

Abstract
The autoimmune destruction of pancreatic β-cells is the hallmark of type 1 diabetes (T1D). Failure of anti-CD3 antibodies to provide long-lasting reversal of T1D and the expression of a natural killer (NK) cell ligand on β-cells suggest that NK cells play a role in disease pathogenesis. Indeed, killing of β-cells by NK cells has been shown to occur, mediated by activation of the NK cell activating receptor, NKp46. α1-Antitrypsin (AAT), an anti-inflammatory and immunomodulatory glycoprotein, protects β-cells from injurious immune responses and is currently evaluated as a therapeutic for recent onset T1D. Although isolated T lymphocytes are not inhibited by AAT, dendritic cells (DC) become tolerogenic in its presence and other innate immune cells become less inflammatory. Yet a comprehensive profile of NK cell responses in the presence of AAT has yet to be described. In the present study, we demonstrate that AAT significantly reduces NK cell degranulation against β-cells, albeit in the whole animal and not in isolated NK cell cultures. AAT-treated mice, and not isolated cultured β-cells, exhibited a marked reduction in NKp46 ligand levels on β-cells. In related experiments, AAT-treated DC exhibited reduced inducible DC-expressed interleukin-15 levels and evoked a weaker NK cell response. NK cell depletion in a T1D mouse model resulted in improved β-cell function and survival, similar to the effects observed by AAT treatment alone; nonetheless, the two approaches were non-synergistic. Our data suggest that AAT is a selective immunomodulator that retains pivotal NK cell responses, while diverting their activities away from islet β-cells.
Keywords: cross-talk, interleukin-15, islets, pancreatic β-cells, type 1 diabetes
Introduction
Type 1 diabetes (T1D) is characterized by the autoimmune destruction of insulin-producing β-cells that reside within pancreatic islets.1 Although predominantly considered the response of the adaptive immune system, several clinical trials have resulted in disappointing outcomes when targeting either CD3+ T lymphocytes or CD20+ B lymphocytes in recent-onset T1D patients.2–4 Hence, multiple innate leucocyte populations are assumed to be important mediators of the underlying autoimmune pathogenesis of T1D.5
Apart from endocrine cells, islets contain up to 3% resident leucocytes, including natural killer (NK) cells. Indeed, NK cells are commonly present in organs affected by autoimmune diseases and, under certain conditions, may attack populations of healthy ‘self’ cells.5,6 Upon activation, NK cells release various cytokines, including the interleukin-15 (IL-15) -inducing cytokine, interferon γ (IFN-γ), and degranulate as part of the process of direct target cell lysis.
Natural killer cells are present in the pancreas of human T1D patients and of non-obese diabetic (NOD) mice,7 both during insulitis and during advanced stages of T1D.8 The number of pancreatic NK cells correlates with severity of disease in NOD mice,5 and depletion of NK cells using α-asialo GM1 antibodies delays the onset of hyperglycaemia in the multiple low-dose streptozotocin (MLD-STZ) model.9 In addition, the activating receptor NKG2D (CD314), expressed on NK cells and CD8+ T cells, has been found to recognize a specific ligand on pancreatic β-cells and to activate autoimmune cytotoxicity.10,11 Gur et al.5 recently described another membrane-associated islet β-cell ligand, recognized by NK cell receptor 1 (NKp46 in humans and NCR1 in mice). Importantly, NKp46 activation was shown to be required for NK cell-mediated β-cell destruction12 and blockade of NKp46 was shown to inhibit the rate and progression of T1D in NOD mice and in the MLD-STZ model using wild-type mice.5
The serine-protease inhibitor α1-antitrypsin (AAT) has recently been suggested as an immunomodulator capable of inducing tolerance to allogeneic islet grafts13,14 and to reduce inflammatory damage to islets,15 and is currently considered for treatment of individuals with recent onset T1D.16–18 The cellular targets of human AAT (hAAT) include neutrophils and macrophages, which are essentially blocked by hAAT, and dendritic cells (DC) and B lymphocytes that turn tolerogenic under hAAT treatment in multiple experimental set-ups;19,20 DC in particular have been found to adopt the semi-mature phenotype upon treatment with hAAT.21 However, isolated T-lymphocyte activities remain intact in the presence of hAAT.14,15 Indeed, while IL-1β, IFN-γ, tumour necrosis factor-α (TNF-α) and several chemokines are significantly inhibited by hAAT, IL-2 levels and related responses persist.14,15 Nevertheless, the mechanism by which hAAT exerts its protective activities on β-cells remains to be clarified.
The present study explores a panel of NK cell activities during hAAT therapy, in the context of autoimmune diabetes, allogeneic reactions, response to β-cells and anti-tumour activities. In particular, we address the possibility that hAAT-treated DC might alter the degree of NK cell activation, in part via the inhibitory activity of hAAT on the IL-15-dependent NK cell activation pathway. Our data suggest that hAAT alters NK cell responses in a highly selective manner.
Materials and methods
Mice
C57BL/6 mice (females, 6–8 weeks old; Harlan, Rehovot, Israel) were used as the source of primary NK cells and bone-marrow-derived dendritic cells (BMDC), as well as for MLD-STZ experiments. DBA/2J mice (females, 6–8 weeks old; Jackson Laboratories, Bar Harbor, ME) were used as the source of primary pancreatic islets. Where indicated, hAAT was injected intraperitoneally (i.p.) at the indicated doses for 3 days unless otherwise specified. Animals were housed in accordance with the guidelines of the Care and Use of Laboratory Animals, Ministry of Health, Israel. Experiments were approved by Ben-Gurion University of the Negev Animal Care and Use Committee.
Cell lines
All cell lines were purchased from the American Type Culture Collection (ATCC; Manassas, VA) and were used within five passages. B16-F10 (murine melanoma cell line, no. CRL-6475), MIN-6 (murine insulin-secreting β-cell line) and YAC1 (murine lymphoma cell line, no. TIB-160), were cultured according to specific ATCC guidelines.
Primary NK cell purification and activation
Natural killer cells were purified from spleens of mice that were treated for 18 hr with poly(I:C) (0·2 mg per mouse i.p.; Sigma-Aldrich, Rehovot, Israel), using negative selection magnetic separation beads (StemCell Technologies, Vancouver, BC, Canada). Cells were then cultured in RPMI-1640 supplemented with 10% fetal bovine serum, 50 U/ml streptomycin/penicillin and 50 μg/ml l-glutamine (all from Biological Industries, Beit Ha’emek, Israel). For NK cell activation experiments, 5 × 104 cells per well in triplicate were cultured at a 1 : 1 ratio with the following target cell populations: YAC1, MIN-6, B16-F10 and dissociated primary pancreatic islet cells. For IFN-γ release assays, 48-hr cell supernatants were examined using specific ELISA (R&D Systems, Minneapolis, MN). For CD107a degranulation assays, co-cultures included α-CD107a antibody (0·1 μg, Biolegend, San Diego, CA) and incubated for 3 hr before flow cytometry analysis.
Primary pancreatic islet isolation and culture studies
Pancreatic islets were purified as described elsewhere.13 Briefly, donor pancreata were inflated with ice-cold collagenase XI (Sigma-Aldrich) and then digested at 37°. Islets (80–350 μm) were collected on a 100-μm sterile nylon cell strainer (Falcon; BD Biosciences Discovery Labware, San Jose, CA) and then gently dissociated with TrypLE enzyme (Gibco, Modi’in, Israel). Cells were treated with hAAT (0·25, 0·5 or 1 mg/ml as indicated, Glassia™, Kamada Ltd., Nes Ziona, Israel).
Generation of BMDC and related activation assays
Tibia and femur bones were thoroughly flushed with PBS, and cells were cultured in RPMI-1640 containing 10% fetal bovine serum, 50 U/ml streptomycin/penicillin, 50 μg/ml l-glutamine and 50 μm β2-mercaptoethanol (Sigma-Aldrich) and supplemented with 20 ng/ml granulocyte–macrophage colony-stimulating factor (Peprotech, Rocky Hills, NJ) and 1 × 105 IU/ml IL-4 (Prospec, East Brunswick, NJ) for 6 days, as described previously.22 BMDC were routinely verified to be ≥ 90% CD11c+ by flow cytometry analysis. For activation experiments, cells were seeded at 2 × 105 per well in 200 μl Iscove’s medium and allowed to adhere, then 0·25, 0·5 or 1 mg/ml hAAT was added, as indicated, for a 24-hr incubation time. Cells were then carefully washed with PBS and recombinant IFN-γ was added (10 ng/ml, R&D Systems) for 24 hr. For IL-15 and IL-15Rα studies, cells were lysed in PBS containing 0·1% nonidet P-40 (Merck Millipore, Darmstadt, Germany) and protease inhibitor cocktail (Sigma-Aldrich). Samples were treated for 5 min with 0·1% SDS at 95°; levels were determined using specific ELISA for each analyte (R&D Systems).
Flow cytometry
In all experiments, cells were incubated in flow cytometry buffer (PBS containing 1% BSA from Biological Industries, Ltd., 0·1% sodium azide and 2 mm EDTA from Sigma-Aldrich). Fc blocking was performed with α-CD16/32 antibody for 20 min at 4° (Biolegend) and staining was performed for 20 min at 4° with the following antibodies: α-GLUT2-phycoerythrin (PE) (bs-0351R) (Bioss, Boston, MA) and α-CD3-AlexaFluor 488 (145-2C11), α-CD107a-allophycocyanin-Cy7 (APC-Cy7) (1D4B), α-NK1.1 Pacific Blue (PK136), α-NK1.1-PE-Cy7 (PK136), α-NKp46-PE (29A1.4), α-CD11c-APC (N418), α-Gr1-PE (RB6-8C5), α-MHC class I-APC (34-1-2S) and α-NKG2D biotin (C7) (followed by secondary staining with PE-conjugated streptavidin), all from Biolegend. Binding of NKp46-Fc (3 μg per sample) was performed as described elsewhere,5 followed by secondary antibody staining with α-human Fc-APC (Cat. 109-136-098, 1 : 200 dilution; Jackson ImmunoResearch, Philadelphia, PA). MHC class I surface expression in B16-F10 cells was evaluated by staining with α-MHC class I antibody, followed by flow cytometry analysis. Samples were read using BD Canto II and data were analysed by Flowjo 7·6·3 software (Flowjo, LLC Data Analysis Software, Ashland, OR).
MLD-STZ and NK cell depletion
Streptozotocin (Sigma-Aldrich) was administered daily at 50 mg/kg i.p, for 5 consecutive days, as described elsewhere.15 Depletion of NK cells was performed using ultrapure α-asialo-GM1 antibody (Biolegend) at 60 μl per mouse, i.p., every 7 days, starting 3 days before initial STZ treatment. NK cell population size was routinely verified to be ≤ 5% of that from untreated animals by flow cytometry analysis. The hAAT was introduced at 1 mg per mouse i.p, starting from 1 day before initial STZ treatment and every 3 days thereafter. For the glucose tolerance test, animals were fasted for 16 hr prior to injection of glucose (1·5 mg/kg, i.p., Sigma-Aldrich). Blood glucose levels were determined at indicated time points using a standard glucometer.
Immunohistochemistry and immunofluorescence
Pancreatic samples were fixed in 10% formalin for 24 hr and embedded in paraffin. After the process of antigen retrieval, slides were blocked with CAS-BLOCK (Invitrogen, Carlsbad, CA). For immunohistochemistry, slides were incubated overnight with NKp46-Fc (8 μg/ml), washed and incubated for 30 min with horseradish peroxidase-conjugated α-human Fc antibody (1/200; Jackson ImmunoResearch, Bar Harbor). Slides were developed with an ImmPACT™ kit (Vector Labs, Burlingame, CA) and counterstained with haematoxylin. For immunofluorescence, slides were incubated overnight with guinea pig α-insulin (1/200; Dako, Glostrup, Denmark), washed and incubated with secondary antibody Cy3-conjugated guinea pig α-IgG (1/200; Jackson ImmunoResearch). Nuclei were stained with DAPI (1/2000; Sigma-Aldrich).
Statistical analysis
Results are expressed as mean ± SEM. Unpaired two tailed Student’s t-test was used to assess differences between treated groups and control. A P-value of ≤ 0·05 was considered significant.
Results
Human AAT reduces DC-derived IL-15 expression levels and subsequent NK cell activation
As hAAT has been shown to skew primed DC towards a tolerogenic phenotype, BMDC were pre-treated with hAAT, washed and stimulated with IFN-γ. Interleukin-15 and IL-15Rα levels were measured in the lysates of activated BMDC. Indeed, as shown in Fig.1(a), IL-15 production was significantly reduced in BMDC pre-treated with hAAT, resulting in non-stimulated IL-15 levels. In contrast, IFN-γ-induced IL-15Rα levels remained unchanged in the presence of hAAT. Pre-treatment of BMDC with various concentrations of hAAT also resulted in lower expression of IL-15, both in non-primed and in IFN-γ-activated BMDC, as shown in Fig.1(b).
Figure 1.
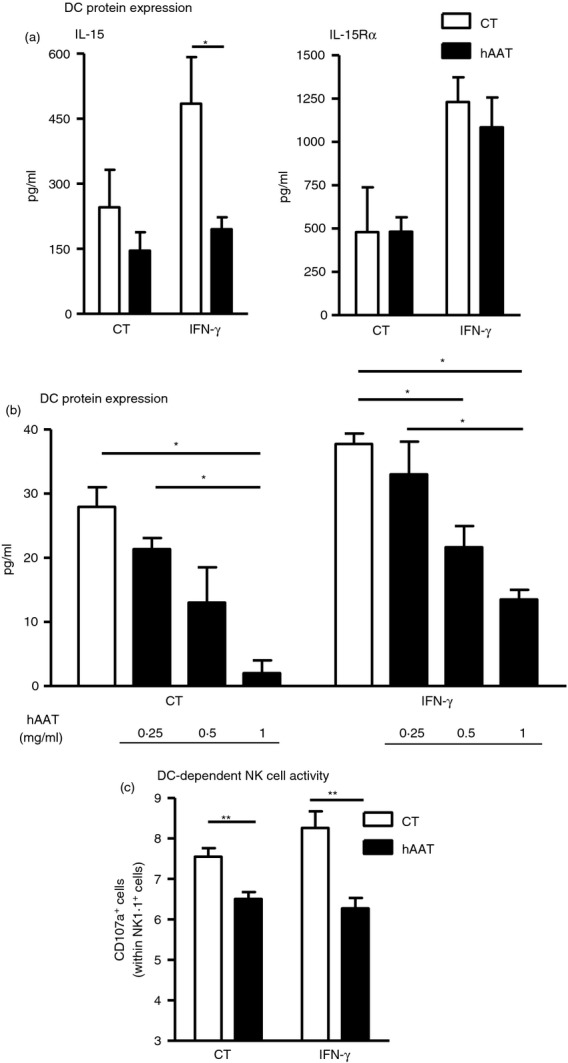
Human α1-antitrypsin (hAAT) reduces interleukin-15 (IL-15) production by dendritic cells (DC) and subsequently alters natural killer (NK) cell activation profiles. (a, b) Bone-marrow-derived DC (BMDC) were pre-treated overnight with hAAT (indicated doses) and then treated with interferon-γ (IFN-γ; 10 ng/ml) for 24 hr. IL-15 and IL-15Rα levels in cell lysates. (c) Poly-I : C treated NK cells were co-cultured with BMDC for 3 hr. CD107a+ out of NK1.1+ cells. Mean ± SEM, *P < 0·05, **P < 0·01.
We next tested whether such hAAT-pre-treated BMDC will also alter NK cell activation profiles. For this, BMDC were pre-treated with hAAT as before and then stimulated with IFN-γ. Poly(I:C)-treated NK cells were then added to washed pre-treated BMDC for 3 hr, and degranulation levels were evaluated by CD107a staining. As shown in Fig.1(c), IFN-γ-stimulated BMDC evoked a mild increase in NK cell degranulation. In contrast, significantly lower NK cell degranulation rates were observed in hAAT-pre-treated BMDC when they were non-stimulated or stimulated (a 14 ± 2% and 24 ± 3·9% reduction in CD107a+ NK cells, respectively).
hAAT alters β-cell-induced NK cell activation
In the context of β-cell injury during T1D, direct recognition of β-cells by NK cells has been proven to be highly significant. To examine whether hAAT treatment affects β-cell recognition by NK cells, poly(I:C)-treated NK cells were co-cultured with islets that were isolated from PBS-treated mice or hAAT-treated mice. Degranulation and IFN-γ release levels by NK cells were measured in comparison to NK cells alone, or in the presence of an agonistic anti-NKp46 antibody. As shown in Fig.2(a), NK cell degranulation was significantly greater when cultured with islets from animals pre-treated with PBS, compared with islets derived from animals pre-treated with hAAT. Nonetheless, when added in vitro, hAAT did not significantly alter the degree of NK cell degranulation.
Figure 2.
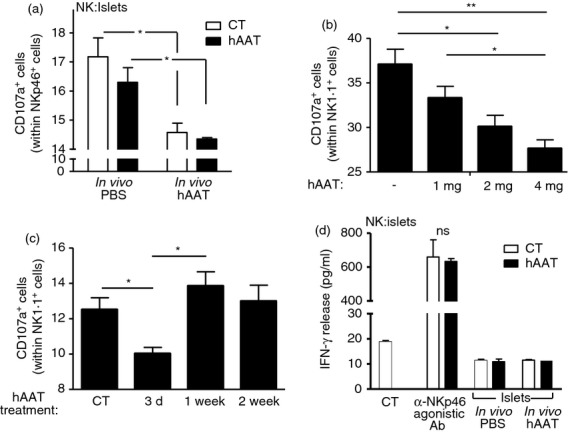
Human α1-antitrypsin (hAAT) alters β-cell-induced natural killer (NK) cell activation. (a–c) Islets were isolated from mice treated with hAAT at the indicated doses or with PBS (n = 4 per group). Twenty dissociated islets per sample were incubated with Poly(I:C)-treated NK cells in the presence or absence of 0·5 mg/ml hAAT. CD107a+ NK cells out of NKp46+ cells. Data representative of four experimental repeats. (d) Islets were isolated from mice treated with hAAT (2 mg per animal) or PBS (n = 3 per group). Twenty dissociated islets per sample in duplicate were incubated with Poly(I:C)-treated NK cells in the presence or absence of 0·5 mg/ml hAAT. Where stated, anti-NKp46 agonist antibody-coated wells were used. Interferon-γ (IFN-γ) release was measured after 48 hr. Data representative of two experimental repeats. Mean ± SEM, *P < 0·05, **P < 0·01.
To evaluate whether hAAT treatment exhibits a concentration-dependent profile, three doses of hAAT were administered to islet donor mice and subsequently islet β-cells derived from treated mice were used for NK cell degranulation assays. As shown in Fig.2(b), higher doses of hAAT resulted in lower levels of NK cell degranulation.
Time-dependent efficacy of hAAT was assayed by administration of uniform doses of hAAT to islet donor mice at different time-points, followed by measurement of NK cell degranulation. As depicted in Fig.2(c), hAAT treatment resulted in reduced levels of NK cell degranulation towards β-cells that were derived from mice treated with hAAT for 3 days, while degranulation levels proved unchanged for β-cells derived from mice treated for either 7 or 14 days.
Regarding the release of IFN-γ, shown in Fig.2(d), ligation of NKp46 by agonistic antibody resulted in a robust release of IFN-γ and was not affected by the presence of hAAT. The presence of islets alone did not evoke IFN-γ release by NK cells, and neither in vivo pre-treatment with hAAT nor in vitro introduction of hAAT affected IFN-γ release.
hAAT reduces membrane-associated NKp46 ligand levels on pancreatic β-cells but not on malignant cells
To examine the effect of hAAT on pancreatic β-cell membrane-associated NKp46 ligand levels, mice were injected with either PBS or hAAT at indicated doses before collection of steady-state pancreatic tissue. As shown in Fig.3(a), islets from PBS-treated mice stained positive with NKp46-Fc soluble receptor. In contrast, islets from hAAT-treated mice displayed markedly reduced staining. No discernible differences in islet morphology were detected, according to haematoxylin & eosin staining, and insulin content appeared uniform. In addition, according to flow cytometry analysis (Fig.3b), while GLUT2-positive β-cells from the PBS group were NKp46 ligand positive, β-cells from the hAAT group displayed 45% less membrane-associated NKp46 levels; GLUT2 levels are shown in the Supporting information (Fig. S1).
Figure 3.

Membrane-associated NKp46 ligand expression levels on pancreatic β-cells. DBA/2 mice were treated with human α1-antitrypsin (hAAT (2 mg per animal) or PBS for 3 days (n = 5 per group). (a) Representative histological images of immunohistochemical staining using soluble NKp46-Fc and horseradish peroxidase-conjugated anti-NKp46-Fc antibody (top), haematoxylin & eosin staining (middle) and anti-insulin immunofluorescence staining (bottom). Islet borders are demarcated by the dashed line. (b, c) NKp46-Fc+ cells derived from primary islets isolated from hAAT-treated (indicated doses) or PBS-treated DBA/2 mice (n = 5 per group). Fold change from PBS group; gated to GLUT2+ cells. Representative overlay is shown. (d) NKp46-Fc+ MIN-6 cells after hAAT treatment (0·5 mg/ml) for 72 hr. (e) NKp46-Fc+ B16-F10 cells after hAAT treatment (0·5 mg/ml) for 24, 48 and 72 hr. (f) Peripheral blood dendritic cells and polymorphonuclear cells were isolated from DBA/2 mice treated with 2 mg hAAT or PBS (n = 3 per group) and stained for NKp46-Fc. Data are representative of two to four experimental repeats. Mean ± SEM, *P < 0·05, **P < 0·01.
To examine whether hAAT displays a dose-dependent effect on NKp46 ligand expression on pancreatic β-cells, islet donor mice were given different doses of hAAT, as indicated in Fig.3(c). Islets were subsequently purified and examined for soluble NKp46-Fc binding capacity. As shown, treatment with higher doses of hAAT resulted in lower binding levels of soluble NKp46-Fc.
In contrast to the findings obtained using primary β-cells, MIN-6 insulinoma cells that were treated with hAAT for 72 hr stained positive for surface NKp46 ligand expression (Fig.3d), as did B16-F10 melanoma cells treated with hAAT for 24, 48 and 72 hr (Fig.3e). Human AAT was also found to allow IFN-γ-induced MHC class I expression, as determined in B16-F10 cells (see Supporting information, Fig. S2).
NKp46 ligand expression was also examined in peripheral blood-derived DC and neutrophils (Fig3f). NKp46 readily bound to neutrophils and was unaffected by in vivo hAAT treatment, while DC stained negative in both control and hAAT-treated groups.
Tumour cell-elicited NK cell activation profiles in the presence of hAAT
We next sought to determine whether membrane expression of the NK cell activating receptors NKp46 and NKG2D was altered by short-term treatment with hAAT. Mice were injected with PBS or hAAT and after 3 days splenic NK cells were examined by flow cytometry analysis. As shown in Fig.4(a), expression of both NKp46 and NKG2D was unchanged. Administration of three different doses of hAAT likewise resulted in unchanged expression levels of both NKp46 and NKG2D, as depicted in Fig.4(b).
Figure 4.
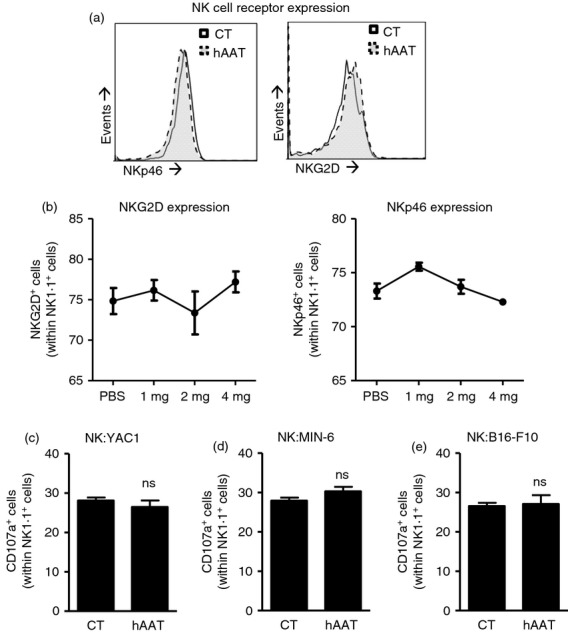
Intact expression of activating receptors and tumour cell-evoked natural killer (NK) cell activation during human α1-antitrypsin (hAAT) treatment. (a, b) C57BL/6 mice were given hAAT at indicated doses or PBS (n = 3 per group). Expression of the membranal activating receptors NKp46 and NKG2D was examined in splenic NK cells. (c–e) Poly(I:C)-treated NK cells were co-cultured with (c) YAC1 cells, (d) MIN-6 insulinoma cells or (e) B16-F10 melanoma cells in the presence or absence of 0·5 mg/ml hAAT for 3 hr. CD107a+ cells out of NK1.1+ cells. Mean ± SEM.
We further examined the effect of hAAT treatment on the general capacity of NK cells to recognize classical target cells. Therefore, poly(I:C)-treated NK cells were co-cultured with a series of tumour cell lines. As expected, upon co-culture with YAC1 cells, NK cell degranulation was evoked, as manifested by CD107a elevation. Comparable levels of CD107a were also found in NK cells treated with hAAT (Fig.4c). A similar pattern of NK activation was also observed when cultured with the MIN-6 insulinoma β-cell line (Fig.4d), and NK cells co-cultured with B16-F10 melanoma cells also exhibited CD107a degranulation levels that were not altered by hAAT (Fig.4e).
NK cell depletion and hAAT do not act synergistically in the MLD-STZ-induced hyperglycaemia model
The possibility that hAAT affords protection to islet β-cells by means of altering NK cell activation profiles was addressed in the MLD-STZ model by comparing the outcomes of hAAT treatment and NK cell depletion; the combination of the two approaches was also examined so as to gather an indication of a mechanism by observing whether these manipulations overlap, or rather exhibit a synergistic behaviour. MLD-STZ-treated mice were injected with suboptimal hAAT (1 mg per mouse) or had undergone NK cell depletion using α-asialo-GM1 antibodies. Blood glucose follow-up was performed and bodyweight loss was recorded (Fig.5). As shown, NK cell depletion partially alleviated the rise in non-fasting blood glucose levels and suboptimal hAAT had no effect on non-fasting blood glucose levels. However, the two approaches diverged in the category of changes in body weight: NK cell depletion was superior in preserving near-normal body weight, and suboptimal hAAT was intermediate in effect. To identify the component of β-cell function in the treatment groups, mice underwent an overnight fast on day 18, glucose was injected i.p. and mice were then tested for glucose clearance. As shown, both approaches displayed a trend for the better in initial glucose clearance (60, 90 and 120 min after challenge). However, while statistical significance arose only at 150 min and 180 min after i.p. glucose challenge, mice treated with hAAT cleared glucose at a significantly superior rate compared with untreated MLD-STZ mice, an outcome that was not observed in the NK cell-depleted group.
Figure 5.
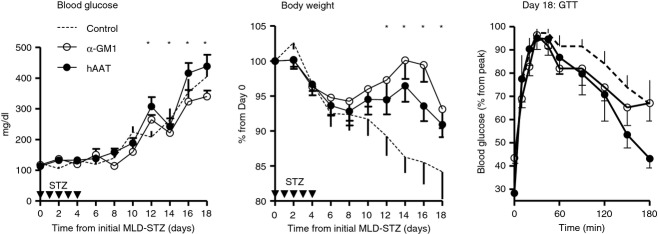
Multiple low-dose streptozotocin (MLD-STZ) : α1-antitrypsin (hAAT) treatment and natural killer (NK) cell depletion. Mice were subjected to MLD-STZ. Groups received either PBS or α-GM1 antibody or hAAT (1 mg per animal). The hAAT treatment started 1 day before STZ injections and α-GM1 was started 3 days before STZ injections. Blood glucose and body weight, mean ± SEM, *P < 0·05 between control and α-GM1 group. GTT, glucose tolerance test, mean ± SEM, *P < 0·05 between control and hAAT group.
The combination of hAAT and NK cell depletion as a single treatment was evaluated (see Supporting information, Fig S3); there was no apparent synergy between the two approaches, the resulting curve in blood glucose levels and body weight overlapped NK cell depletion results, and the curve in the glucose tolerance test assay overlapped hAAT treatment outcomes.
Discussion
Clinical application of hAAT therapy for several autoimmune pathologies has recently been suggested, prompting the study of its effect on various immune cell populations. However, a comprehensive study of hAAT regulation of NK cell activities has yet to be performed, despite evidence for the involvement of NK cells in islet injury during autoimmune diabetes, and the established beneficial effect of hAAT on islet survival under comparable conditions.
As hAAT allows isolated T-lymphocyte responses, it was not unexpected to find that NK cell responses remain intact in the presence of hAAT. Yet in vivo outcomes led us to conclude that NK cell responses are indirectly altered by hAAT towards protection of β-cells. Hence, we hypothesized that hAAT may regulate DC : NK cell interactions.
Dendritic cell activities are known to be modified by hAAT towards a tolerogenic profile.21 Our findings suggest that hAAT indirectly down-regulates NK cell activation upon encounter with hAAT-pre-treated DC, possibly by reduced membrane-bound IL-15 levels. In parallel, despite being an inducible receptor, IL-15Rα expression was unchanged by hAAT. Interleukin-15 cross-presentation is a critical component of DC-mediated activation of NK cells, as well as a potent driver of allograft rejection23 and islet injury.24 These data support an indirect inhibitory effect of hAAT on NK cells, as they appear to be functional but are less cytotoxic when primed by DC that fall short of complete inflammatory maturation. Hence, our findings offer a novel immunological mechanism by which hAAT may act to protect β-cells from immune responses in autoimmune diabetes and allograft transplantation.
Time- and dose-dependency experiments proved informative: short-term in vivo hAAT monotherapy resulted in reduced NK cell responses against islets in a dose-dependent manner. The timeframe required for hAAT to exert a protective effect on β-cells was determined to be < 3 days, which confirmed other in vivo studies in which effective hAAT therapy requires administration of hAAT every 3 days. We further examined expression and specific activation of the NKp46 receptor, and found that they are unaffected by hAAT across several doses. Hence, NKp46 expression appears not to be the mode of action employed by hAAT when modifying responses towards β-cells; rather, selective down-regulation of NKp46 ligand expression by β-cells takes place, as seen during short-term hAAT treatment. The identity of the β-cell-expressed NKp46 ligand, its half-life and their regulations are unknown; further investigation is required to identify the association of these pathways with hAAT.
Inhibition of tumour growth by hAAT was previously demonstrated using nude mice.20 The present study is the first to examine outcomes of hAAT treatment in an intact immune system response to cancer targets. The selectivity of reduction in NK cell responses against β-cells was addressed by evaluating NK cell activity towards classic tumour targets. Three lineages of tumour origin were examined: MIN-6 cells that represent insulinoma, B16-F10 cells that represent invasive melanoma and YAC1 cells that represent T-cell lymphoma. Findings support the conclusion that hAAT allows NK cell activation and NKp46 ligand expression in the aforementioned target cells. Similarly, inducible MHC class I expression on B16-F10 cells was preserved. Hence, one may conclude that hAAT allows anti-tumour NK cell responses in conjugation with protecting β-cells, as opposed to the implications of protective immunosuppression.
The MLD-STZ model was used to examine whether the reported protection of hAAT on islet β-cells might be NK cell-dependent. Since the effect of hAAT in this model has been previously established using 2 mg hAAT per mouse, we introduced a suboptimal dose of hAAT with the motivation of exposing potential synergy between hAAT treatment and NK cell depletion. Several benefits were observed in mice treated by both approaches, albeit to different degrees. NK cell depletion was superior in controlling blood glucose, although one should consider that the present comparison refers to a suboptimal dose of hAAT. Similarly, prevention of weight loss appears to be an outcome of NK cell depletion more than hAAT therapy. That said, 18 days into the model, and despite the depleted group displaying lower blood glucose levels, it was the hAAT group that displayed a significantly more pronounced clearance of glucose. One might consider that the model initially addresses β-cell function, in which stage both groups show benefit; the model later addresses peripheral glucose control; hAAT, in a suboptimal dose, exceeded NK cell depletion in facilitating peripheral glucose clearance at this stage. This distinction between the two approaches signifies the multiple attributes of hAAT in controlling systemic inflammation, compared with the isolated removal of NK cells.
Taken together, our results depict an immunomodulatory function for hAAT in the context of β-cell protection with relevance to DC-regulated activation of NK cells and by reduction of membrane-associated NKp46 ligand expression on β-cells. Our findings provide mechanistic evidence for the marked safety profile of long-term hAAT therapy, yet further studies are required to fully clarify the mechanism by which hAAT selectively decreases NKp46 ligand expression on β-cells, and to identify the potential significance of hAAT-induced modification of the critical DC : NK cell cross-talk.
Acknowledgments
Ofer Guttman performed the experiments, designed the study and wrote the paper. Rami Yossef performed the experiments, designed the study and wrote the paper. Gabriella Freixo-Lima performed the experiments. Peleg Rider designed the study and wrote the paper. Angel Porgador designed the study and wrote the paper. Eli C. Lewis designed the study and wrote the paper. This study was supported by the Juvenile Diabetes Research Foundation (JDRF) and Israel Science Foundation (ISF) – JDRF Joint Program in Type I Diabetes Research. We thank Dr Yaron Carmi for his instrumental help in critical reading of the manuscript, and Mrs Valeria Frishman for her excellent technical assistance.
Glossary
- AAT
α1-antitrypsin
- APC
allophycocyanin
- BMDC
bone-marrow-derived DC
- DC
dendritic cell
- IFN
interferon
- IL
interleukin
- i.p.
intraperitoneal
- MLD-STZ
multiple low-dose streptozotocin
- NK
natural killer
- NOD
non-obese diabetic
- PE
phycoerythrin
- T1D
type 1 diabetes
- TNF
tumour necrosis factor
Disclosures
The authors declare no financial conflict of interest.
Supporting Information
Figure S1. Association between GLUT2 and NKp46 ligand surface expression levels in pancreatic β-cells. DBA/2 mice were treated with human α1-antitrypsin (hAAT; 2 mg per animal) or PBS for 3 days (n = 5 per group). Fold change mean fluorescence intensity for GLUT2 staining; representative overlay. Mean ± SEM, *P < 0·05.
Figure S2. B16-F10 cells (1 × 105 cells per well, in triplicate) were treated with α1-antitrypsin (0·5 mg/ml) for 24 hr, washed and treated with interferon-γ (200 U/ml) for 24 hr. MHC class Ihigh B16-F10 cells (% of total). Mean ± SEM.
Figure S3. Multiple low-dose streptozotocin (MLD-STZ): α1-antitrypsin (hAAT) treatment combined with natural killer (NK) cell depletion. Mice were subjected to MLD-STZ. Groups received either PBS or α-GM1 antibody or hAAT (1 mg per animal). The hAAT treatment started 1 day before STZ injections and α-GM1 was started 3 days before STZ injections. Blood glucose and body weight; mean ± SEM, *P < 0·05 between control and α-GM1 + hAAT group. GTT, glucose tolerance test; mean ± SEM.
References
- Paik SG, Fleischer N, Shin SI. Insulin-dependent diabetes mellitus induced by subdiabetogenic doses of streptozotocin: obligatory role of cell-mediated autoimmune processes. Proc Natl Acad Sci USA. 1980;77:6129–33. doi: 10.1073/pnas.77.10.6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludvigsson J, Faresjo M, Hjorth M, et al. GAD treatment and insulin secretion in recent-onset type 1 diabetes. N Engl J Med. 2008;359:1909–20. doi: 10.1056/NEJMoa0804328. [DOI] [PubMed] [Google Scholar]
- Kaufman A, Herold KC. Anti-CD3 mAbs for treatment of type 1 diabetes. Diabetes Metab Res Rev. 2009;25:302–6. doi: 10.1002/dmrr.933. [DOI] [PubMed] [Google Scholar]
- von Herrath M, Peakman M, Roep B. Progress in immune-based therapies for type 1 diabetes. Clin Exp Immunol. 2013;172:186–202. doi: 10.1111/cei.12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gur C, Porgador A, Elboim M, et al. The activating receptor NKp46 is essential for the development of type 1 diabetes. Nat Immunol. 2010;11:121–8. doi: 10.1038/ni.1834. [DOI] [PubMed] [Google Scholar]
- Caligiuri MA. Human natural killer cells. Blood. 2008;112:461–9. doi: 10.1182/blood-2007-09-077438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delovitch TL, Singh B. The nonobese diabetic mouse as a model of autoimmune diabetes: immune dysregulation gets the NOD. Immunity. 1997;7:727–38. doi: 10.1016/s1074-7613(00)80392-1. [DOI] [PubMed] [Google Scholar]
- Dotta F, Censini S, van Halteren AG, et al. Coxsackie B4 virus infection of β cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proc Natl Acad Sci USA. 2007;104:5115–20. doi: 10.1073/pnas.0700442104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama T, Watanabe K, Yanagawa T, et al. The suppressive effect of anti-asialo GM1 antibody on low-dose streptozotocin-induced diabetes in CD-1 mice. Diabetes Res. 1991;16:171–5. [PubMed] [Google Scholar]
- Ogasawara K, Hamerman JA, Ehrlich LR, Bour-Jordan H, Santamaria P, Bluestone JA, Lanier LL. NKG2D blockade prevents autoimmune diabetes in NOD mice. Immunity. 2004;20:757–67. doi: 10.1016/j.immuni.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Ogasawara K, Hamerman JA, Hsin H, et al. Impairment of NK cell function by NKG2D modulation in NOD mice. Immunity. 2003;18:41–51. doi: 10.1016/s1074-7613(02)00505-8. [DOI] [PubMed] [Google Scholar]
- Gur C, Enk J, Kassem SA, et al. Recognition and killing of human and murine pancreatic β cells by the NK receptor NKp46. J Immunol. 2011;187:3096–103. doi: 10.4049/jimmunol.1101269. [DOI] [PubMed] [Google Scholar]
- Lewis EC, Mizrahi M, Toledano M, et al. α1-Antitrypsin monotherapy induces immune tolerance during islet allograft transplantation in mice. Proc Natl Acad Sci USA. 2008;105:16236–41. doi: 10.1073/pnas.0807627105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis EC, Shapiro L, Bowers OJ, Dinarello CA. α1-antitrypsin monotherapy prolongs islet allograft survival in mice. Proc Natl Acad Sci USA. 2005;102:12153–8. doi: 10.1073/pnas.0505579102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Lu Y, Campbell-Thompson M, Spencer T, Wasserfall C, Atkinson M, Song S. α1-antitrypsin protects β-cells from apoptosis. Diabetes. 2007;56:1316–23. doi: 10.2337/db06-1273. [DOI] [PubMed] [Google Scholar]
- Lewis EC. Expanding the clinical indications for α1-antitrypsin therapy. Mol Med. 2012;18:957–70. doi: 10.2119/molmed.2011.00196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, Qiao F, Atkinson C, Holers VM, Tomlinson S. A complement C3 inhibitor specifically targeted to sites of complement activation effectively ameliorates collagen-induced arthritis in DBA/1J mice. J Immunol. 2007;179:7860–7. doi: 10.4049/jimmunol.179.11.7860. [DOI] [PubMed] [Google Scholar]
- Andre F, Schartz NE, Chaput N, Flament C, Raposo G, Amigorena S, Angevin E, Zitvogel L. Tumor-derived exosomes: a new source of tumor rejection antigens. Vaccine. 2002;20(Suppl 4):A28–31. doi: 10.1016/s0264-410x(02)00384-5. [DOI] [PubMed] [Google Scholar]
- Huang H, Campbell SC, Bedford DF, et al. Peroxisome proliferator-activated receptor γ ligands improve the antitumor efficacy of thrombospondin peptide ABT510. Mol Cancer Res. 2004;2:541–50. [PubMed] [Google Scholar]
- Huang H, Campbell SC, Nelius T, Bedford DF, Veliceasa D, Bouck NP, Volpert OV. α1-antitrypsin inhibits angiogenesis and tumor growth. Int J Cancer. 2004;112:1042–8. doi: 10.1002/ijc.20494. [DOI] [PubMed] [Google Scholar]
- Ozeri E, Mizrahi M, Shahaf G, Lewis EC. α1 antitrypsin promotes semimature, IL-10-producing and readily migrating tolerogenic dendritic cells. J Immunol. 2012;189:146–53. doi: 10.4049/jimmunol.1101340. [DOI] [PubMed] [Google Scholar]
- Fernandez NC, Lozier A, Flament C, et al. Dendritic cells directly trigger NK cell functions: cross-talk relevant in innate anti-tumor immune responses in vivo. Nat Med. 1999;5:405–11. doi: 10.1038/7403. [DOI] [PubMed] [Google Scholar]
- Kroemer A, Xiao X, Degauque N, Edtinger K, Wei H, Demirci G, Li XC. The innate NK cells, allograft rejection, and a key role for IL-15. J Immunol. 2008;180:7818–26. doi: 10.4049/jimmunol.180.12.7818. [DOI] [PubMed] [Google Scholar]
- Cardozo AK, Proost P, Gysemans C, Chen MC, Mathieu C, Eizirik DL. IL-1β and IFN-γ induce the expression of diverse chemokines and IL-15 in human and rat pancreatic islet cells, and in islets from pre-diabetic NOD mice. Diabetologia. 2003;46:255–66. doi: 10.1007/s00125-002-1017-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Association between GLUT2 and NKp46 ligand surface expression levels in pancreatic β-cells. DBA/2 mice were treated with human α1-antitrypsin (hAAT; 2 mg per animal) or PBS for 3 days (n = 5 per group). Fold change mean fluorescence intensity for GLUT2 staining; representative overlay. Mean ± SEM, *P < 0·05.
Figure S2. B16-F10 cells (1 × 105 cells per well, in triplicate) were treated with α1-antitrypsin (0·5 mg/ml) for 24 hr, washed and treated with interferon-γ (200 U/ml) for 24 hr. MHC class Ihigh B16-F10 cells (% of total). Mean ± SEM.
Figure S3. Multiple low-dose streptozotocin (MLD-STZ): α1-antitrypsin (hAAT) treatment combined with natural killer (NK) cell depletion. Mice were subjected to MLD-STZ. Groups received either PBS or α-GM1 antibody or hAAT (1 mg per animal). The hAAT treatment started 1 day before STZ injections and α-GM1 was started 3 days before STZ injections. Blood glucose and body weight; mean ± SEM, *P < 0·05 between control and α-GM1 + hAAT group. GTT, glucose tolerance test; mean ± SEM.