

Abstract
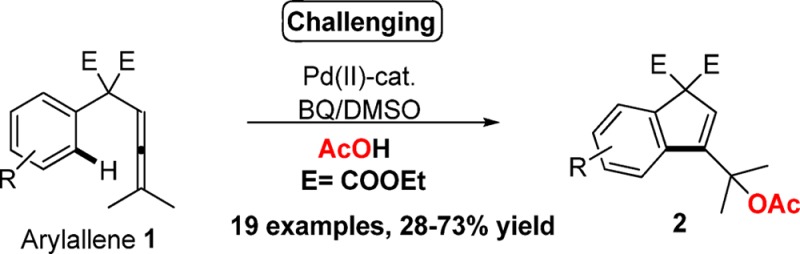
Herein we report an example of tandem oxidative acetoxylation/carbocyclization of arylallenes 1 using Pd(OAc)2. The catalytic protocol is highly selective and provides access to new C–C and C–O bonds leading to a carbocyclization. The reaction proceeds via C–H activation by Pd. Mechanistic investigations show that the C–H activation is not the rate-limiting step and indicate that the reaction proceeds via acetoxylation of the allene.
Transition-metal-catalyzed C–H bond functionalization for construction of new C–C bonds in a selective and controlled manner is an actual challenge in organic synthesis and has received significant attention in recent years.1 Despite notable progress in this area, more practical and general applications of aryl C–H bond activation still rely on the assistance of a heteroatom-based neighboring functional group, which often is difficult to remove or modify.1 However, an atom-economic approach would be to utilize a directing group directly involved in the functionalization in a tandem fashion, which intramolecularly induces an activation of the ortho arene C–H bond and leads to a carbocyclization.2
Transition-metal-catalyzed oxidative carbocyclizations have been identified as potential technologies for designing more complex structures occurring in various biologically active natural products and pharmaceuticals.3 During the past decade, we have developed a number of highly regio- and stereoselective palladium-catalyzed oxidative carbocyclizations of enallenes, dienallenes, allenynes, and enynes.4,5
Some time ago we reported on the intramolecular Pd(II)-catalyzed oxidative carbocyclizations of dienallenes for the stereospecific formation of bicyclic systems (Scheme 1a).5c,6 A detailed mechanistic study revealed the participation of a (π-allyl)palladium species, which is attacked by a nucleophile to furnish the product. More recently we have also developed a novel Pd(II)-catalyzed oxidative acetoxylation/carbocyclization protocol for allenynes to form acetoxylated vinylallenes in a one pot operation (Scheme 1b).7
Scheme 1. Pd-Catalyzed Intramolecular Oxidative Acetoxylation/Carbocyclization of Dienallenes, Allenynes, and Arylallenes.
To date, no carbocyclization process8 is known where an allene moiety acts as directing group for intramolecular aryl sp2 C–H bond activation. We have now developed an oxidative palladium-catalyzed protocol for tandem acetoxylation/carbocyclization of arylallenes 1, where the allene moiety forms a new C–C bond with the aryl via a sp2 C–H bond activation (Scheme 1c).9
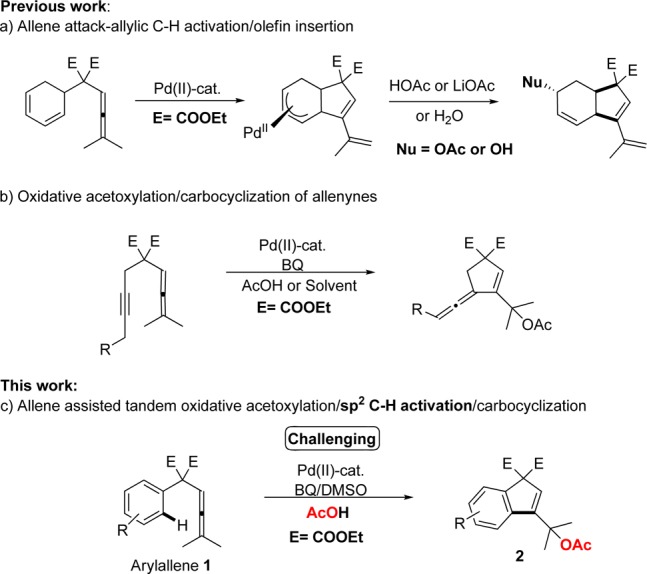
In our initial investigations we choose arylallene 1a as model substrate for the tandem carbocyclization/acetoxylation reactions using 5 mol % of Pd(OAc)2 in the presence of 1.5 equiv of p-benzoquinone (BQ) in acetic acid at 60 °C. Under these conditions the desired carbocyclization product was obtained in 24% yield (Table 1, entry 1). An increase of the catalyst loading to 10 mol % of Pd(OAc)2 proved beneficial and afforded a 45% yield of 2a along with 18% yield of dimer 3a (Table 1, entry 2).
Table 1. Optimization Studies for Palladium(II)-Catalyzed Oxidative Carbocyclizationsa.
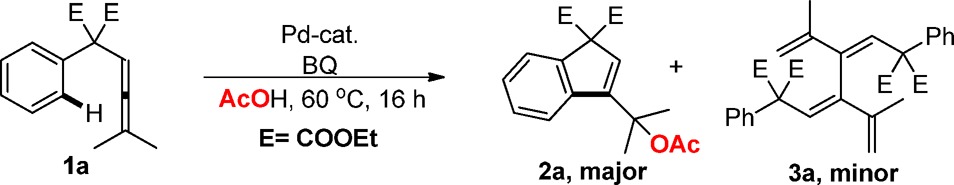
Reaction conditions: 1a (0.1 mmol), Pd-cat (5–10 mol %), additive (0.2 equiv), AcOH (1 mL), 60 °C.
Yield was determined by 1H NMR spectroscopy using mesitylene as internal standard. Parentheses represent the isolated yield. Product ratio was determined by 1H NMR spectroscopy using mesitylene as internal standard.
White catalyst.
Reaction at 25 °C.
Reaction at 40 °C.
Reaction at 80 °C. L = triphenylphosphine, L1 = acridine. BPA = BINOL−racemic phosphoric acid.
We next studied the influence of different Pd-salts and additives on the model reaction. Use of other Pd(II)-salts, such as the White catalyst ([1,2-bis(phenylsulfinyl)ethane]palladium acetate), PdCl2, or Pd(TFA)2 did not lead to any improvement of the yield of 2a, whereas Pd(acac)2 resulted in a promising yield of 46% of 2a together with 16% yield of dimer 3a (Table 1, entries 3–6). We therefore studied the influence of different additives on the carbocyclization reaction using Pd(OAc)2 as catalyst. Application of acridine (L1), triphenylphosphine (L), or racemic BINOL−phosphoric acid (BPA) inhibited the reaction (Table 1, entries 7–9, see Supporting Information (SI) for details).
On the other hand, the use of 2 equiv of dimethyl sulfoxide (DMSO) as additive in combination with Pd(OAc)2 enhanced the formation of product 2a (60% yield) while inhibiting the formation of the dimeric byproduct 3a (Table 1, entries 10 and 11). Various electronically and sterically different DMSO derivatives were tested, which showed that DMSO was the best sulfoxide for this carbocyclization reaction (Table S1). The oxidant also plays a crucial role for the selective Pd-catalyzed carbocyclizations. The use of different oxidants such as PhI(OAc)2, a series of substituted benzoquinones, or various metal salts (CuCl2, Cu(OAc)2, AgOAc, and Ag2O) in the presence of catalytic amounts of Pd(OAc)2 proved to be inefficient (Table S3). These studies show that BQ is the optimal oxidant and use of 1.7 equiv of BQ in combination with 2 equiv of DMSO and 10 mol % of Pd(OAc)2 in acetic acid afforded 62% isolated yield of 2a (Table 1, entry 12). The use of Pd(acac)2 as catalyst under these conditions gave a lower yield (Table 1, entry 13).
Surprisingly, under the optimal reaction conditions, examination of different cosolvents (THF, 1,4-dioxane, 1,2-dichloroethane, acetonitrile and toluene) showed that the use of acetic acid as the sole solvent is crucial for the carbocyclization/acetoxylation of 1a to occur (Table 1, entry 12 and Table S5).
Variation of the reaction temperature proved that 60 °C is the optimal temperature for this protocol (Table 1, entries, 12, 14–16). Control experiments showed that removal of either Pd(OAc)2 or BQ completely stopped the reaction (Table 1, entries 17–18).
After having established the optimized reaction condition, we next explored the scope of the Pd(II)-catalyzed tandem acetoxylation/carbocyclizations of arylallenes 1 (Table 2). Arylallenes with m-Me- or p-Me-substituents on the aromatic ring afforded 56 and 62% yield of 2c and 2d, respectively. However, o-Me-substituted arylallene resulted in a lower yield of 2b, which may be due to steric effects. The introduction of electron-donating substituents led to a decrease in the efficiency of the reaction (2e, 2f, and 2g vs 2a). Interestingly, in the case of m-OMe-substituted arylallene a mixture of two different regioisomers in a 6.5:1 ratio was formed. Coordination of the neighboring OMe-group should facilitate the formation of the intermediate Pd-species that leads to 2e.
Table 2. Pd(II)-Catalyzed Tandem Oxidative Acetoxylation/Carbocyclization of Arylallenesa,b.
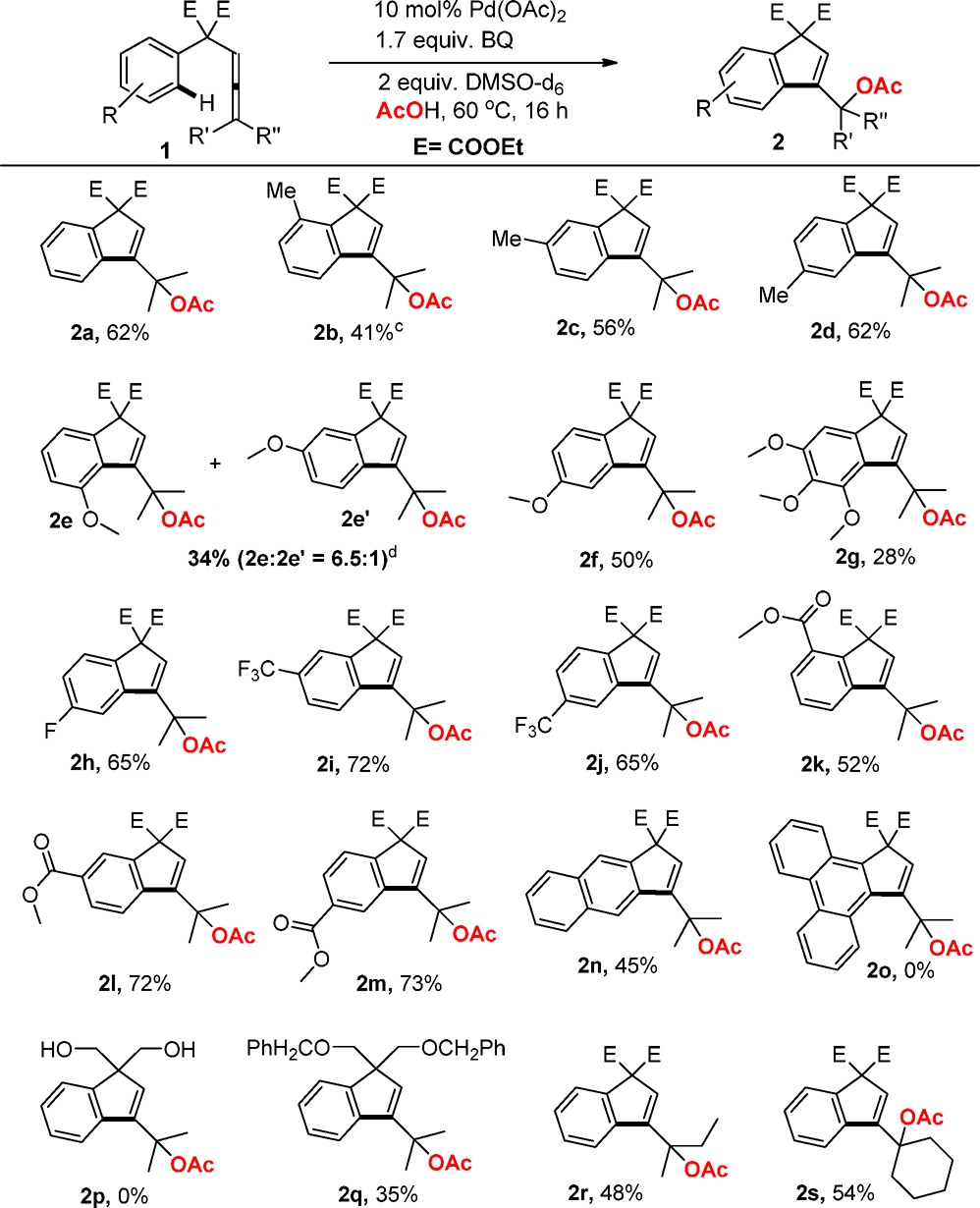
Reaction conditions: 1a (0.2 mmol), Pd-cat (10 mol %), DMSO-d6 (0.4 equiv), BQ (1.7 equiv), AcOH (2 mL), 60 °C.
Isolated Yield.
Pd-cat (20 mol %) was used.
Product ratio was determined by 1H NMR.
Arylallenes with electron-withdrawing substituents on the aromatic ring such as F, CF3, or an ester substituent improved the catalytic performance of the system leading to yields up to 73% (2h–2m). The reaction of naphthalene-derived substrate 1n afforded 45% isolated yield of 2n, whereas, the analogous phenanthrene derivative 1o was completely unreactive (2o). Furthermore, reaction of functionalized arylallene with dibenzyl ether (1q) resulted in a moderate yield of the carbocyclization product 2q, while the corresponding diol derivative 2p was found to be unreactive, probably due to coordination of the free alcohols to Pd. Also other substituents on the allene moiety worked (1r and 1s) and 2r and 2s were obtained in 48 and 54% yield, respectively.
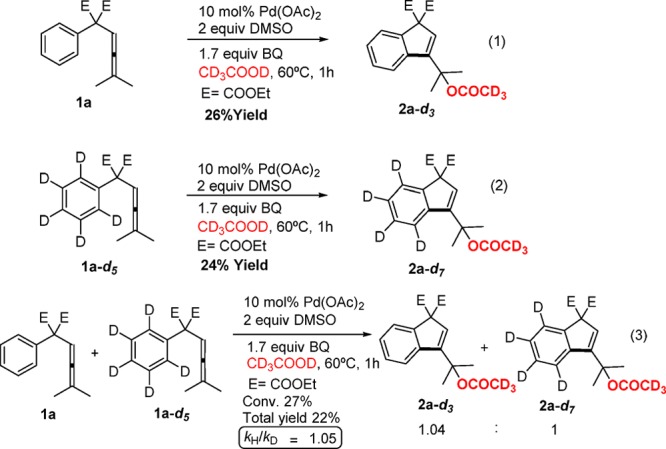
To gain more insight into the mechanism of the carbocyclization reaction, deuterium kinetic isotope effect (KIE) studies were performed.10 To investigate whether there is a deuterium KIE, we initially studied the parallel oxidative carbocyclization reactions of arylallenes 1a and 1a-d5 under the standard catalytic conditions of Table 2, for 1 h. The progress of the reactions was monitored by 1H NMR in different time intervals (see SI for details), and for these studies deuterated acetic acid (CD3COOD) had to be used as solvent. The individual parallel experiments revealed that 1a and 1a-d5 react with almost equal rates (kH/kD ≈ 1, kH/kD < 1.2, Scheme 2 and SI). These experimental findings prove that C–H bond breaking is not the rate-determining step in the oxidative carbocyclization reaction. The lack of kinetic isotopic effect is in contrast to the large isotope effect usually observed in similar kinds of C–H activation processes reported in the literature.11 This observation suggests that the present tandem acetoxylation/carbocyclization follows a different mechanism compared to those previously reported. A competitive experiment was also performed using a 1:1 mixture of 1a and 1a-d5 under the optimized reaction conditions in deuterated acetic acid. The reaction was monitored by 1H NMR for 1 h, and the observed product ratio 2a-d3/2a-d7 was 1.04/1 at 22% combined yield which gives kH/kD = 1.05 (Scheme 2). To rule out a reversible C–H bond activation1a with an equilibrium isotope effect, we showed that no hydrogen–deuterium exchange was observed when nondeuterated acetic acid (CH3COOH) was used as solvent in the acetoxylation/carbocyclization of the deuterated substrate 1a-d5 under the standard conditions. The negligible isotope effect and the lack of deuterium scrambling show that there must be an irreversible step before the C–H bond cleavage.
Scheme 2. Kinetic Studies on the Pd-Catalyzed Oxidative Acetoxylation/Carbocyclization of Arylallenes.
To exclude the possibility of the involvement of the dimethyl unit of the allene in the carbocyclization process, the hexadeuterated arylallene 1a-d6 and 1a were allowed to react in separate experiments. As expected, no deuterium isotope effect was observed showing that C–H bond breaking is not involved in the carbocyclization process (see SI). A competitive experiment using a 1:1 mixture of 1a and 1a-d6 under the standard reaction conditions gave kH/kD = 1.2 (Scheme 3).
Scheme 3. Kinetic Studies on the Pd-Catalyzed Oxidative Acetoxylation/Carbocyclization of Arylallenes.
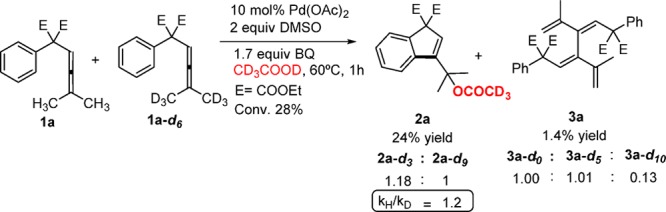
Interestingly, there was a large deuterium isotope effect for the formation of the dimer in the competitive experiment in Scheme 3. The dimers 3a-d0, 3a-d5, and 3a-d10 were formed in a ratio of 1.00:1.01:0.13, and this ratio gives an isotope effect kH/kD = 7.2 ± 0.8 for the first step of the dimer formation (A → D, Scheme 4).12
Scheme 4. Plausible Mechanism for the Pd-Catalyzed Tandem Oxidative Acetoxylation/Carbocyclization of Arylallene.

On the basis of the above experimental findings, a probable mechanism for the tandem acetylation/carbocyclizations of arylallene 1 is proposed in Scheme 4. Initial allene activation by Pd(OAc)2 is expected to form intermediate chelate complex A, which is followed by nucleophilic attack by acetate on the coordinated allene. This attack leads to formation of vinyl Pd(II)-intermediate B. Subsequent acetate-assisted C–H activation1a of the ortho arene C–H bond via TSB–C with concomitant loss of HOAc would generate intermediate C. A reductive elimination from C would give product 2 and Pd(0). The latter is reoxidized to Pd(II) by BQ, which closes the catalytic cycle. The observation that electron-deficient aryls react faster than electron-rich ones is in accordance with a C–H activation via a concerted metalation-deprotonation pathway.1d,13,14
This mechanism is supported by the fact that no KIE was observed according to Scheme 2. Also, allene attack on Pd(II) in intermediate A, would generate vinyl Pd(II)-intermediate D. Insertion of the allene of another molecule of 1 into the palladium−carbon bond of D would give a (π-allyl)palladium intermediate (see Scheme S1), which on β-elimination would generate dimer 3. Apparently, intermediate D undergoes dimerization faster than cyclization.
According to the mechanism in Scheme 4 there are two competing pathways leading to either product 2 or dimer 3. To validate the presence of competing pathways, the carbocyclization reaction of 1a-d6 was run under the standard conditions of Table 2. The product 2a-d6 was obtained in 83% NMR yield, and the formation of dimer was suppressed (<1% dimer 3a-d10) (see SI).
In conclusion, we have developed a novel and efficient oxidative Pd(II)-catalyzed tandem acetoxylation/carbocyclization of arylallenes to access synthetically important functionalized indene frameworks. Notably, it was found that the allene moiety can function as a potential directing group and assist the activation for a challenging ortho arene C–H bond activation in a tandem carbocyclization process. Measurements of deuterium kinetic isotope effects suggest that C–H bond breaking is not the rate-limiting step and that the acetoxypalladation of the allene moiety is the first irreversible step. Further studies on the tandem carbocyclization mechanism are ongoing in our laboratory and will be reported in due course.
Acknowledgments
Financial support from the European Research Council (ERC AdG 247014), the Swedish Research Council, and the Berzelius Center EXSELENT is gratefully acknowledged.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b06068.
Experimental details (PDF)
Author Contributions
† These authors contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- For selected recent reviews on C-H activation, see:; a Ackermann L. Acc. Chem. Res. 2014, 47, 281. 10.1021/ar3002798. [DOI] [PubMed] [Google Scholar]; b Arockiam P. B.; Bruneau C.; Dixneuf P. H. Chem. Rev. 2012, 112, 5879. 10.1021/cr300153j. [DOI] [PubMed] [Google Scholar]; c Kuhl N.; Hopkinson M. N.; Wencel-Delord J.; Glorius F. Angew. Chem., Int. Ed. 2012, 51, 10236. 10.1002/anie.201203269. [DOI] [PubMed] [Google Scholar]; d Ackermann L. Chem. Rev. 2011, 111, 1315. 10.1021/cr100412j. [DOI] [PubMed] [Google Scholar]; e Lyons T. W.; Sanford M. S. Chem. Rev. 2010, 110, 1147. 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Chen X.; Engle K. M.; Wang D.-H.; Yu J.-Q. Angew. Chem., Int. Ed. 2009, 48, 5094. 10.1002/anie.200806273. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Beccalli E. M.; Broggini G.; Martinelli M.; Sottocornola S. Chem. Rev. 2007, 107, 5318. 10.1021/cr068006f. [DOI] [PubMed] [Google Scholar]; h Ritleng V.; Sirlin C.; Pfeffer M. Chem. Rev. 2002, 102, 1731. 10.1021/cr0104330. [DOI] [PubMed] [Google Scholar]
- For selected reviews involving palladium-catalyzed carbocyclization reactions, see:; a Ojima I.; Tzamarioudaki M.; Li Z.; Donovan R. J. Chem. Rev. 1996, 96, 635. 10.1021/cr950065y. [DOI] [PubMed] [Google Scholar]; b Dns F.; Prez-Luna A.; Chemla F. Chem. Rev. 2010, 110, 2366. 10.1021/cr800420x. [DOI] [PubMed] [Google Scholar]; c Yeung C. S.; Dong V. M. Chem. Rev. 2011, 111, 1215. 10.1021/cr100280d. [DOI] [PubMed] [Google Scholar]; d Deng Y.; Persson A. K. Å.; Bäckvall J.-E. Chem. - Eur. J. 2012, 18, 11498. 10.1002/chem.201201494. [DOI] [PubMed] [Google Scholar]; e Lechel T.; Pfrengle F.; Reissig H.-U.; Zimmer R. ChemCatChem 2013, 5, 2100. 10.1002/cctc.201200875. [DOI] [Google Scholar]
- For selected applications, see:; a Nakanishi M.; Mori M. Angew. Chem., Int. Ed. 2002, 41, 1934.. [DOI] [PubMed] [Google Scholar]; b Baran P. S.; Corey E. J. J. Am. Chem. Soc. 2002, 124, 7904. 10.1021/ja026663t. [DOI] [PubMed] [Google Scholar]; c Garg N. K.; Caspi D. D.; Stoltz B. M. J. Am. Chem. Soc. 2004, 126, 9552. 10.1021/ja046695b. [DOI] [PubMed] [Google Scholar]; d Li M.; Dixon D. J. Org. Lett. 2010, 12, 3784. 10.1021/ol101425y. [DOI] [PubMed] [Google Scholar]; e Sui X.; Zhu R.; Li G.; Ma X.; Gu Z. J. Am. Chem. Soc. 2013, 135, 9318. 10.1021/ja404494u. [DOI] [PubMed] [Google Scholar]
- a Franzén J.; Bäckvall J.-E J. Am. Chem. Soc. 2003, 125, 6056. 10.1021/ja029505a. [DOI] [PubMed] [Google Scholar]; b Deng Y.; Bartholomeyzik T.; Persson A. K. Å.; Sun J.; Bäckvall J.-E. Angew. Chem., Int. Ed. 2012, 51, 2703. 10.1002/anie.201107592. [DOI] [PubMed] [Google Scholar]; c Bartholomeyzik T.; Mazuela J.; Deng Y.; Bäckvall J.-E. Angew. Chem., Int. Ed. 2014, 53, 8696. 10.1002/anie.201404264. [DOI] [PubMed] [Google Scholar]
- a Deng Y.; Bartholomeyzik T.; Bäckvall J.-E. Angew. Chem., Int. Ed. 2013, 52, 6283. 10.1002/anie.201301167. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Persson A. K. Å.; Jiang T.; Johnson M. T.; Bäckvall J.-E. Angew. Chem., Int. Ed. 2011, 50, 6155. 10.1002/anie.201008032. [DOI] [PubMed] [Google Scholar]; c Piera J.; Persson A.; Caldentey X.; Bäckvall J.-E. J. Am. Chem. Soc. 2007, 129, 14120. 10.1021/ja075488j. [DOI] [PubMed] [Google Scholar]
- a Karlsson E. A.; Bäckvall J.-E. Chem. - Eur. J. 2008, 14, 9175. 10.1002/chem.200801294. [DOI] [PubMed] [Google Scholar]; b Löfstead J.; Franzén J.; Bäckvall J.-E. J. Org. Chem. 2001, 66, 8015. 10.1021/jo0157324. [DOI] [PubMed] [Google Scholar]
- Deng Y.; Bäckvall J.-E. Angew. Chem., Int. Ed. 2013, 52, 3217. 10.1002/anie.201208718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected recent oxidative carbocyclization reactions by other groups, see:; a Yip K.-T.; Yang D. Org. Lett. 2011, 13, 2134. 10.1021/ol2006083. [DOI] [PubMed] [Google Scholar]; b Matsuura B. S.; Condie A. G.; Buff R. C.; Karahalis G. J.; Stephenson C. R. J. Org. Lett. 2011, 13, 6320. 10.1021/ol202881q. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Wu T.; Mu X.; Liu G. Angew. Chem., Int. Ed. 2011, 50, 12578. 10.1002/anie.201104575. [DOI] [PubMed] [Google Scholar]; d Mu X.; Wu V.; Wang H.; Guo Y.; Liu G. J. Am. Chem. Soc. 2012, 134, 878. 10.1021/ja210614y. [DOI] [PubMed] [Google Scholar]; e Zhu R.; Buchwald S. L. Angew. Chem., Int. Ed. 2012, 51, 1926. 10.1002/anie.201108129. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Wei Y.; Deb I.; Yoshikai N. J. Am. Chem. Soc. 2012, 134, 9098. 10.1021/ja3030824. [DOI] [PubMed] [Google Scholar]
- For a related Rh-catalyzed C–H activation without any heteroatom bond in the ortho position see:Kawaguchi Y.; Yasuda S.; Kaneko A.; Oura Y.; Mukai C. Angew. Chem., Int. Ed. 2014, 53, 7608. 10.1002/anie.201403990. [DOI] [PubMed] [Google Scholar]
- Simmons E. M.; Hartwig J. F. Angew. Chem., Int. Ed. 2012, 51, 3066. 10.1002/anie.201107334. [DOI] [PubMed] [Google Scholar]
- a Boele M. D. K.; van Strijdonck G. P. F.; de Vries A. H. M.; Kamer P. C. J.; de Vries J. G.; van Leeuwen P. W. N. M. J. Am. Chem. Soc. 2002, 124, 1586. 10.1021/ja0176907. [DOI] [PubMed] [Google Scholar]; b Yokota T.; Tani M.; Sakaguchi S.; Ishii Y. J. Am. Chem. Soc. 2003, 125, 1476. 10.1021/ja028903a. [DOI] [PubMed] [Google Scholar]; c Engle K. M.; Wang D.-H.; Yu J.-Q. J. Am. Chem. Soc. 2010, 132, 14137. 10.1021/ja105044s. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Babu B. P.; Meng X.; Bäckvall J. E. Chem. - Eur. J. 2013, 19, 4140. 10.1002/chem.201300100. [DOI] [PubMed] [Google Scholar]
- We have assumed that step A → D is irreversible. For the calculation of the isotope effect see the SI.
- Lapointe D.; Fagnou K. Chem. Lett. 2010, 39, 1118. 10.1246/cl.2010.1118. [DOI] [Google Scholar]
- It is assumed that benzoquinone primarily acts as Pd(0) oxidant in the catalytic cycle. However, it is likely that BQ coordinates to Pd(II) in diorgano-Pd(II) intermediate C triggering the reductive elimination. For relevant references see:; a Trejos A.; Fardost A.; Yahiaoui S.; Larhed M. Chem. Commun. 2009, 7587. 10.1039/b918358b. [DOI] [PubMed] [Google Scholar]; b Sköld C.; Kleimark J.; Trejos A.; Odell L. R.; Lill S. O. N.; Norrby P.-O.; Larhed M. Chem. - Eur. J. 2012, 18, 4714. 10.1002/chem.201102678. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.