

Abstract
Adverse drug events (ADEs) are a common cause of patient morbidity and mortality and are classically thought to result, in part, from variation in expression and activity of hepatic enzymes of drug metabolism. It is now known that alterations in the expression of genes that encode drug- and bile-acid–transporter proteins in both the gut and liver play a previously unrecognized role in determining patient drug response and eventual clinical outcome. Four nuclear receptor (NR) superfamily members, including pregnane X receptor (PXR, NR1I2), constitutive androstane receptor (NR1I3), farnesoid X receptor (NR1H4), and vitamin D receptor (NR1I1), play pivotal roles in drug- and bile-acid– activated programs of gene expression to coordinately regulate drug- and bile-acid transport activity in the intestine and liver. This review focuses on the NR-mediated gene activation of drug and bile-acid transporters in these tissues as well as the possible underlying molecular mechanisms.
Keywords: Pregnane x receptor, constitutive androstane receptor, farnesoid x receptor, vitamin D receptor, cholestasis, inflammation, liver disease, adverse drug events, drug interactions
Introduction
It is well recognized that a significant portion of adverse drug events (ADEs) in the clinical setting are the result of the atypical metabolism of drugs (Wilkinson, 2005). A more-recent thrust of research has focused on the identification of aberrant transport of clinically prescribed drugs resulting from alterations in expression and activity of drug- and bile-acid–transporter proteins in the gut and liver (Tirona, 2011). It is worth noting that many of the metabolic enzymes and transporter proteins that act on drugs also metabolize and transport bile acids into and out of these tissues. Moreover, a small group of ligand-activated transcription factors that “sense” the presence of drugs and bile acids has coevolved to regulate their expression and maintain a level of homeostasis.
The molecular mechanisms governing the drug- and bile-acid–inducible expression of genes that encode drug- and bile-acid–transporter proteins are well described, and it is now clear that several members of the nuclear receptor (NR)1 subfamily of the NR superfamily are key mediators of their expression in the enterohepatic system. Perturbations in the lines of communication between these two tissues during pathological conditions lead to the aberrant transport and metabolism of clinically prescribed drugs in patients. In particular, four different ligand-activated NR transcription factors, including pregnane X receptor (PXR, NR1I2), constitutive androstane receptor (CAR, NR1I3), farnesoid X receptor (FXR, NR1H4), and vitamin D receptor (VDR, NR1I1), directly regulate the inducible expression of genes in enterohepatic tissue that encode pivotal drug- and bile-acid– transporter proteins (Figure 1).
Figure 1.

NRs act as sensors in the regulation of enterohepatic drug transporters. Drugs and bile acids enter enterohepatic circulation by intestinal uptake transporters (e.g., ASBT). In many cases, these molecules are immediately transported back into the intestinal lumen by apical efflux transporters (e.g., MDR1, MRP2, and BCRP). Molecules that remain within the enterocyte can either serve as ligands to resident NRs, such as PXR, CAR, VDR, and FXR (bottom figure), or are effluxed into the portal circulation by sinusoidal efflux transporters (e.g., MRP3). After efflux to the portal vein, drugs and bile acids travel to the liver, where they are transported into hepatocytes by sinusoidal uptake transporters (e.g., OATPs and NTCP). Once inside hepatocytes, these molecules are either excreted into the bile canaliculus by canalicular efflux transporters (e.g., BSEP, MDR1, BCRP, and so forth) or serve as ligands to resident NRs, such as PXR, CAR, and FXR (top figure). Activation of these NRs in both the liver and intestine leads to their heterodimerization with RXRα. This NR/RXRα heterodimer, which is now bound to chromatin, initiates transcription of NR-specific target genes, including phase I and II drug-metabolizing enzymes as well as the drug transporters involved in uptake and efflux of NR-specific ligands (e.g., bile acids, drugs, and so forth). This drug-and bile-acid–induced regulation allows the enterohepatic system to maintain a homeostatic level of bile acids, drugs, and xenobiotics through feedback regulation that protects the body from potential toxic events.
This review considers the coordinate regulation of both drug and bile-acid transporters by members of the NR superfamily and highlights recent research efforts that focus on the molecular mechanisms underlying the gene expression that directly affect the uptake and efflux of drugs and bile acids in these tissues.
NR regulation of genes encoding drug and bile-acid transporters
Among the NR target genes that encode transporter proteins considered in this review, most have been characterized as transporters of both endogenous and xenobiotic compounds and can be classified as either uptake or efflux transporters. The uptake transporters discussed are members of the solute carrier organic anion transporter (SLCO) family (organic anion transporter polypetides; OATPs) or the solute carrier (SLC) family [sodium taurocholate cotransporting polypeptide (NTCP) and apical sodium-dependent bile-acid transporter (ASBT)]. These transporters have a vast array of endo- and exogenous substrates and function in the cellular uptake of organic ions, xenobiotics, and bile acids from the lumen of the intestine and from portal circulation into hepatocytes (Table 1). The efflux transporters that will be discussed are mainly members of the adenosine triphosphate (ATP)-binding cassette (ABC) superfamily, with the exception of organic solute transporters (OSTs), and function in the efflux and excretion of many of the same xenobiotics, as well as endogenous molecules that are substrates for uptake transporters (Table 2). The coordinating uptake and efflux functions of these transporters allow the body to maintain a level of homeostasis by regulating the expression of the genes that encode these transporters under healthy physiological and pathological states. This section discusses the regulation of uptake and efflux transporters by the previously mentioned members of the NR superfamily. This review focuses on regulation of drug and bile-acid transporters in relevant human model systems; however, wherever distinct species differences exist, we include references to the appropriate rodent systems.
Table 1.
Enterohepatic localization, transporter function, gene and corresponding protein name, and NRs that regulate the drug- and bile-acid–uptake transporters discussed in this review.
| Gene | Protein | Function | Endogenous substrates | Exogenous substrates | Enterohepatic location | NR regulators |
|---|---|---|---|---|---|---|
| SLCO1A2 | OATP1A1 | Na+-independent uptake | Bile salts, steroid conjugates, prostaglandin E2, and thyroid hormones T4, T3, and rT3 (Hagenbuch and Meier, 2004). For complete list, see Hagenbuch and Meier (2004) | Rocuronium, BQ-123, CRC-220, fexofenadine, deltorphin II, MRI contrast, and microcystins (Hagenbuch and Meier, 2004). For complete list, see Hagenbuch and Meier (2004) | Apical membrane of enterocytes and basolateral membrane of hepatocytes (Glaeser et al., 2007; Klaassen and Lu, 2008) | PXR VDR |
| SLCO1B1 | OATP1B1 | Na+-independent uptake | Cholate, thyroxine, bilirubin, leukotriene C4 and E4, and estradiol 17β-glucuronide (Meyer zu Schwabedissen and Kim, 2009). For complete list, see Hagenbuch and Meier (2004) | Atorvastatin, cerivastatin, fluvastatin, rifampin, enalapril, methotrexate, and olmesartan (Meyer zu Schwabedissen and Kim, 2009). For complete list, see Hagenbuch and Meier (2004) | Basolateral membrane of hepatocytes (Klaassen and Lu, 2008) | PXR FXR |
| SLCO1B3 | OATP1B3 | Na+-independent uptake | Cholate, thyroxine, bilirubin, leukotriene C4, dihydroepiandrosterone 3-sulfate, and estrone 3-sulfate (Meyer zu Schwabedissen and Kim, 2009). For complete list, see Meyer zu Schwabedissen and Kim (2009) | Docetaxel, enalapril, erythromycin, fexofenadine, fluvastatin, methotrexate, rifampin, and paclitaxel (Meyer zu Schwabedissen and Kim, 2009). For complete list, see Meyer zu Schwabedissen and Kim (2009) | Basolateral membrane of hepatocytes (Klaassen and Lu, 2008) | PXR FXR |
| SCL10A1 | NTCP | Na+-dependent uptake | Conjugated and unconjugated bile salts (Hagenbuch and Dawson, 2004) and estrone 3-sulfate (Schroeder et al., 1998) | Rosuvastatin (Ho et al., 2006) and micafungin (Yanni et al., 2010) | Basolateral membrane of hepatocytes (Klaassen and Lu, 2008) | FXR |
| SLC10A2 | ASBT | Na+-dependent uptake | Conjugated and unconjugated bile acids (Hagenbuch and Dawson, 2004; Dawson, 2011) | None identified (Dawson, 2011) | Apical membrane of enterocytes (Klaassen and Lu, 2008) | FXR |
Also included are some (not all) substrates of each transporter.
MRI, magnetic resonance imaging.
Table 2.
Enterohepatic localization, transporter function, gene and corresponding protein name, and NRs that regulate the drug and bile-acid efflux transporters discussed in this review.
| Gene | Protein | Function | Endogenous substrates | Exogenous substrates | Enterohepatic location | NR regulators |
|---|---|---|---|---|---|---|
| ABCB1 | MDR1/P-gp | ATP-dependent efflux | Vinblastine, paclitaxel, doxorubicin, etoposide, verapamil, digoxin, cyclosporine A (Klaassen and Lu, 2008), antiepileptics (Zhang et al., 2012), rifampicin, dexamethasone, and ritonavir (Cascorbi, 2011). For complete list, see Cheng and Klaassen (2006) | Apical membrane of enterocytes and hepatocytes (Klaassen and Lu, 2008; Cascorbi, 2011) | PXR VDR CAR |
|
| OSTα/OSTβ | OSTα/OSTβ | Efflux | Bile acids (Dawson et al., 2005). estrone 3-sulfate, and prostaglandins (Klaassen and Aleksunes, 2010) | Digoxin (Klaassen and Aleksunes, 2010) | Basolateral membrane of hepatocytes and enterocytes (Klaassen and Lu, 2008) | FXR VDR |
| ABCC2 | MRP2 | Efflux | Bilirubin, leukotriene C4, estrone 3-sulfate, and cholecystokinin (Keppler, 2011) | Ochratoxin A (Keppler, 2011), anthracyclines, Vinca alkaloids, methotrexate, and HIV protease inhibitors (Meijerman et al., 2008) | Apical membrane of hepatocyte and enterocyte (Klaassen and Lu, 2008) | PXR FXR CAR |
| ABCC3 | MRP3 | Efflux | leukotriene C4, bilirubin, dehydroepiandrosterone 3-sulfate, and cholylglycine (Keppler, 2011) bile acids | Methotrexate, etopiside, taniposide, and epipodophyllotoxins (Meijerman et al., 2008) | Basolateral membrane of enterocytes and hepatocytes (Klaassen and Lu, 2008) | PXR FXR CAR |
| ABCC4 | MRP4 | Efflux | Leukotrienes, Prostaglandins, Thromboxane B2, dehydroepiandrosterone 3-sulfate, and conjugated bile salts (Keppler, 2011) | Nucleotide analogs and methotrexate (Meijerman et al., 2008) | Basolateral membrane of hepatocytes (Klaassen and Lu, 2008) | CAR |
| ABCG2 | BCRP | Efflux | dehydroepiandrosterone sulfate, estradiol-17β-glucuronide, and estrone 3-sulfate (Klaassen and Aleksunes, 2010) | Fluoroquinolones, statins, anticancer agents, and furosemide. For complete list, see Denson et al. (2001) | Apical membrane of hepatocytes and enterocytes (Klaassen and Lu, 2008) | PXR CAR |
| ABCB11 | BSEP | Efflux | Conjugated and unconjugated bile acids | Apical membrane of hepatocytes (Klaassen and Lu, 2008) | FXR |
Also included are some (not all) substrates of each transporter.
Uptake transporters
Organic anion transporting polypeptide family member 1A2 (OATP1A2)
OATP1A2 is responsible for the uptake of a wide variety of endo- and exogenous substrates from the intestinal lumen as well as from the portal circulation. Several reports indicate that the expression of OATP1A2 is induced by PXR activators in breast cancer cells and cultured hepatocytes (Meyer zu Schwabedissen et al., 2008; Miki et al., 2006; Oscarson et al., 2006). Using human-derived cell lines (MCF-7, T47-D, HeLa, and HepG2), as well as malignant and adjacent nonmalignant human breast tissue samples, promoter analysis indicates that this effect is the result of direct regulation by PXR (Meyer zu Schwabedissen et al., 2008). Direct interaction between PXR, a major xenobiotic sensing NR, and the OATP1A2 promoter indicates the possibility of a regulatory pathway that is designed to increase the uptake of xenobiotics, therefore increasing the body’s ability to conjugate and excrete drugs. Recent reports also implicate VDR in the ligand-dependent transactivation of OATP1A2 using Caco-2 cells derived from human colon carcinoma. These studies show a significant increase in the expression of OATP1A2 messenger RNA (mRNA) after 24-hour treatment with 500 nM of vitamin D3 as well as after treatment of 50 μM of lithocholic acid (LCA), a VDR agonist, which was attenuated after small interfering RNA (siRNA)-mediated knockdown of VDR. This study also demonstrated an increase in OATP1A2 protein levels after 8 hours of treatment with vitamin D3 (Eloranta et al., 2012). Taken together, these data strongly implicate VDR in the regulation of OATP1A2 in a manner that parallels PXR-mediated regulation in human model systems.
OATP1B1
OATP1B1 is also involved in the uptake of numerous endogenous substrates as well as several clinically important drugs, such as statins, olmesartan, and methotrexate, from the portal circulation (Hirano et al., 2004). Because OATP1B1 is a high-affinity sinusoidal transporter for rifampicin, a well-known prototypical PXR activator, this highlights a unique mode of interplay by serving as the rate-limiting determinant of PXR-mediated gene activation (Gui et al., 2008; Tirona et al., 2003). Although there are not strong data implicating PXR in direct gene regulation of OATP1B1, two reports suggest that treatment of seven independent human hepatocyte cultures with PXR ligands, such as rifampicin, led to a moderate increase in mRNA expression (Jigorel et al., 2006; Oscarson et al., 2006). Using the human hepatoma Huh-7 cell line, FXR has been clearly demonstrated to be a direct, major regulator of OATP1B1 expression in response to treatment with the human FXR bile-acid activator, chenodeoxycholic acid (CDCA) (Meyer Zu Schwabedissen et al., 2010). The fact that many substrates of OATP1B1 are also ligands for the NRs that regulate expression of this transporter reveals a feedback loop that functions to maintain the homeostatic condition. It is interesting to note that atorvastatin, cerivastatin, and fluvastatin, which are all OATP1B1 substrates, can also activate CAR (Meyer zu Schwabedissen and Kim, 2009). Though there is currently no direct evidence supporting the regulation of OATP1B1 by CAR, it is formally possible that this uptake transporter is regulated in an overlapping, yet distinct, manner by CAR, PXR, and FXR cross-talk.
OATP1B3
OATP1B3 is another liver-specific member of the organic anion transporter family that mediates the sodium-independent uptake of many xenobiotic compounds. The OATP1B3 transporter protein also plays a critical role in bile-acid and bilirubin transport. In fact, mutations in this gene are a cause of Rotor type hyperbilirubinemia, a disease that leads to an increase in conjugated bilirubin in serum (van de Steeg et al., 2012). The bile acid, CDCA, and strong activator of human FXR induces the expression of OATP1B3 promoter activity in human hepatoma HepG2 and Huh7 cell lines, which is consistent with this OATP bile-acid–transporting function (Jung et al., 2002b). This same study showed that direct binding of FXR to an identified FXR-response element in the upstream promoter region of the gene encoding OATP1B3 is required for induction by bile acids in these human cell lines. Two separate reports indicated that treatment of human hepatocytes (Jigorel et al., 2006) or liver slices (Olinga et al., 2008) with rifampicin, a strong human PXR activator, reduced the expression level of OATP1B3. It is important to note that although both OATP1B1 and OATP1B3 exhibit similar substrate selectivities and are similarly localized on the basolateral membrane of hepatocytes, they are regulated in an opposing manner after activation of human PXR with rifampicin. The molecular mechanisms that produce downregulation of OATP1B3 are not currently known, though competition for transcription-factor–associated coactivator proteins represents a potential mechanism. The functional and biological significance of opposing regulation of these two OATP family members after PXR activation also remains to be determined; however, it is tempting to speculate that this is a physiological compensatory mechanism used to maintain xenobiotic homeostasis during pharmacological treatment with PXR agonists.
NTCP
NTCP, sometimes called the liver bile-acid transporter, is expressed in the basolateral membrane of hepatocytes, where it functions as the primary uptake transporter of conjugated and unconjugated bile acids under healthy physiological conditions. When cholestatic conditions are experimentally produced using common bile-duct ligation (BDL) in rats and bile acids accumulate in serum and liver, NTCP expression is rapidly downregulated (Gartung et al., 1996). This may be a mechanism that limits the hepatic uptake of bile acids and prevents subsequent hepatotoxicity. Downregulation of NTCP in primary rat hepatocytes, as well as in human HepG2 and monkey Cos1 cell lines, is mediated by an indirect mechanism involving bile acid/FXR-mediated induction of a dominant-negative atypical NR called small heterodimer partner (SHP) (Denson et al., 2001). In this way, induction of SHP expression by FXR is thought to downregulate NR-mediated transactivation of the promoters of key genes whose gene products are pivotal regulators of bile-acid homeostasis, including NTCP, as well as cholesterol 7α hydroxylase (CYP7A1), the rate-limiting enzyme in the conversion of cholesterol to bile acids (Denson et al., 2001; Goodwin et al., 2000).
ASBT
The intestinal counterpart to NTCP is the ileal sodium/bile-acid cotransporter (ASBT or IBAT). This transporter is the primary mediator of bile-acid uptake from the lumen of the intestine. Because bile acids are the catabolic product of cholesterol metabolism, both NTCP and ASBT are critically important for cholesterol homeostasis. In humans, a loss-of-function mutation in SLC10A2, the gene that encodes ASBT, causes fatty diarrhea, fat malabsorption, and malnutrition (Oelkers et al., 1997; Wong et al., 1995). As with NTCP, activation of FXR with CDCA in the Caco-2 cell line indirectly induces negative feedback regulation of human ASBT by an FXR-mediated/SHP-dependent effect upon expression of ASBT in the intestine (Neimark et al., 2004). Though there is no biochemical evidence implicating PXR and CAR in the direct regulation of human ASBT, it has been shown that atorvastatin delivered to mice in a 0.1% enriched diet over 7 days significantly increased the expression of mouse Asbt mRNA (Wagner et al., 2005). Although it is possible that atorvastatin directly activates PXR/CAR to upregulate the expression of Asbt, a more-likely explanation comes from the fact that this statin upregulates the expression of the NR family member, peroxisome proliferator-activated receptor alpha (PPAR-α) (Roglans et al., 2002), which is a potent, direct regulator of ASBT in human model systems (Jung et al., 2002a).
Efflux transporters
Multidrug Resistance Protein 1/P-Glycoprotein (MDR1/P-gp)
MDR1, also commonly known as P-gp, is a major efflux transporter that is primarily responsible for the rapid “inside-out” efflux of a vast majority of xenobiotics. Therefore, MDR1/P-gp plays a major role in decreased drug accumulation in multidrug-resistant cells and is associated with drug bioavailability (Kurata et al., 2002; Siegmund et al., 2002a, 2002b). Overexpression of MDR1/P-gp mediates the development of resistance to anticancer drugs in certain tumor types (Juranka et al., 1989). NR-mediated regulation of MDR1/P-gp by PXR was demonstrated approximately 20 years ago. Data published by Schuetz et al. showed that treatment of a colon adenocarcinoma cell line with several clinically prescribed drugs, including the PXR activator, rifampicin, was found to increase the expression and activity of MDR1/P-gp (Schuetz et al., 1996). Another study showed an increase in MDR1/P-gp protein expression, which directly corresponded with a decrease in levels of digoxin, a MDR1/P-gp substrate, upon administration of rifampin, a strong PXR agonist (Greiner et al., 1999). Because of the overlapping regulatory functions of PXR and CAR, it was also hypothesized that CAR may regulate the expression of MDR1/P-GP. Subsequent studies found that NRs PXR and CAR directly regulate expression of the MDR1 gene through an immediate upstream promoter element exclusively in the intestine (Burk et al., 2005; Geick et al., 2001). A more-recent study showed induction of MDR1/P-gp mRNA expression after treatment with rifampicin and phenobarbital, a CAR agonist, in primary cultures of human hepatocytes (Jigorel et al., 2006). There is also evidence implicating VDR in the regulation of MDR1/P-gp. One study showed that treatment with vitamin D induced high levels of expression of MDR1/P-gp in the intestinal adenocarcinoma cell line, Caco-2 (Schmiedlin-Ren et al., 1997). A more-recent study showed significant induction of MDR1/P-gp protein and mRNA expression after treatment with 100 nM of 1α,25-dihydroxyvitamin D3 (1,25(OH)2D3) in the human colon carcinoma cell line, LS180 (Aiba et al., 2005). Successive studies from the same research group identified multiple functional VDR response elements in the promoter of the human MDR1 gene, which collectively contributed to gene expression (Saeki et al., 2008).
OSTα/OSTβ
Organic solute transporters -alpha and -beta (OSTα/OSTβ) form a heterodimeric efflux transporter that is expressed at relatively high levels on the basolateral membranes of enterohepatic tissue. Its primary function is the efflux of bile acids into the portal circulation from both enterocytes and hepatocytes, making it an essential mediator of bile-acid homeostasis in the enterohepatic circulation (Ballatori et al., 2005; Dawson et al., 2005). The regulatory effects of OSTα/OSTβ on bile-acid homeostasis make this transporter a plausible target of FXR-mediated regulation, and there are many studies to support this. One study showed induction of human OSTα/OSTβ mRNA in two different human hepatoma cell lines (Huh7 and HepG2) after treatment of cells with both endogenous (50 μM of CDCA) and exogenous (200 nM of GW4064) FXR ligands. This induction is directly attributed to FXR because of the fact that knockdown of FXR using siRNA attenuated the ligand-mediated induction (Landrier et al., 2006). Another study performed by a separate research group showed induction of OSTα/OSTβ mRNA after 24-hour treatment with CDCA in the human hepatoma HepG2 cell line, HepG2. The same group showed significant increases in protein and mRNA levels of OSTα and OSTβ in patients with primary biliary cirrhosis, a disease resulting in blockage of bile flow caused by chronic inflammation of the bile duct. There is also evidence to support the regulation of OSTα/OSTβ by VDR. One study found that treatment with 50 μM of CDCA and 5–10 μM of LCA, both of which are VDR agonists, increased the expression of OSTα/OSTβ in human ileum and liver slices (Khan et al., 2009). However, because of the fact that 1,25(OH)2D3 had no effect on transporter expression, and that both CDCA and LCA are also agonists of FXR, it is unclear whether this increase can be completely attributed to VDR or is perhaps a ligand-specific effect.
Multidrug resistance-associated protein 2 (MRP2)
MRP2 is an efflux transporter protein responsible for the removal of conjugated xenobiotics and bile acids from the liver into bile. Because of its wide substrate selectivity and pivotal excretory function on apical membranes in hepatocytes, it is interesting to note that expression of this efflux transporter is coordinately regulated by multiple NRs that function as both xenobiotic and bile-acid sensors. Regulation of MRP2 expression by PXR, FXR, and CAR was originally described in human, mouse, and rat hepatocytes in 2002. This study showed induction of MRP2 mRNA after treatment with agonists of FXR, PXR, and CAR (Kast et al., 2002). Additional studies implicated both PXR and FXR as positive regulators of MRP2 expression and activity in the intestine (Cheng and Klaassen, 2006; Zollner et al., 2003). A recent study revealed that ortho-aminoazotoluene, a possible (class 2B) human carcinogen, activates CAR to upregulate MRP2 expression in mouse liver in a CAR-dependent manner (Smetanina et al., 2011). Though there is no direct evidence showing VDR-mediated regulation of human MRP2, a recent study shows a significant increase in rat Mrp2 protein expression after intraperitoneal injection of 1,25(OH)2D3 at a dose of 0.64 nmol/kg/day (Chow et al., 2010). Taken together, these studies suggest that PXR, CAR, VDR, and FXR upregulate protein and mRNA expression of MRP2 in response to distinct ligands to increase rate of efflux and/or biliary elimination of numerous organic anions, thus providing a mechanism to enhance excretion of these compounds from the body.
MRP3
MRP3 is an efflux transporter that functions in the removal of exo- and endogenous compounds from hepatocytes into the blood. Though the tissue localization and transporter function of MRP3 is well-known, its specific substrates have only recently been well characterized. They include methotrexate, acetaminophen (APAP) glucuronide, APAP sulphate, bilirubin, estradiol-17β-glucuronide, ritonavir, sequinavir, conjugated cholic acid (CA), and several other endogenous and xenobiotic compounds (Keppler, 2011; Meijerman et al., 2008). It has been shown that treatment with 5–10 μM of LCA significantly induces the expression of MRP3 mRNA in human ileum slices, implicating a regulatory role for FXR and/or VDR at the transcriptional level (Khan et al., 2011). Regulation of MRP3 expression by PXR and CAR in the liver was demonstrated in mice (Staudinger et al., 2003), and a subsequent study further implicated a complimentary and combined role for PXR, FXR, and CAR in protecting against bile-acid–induced hepatotoxicity, in part resulting from induction of MRP3 expression and activity in the liver (Guo et al., 2003). Robust information demonstrating the transcriptional regulation of human MRP3 by PXR is currently lacking. However, one study examined the relationship between PXR and MRP3 in human colon cancer chemoresistance. The investigators used reverse-transcriptase polymerase chain reaction and western blot analysis to demonstrate that mRNA expression and protein levels of PXR and MRP3 were much higher in colon cancer tissues than that found in non-neoplastic tissues (Jiang et al., 2009). More research is necessary to determine the extent to which this transporter protein is differentially regulated by NRs in human liver and intestine.
MRP4
MRP4 is an efflux transporter that transports several clinically prescribed drugs, such as adefovir, cefazolin, ceftizoxime, furosemide, irinotecan, leucovorin, methotrexate, and topotecan. Conjugated divalent sulphated and monovalent bile acids are also substrates for this transporter (Keppler, 2011; Meijerman et al., 2008). It has been shown that expression of MRP4 undergoes adaptive upregulation in response to cholestatic injury or bile-acid feeding (Zollner et al., 2003), and CAR has been implicated in the regulation of MRP4 in several studies. One study performed using CAR knockout mice indicated that CAR is required for induction of MRP4 in the liver after treatment with 1,4-bis-[2-(3,5-dichloropyridyloxy)]benzene, 3,3′,5,5′-tetrachloro-1,4-bis(pyridyloxy)benzene (Aleksunes and Klaassen, 2012), followed by experimental data from another research group demonstrating that CAR activators increase MRP4 expression in primary human hepatocytes and HepG2 cells (Assem et al., 2004). Chai et al. demonstrated a significant increase in MRP4 mRNA levels in patients suffering from obstructive cholestasis. This same investigation established a statistically positive correlation between MRP4 mRNA levels and CAR protein levels, suggesting that CAR may positively regulate MRP4 transcriptional activity in an effort to increase bile-acid efflux in hepatic injury (Chai et al., 2011). A different group demonstrated an increase in MRP4 mRNA in patients suffering from alcoholic liver disease, chronic hepatitis C infection, and primary sclerosing cholangitis, which correlated with a decrease in CAR, PXR, and RXRα mRNA levels. Data pertaining to CAR mRNA levels are consistent with those found in the previously discussed research by Chai et al., but protein expression levels were not examined in this study. More recently, a negative role for FXR in the regulation of MRP4 has been hypothesized. Renga et al. demonstrated that FXR negatively regulates MRP4 both in vivo and in vitro. Data from this study showed that treatment of HepG2 cells with CDCA, CA, and the synthetic FXR activator, GW4064, suppressed basal mRNA levels of MRP4 and inhibits the CAR-mediated induction of MRP4 expression by 6-(4-chlorophenyl)imidazol[2,1-b] [1,3]thiazole-5-carbaldehyde-O-(3,4-dicholorobenzyl) oxime. The investigators also demonstrate that deletion of FXR in vivo leads to an increase in basal expression levels of MRP4 (Renga et al., 2011).
Breast Cancer Resistance Protein (BCRP)
BCRP functions as a xenobiotic transporter to mediate excretion of its substrates and thus influences the pharmacokinetics of many drugs. For example, BCRP serves as a cellular defense mechanism in response to mitoxantrone, anthracycline, and methotrexate exposure in multiple cell lines (Allen and Schinkel, 2002; Doyle and Ross, 2003). BRCP also transports sulfoconjugated organic anions, such as estrone 3-sulfate. One of the main expression sites for BCRP is the placenta, where it is expressed at the apical membrane of syncytiotrophoblasts. In the placenta, BCRP is likely involved in the transport of its substrates from fetal to maternal blood, thereby playing a protective role for the fetus by transporting toxic compounds from fetal circulation into maternal circulation for eventual excretion. Conflicting information exists regarding the regulation of BCRP by PXR and CAR. Early studies using primary cultures of human hepatocytes indicated that PXR activators increased the expression of BCRP (Jigorel et al., 2006; Oscarson et al., 2006). More-recent studies indicate that PXR and CAR activators do not regulate the expression of this transporter in rodent liver (Aleksunes and Klaassen, 2012). Mice engineered to lack PXR exhibit increased expression of BCRP in the placenta, though it is not clear whether this is a direct effect, because PCN did not induce BCRP expression (Gahir and Piquette-Miller, 2011). Therefore, more research is necessary to determine the extent to which this transporter is under the regulation of NRs in hepatocytes and placenta.
Bile Salt Excretory Pump (BSEP)
BSEP is the major canalicular (apical) bile-salt export pump in mammals. Mutations in the gene that encodes this transporter will cause progressive familial intrahepatic cholestasis, a severe, often lethal disease characterized by cholestatic liver disease and eventual liver failure beginning from early infancy. The primary NR regulator of BSEP is FXR. FXR has been shown to directly regulate the expression of BSEP by binding to its proximal promoter and is transactivated by the FXR activator, CDCA (Ananthanarayanan et al., 2001; Schuetz et al., 2001). Although PXR, CAR, and VDR are not known to directly regulate BSEP expression in hepatocytes, more research is necessary to determine the interactions between these signaling pathways, because treatment with PXR ligands causes a significant decrease in expression of BSEP in primary cultures of human hepatocytes (Jigorel et al., 2006). The NR superfamily member liver receptor homolog 1 (LRH-1) is required for maximal hepatic expression of CYP7A1 and was recently shown to play a supporting role, together with FXR, in maintaining hepatic bile-acid levels by coordinately regulating both CYP7A1 and BSEP expression and activity for bile-acid synthesis and elimination (Song et al., 2008). Interestingly, SHP-mediated repression of BSEP was subservient to FXR-mediated activation toward BSEP, thus bile acids downregulate CYP7A1 to decrease bile-acid production, whereas, at the same time, they increase BSEP expression and activity to increase bile-acid elimination from the liver. Differential downregulation of liver-enriched FXR-target genes can likely be explained by differences in cell-signaling pathways and the presence of a newly discovered gut-liver axis.
NR cross-talk and disruption in the gut-liver axis
It is physiologically important to maintain strict control of serum bile-acid levels because accumulation of these compounds in blood and/or cells can produce profound hepatotoxicity. Moreover, infection with certain viruses and bacteria can produce severe liver disease that is often a result of chronic inflammation, which can inhibit the normal function of regulatory NRs. Communication between NRs can have differential effects on the genes being regulated, thus understanding the cross-talk that occurs between NRs is critical in the discovery of novel strategies to combat pathological conditions and ADEs in the clinic as well as in the development of new pharmaceutical agents. Below, we focus on the cross-talk between the previously discussed regulatory NRs as well an emerging molecular circuit involving NR cross-talk that is highly implicated in drug- and bile-acid transporter regulation.
NR cross-talk in enterohepatic tissue
NR cross-talk in relation to the regulation of enterohepatic transporters can come in several forms: competitive cross-talk; inhibitory cross-talk; and noncompetitive cross-talk. This is a relatively new field, but there are still multiple studies to support all three ideas. Multiple studies have shown that PXR, CAR, and VDR all compete for the same response elements on the promoter of CYP2A4 (Pascussi et al., 2005; Moreau et al., 2007), demonstrating the notion of competitive NR cross-talk. Another research group has also suggested the concept of competitive cross-talk between CAR and FXR. Analysis of the MRP4 promoter revealed the presence of a CAR-responsive element embedded within the FXR response element, suggesting that FXR and CAR compete for binding and that FXR could hinder the protective effects modulated by CAR activation in disease states (Renga et al., 2011). There is also evidence that supports inhibitory cross-talk, in which the activation of one NR leads to the suppression of another. One study showed that PXR transcriptional activity is hindered by the presence of SHP, an NR that is upregulated in the FXR/SHP-mediated regulatory mechanism (Ourlin et al., 2003). Noncompetitive cross-talk often occurs between CAR and PXR. These two NRs are closely related and have a large subset of overlapping ligands. Studies show that in the absence of both PXR and CAR, the CAR ligand, Phenobarbital, will activate another liver-enriched NR: PPAR-α (Tamasi et al., 2009). NR cross-talk is an important part in the regulation of transporters as well as in the understanding of xenobiotic uptake, metabolism, and clearance. The fact that different NRs can regulate distinct and overlapping groups of target genes and can be activated by common, as well as individual, ligands suggests that NRs communicate to simultaneously maintain homeostatic conditions, depending upon cellular needs. These cellular needs could include xenobiotic uptake and clearance, compensation for a defective protein or regulatory cascade, or protection from drug and bile-acid accumulation. The fact that NR cross-talk aids in the regulation of xenobiotic clearance is an important notion in the effort of researchers to eliminate ADEs and drug-drug interactions (DDIs) in clinical and personal settings. According to the IMS Institution for Healthcare Informatics, simvastatin was the second-most prescribed medication (94.1 million prescriptions) in early 2011 (http://www.webmd.com/news/20110420/the-10-most-prescribed-drugs), and, as shown in Tables 1 and 2, it is also a substrate for previously discussed uptake and efflux transporters as well as certain NRs. This one medication is known to have 213 drug interactions, ranging from slight to severe, in the healthy individual (http://www.drugs.com/drug-interactions/simvastatin.html). The fact that this popular medication is also known to have potentially life-threatening hepatotoxic effects, if drug metabolism by the liver is even slightly compromised, also furthers the need to elucidate the full mechanism behind NR cross-talk and transporter regulation. Discovering the mechanism by which the body partially or totally compensates for the loss or malfunction of a xenobiotic transporter or regulatory NR in both healthy and pathological states could possibly aid in the development of novel strategies to prevent ADEs in the clinical setting.
An FXR/SHP-mediated mechanism of drug- and bile-acid–transporter regulation
A prime example of NR communication is presented in the FXR-SHP regulatory pathway. In the liver and intestine, activation of FXR induces expression of SHP. The SHP protein, in turn, represses expression of CYP7A1 by inhibiting the activity of LRH-1 (Figure 2). This FXR-mediated regulatory cascade provides, in part, the molecular basis for the coordinate suppression of CYP7A1 and other genes involved in bile-acid biosynthesis and transport including, NTCP (Goodwin et al., 2000; Lu et al., 2000). Consistent with this model, mice genetically engineered to lack the SHP gene exhibited defects in bile-acid homeostasis and failed to repress CYP7A1 expression in response to a pharmacological FXR activator, GW4064 (Wang et al., 2002). However, repression of CYP7A1 expression is retained in SHP-null mice fed bile-acid activators (e.g., CA) of FXR, suggesting an intestine-specific effect. Other studies showed that BDL, or decreased bile acids in the intestine, paradoxically increased CYP7A1 in livers of rats (Dueland et al., 1991; Gustafsson, 1978). Hepatic bile acid levels increase under these conditions, thus further suggesting a role for the intestine in feedback repression of bile-acid synthesis. Still, other studies have shown that direct administration of bile acids into the intestine inhibited CYP7A1 expression; in contrast, administration of bile acids intravenously or directly into the portal circulation did not (Nagano et al., 2004; Pandak et al., 1991). Collectively, these observations led to the hypothesis that a functionally redundant pathway for CYP7A1 repression, involving a previously unknown gut-liver–signaling pathway, exists. Recently, this hypothesis was proven true and resulted in the identification of a novel gut-liver axis involving bile-acid–mediated induction of the expression of fibroblast growth factor family member 15 (FGF15) (Reviewed in Potthoff et al., 2012).
Figure 2.

Model for the FXR-mediated feed-forward and feedback regulation effects on gene expression. Feed-forward regulation by FXR in the liver and intestine is mediated by bile-acid activators of this NR superfamily member, primarily CA, and CDCA. Activated FXR, in turn, positively regulates the expression of FXR target genes that encode both drug- and bile-acid–transporter proteins OATP1B1, OATP1B3, OSTα/β, and BSEP. These transporter proteins function in the uptake and eventual elimination of drugs and bile acids from enterocytes and hepatocytes, hence the presence of excess bile acids in these tissues upregulates their own excretion and efflux. Feedback inhibition of primary bile-acid–uptake transporters ASBT and NTCP, in enterocytes and hepatocytes, respectively, is accomplished through a regulatory circuit that involves FXR-mediated induction of the expression and activity of SHP. The increased levels of the SHP nuclear receptor, which functions in a dominant-negative manner because of the lack of a functional DNA-binding domain, interacts with LRH-1 to repress the expression of genes encoding CYP7A1 and CYP8B1, enzymes centrally involved in the de novo production of additional bile acids from endogenous excess cholesterol. Increased SHP protein also represses the expression of its own gene through sequestration of LRH-1 from its own promoter. Thus, both synthesis of new bile acids from cholesterol, as well as uptake of old bile acids, is downregulated simultaneously to prevent the accumulation of excess bile acids in enterohepatic circulation.
Expression of FGF15 is stimulated in the small intestine by bile-acid activation of FXR and acts in an endocrine manner to repress CYP7A1 expression in the liver through a mechanism that involves FGF receptor 4 (FGFR4) and SHP (Figure 3). Mice lacking the gene encoding FGF15 exhibited increased hepatic CYP7A1 mRNA and protein levels and corresponding increases in CYP7A1 enzyme activity and fecal bile-acid excretion (Inagaki et al., 2005). These studies defined FGF15 and FGFR4 as key components of a gut-liver axis that synergizes with SHP to regulate bile-acid synthesis by CYP7A1. Interestingly, activation of the VDR in mice produced similar effects and has been attributed to increased intestinal absorption of bile acids through increased ASBT-mediated transport of bile acids in the ileum and subsequent elevated levels of FGF15, events that led to the activation of FXR in the liver (Chow et al., 2009). FXR-mediated induction of BSEP removes bile acids from the liver (Ananthanarayanan et al., 2001), whereas hepatic uptake of bile acids is slowed by SHP-mediated repression of NTCP (Denson et al., 2001). Thus, during cholestasis, the liver is protected from injury by potentially toxic levels of accumulating bile acids in serum. Future research should focus on determining the extent to which FGF-mediated repressive signaling pathways alter expression levels of other drug- and bile-acid–transporter proteins in the liver and intestine, especially during pathological conditions, such cholestasis and inflammatory liver and bowel disease.
Figure 3.
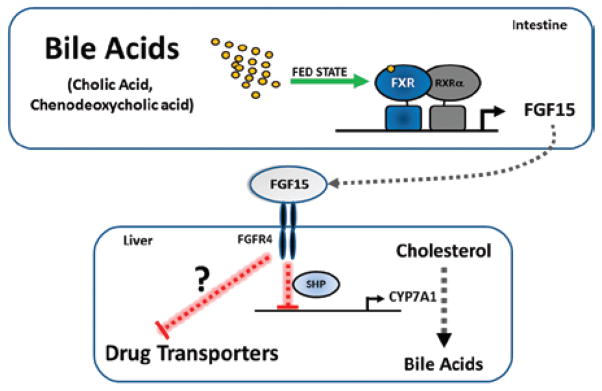
Model for the gut-liver axis that involves the FGF15/19-signaling mechanism. Endocrine actions of FGF15. Expression of the gene encoding FGF15 is induced in the small intestine by bile acids acting on the FXR/RXRα heterodimer. Subsequently, FGF15 is secreted into the enterohepatic circulation to act in an endocrine manner by binding a receptor on hepatocytes called FGFR4. The activated FGFR4 receptor signals the liver to repress CYP7A1 expression and activity. The CYP7A1 enzyme is the rate-limiting step in the synthesis of bile acids from cholesterol. In this manner, FGF15 (FGF19 in humans) suppresses de novo synthesis of bile acids from cholesterol through a mechanism that requires SHP. The extent to which this mechanism functions to downregulate drug-and bile-acid–transporter proteins in hepatocytes is currently unknown, but is a likely fruitful area of future research.
Conclusions
Although the role of liver- and gut-enriched NR proteins PXR, CAR, FXR, and VDR in the regulation of drug-metabolizing enzymes has been well established, the challenge remains to accurately assess the role of these ligand-activated transcription factors in the regulation of drug and bile-acid transporters in these tissues during specific pathological conditions. Recent advances in knowledge regarding identification of ligands for these important NRs, as well as identification of novel regulatory circuits that coregulate drug-metabolizing enzymes and transporter proteins to affect both drug and bile-acid disposition, are positive steps toward providing novel paradigms that can be exploited clinically. The ability to better understand how these molecular circuits function during disease states is likely to provide opportunities to develop novel strategies for targeting NRs during certain pathologies, such as cholestatic liver disease and inflammatory bowel or liver disease. For example, a recent study used combination bezafibrate/ursodeoxycholic acid (UCDA) therapy as an experimental treatment of patients presenting with cholestatic liver disease resulting from early primary biliary cirrhosis (Honda et al., 2012). In addition to the significant improvement of serum biliary enzymes, immunoglobulin M, cholesterol, and triglyceride concentrations in patients treated with this combination therapy, a reduction of 7α-hydroxy-4-cholesten-3-one, a marker of bile-acid synthesis, and increase of 4β-hydroxycholesterol, a marker of CYP3A4/5 activity, were observed. Moreover, using cell-based analysis, the investigators of this study observed a downregulation of CYP7A1, CYP27A1, and NTCP and an upregulation of CYP3A4, MDR1, and MRP2 after combination bezafibrate/UCDA treatment.
An appreciation for the existence of a gut-liver axis that regulates bile-acid disposition is a major leap forward in our understanding of the biological links between drug and bile-acid transport and drug metabolism in these tissues. It remains to be immediately determined whether drug- and bile-acid–transporter proteins are under the same FGF15/19-mediated regulation as enzymes of bile-acid synthesis in these tissues. In the future, patients who present with cholestatic liver disease and inflammatory bowel or Crohn’s disease will likely benefit from these discoveries as new strategies for targeting these diseases emerge. We are just beginning to understand the relationship between NRs and drug and bile-acid metabolism and transport in these tissues, and until a thorough understanding of these relationships is achieved we will likely be unable to significantly diminish the occurrence of ADEs, DDI, and disease-drug interaction. We should look forward to maximizing the pharmacological opportunities that are likely to be presented from this novel thrust of research.
Footnotes
Declaration of interest
The authors declare no financial conflicts of interest. The authors alone are responsible for the content and writing of this paper.
References
- Aiba T, Susa M, Fukumori S, Hashimoto Y. The effects of culture conditions on CYP3A4 and MDR1 mRNA induction by 1alpha,25-dihydroxyvitamin D(3) in human intestinal cell lines, Caco-2 and LS180. Drug Metab Pharmacokinet. 2005;20:268–274. doi: 10.2133/dmpk.20.268. [DOI] [PubMed] [Google Scholar]
- Aleksunes LM, Klaassen CD. Coordinated regulation of hepatic phase I and II drug-metabolizing genes and transporters using AhR-, CAR-, PXR-, PPARalpha-, and Nrf2-null mice. Drug Metab Dispos. 2012;40:1366–1379. doi: 10.1124/dmd.112.045112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen JD, Schinkel AH. Multidrug resistance and pharmacological protection mediated by the breast cancer resistance protein (BCRP/ABCG2) Mol Cancer Ther. 2002;1:427–434. [PubMed] [Google Scholar]
- Ananthanarayanan M, Balasubramanian N, Makishima M, Mangelsdorf DJ, Suchy FJ. Human bile salt export pump promoter is transactivated by the farnesoid X receptor/bile acid receptor. J Biol Chem. 2001;276:28857–28865. doi: 10.1074/jbc.M011610200. [DOI] [PubMed] [Google Scholar]
- Assem M, Schuetz EG, Leggas M, Sun D, Yasuda K, Reid G, et al. Interactions between hepatic Mrp4 and Sult2a as revealed by the constitutive androstane receptor and Mrp4 knockout mice. J Biol Chem. 2004;279:22250–22257. doi: 10.1074/jbc.M314111200. [DOI] [PubMed] [Google Scholar]
- Ballatori N, Christian WV, Lee JY, Dawson PA, Soroka CJ, Boyer JL, et al. OSTalpha-OSTbeta: a major basolateral bile acid and steroid transporter in human intestinal, renal, and biliary epithelia. Hepatology. 2005;42:1270–1279. doi: 10.1002/hep.20961. [DOI] [PubMed] [Google Scholar]
- Burk O, Arnold KA, Geick A, Tegude H, Eichelbaum M. A role for constitutive androstane receptor in the regulation of human intestinal MDR1 expression. Biol Chem. 2005;386:503–513. doi: 10.1515/BC.2005.060. [DOI] [PubMed] [Google Scholar]
- Cascorbi I. P-glycoprotein: tissue distribution, substrates, and functional consequences of genetic variations. Handb Exp Pharmacol. 2011:261–283. doi: 10.1007/978-3-642-14541-4_6. [DOI] [PubMed] [Google Scholar]
- Chai J, Luo D, Wu X, Wang H, He Y, Li Q, et al. Changes of organic anion transporter MRP4 and related nuclear receptors in human obstructive cholestasis. J Gastrointest Surg. 2011;15:996–1004. doi: 10.1007/s11605-011-1473-2. [DOI] [PubMed] [Google Scholar]
- Cheng X, Klaassen CD. Regulation of mRNA expression of xenobiotic transporters by the pregnane x receptor in mouse liver, kidney, and intestine. Drug Metab Dispos. 2006;34:1863–1867. doi: 10.1124/dmd.106.010520. [DOI] [PubMed] [Google Scholar]
- Chow EC, Maeng HJ, Liu S, Khan AA, Groothuis GM, PANG KS. 1alpha,25-Dihydroxyvitamin D(3) triggered vitamin D receptor and farnesoid X receptor-like effects in rat intestine and liver in vivo. Biopharm Drug Dispos. 2009;30:457–475. doi: 10.1002/bdd.682. [DOI] [PubMed] [Google Scholar]
- Chow EC, Sun H, Khan AA, Groothuis GM, Pang KS. Effects of 1alpha,25-dihydroxyvitamin D3 on transporters and enzymes of the rat intestine and kidney in vivo. Biopharm Drug Dispos. 2010;31:91–108. doi: 10.1002/bdd.694. [DOI] [PubMed] [Google Scholar]
- Dawson PA. Role of the intestinal bile acid transporters in bile acid and drug disposition. Handb Exp Pharmacol. 2011:169–203. doi: 10.1007/978-3-642-14541-4_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson PA, Hubbert M, Haywood J, Craddock AL, Zerangue N, Christian WV, et al. The heteromeric organic solute transporter alpha-beta, Ostalpha-Ostbeta, is an ileal basolateral bile acid transporter. J Biol Chem. 2005;280:6960–6968. doi: 10.1074/jbc.M412752200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denson LA, Sturm E, Echevarria W, Zimmerman TL, Makishima M, Mangelsdorf DJ, et al. The orphan nuclear receptor, shp, mediates bile acid-induced inhibition of the rat bile acid transporter, ntcp. Gastroenterology. 2001;121:140–147. doi: 10.1053/gast.2001.25503. [DOI] [PubMed] [Google Scholar]
- Doyle LA, Ross DD. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2) Oncogene. 2003;22:7340–7358. doi: 10.1038/sj.onc.1206938. [DOI] [PubMed] [Google Scholar]
- Dueland S, Reichen J, Everson GT, Davis RA. Regulation of cholesterol and bile acid homoeostasis in bile-obstructed rats. Biochem J. 1991;280(Pt 2):373–377. doi: 10.1042/bj2800373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eloranta JJ, Hiller C, Juttner M, Kullak-Ublick GA. The SLCO1A2 gene, encoding human organic anion-transporting polypeptide 1A2, is transactivated by the vitamin D receptor. Mol Pharmacol. 2012;82:37–46. doi: 10.1124/mol.112.077909. [DOI] [PubMed] [Google Scholar]
- Gahir SS, Piquette-Miller M. Gestational and pregnane X receptor-mediated regulation of placental ATP-binding cassette drug transporters in mice. Drug Metab Dispos. 2011;39:465–471. doi: 10.1124/dmd.110.034983. [DOI] [PubMed] [Google Scholar]
- Gartung C, Ananthanarayanan M, Rahman MA, Schuele S, Nundy S, Soroka CJ, et al. Down-regulation of expression and function of the rat liver Na+/bile acid cotransporter in extrahepatic cholestasis. Gastroenterology. 1996;110:199–209. doi: 10.1053/gast.1996.v110.pm8536857. [DOI] [PubMed] [Google Scholar]
- Geick A, Eichelbaum M, Burk O. Nuclear receptor response elements mediate induction of intestinal MDR1 by rifampin. J Biol Chem. 2001;276:14581–14587. doi: 10.1074/jbc.M010173200. [DOI] [PubMed] [Google Scholar]
- Glaeser H, Bailey DG, Dresser GK, Gregor JC, Schwarz UI, McGrath JS, et al. Intestinal drug transporter expression and the impact of grapefruit juice in humans. Clin Pharmacol Ther. 2007;81:362–370. doi: 10.1038/sj.clpt.6100056. [DOI] [PubMed] [Google Scholar]
- Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000;6:517–526. doi: 10.1016/s1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- Greiner B, Eichelbaum M, Fritz P, Kreichgauer HP, von Richter O, Zundler J, et al. The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin. J Clin Invest. 1999;104:147–153. doi: 10.1172/JCI6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui C, Miao Y, Thompson L, Wahlgren B, Mock M, Stieger B, et al. Effect of pregnane X receptor ligands on transport mediated by human OATP1B1 and OATP1B3. Eur J Pharmacol. 2008;584:57–65. doi: 10.1016/j.ejphar.2008.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo GL, Lambert G, Negishi M, Ward JM, Brewer HB, Jr, Kliewer SA, et al. Complementary roles of farnesoid X receptor, pregnane X receptor, and constitutive androstane receptor in protection against bile acid toxicity. J Biol Chem. 2003;278:45062–45071. doi: 10.1074/jbc.M307145200. [DOI] [PubMed] [Google Scholar]
- Gustafsson J. Effect of biliary obstruction on 26-hydroxylation of C27-steroids in bile acid synthesis. J Lipid Res. 1978;19:237–243. [PubMed] [Google Scholar]
- Hagenbuch B, Dawson P. The sodium bile salt cotransport family SLC10. Pflugers Arch. 2004;447:566–570. doi: 10.1007/s00424-003-1130-z. [DOI] [PubMed] [Google Scholar]
- Hagenbuch B, Meier PJ. Organic anion transporting polypeptides of the OATP/SLC21 family: phylogenetic classification as OATP/SLCO superfamily, new nomenclature and molecular/functional properties. Pflugers Arch. 2004;447:653–665. doi: 10.1007/s00424-003-1168-y. [DOI] [PubMed] [Google Scholar]
- Hirano M, Maeda K, Shitara Y, Sugiyama Y. Contribution of OATP2 (OATP1B1) and OATP8 (OATP1B3) to the hepatic uptake of pitavastatin in humans. J Pharmacol Exp Ther. 2004;311:139–146. doi: 10.1124/jpet.104.068056. [DOI] [PubMed] [Google Scholar]
- Ho RH, Tirona RG, Leake BF, Glaeser H, Lee W, Lemke CJ, WANG Y, KIM RB. Drug and bile acid transporters in rosuvastatin hepatic uptake: function, expression, and pharmacogenetics. Gastroenterology. 2006;130:1793–1806. doi: 10.1053/j.gastro.2006.02.034. [DOI] [PubMed] [Google Scholar]
- Honda A, Ikegami T, Nakamuta M, Miyazaki T, Iwamoto J, Hirayama T, et al. Anticholestatic effects of bezafibrate in patients with primary biliary cirrhosis treated with ursodeoxycholic acid. Hepatology. 2012 Aug 22; doi: 10.1002/hep.26018. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217–225. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Jiang H, Chen K, He J, Pan F, Li J, Chen J, et al. Association of pregnane X receptor with multidrug resistance-related protein 3 and its role in human colon cancer chemoresistance. J Gastrointest Surg. 2009;13:1831–1838. doi: 10.1007/s11605-009-0964-x. [DOI] [PubMed] [Google Scholar]
- Jigorel E, Le Vee M, Boursier-Neyret C, Parmentier Y, Fardel O. Differential regulation of sinusoidal and canalicular hepatic drug transporter expression by xenobiotics activating drug-sensing receptors in primary human hepatocytes. Drug Metab Dispos. 2006;34:1756–1763. doi: 10.1124/dmd.106.010033. [DOI] [PubMed] [Google Scholar]
- Jung D, Fried M, Kullak-Ublick GA. Human apical sodium-dependent bile salt transporter gene (SLC10A2) is regulated by the peroxisome proliferator-activated receptor alpha. J Biol Chem. 2002a;277:30559–30566. doi: 10.1074/jbc.M203511200. [DOI] [PubMed] [Google Scholar]
- Jung D, Podvinec M, Meyer UA, Mangelsdorf DJ, Fried M, Meier PJ, et al. Human organic anion transporting polypeptide 8 promoter is transactivated by the farnesoid X receptor/bile acid receptor. Gastroenterology. 2002b;122:1954–1966. doi: 10.1053/gast.2002.33583. [DOI] [PubMed] [Google Scholar]
- Juranka PF, Zastawny RL, Ling V. P-glycoprotein: multidrug-resistance and a superfamily of membrane-associated transport proteins. FASEB J. 1989;3:2583–2592. doi: 10.1096/fasebj.3.14.2574119. [DOI] [PubMed] [Google Scholar]
- Kast HR, Goodwin B, Tarr PT, Jones SA, Anisfeld AM, Stoltz CM, et al. Regulation of multidrug resistance-associated protein 2 (ABCC2) by the nuclear receptors pregnane X receptor, farnesoid X-activated receptor, and constitutive androstane receptor. J Biol Chem. 2002;277:2908–2915. doi: 10.1074/jbc.M109326200. [DOI] [PubMed] [Google Scholar]
- Keppler D. Multidrug resistance proteins (MRPs, ABCCs): importance for pathophysiology and drug therapy. Handb Exp Pharmacol. 2011:299–323. doi: 10.1007/978-3-642-14541-4_8. [DOI] [PubMed] [Google Scholar]
- Khan AA, Chow EC, Porte RJ, Pang KS, Groothuis GM. Expression and regulation of the bile acid transporter, OSTalpha-OSTbeta in rat and human intestine and liver. Biopharm Drug Dispos. 2009;30:241–258. doi: 10.1002/bdd.663. [DOI] [PubMed] [Google Scholar]
- Khan AA, Chow EC, Porte RJ, Pang KS, Groothuis GM. The role of lithocholic acid in the regulation of bile acid detoxication, synthesis, and transport proteins in rat and human intestine and liver slices. Toxicol In Vitro. 2011;25:80–90. doi: 10.1016/j.tiv.2010.09.011. [DOI] [PubMed] [Google Scholar]
- Klaassen CD, Aleksunes LM. Xenobiotic, bile acid, and cholesterol transporters: function and regulation. Pharmacol Rev. 2010;62:1–96. doi: 10.1124/pr.109.002014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaassen CD, Lu H. Xenobiotic transporters: ascribing function from gene knockout and mutation studies. Toxicol Sci. 2008;101:186–196. doi: 10.1093/toxsci/kfm214. [DOI] [PubMed] [Google Scholar]
- Kurata Y, Ieiri I, Kimura M, Morita T, Irie S, Urae A, et al. Role of human MDR1 gene polymorphism in bioavailability and interaction of digoxin, a substrate of P-glycoprotein. Clin Pharmacol Ther. 2002;72:209–219. doi: 10.1067/mcp.2002.126177. [DOI] [PubMed] [Google Scholar]
- Landrier JF, Eloranta JJ, Vavricka SR, Kullak-Ublick GA. The nuclear receptor for bile acids, FXR, transactivates human organic solute transporter-alpha and -beta genes. Am J Physiol Gastrointest Liver Physiol. 2006;290:G476–G485. doi: 10.1152/ajpgi.00430.2005. [DOI] [PubMed] [Google Scholar]
- Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, Auwerx J, et al. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell. 2000;6:507–515. doi: 10.1016/s1097-2765(00)00050-2. [DOI] [PubMed] [Google Scholar]
- Meijerman I, Beijnen JH, Schellens JH. Combined action and regulation of phase II enzymes and multidrug resistance proteins in multidrug resistance in cancer. Cancer Treat Rev. 2008;34:505–520. doi: 10.1016/j.ctrv.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Meyer zu Schwabedissen HE, Bottcher K, Chaudhry A, Kroemer HK, Schuetz EG, Kim RB. Liver X receptor alpha and farnesoid X receptor are major transcriptional regulators of OATP1B1. Hepatology. 2010;52:1797–1807. doi: 10.1002/hep.23876. [DOI] [PubMed] [Google Scholar]
- Meyer zu Schwabedissen HE, Kim RB. Hepatic OATP1B transporters and nuclear receptors PXR and CAR: interplay, regulation of drug disposition genes, and single nucleotide polymorphisms. Mol Pharm. 2009;6:1644–1661. doi: 10.1021/mp9000298. [DOI] [PubMed] [Google Scholar]
- Meyer zu Schwabedissen HE, Tirona RG, Yip CS, Ho RH, Kim RB. Interplay between the nuclear receptor pregnane X receptor and the uptake transporter organic anion transporter polypeptide 1A2 selectively enhances estrogen effects in breast cancer. Cancer Res. 2008;68:9338–9347. doi: 10.1158/0008-5472.CAN-08-0265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki Y, Suzuki T, Kitada K, Yabuki N, Shibuya R, Moriya T, et al. Expression of the steroid and xenobiotic receptor and its possible target gene, organic anion transporting polypeptide-A, in human breast carcinoma. Cancer Res. 2006;66:535–542. doi: 10.1158/0008-5472.CAN-05-1070. [DOI] [PubMed] [Google Scholar]
- Moreau A, Maurel P, Vilarem MJ, Pascussi JM. Constitutive androstane receptor-vitamin D receptor crosstalk: consequence on CYP24 gene expression. Biochem Biophys Res Commun. 2007;360:76–82. doi: 10.1016/j.bbrc.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Nagano M, Kuroki S, Mizuta A, Furukawa M, Noshiro M, Chijiiwa K, et al. Regulation of bile acid synthesis under reconstructed enterohepatic circulation in rats. Steroids. 2004;69:701–709. doi: 10.1016/j.steroids.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Neimark E, Chen F, Li X, Shneider BL. Bile acid-induced negative feedback regulation of the human ileal bile acid transporter. Hepatology. 2004;40:149–156. doi: 10.1002/hep.20295. [DOI] [PubMed] [Google Scholar]
- Oelkers P, Kirby LC, Heubi JE, Dawson PA. Primary bile acid malabsorption caused by mutations in the ileal sodium-dependent bile acid transporter gene (SLC10A2) J Clin Invest. 1997;99:1880–1887. doi: 10.1172/JCI119355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olinga P, Elferink MG, Draaisma AL, Merema MT, Castell JV, Perez G, et al. Coordinated induction of drug transporters and phase I and II metabolism in human liver slices. Eur J Pharm Sci. 2008;33:380–389. doi: 10.1016/j.ejps.2008.01.008. [DOI] [PubMed] [Google Scholar]
- Oscarson M, Zanger UM, Rifki OF, Klein K, Eichelbaum M, Meyer UA. Transcriptional profiling of genes induced in the livers of patients treated with carbamazepine. Clin Pharmacol Ther. 2006;80:440–456. doi: 10.1016/j.clpt.2006.08.013. [DOI] [PubMed] [Google Scholar]
- Ourlin JC, Lasserre F, Pineau T, Fabre JM, Sa-Cunha A, Maurel P, et al. The small heterodimer partner interacts with the pregnane X receptor and represses its transcriptional activity. Mol Endocrinol. 2003;17:1693–1703. doi: 10.1210/me.2002-0383. [DOI] [PubMed] [Google Scholar]
- Pandak WM, Li YC, Chiang JY, Studer EJ, Gurley EC, Heuman DM, et al. Regulation of cholesterol 7 alpha-hydroxylase mRNA and transcriptional activity by taurocholate and cholesterol in the chronic biliary diverted rat. J Biol Chem. 1991;266:3416–3421. [PubMed] [Google Scholar]
- Pascussi JM, Robert A, Nguyen M, Walrant-Debray O, Garabedian M, Martin P, et al. Possible involvement of pregnane X receptor-enhanced CYP24 expression in drug-induced osteomalacia. J Clin Invest. 2005;115:177–186. doi: 10.1172/JCI21867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potthoff MJ, Kliewer SA, Mangelsdorf DJ. Endocrine fibroblast growth factors 15/19 and 21: from feast to famine. Genes Dev. 2012;26:312–324. doi: 10.1101/gad.184788.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renga B, Migliorati M, Mencarelli A, Cipriani S, D’Amore C, Distrutti E, et al. Farnesoid X receptor suppresses constitutive androstane receptor activity at the multidrug resistance protein-4 promoter. Biochim Biophys Acta. 2011;1809:157–165. doi: 10.1016/j.bbagrm.2011.01.008. [DOI] [PubMed] [Google Scholar]
- Roglans N, Sanguino E, Peris C, Alegret M, Vazquez M, Adzet T, DIAZ C, et al. Atorvastatin treatment induced peroxisome proliferator-activated receptor alpha expression and decreased plasma nonesterified fatty acids and liver triglyceride in fructose-fed rats. J Pharmacol Exp Ther. 2002;302:232–239. doi: 10.1124/jpet.302.1.232. [DOI] [PubMed] [Google Scholar]
- Saeki M, Kurose K, Tohkin M, Hasegawa R. Identification of the functional vitamin D response elements in the human MDR1 gene. Biochem Pharmacol. 2008;76:531–542. doi: 10.1016/j.bcp.2008.05.030. [DOI] [PubMed] [Google Scholar]
- Schmiedlin-Ren P, Thummel KE, Fisher JM, Paine MF, Lown KS, Watkins PB. Expression of enzymatically active CYP3A4 by Caco-2 cells grown on extracellular matrix-coated permeable supports in the presence of 1alpha,25-dihydroxyvitamin D3. Mol Pharmacol. 1997;51:741–754. doi: 10.1124/mol.51.5.741. [DOI] [PubMed] [Google Scholar]
- Schroeder A, Eckhardt U, Stieger B, Tynes R, Schteingart CD, Hofmann AF, et al. Substrate specificity of the rat liver Na(+)-bile salt cotransporter in Xenopus laevis oocytes and in CHO cells. Am J Physiol. 1998;274:G370–G375. doi: 10.1152/ajpgi.1998.274.2.G370. [DOI] [PubMed] [Google Scholar]
- Schuetz EG, Beck WT, Schuetz JD. Modulators and substrates of P-glycoprotein and cytochrome P4503A coordinately up-regulate these proteins in human colon carcinoma cells. Mol Pharmacol. 1996;49:311–318. [PubMed] [Google Scholar]
- Schuetz EG, Strom S, Yasuda K, Lecureur V, Assem M, Brimer C, et al. Disrupted bile acid homeostasis reveals an unexpected interaction among nuclear hormone receptors, transporters, and cytochrome P450. J Biol Chem. 2001;276:39411–39418. doi: 10.1074/jbc.M106340200. [DOI] [PubMed] [Google Scholar]
- Siegmund W, Altmannsberger S, Paneitz A, Hecker U, Zschiesche M, Franke G, et al. Effect of levothyroxine administration on intestinal P-glycoprotein expression: consequences for drug disposition. Clin Pharmacol Ther. 2002a;72:256–264. doi: 10.1067/mcp.2002.126706. [DOI] [PubMed] [Google Scholar]
- Siegmund W, Ludwig K, Giessmann T, Dazert P, Schroeder E, Sperker B, et al. The effects of the human MDR1 genotype on the expression of duodenal P-glycoprotein and disposition of the probe drug talinolol. Clin Pharmacol Ther. 2002b;72:572–583. doi: 10.1067/mcp.2002.127739. [DOI] [PubMed] [Google Scholar]
- Smetanina MA, Pakharukova MY, Kurinna SM, Dong B, Hernandez JP, Moore DD, et al. Ortho-aminoazotoluene activates mouse constitutive androstane receptor (mCAR) and increases expression of mCAR target genes. Toxicol Appl Pharmacol. 2011;255:76–85. doi: 10.1016/j.taap.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Kaimal R, Yan B, Deng R. Liver receptor homolog 1 transcriptionally regulates human bile salt export pump expression. J Lipid Res. 2008;49:973–984. doi: 10.1194/jlr.M700417-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudinger JL, Madan A, Carol KM, Parkinson A. Regulation of drug transporter gene expression by nuclear receptors. Drug Metab Dispos. 2003;31:523–527. doi: 10.1124/dmd.31.5.523. [DOI] [PubMed] [Google Scholar]
- Tamasi V, Juvan P, Beer M, Rozman D, Meyer UA. Transcriptional activation of PPARalpha by phenobarbital in the absence of CAR and PXR. Mol Pharm. 2009;6:1573–1581. doi: 10.1021/mp9001552. [DOI] [PubMed] [Google Scholar]
- Tirona RG. Molecular mechanisms of drug transporter regulation. Handb Exp Pharmacol. 2011:373–402. doi: 10.1007/978-3-642-14541-4_10. [DOI] [PubMed] [Google Scholar]
- Tirona RG, Leake BF, Wolkoff AW, Kim RB. Human organic anion transporting polypeptide-C (SLC21A6) is a major determinant of rifampin-mediated pregnane X receptor activation. J Pharmacol Exp Ther. 2003;304:223–228. doi: 10.1124/jpet.102.043026. [DOI] [PubMed] [Google Scholar]
- van de Steeg E, Stránecký V, Hartmannová H, Nosková L, Hřebíček M, Wagenaar E, et al. Complete OATP1B1 and OATP1B3 deficiency causes human Rotor syndrome by interrupting conjugated bilirubin reuptake into the liver. J Clin Invest. 2012;122:519–528. doi: 10.1172/JCI59526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner M, Halilbasic E, Marschall HU, Zollner G, Fickert P, Langner C, et al. CAR and PXR agonists stimulate hepatic bile acid and bilirubin detoxification and elimination pathways in mice. Hepatology. 2005;42:420–430. doi: 10.1002/hep.20784. [DOI] [PubMed] [Google Scholar]
- Wang L, Lee YK, Bundman D, Han Y, Thevananther S, Kim CS, et al. Redundant pathways for negative feedback regulation of bile acid production. Dev Cell. 2002;2:721–731. doi: 10.1016/s1534-5807(02)00187-9. [DOI] [PubMed] [Google Scholar]
- Wilkinson GR. Drug metabolism and variability among patients in drug response. New Engl J Med. 2005;352:2211–2221. doi: 10.1056/NEJMra032424. [DOI] [PubMed] [Google Scholar]
- Wong MH, Oelkers P, Dawson PA. Identification of a mutation in the ileal sodium-dependent bile acid transporter gene that abolishes transport activity. J Biol Chem. 1995;270:27228–27234. doi: 10.1074/jbc.270.45.27228. [DOI] [PubMed] [Google Scholar]
- Yanni SB, Augustijns PF, Benjamin DK, Jr, Brouwer KL, Thakker DR, Annaert PP. In vitro investigation of the hepatobiliary disposition mechanisms of the antifungal agent micafungin in humans and rats. Drug Metab Dispos. 2010;38:1848–1856. doi: 10.1124/dmd.110.033811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Kwan P, Zuo Z, Baum L. The transport of antiepileptic drugs by P-glycoprotein. Adv Drug Deliv Rev. 2012;64:930–942. doi: 10.1016/j.addr.2011.12.003. [DOI] [PubMed] [Google Scholar]
- Zollner G, Fickert P, Fuchsbichler A, Silbert D, Wagner M, Arbeiter S, et al. Role of nuclear bile acid receptor, FXR, in adaptive ABC transporter regulation by cholic and ursodeoxycholic acid in mouse liver, kidney, and intestine. J Hepatol. 2003;39:480–488. doi: 10.1016/s0168-8278(03)00228-9. [DOI] [PubMed] [Google Scholar]