

Abstract
Airway remodelling contributes to increased mortality in asthma. We have reported that triptolide can inhibit airway remodelling in a mouse asthma model. In this study, we aimed to investigate the effect of triptolide on proliferation and migration of airway smooth muscle cells (ASMC), and the possible mechanism. Rat ASMC were cultured and synchronized, then pre-treated with different concentrations of triptolide before being stimulated by transforming growth factor-β1 (TGF-β1). Cell proliferation was evaluated by cell counting and MTT assay. Flow cytometry was used to study the influence of triptolide on the cell cycle. Migration was measured by Transwell analysis. Signal proteins [nuclear factor-κB (NF-κB) p65 and extracellular signal-regulated kinase 1/2 (ERK1/2)] were detected by Western blotting. A lactate dehydrogenase releasing test and flow cytometry analysis of apoptosis were also performed to explore the potential cytotoxic or pro-apoptotic effects of triptolide. Triptolide significantly inhibited TGF-β1-induced ASMC proliferation and migration (P < 0·05). The cell cycle was dose-dependently blocked at G1/S-interphase by triptolide. Western blotting analysis showed that TGF-β1-induced NF-κB p65 phosphorylation was inhibited by triptolide pre-treatment, but ERK1/2 was not affected. No cytotoxic or pro-apoptotic effects were detected under the concentration of triptolide that was used. Triptolide may function as an inhibitor of asthma airway remodelling by suppressing ASMC proliferation and migration through inactivation of the NF-κB pathway.
Keywords: airway smooth muscle cells, asthma, nuclear factor-κB, triptolide
Introduction
Asthma is a chronic inflammatory disorder of the airways in which many cells and cellular elements contribute to its pathophysiological processes. The frequent occurrence of injury and repair initiated by chronic inflammation could lead to the structural changes in airway, collectively termed as airway remodelling. Airway remodelling is characterized by airway wall thickening, sub-epithelial fibrosis, increased smooth muscle mass, angiogenesis and increased mucous glands.1,2 Among these, the proliferation and migration of airway smooth muscle cells (ASMC) is a key factor.3
The treatment strategy for asthma mainly consists of the use of bronchodilators and anti-inflammatory drugs (such as β2-agonists, theophylline, anti-cholinergics, corticosteroids, H1-anti-histamine and anti-leukotrienes). However, approximately 5% of patients do not respond to these therapies;4–6 moreover, none of the current treatments can effectively retard the ongoing process of airway remodelling. So, new drugs which can block these processes are urgently needed.
Triptolide (PG-490), which is a diterpenoid triepoxide purified from a Chinese herb Tripterygium wilfordii Hook F, is the major component responsible for the immunosuppressive and anti-inflammatory effects of T. wilfordii. Besides, triptolide has the effects of inhibiting proliferation and inducing apoptosis.7,8 We confirmed that triptolide inhibited airway remodelling in ovalbumin-sensitized mice in our previous work.9 However, the mechanism of how triptolide affects airway remodelling remains unclear.
It has been shown that triptolide inhibits the activity of nuclear factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK).10,11 Nuclear factor-κB is a pleiotropic transcription factor that plays an important role in regulating the expression of multiple genes involved in immune responses; MAPK is also important in the regulation of cell growth and differentiation in the control of cellular responses to stresses and cytokines.12,13 Rong Tao et al. reported that triptolide inhibited rat vascular smooth muscle cell proliferation and cell cycle progression via attenuating phosphorylation of extracellular signal-regulated protein kinase 1/2 (ERK1/2), which is a major member of MAPK.14 It is therefore reasonable to propose that triptolide treatment may inhibit proliferation of ASMC through various signalling pathways including NF-κB and ERK1/2.
Transforming growth factor β1 (TGF-β1), a key mediator, has been shown to participate in the development of lung fibrosis in patients with severe asthma in previous studies.15,16 It can also stimulate the proliferation and migration of smooth muscle cells in vitro.17,18 Therefore TGF-β1 stimulation can be used in vitro as a model of inflammation and remodelling, because of its central effects in these processes.19
The purpose of this study was to investigate the anti-proliferation and anti-migration effects of triptolide on primary cultured ASMC induced by TGF-β1, and to explore the possible molecular mechanism.
Materials and methods
Chemicals and reagents
The following drugs and chemicals were purchased commercially and used: crystalline triptolide (PG490, molecular weight 360·40, purity 98%) was purchased from Sigma (St Louis, MO). Triptolide was dissolved in DMSO (MP biomedicals, Santa Ana, CA) and the stock solutions (1 mg/ml) were stored at −20°. Triptolide was freshly diluted to the indicated concentration with culture medium before use in experiments. The DMSO concentration in the medium did not exceed 0·1%. CHO-derived recombinant human TGF-β1 was purchased from R&D Systems (Minneapolis, MN). RIPA buffer and antibodies against p44/42 MAPK, Phospho-p44/42 MAPK (Thr202/Tyr204), NF-κB p65, phospho-NF-κB p65 (Ser536), GAPDH were purchased from Cell Signaling Technology (Danvers, MA). Antibodies against α-actin (Thermo Scientific IHC, Fremont, CA) were also obtained. The cell culture materials and fetal bovine serum (FBS) were obtained from Gibco/Life Technologies (Grand Island, NY). An enhanced chemiluminescence reagent kit was purchased from Millipore (Boston, MA). Cell Proliferation Kit I (MTT), Cytotoxicity Detection Kit (lactate dehydrogenase; LDH), bicinchoninic acid protein measurement kit, proteinase inhibitor cocktail tablets and phosphatase inhibitor cocktail tablets were from Roche (Basel, Switzerland). Annexin V-FITC/PI Apoptosis Detection Kit was from Invitrogen (Carlsbad, CA). All other chemicals used were of the highest grade available.
Culture of ASMC
Adult 6- to 8 week-old male Sprague-Dawley rats were obtained from the Centre of Animal Experiments of Sun Yat-sen University (Certificate of Conformity: Guangdong Experimental Animal Testing by certificate No. 2006A059). All the experiments were performed in accordance with the regulations of the Centre of Animal Experiments of Sun Yat-sen University. Ethical approval for this investigation was obtained from the research ethics committee, Sun Yat-sen University. ASMC were prepared by the explant method from tracheae and bronchi of Sprague-Dawley rats. Briefly, rats were killed by cervical dislocation, then the tracheae and bronchi were freed of lung tissue, bronchial vessels, connective tissue and adherent fat, the endothelial cell layer of the intima was also removed, and the remaining tissue was cut into c. 1 × 1×1 mm3 cubes. They were then placed in Dulbecco’s modified Eagle’s medium (DMEM) containing 20% FBS, 100 U/ml penicillin and 100 mg/ml streptomycin in a humidified atmosphere of 5% CO2 incubator at 37°. ASMC were identified by the typical ‘hill and valley’ growth pattern and immunocytochemistry staining of α-smooth muscle actin. Confluent cells at passage numbers 4–6 were used for the experiments.
Immunocytochemistry
ASMC were seeded onto sterile glass coverslips and grown to 70% confluence. Cells were then fixed in 4% paraformaldehyde for 20 min. Immunolabelling was performed with mouse anti-rat α-actin antibody at 4° overnight. Cells were incubated with biotin-labelled rabbit anti-mouse IgG as secondary antibody. The signals were detected by adding fast red, and then cells were counterstained with modified Mayer haematoxylin. Coverslips were mounted with crystal mount and visualized with AxioVision software (Carl Zeiss, Inc, Thornwood, NY).
Cell counting
Proliferation of ASMC was measured by counting cell number. Cells were seeded at a concentration of 2 × 104 cells/well in six-well plates and grown in DMEM containing 10% FBS for 24 hr. Then cells were starved in serum-free medium overnight and were pre-incubated with triptolide (0, 1, 2·5, 10 and 50 nm) and vehicle DMSO 30 min before stimulation with TGF-β1 (10 ng/ml) for the times indicated. The cells were counted on a haemocytometer at 24, 48 and 72 hr after treatment with TGF-β1. Experiments were repeated three times.
MTT cell proliferation assay
Proliferation of ASMC was also measured by MTT assay. Cells were seeded at a concentration of 5 × 103 cells/well in a 96-well plate and grown in DMEM containing 10% FBS for 24 hr. Then cells were treated in the same way as described above. At the end of the each culture time-point, cells were incubated with 5 mg/ml MTT in PBS for 4 hr at 37° in 5% CO2. Then 150 μl dissolving reagent DMSO was added to dissolve the formazan crystals. The optical density (OD) was determined using an ELISA plate reader (MN 3663; Molecular Devices, Sunnyvale, CA) with a reference wave length of 490 nm. The initial absorbance before triptolide treatment was also measured.
LDH releasing cytotoxic assay
To evaluate the potential cytotoxic effect of triptolide, an LDH-releasing test was also performed using a cytotoxicity detection kit (Roche). Cells were treated in the same way as described above. Six hours after stimulated with TGF-β1, cytotoxic assay was performed according to the manufacturer’s instructions. LDH released from ASMC was determined in cell culture media as a standard marker of cytotoxicity and cellular breakdown. The rate of NAD+ reduction was monitored by ELISA reader at 490 nm. The cytotoxicity results were expressed as a percentage of total ASMC content of LDH, determined by cell lysis with cell lysis solution from the kit.
Flow cytometry analysis of cell cycle and apoptosis
To estimate the proportions of cells in various phases of the cell cycle, cellular DNA contents were measured by flow cytometry (FACS). Twenty-four hours after treating with triptolide and TGF-β1 as described above, cells were trypsinized. The pellets were suspended in PBS and washed twice. The cells were then resuspended in PBS. The cell number was adjusted to 1 × 106 using PBS and centrifuged at 150 g for 10 min. The pellet was suspended in 70% ethanol and fixed overnight at 4°. The fixed cells were briefly vortexed and centrifuged at 300 g for 5 min. The ethanol was discarded and the pellets were stained with 0·5 ml propidium iodide (PI) solution (50 μg/ml PI in sample buffer containing 100 μg/ml of RNase A). After incubation at 4° overnight, PI-stained nuclei were analysed by flow cytometry (FACScalibur; BD Biosciences, Heidelberg, Germany). The rate of the cell cycle within G0/G1, S and G2/M phase was determined by analysis with the computer program ModFit LT V3.3.11 (Verity Software House, Topsham, ME).
In addition, flow cytometry analysis of apoptosis was performed to detect the possible pro-apoptotic effect of triptolide. The above-mentioned cell samples were trypsinized and then centrifuged at 150 g for 5 min. The cells were resuspended with 500 ml binding buffer at a concentration of 106 cells/ml, after washing twice with PBS at 150 g for 5 min. Then, 5 ml FITC-conjugated Annexin V and 5 ml PI were added to the cells and incubated at room temperature for 15 min in the dark. The samples were analysed within 1 h post-staining. Experiments were repeated three times.
Transwell analysis
Transwell analysis was conducted after cells were harvested with trypsin and resuspended (1·0 × 106 cells/ml) in serum-free growth medium. Cells were pre-treated with triptolide and vehicle for 30 min using the indicated concentrations and subsequently stimulated with TGF-β1 (10 ng/ml). After 24 hr of incubation at 37°, the membranes were removed, the cells on the upper side were scraped off, and cells that migrated to the lower side of the membrane were fixed with 4% polyoxymethylene and stained by crystal violet. The number of cells was counted in five random fields under 200 × magnification, and the means were calculated. The experiments were repeated three times.
Western blot analysis
Thirty minutes after triptolide and TGF-β1 stimulation, cells were harvested on ice, lysed with RIPA buffer containing 20 mm Tris–HCl (pH 7·5), 150 mm NaCl, 1 mm Na2 EDTA, 1 mm EGTA, 1% Nonidet P-40,1% sodium deoxycholate, 2·5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm Na3VO4 and 1 μg/ml leupeptin. Proteinase inhibitor cocktail tablets and phosphatase inhibitor cocktail tablets were also added. The bicinchoninic acid method was used to determine the protein concentrations. An equal amount of protein was separated by 10% SDS–PAGE and transferred to a PVDF membrane. The PVDF membrane was then blocked for 1 hr at room temperature in 5% non-fat dried milk and incubated overnight at 4° with the special antibody. After washing in TBS-T solution, membranes were incubated further with horseradish peroxidase-conjugated IgG secondary antibody. The blots were then washed three times in TBS-T, and antibody-bound protein was visualized with an enhanced chemiluminescense kit.
Statistical analysis
All experiments were performed at least three times. Data were analysed using GraphPad Prism (version 5, GraphPad Software, San Diego, CA). Results are expressed as mean ± SEM. One-way analysis of variance and Dunnett’s multiple comparison test were used. P-values were two-sided and subject to a significance level of 5%.
Result
Identification of primary rat airway smooth muscle cells
Primary cultures of ASMC obtained from tissue explants were identified by their characteristic ‘hill and valley’ appearance under a phase-contrast microscope and positive immunofluorescence staining of α-smooth muscle actin.20 Both results showed that all the cells were smooth muscle cells and the cell morphology was identical (Fig.1).
Figure 1.
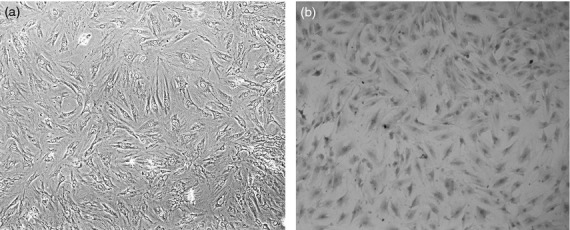
Culture and identification of airway smooth muscle cells (ASMC) isolated from rat. (a) ASMC displayed the characteristic ‘hill and valley’ appearance (phase contrast microscope ×100). (b) Positive expression of α-actin in ASMC (immunocytochemical staining × 100).
Triptolide inhibited TGF-β1 induced ASMC proliferation without apparent cytotoxic effect
To evaluate the effect of triptolide on ASMC proliferation, cell counting and MTT cell proliferation assays were performed. As shown in Fig.2(a–c), at 0 hr, cell numbers and OD490 values were at the same level (P > 0·05). After 24 hr, TGF-β1 control group showed significantly larger cell numbers than the untreated group. At the same time, triptolide at the concentration of 2·5 nm or higher can significantly and dose dependently inhibit the effect of TGF-β1 (P < 0·05). Triptolide at 50 nm completely blocked this process.
Figure 2.

Triptolide inhibited transforming growth factor-β1 (TGF-β1) -induced airway smooth muscle cell (ASMC) proliferation without apparent cytotoxic effect. Triptolide caused a time- and dose-dependent inhibition of ASMC proliferation without distinct cytotoxic effect. ASMC were cultured with triptolide at different doses 30 min before TGF-β1 stimulation. Cell proliferation was examined at 24, 48 and 72 hr after TGF-β1 stimulation; lactate dehydrogenase (LDH) releasing test was preformed 6 hr after TGF-β1 stimulation. (a) and (b) Results of cell counting. The cell numbers of viable cells estimated by haemocytometer at different time are shown. (c) Results of MTT assay. The OD490 values of six samples carried in triplicate are shown. (d) Results of LDH releasing test. Data were expressed as percentage of total ASMC content of LDH, determined by cell lysis. Effects of triptolide on LDH release from ASMC reached no statistical significance. n = 4 experiments in each group. All data are expressed as mean ± SEM (#P < 0·05, ##P < 0·01, ###P < 0·001 versus untreated group; *P < 0·05, **P < 0·01, ***P < 0·001 versus TGF control group at the same point of time). The cell numbers and OD490 values of 0 hr is the initial data before cells were pre-treated with triptolide.
At the same time, we proved that the effect of triptolide on cell number was not the result of cytotoxicity, as the LDH released from the ASMC of triptolide-treated groups reached no statistical significance compared with control groups as shown in Fig.2(d) (P > 0·05).
Effect of triptolide on cell cycle and apoptosis
The effects of triptolide on cell cycle progression were explored (Fig.3a, b).Treatment with TGF-β1 markedly decreased the percentage of ASMC at G0/G1 phase and correspondingly increased their percentage at the S and G2/M phases. Triptolide significantly blocked this event (P < 0·05). The results for the TGF + Triptolide 50 nm group showed no difference compared with the untreated group (P > 0·05). These data suggest a dose-dependent accumulation of triptolide-treated cells at the G1/S interphase, with 50 nm triptolide leading to a complete block in cell cycle progression.
Figure 3.

FACS analysis of cell cycle and apoptosis. Transforming growth factor-β1 (TGF-β1) -stimulated cells that were pre-treated with triptolide were cultured for 24 hr. After that, cell cycle was determined by individual nuclear DNA content reflected by fluorescence intensity of incorporated propidium iodide. Apoptosis was determined by Annexin V FITC and PI staining. (a) Images show one representative profile of cell cycle distribution in three independent experiments. (b) The percentage of cell cycle distribution after the indicated treatment. Cells with single and doubled DNA contents were defined in G0/G1 and G2/M phase, respectively, and were classified to S phase with DNA content between them. Data were analysed automatically using computer software and are presented as the mean ± SEM (#P < 0·05, ##P < 0·01, ###P < 0·001, versus untreated group; *P < 0·05, **P < 0·01, ***P < 0·001 versus TGF control group). (c) Images show one representative profile of cell apoptosis rate in three independent experiments. The early and late apoptosis was quantified and indicated in UL and LR gates, respectively. The critical values were determined by the control samples without any staining. (d) Date of apoptosis rates of cells (cells in LR gate) is shown. There was no statistical significance among groups.
The representative results of apoptosis analysis are shown in Fig.3(c). Neither TGF-β1 nor triptolide can affect the apoptosis rate of ASMC, because percentages of cells in the upper left and lower right quadrants, which represent early and late apoptotic cells, respectively, reached no statistical significance among groups (partly shown in Fig.3d).
Triptolide inhibited TGF-β1-induced ASMC migration
The effect of triptolide on TGF-β1-induced cell migration was evaluated by the cell migration assay. The image and data show that TGF-β1 can dramatically stimulate the migration of ASMC, and pre-treatment with triptolide at the concentration of 2·5 nm or higher significantly attenuated this process (P < 0·05) (Fig.4).
Figure 4.
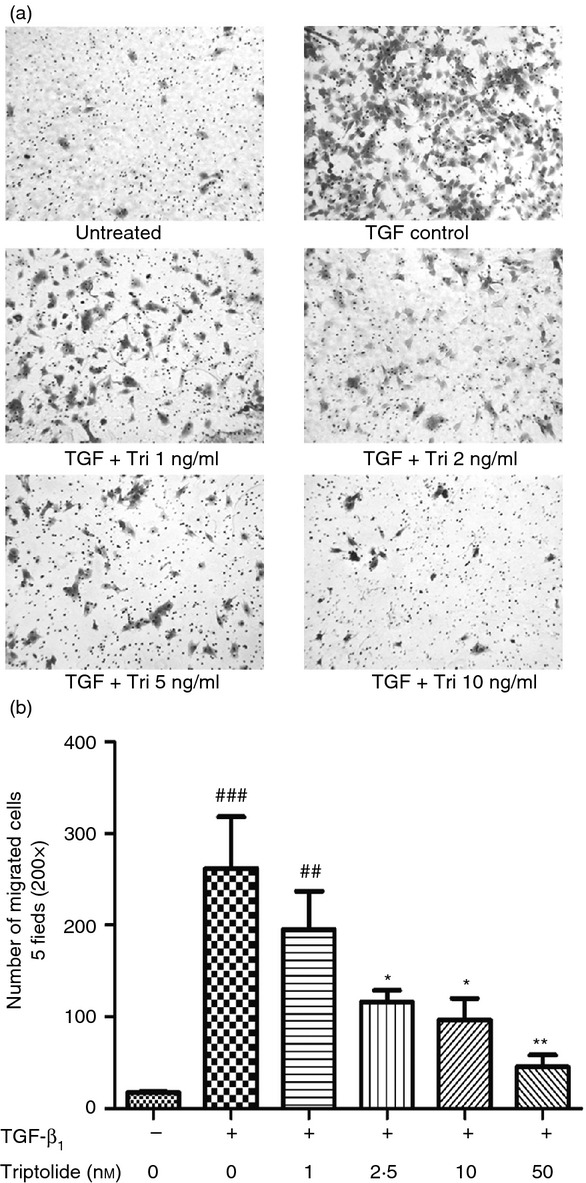
Triptolide inhibited transforming growth factor-β1 (TGF-β1) induced airway smooth muscle cell (ASMC) migration. Twenty-four hours after treatment with/without TGF-β1 and triptolide, the cells that migrated to the lower side of the membrane were fixed and stained with crystal violet. Then they were counted in five random fields under 200 × magnification. Numbers of migrated cells were counted. (a) Representative examples of each group (200 × magnification). (b) Numbers of migrated cells. Data are expressed as mean ± SEM, (##P < 0·01, ###P < 0·001 versus untreated group; *P < 0·05, **P < 0·01 versus TGF control group). The experiments were repeated three times.
Effect of triptolide on ERK1/2 and NF-κB
As shown in Fig.5, triptolide can significantly inhibit the phosphorylation of NF-κB p65 at the concentration of 2·5 nm or higher (P < 0·05). It seems that the phosphorylation level of the TGF control group is higher than of the untreated group but they did not reach statistical significance (P > 0·05), possibly due to the limiting of sample size. In the ERK1/2 pathway, TGF-β1 stimulated the phosphorylation of ERK1/2 dramatically (P < 0·05). However, the phosphorylation level of ERK1/2 remained unchanged despite different concentrations of triptolide used for pre-treatment (P > 0·05).
Figure 5.
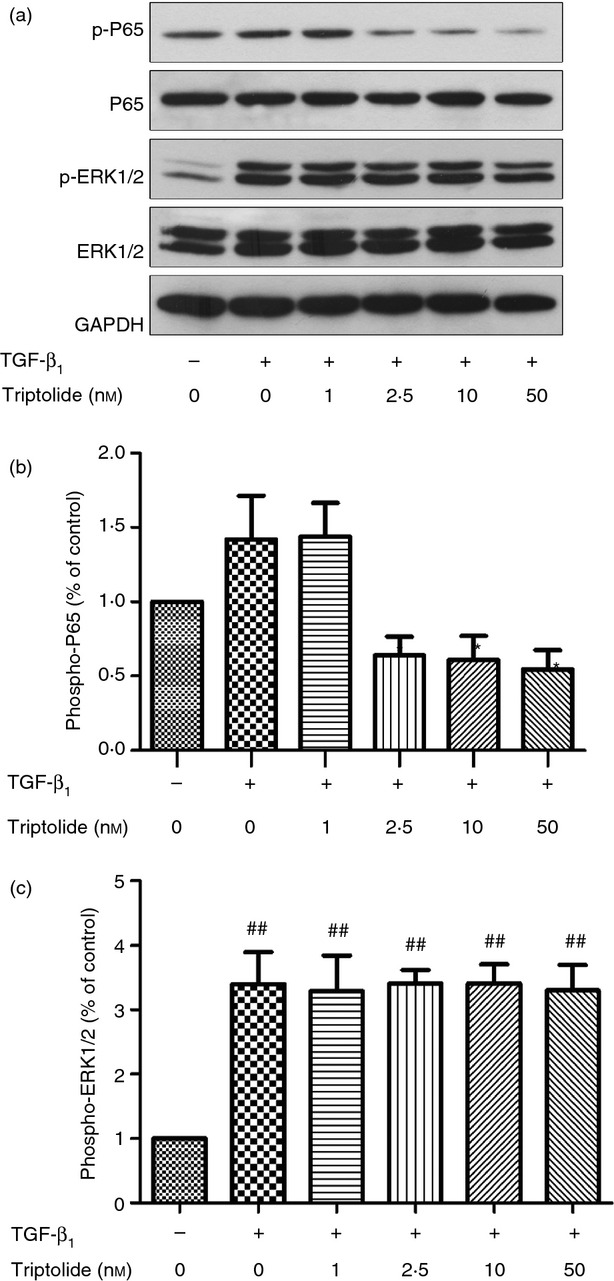
Western blot analysis of phosphorylation of nuclear factor-κB (NF-κB) p65 and extracellular signal-regulated kinase (ERK) 1/2. Arrested airway smooth muscle cells (ASMC) were pre-treated with/without various concentrations of triptolide for 30 min, and stimulated with 10 ng/ml transforming growth factor-β1 (TGF-β1) consequently. Thirty minutes later, whole cell lysate was extracted and immunoblotted with antibodies specific for NF-κB p65 and ERK1/2. The band intensities of phosphoproteins were normalized against total NF-κB p65 and ERK1/2 and expressed as fold increase over the untreated group. Data are means ± SEM of three independent experiments (*P < 0·05 compared with TGF control group; ##P < 0·01, versus untreated group).
Discussion
Our study has proved that triptolide significantly inhibited TGF-β1-induced ASMC proliferation and migration without apparent cytotoxic or pro-apoptotic effects. The cell cycle was blocked at G1/S-interphase by triptolide, and was dose dependent. In addition, TGF-β1-induced NF-κB p65 phosphorylation can be inhibited by triptolide pre-treatment, but the ERK1/2 pathway was not affected.
Triptolide-containing T. wilfordii has been used in traditional Chinese medicine for more than two centuries to treat a variety of autoimmune and inflammatory diseases. Molecular weight of triptolide is 360, the formula is C20H24O6.. Three epoxy groups and a five-unsaturated lactone ring are the structural basis of triptolide, which play a variety of biological roles. It inhibits several pro-inflammatory cytokines and adhesion molecules, which are important mediators of many autoimmune diseases. Triptolide has been shown to be safe and clinically beneficial in several autoimmune diseases, such as rheumatoid arthritis and asthma. We previously confirmed that triptolide inhibited pulmonary inflammation in patients with steroid-resistant asthma.21 Triptolide or its derivative has also been shown to be effective in the treatment of lung inflammation and fibrosis in animal models.11,22 Besides, triptolide has been reported to inhibit proliferation and induce apoptosis of cancer cells in vitro and reduce the growth and metastases of tumours in vivo.23,24 Here, we found that triptolide inhibited TGF-β1-induced proliferation of ASMC, which are key processes underlying the formation of airway remodelling in asthma. Consequently, it is a potential effective therapeutic strategy for the treatment of airway remodelling in asthma.
In our study, the anti-proliferative effect of triptolide was associated with an accumulation of cells in G0/G1 phase of the cycle as revealed by flow cytometry. Treatment with TGF-β1 markedly decreased the per cent of ASMC at G0/G1 phase and correspondingly increased their percentages at S and G2/M phases. Triptolide blocked this event. Although some research has reported the cytotoxic and pro-apoptotic effects of triptolide on cancer cells, such effects were not observed in our study because the results of flow cytometry apoptosis analysis had no significant difference among groups. This is just because in researches focused on anti-cancer effects, the concentration of triptolide was thousands of times higher than what we used in this study.
In this study, TGF-β1 was used to induce ASMC proliferation and migration. TGF-β1 binds to two serine/threonine kinase receptors which consist of TGF-βRI and TGF-βRII. When a ligand binds, TGF-βRII phosphorylates TGF-βRI and activates Smad-dependent intracellular signalling pathways and so leads to expression of several genes. In addition to activation of Smad-dependent pathways, TGF-β1 can affect several signal transduction pathways in a Smad-independent manner, such as ERK1/2 and NF-κB. Our study has shown that triptolide pre-treatment inhibited proliferation and migration of ASMC, possibly through inhibiting NF-κB activation, but not ERK1/2. NF-κB is a transcription factor that has multiple regulatory roles. Liu’s work suggested that triptolide inhibits dendritic cell migration through decreasing COX-2 and CCR7 expression via NF-κB signalling pathways.25 Many studies have confirmed that NF-κB is over-activated in asthma patients.26,27 Also, Mo’s group reported that triptolide may decrease the proliferation of ASMC by inhibiting the expression of NF-κB and Bcl-2 in rats.28 During the pathogenesis of asthma, NF-κB may act in different aspect of airway remodelling, such as regulating ASMC proliferation and their expressions of inflammatory cytokines.29,30 The precise mechanism bu which triptolide inhibited the activation of NF-κB is not clearly understood. One explanation is that triptolide could suppress the production of reactive oxygen species by TGF-β1. Reactive oxygen species are known to activate NF-κB through the phosphorylation of p65.10
MAPK (especially ERK1/2) are also an important intracellular transduction pathway in the pathophysiological process of asthma. But the effects of triptolide on this pathway are controversial. It has been found that the immune suppressive and anti-tumour effects of triptolide were mediated at least partly by suppression of MAPK and PI-3 kinase gene expression.31,30 However, some studies reported that the anti-inflammatory or anti-proliferative effects of triptolide are independent of the ERK pathway.32,33 Koo et al. reported that triptolide inhibits the proliferation of immortalized HT22 hippocampal cells via persistent activation of ERK1/2 by down-regulating MAPK phosphatase-1 expression.34 The conflicting results suggest the possibility that triptolide may affect different upstream factors of ERK1/2 in different kinds of cells, instead of acting directly on ERK1/2. The underling mechanisms by which triptolide affects ASMC, especially its effect on different signal pathway, still need further exploration.
In summary, our study for the first time demonstrated that triptolide is able to inhibit ASMC proliferation induced by TGF-β1. The anti-proliferative effect is at least partly mediated by inhibition of the NF-κB pathway. The results have provided evidence that triptolide may be an effective candidate for the systemic therapy of asthma airway remodelling. However, to fully elucidate the biological activity of triptolide on ASMC, the respiratory and immune system, and eventually the whole organism, further studies are needed.
Acknowledgments
We thank Wei Jing for technical support for the flow cytometry. We also thank the Centre of Animal Experiments of Sun Yat-sen University for the experimentation on animals. This work was supported by the National Natural Science Foundation of China (81070027) and by the Guangdong Natural Science Foundation (S2011010004278).
Glossary
- ASMC
airway smooth muscle cells
- DMEM
Dulbecco’s modified Eagle’s medium
- ERK
extracellular regulated protein kinases
- FBS
fetal bovine serum
- LDH
lactate dehydrogenase
- MAPK
mitogen-activated protein kinase
- NF-κB
nuclear factor-κB
- PI
propidium iodide
- TGF-β1
transforming growth factor-β1
Disclosures
The authors declare no financial or commercial conflict of interest.
References
- Hirota N, Martin JG. Mechanisms of airway remodeling. Chest. 2013;144:1026–32. doi: 10.1378/chest.12-3073. [DOI] [PubMed] [Google Scholar]
- Berair R, Saunders R, Brightling CE. Origins of increased airway smooth muscle mass in asthma. BMC Med. 2013;11:145. doi: 10.1186/1741-7015-11-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash YS. Airway smooth muscle in airway reactivity and remodeling: what have we learned? Am J Physiol Lung Cell Mol Physiol. 2013;305:L912–33. doi: 10.1152/ajplung.00259.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson DS, Campbell DA, Durham SR, Pfeffer J, Barnes PJ, Chung KF. Systematic assessment of difficult-to-treat asthma. Eur Respir J. 2003;22:478–83. doi: 10.1183/09031936.03.00017003. [DOI] [PubMed] [Google Scholar]
- Adams A, Saglani S. Difficult-to-treat asthma in childhood. Paediatr Drugs. 2013;15:171–9. doi: 10.1007/s40272-013-0025-5. [DOI] [PubMed] [Google Scholar]
- Busse W, Banks-Schlegel S, Noel P, Ortega H, Taggart V, Elias J. Future research directions in asthma: an NHLBI Working Group report. Am J Respir Crit Care Med. 2004;170:683–90. doi: 10.1164/rccm.200311-1539WS. [DOI] [PubMed] [Google Scholar]
- Zhao F, Huang W, Ousman T, et al. Triptolide induces growth inhibition and apoptosis of human laryngocarcinoma cells by enhancing p53 activities and suppressing E6-mediated p53 degradation. PLoS ONE. 2013;8:e80784. doi: 10.1371/journal.pone.0080784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q. Triptolide and its expanding multiple pharmacological functions. Int Immunopharmacol. 2011;11:377–83. doi: 10.1016/j.intimp.2011.01.012. [DOI] [PubMed] [Google Scholar]
- Chen M, Lv Z, Jiang S. The effects of triptolide on airway remodelling and transforming growth factor-β1/Smad signalling pathway in ovalbumin-sensitized mice. Immunology. 2011;132:376–84. doi: 10.1111/j.1365-2567.2010.03392.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Guo X, Mathew S, Armesilla AL, Cassidy J, Darling JL, Wang W. Triptolide simultaneously induces reactive oxygen species, inhibits NF-κB activity and sensitizes 5-fluorouracil in colorectal cancer cell lines. Cancer Lett. 2010;291:200–8. doi: 10.1016/j.canlet.2009.10.013. [DOI] [PubMed] [Google Scholar]
- Hoyle GW, Hoyle CI, Chen J, Chang W, Williams RW, Rando RJ. Identification of triptolide, a natural diterpenoid compound, as an inhibitor of lung inflammation. Am J Physiol Lung Cell Mol Physiol. 2010;298:L830–6. doi: 10.1152/ajplung.00014.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011;75:50–83. doi: 10.1128/MMBR.00031-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao R, Lu L, Zhang R, Hu J, Ni J, Shen W. Triptolide inhibits rat vascular smooth muscle cell proliferation and cell cycle progression via attenuation of ERK1/2 and Rb phosphorylation. Exp Mol Pathol. 2011;90:137–42. doi: 10.1016/j.yexmp.2010.12.001. [DOI] [PubMed] [Google Scholar]
- Makinde T, Murphy RF, Agrawal DK. The regulatory role of TGF-β in airway remodeling in asthma. Immunol Cell Biol. 2007;85:348–56. doi: 10.1038/sj.icb.7100044. [DOI] [PubMed] [Google Scholar]
- Brown SD, Baxter KM, Stephenson ST, Esper AM, Brown LA, Fitzpatrick AM. Airway TGF-β1 and oxidant stress in children with severe asthma: association with airflow limitation. J Allergy Clin Immunol. 2012;129:388–396. doi: 10.1016/j.jaci.2011.11.037. 391–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Wang Q, Fei T, Han JD, Chen YG. MCP-1 mediates TGF-β-induced angiogenesis by stimulating vascular smooth muscle cell migration. Blood. 2007;109:987–94. doi: 10.1182/blood-2006-07-036400. [DOI] [PubMed] [Google Scholar]
- Chen G, Khalil N. TGF-β1 increases proliferation of airway smooth muscle cells by phosphorylation of MAP kinases. Respir Res. 2006;7:2. doi: 10.1186/1465-9921-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ierodiakonou D, Postma DS, Koppelman GH, Gerritsen J, Ten HN, Timens W, Boezen HM, Vonk JM. TGF-β1 polymorphisms and asthma severity, airway inflammation, and remodeling. J Allergy Clin Immunol. 2013;131:582–5. doi: 10.1016/j.jaci.2012.08.013. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Zhai YL, Iinuma M, Horiuchi A, Nikaido T, Fujii S. Effects of a GnRH analogue on human smooth muscle cells cultured from normal myometrial and from uterine leiomyomal tissues. Mol Hum Reprod. 1997;3:91–9. doi: 10.1093/molehr/3.2.91. [DOI] [PubMed] [Google Scholar]
- Jiang SP, Liang RY, Yang L, Yang L, Zhang W, Lü ZQ. Effects of triptolide on serum cytokine levels symptoms and pulmonary function in patients with steroid-resistant asthma. Chin J Pathophysiol. 2006;22:1571–4. [Google Scholar]
- Krishna G, Liu K, Shigemitsu H, Gao M, Raffin TA, Rosen GD. PG490-88, a derivative of triptolide, blocks bleomycin-induced lung fibrosis. Am J Pathol. 2001;158:997–1004. doi: 10.1016/S0002-9440(10)64046-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Liu R, Yang Y, Huang Y, Li X, Liu R. Triptolide-induced in vitro and in vivo cytotoxicity in human breast cancer stem cells and primary breast cancer cells. Oncol Rep. 2014;31:2181–6. doi: 10.3892/or.2014.3115. [DOI] [PubMed] [Google Scholar]
- Wu PP, Liu KC, Huang WW, Ma CY, Lin H, Yang JS, Chung JG. Triptolide induces apoptosis in human adrenal cancer NCI-H295 cells through a mitochondrial-dependent pathway. Oncol Rep. 2011;25:551–7. doi: 10.3892/or.2010.1080. [DOI] [PubMed] [Google Scholar]
- Liu Q, Chen T, Chen G, Li N, Wang J, Ma P, Cao X. Immunosuppressant triptolide inhibits dendritic cell-mediated chemoattraction of neutrophils and T cells through inhibiting Stat3 phosphorylation and NF-κB activation. Biochem Biophys Res Commun. 2006;345:1122–30. doi: 10.1016/j.bbrc.2006.05.024. [DOI] [PubMed] [Google Scholar]
- Ventura I, Vega A, Chamorro C, et al. Allergen immunotherapy decreases LPS-induced NF-κB activation in neutrophils from allergic patients. Pediatr Allergy Immunol. 2014;25:129–35. doi: 10.1111/pai.12145. [DOI] [PubMed] [Google Scholar]
- Gagliardo R, Chanez P, Profita M, et al. IκB kinase-driven nuclear factor-κB activation in patients with asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2011;128:635–45. doi: 10.1016/j.jaci.2011.03.045. [DOI] [PubMed] [Google Scholar]
- Mo BW, Wei JH, Huang JW, et al. Effect of triptolide on airway smooth muscle proliferation and the expression of nuclear factor-κB, Bcl-2 in asthmatic rats. Zhongguo Ying Yong Sheng Li Xue Za Zhi. 2010;26:385–90. [PubMed] [Google Scholar]
- Ying-fang S, Jing-fang H, Huan-zhang L, Hao-wen Q. Effect of platelet-activating factor on cell proliferation and NF-κB activation in airway smooth muscle cells in rats. Indian J Med Res. 2007;126:139–45. [PubMed] [Google Scholar]
- Zhao JP, Gao M, Ye YJ, Hu WH, Zhou ZG, Hu HL. Regulation of rat airway smooth muscle cell proliferation by mitochondrial ATP-sensitive K+ channel in asthmatic rats. Sheng Li Xue Bao. 2009;61:65–71. [PubMed] [Google Scholar]
- Du ZY, Li XY, Li YC, Wang SY. Analysis of triptolide-regulated gene expression in Jurkat cells by complementary DNA microarray. Acta Pharmacol Sin. 2003;24:864–72. [PubMed] [Google Scholar]
- Kimura K, Nomi N, Yan ZH, Orita T, Nishida T. Inhibition of poly(I:C)-induced matrix metalloproteinase expression in human corneal fibroblasts by triptolide. Mol Vis. 2011;17:526–32. [PMC free article] [PubMed] [Google Scholar]
- Chang HJ, Kim MH, Baek MK, Park JS, Chung IJ, Shin BA, Ahn BW, Jung YD. Triptolide inhibits tumor promoter-induced uPAR expression via blocking NF-κB signaling in human gastric AGS cells. Anticancer Res. 2007;27:3411–7. [PubMed] [Google Scholar]
- Koo HS, Kang SD, Lee JH, Kim NH, Chung HT, Pae HO. Triptolide inhibits the proliferation of immortalized HT22 hippocampal cells via persistent activation of extracellular signal-regulated kinase-1/2 by down-regulating mitogen-activated protein kinase phosphatase-1 expression. J Korean Neurosurg Soc. 2009;46:389–96. doi: 10.3340/jkns.2009.46.4.389. [DOI] [PMC free article] [PubMed] [Google Scholar]