. Characterization of the insulin-like growth factor axis in a human hepatoma cell line (PLC) Carcinogenesis. 1998;19:2121–8. doi: 10.1093/carcin/19.12.2121. [DOI] [PubMed] [Google Scholar]
- 46.Wu J, Zhu AX. Targeting insulin-like growth factor axis in hepatocellular carcinoma. J Hematol Oncol. 2011;4:30. doi: 10.1186/1756-8722-4-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pal S, Shankar BS, Sainis KB. Cytokines from the tumor microenvironment modulate sirtinol cytotoxicity in A549 lung carcinoma cells. Cytokine. 2013;64:196–207. doi: 10.1016/j.cyto.2013.07.029. [DOI] [PubMed] [Google Scholar]
- 48.Sun Y, Zheng S, Torossian A, Speirs CK, Schleicher S, Giacalone NJ, et al. Role of insulin-like growth factor-1 signaling pathway in cisplatin-resistant lung cancer cells. Int J Radiat Oncol Biol Phys. 2012;82:e563–72. doi: 10.1016/j.ijrobp.2011.06.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Juan HC, Tsai HT, Chang PH, Huang CY, Hu CP, Wong FH. Insulin-like growth factor 1 mediates 5-fluorouracil chemoresistance in esophageal carcinoma cells through increasing survivin stability. Apoptosis. 2011;16:174–83. doi: 10.1007/s10495-010-0555-z. [DOI] [PubMed] [Google Scholar]
- 50.Zhang YW, Yan DL, Wang W, Zhao HW, Lu X, Wu JZ, et al. Knockdown of insulin-like growth factor I receptor inhibits the growth and enhances chemo-sensitivity of liver cancer cells. Curr Cancer Drug Targets. 2012;12:74–84. doi: 10.2174/156800912798888974. [DOI] [PubMed] [Google Scholar]
- 51.Eckstein N, Servan K, Hildebrandt B, Pölitz A, von Jonquières G, Wolf-Kümmeth S, et al. Hyperactivation of the insulin-like growth factor receptor I signaling pathway is an essential event for cisplatin resistance of ovarian cancer cells. Cancer Res. 2009;69:2996–3003. doi: 10.1158/0008-5472.CAN-08-3153. [DOI] [PubMed] [Google Scholar]
- 52.Gao F, Liang B, Reddy ST, Farias-Eisner R, Su X. Role of inflammation-associated microenvironment in tumorigenesis and metastasis. Curr Cancer Drug Targets. 2014;14:30–45. doi: 10.2174/15680096113136660107. [DOI] [PubMed] [Google Scholar]