

Abstract
Background
Lophophora williamsii (commonly named peyote) is a small, spineless cactus with psychoactive alkaloids, particularly mescaline. Peyote utilizes crassulacean acid metabolism (CAM), an alternative form of photosynthesis that exists in succulents such as cacti and other desert plants. Therefore, its transcriptome can be considered an important resource for future research focused on understanding how these plants make more efficient use of water in marginal environments and also for research focused on better understanding of the overall mechanisms leading to production of plant natural products and secondary metabolites.
Results
In this study, two cDNA libraries were generated from L. williamsii. These libraries, representing buttons (tops of stems) and roots were sequenced using different sequencing platforms (GS-FLX, GS-Junior and PGM, respectively). A total of 5,541,550 raw reads were generated, which were assembled into 63,704 unigenes with an average length of 564.04 bp. A total of 25,149 unigenes (62.19 %) was annotated using public databases. 681 unigenes were found to be differentially expressed when comparing the two libraries, where 400 were preferentially expressed in buttons and 281 in roots. Some of the major alkaloids, including mescaline, were identified by GC-MS and relevant metabolic pathways were reconstructed using the Kyoto encyclopedia of genes and genomes database (KEGG). Subsequently, the expression patterns of preferentially expressed genes putatively involved in mescaline production were examined and validated by qRT-PCR.
Conclusions
High throughput transcriptome sequencing (RNA-seq) analysis allowed us to efficiently identify candidate genes involved in mescaline biosynthetic pathway in L. williamsii; these included tyrosine/DOPA decarboxylase, hydroxylases, and O-methyltransferases. This study sets the theoretical foundation for bioassay design directed at confirming the participation of these genes in mescaline production.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1821-9) contains supplementary material, which is available to authorized users.
Background
The “Peyote” is a small, fleshy cactus without spines that grows wild in the Mexican highlands and in the arid regions of South-western United States [1]. Peyote belongs to the genus Lophophora, which includes two species, L. williamsii and L. diffusa [1]. This plant is capable of producing large amounts of alkaloids with psychotropic activity, such as β-phenylethylamine (class I) or tetrahydroisoquinoline (class II), which are derived from the amino acid tyrosine [2, 3]. The function of most alkaloids in plants is unclear and their importance in metabolism is highly controversial.
Since the identification of morphine in 1806, contained in opium poppy (Papaver somniferum), more than ten thousand alkaloids with different properties and a variety of biological activities have been isolated from plants. Alkaloids are heterocyclic compounds that contain a nitrogen atom. The position of the nitrogen atom in the hydrocarbon ring varies among different alkaloids and different plant families. The levels of alkaloids in plants also vary from trace amounts to up to 10 % of dry weight, and a single plant species might contain over one hundred of different types. Most alkaloids are highly toxic and therefore have the potential to function in the chemical defense arsenal of plants against attack by herbivores and microorganisms. For example, nicotine (present in tobacco leaves) inhibits the growth of tobacco hornworm larvae, and the purified compound can also be applied as an effective insecticide in greenhouses. In addition, alkaloids have been suggested to serve as a storage form of nitrogen or as protectants against damage by ultraviolet light [4]. Alkaloids have traditionally been of great interest to humans because of their pronounced physiological and medical properties (e.g. morphine, atropine, quinine, caffeine and nicotine) [5–7]. Peyote mescaline (3,4,5-trimethoxyphenethylamine) is a class I hallucinogenic alkaloid, and although it is chemically unrelated to lysergic acid diethyl amide (LSD), the hallucinogenic effects of mescaline are similar to those of LSD, albeit longer lasting [8]. For centuries, North American indigenous peoples have used mescaline as a medicine, and as a part of hallucinogenic religious sacrament [9]. The ceremonial use of peyote alkaloids has masked and mythologized the potential use of peyote in modern medicine. For example, some of the illnesses treated with peyote by Mexican Natives are tuberculosis, pneumonia, scarlet fever, intestinal ills, diabetes, rheumatic pains, colds, grippe, fevers, and venereal diseases, which is why peyote is officially listed in the Mexican pharmacopoeia [10].
Although L. williamsii is morphologically similar to L. diffusa, these two species differ in stem color, the presence or absence of ribs and furrows across stems, and the color of flowers. While L. williamsii has blue-green or sometimes reddish-green stems with ribs and furrows and the flower is usually pink, L. diffusa shows yellowish-green stems without ribs and the flower is usually white. Additionally, it has been proposed that L. williamsii specimens can be classified into two groups according to: (i) differences in button morphology, such as size of protuberances on the epidermis of stem, (ii) levels of mescaline, and (iii) differences between their chloroplast trnL/trnF regions [11].
Next-generation sequencing technologies, such GS-FLX (also named 454) and PGM (also named Ion torrent) pyrosequencing, have the potential to dramatically increase the availability of sequence data in non-model plants that lack complete genome sequence information. In order to identify genes with potential relevance in mescaline biosynthetic pathways, we sequenced and assembled 307.2 Mpb of L. williamsii transcriptome data using both the GS-FLX and PGM pyrosequencing platforms. Additionally, using gas chromatography–mass spectrometry analysis (GC–MS), we provide evidence that some alkaloids present in peyote are confined to specific organs; for example, mescaline was only detected in the peyote buttons (tops of stems) but not in roots. This work provides one of the first catalogs of genes present in a medicinally relevant member of the Cactaceae family, a horticulturally important group of plants.
Results and discussion
Mescaline content analysis
In order to perform transcriptome analysis of mescaline-producing cacti, we collected samples in Cuatro Cienegas, Coahuila, Mexico. To confirm the identity of the collected samples, the intergenic spacer region trnL/trnF sequence was amplified using previously reported primers [11] and sequenced (ABI PRISM 3730xl). The sequence obtained was 879 bp, corresponding to the typical size reported for group 1 of L. williamsii species, which contain mescaline [11] (Additional file 1). To explore the content of alkaloids present in L. williamsii, a standard alkaloid extraction procedure (see methods) was carried out for the roots and buttons (tops of stems) from one of the peyote plants collected. The extracts were then analyzed by gas chromatography–mass spectrometry (GC-MS). Several compounds were then reliably identified by comparison against internal library spectra (Additional file 2: Figure S1 and Additional file 3: Table S1). Some of the detected compounds include hordenine, N-methylmescaline, N-acetylmescaline, pellotine, anhalonine, anhalidine, anhalonidine, O-methylanhalonidine, and lophophorine. Not all of these substances exhibit psychopharmacological activity when administered singly, but in combination, they apparently potentiate the effects of the mescaline and definitely alter some characteristics for the experience. Alkaloids such as lophophorine can be detected in extracts derived from both button and roots while some others like hordenine, which possess antibacterial properties, presumably because of their phenolic function [12], were detected only in roots (Additional file 3: Table S1). Mescaline, with retention time ≈ 8.36, was detected in buttons, but barely present in root extracts, to levels of 15.68 and 0.05 % respectively. These results show that mescaline accumulation is almost totally confined to peyote buttons.
Sequencing and de novo assembly of the L. williamsii transcriptome
In order to explore the peyote transcriptome, we synthesized cDNA from pooled RNAs isolated from both roots and buttons. The cDNAs were used to construct sequencing libraries for two 454 Genome Sequencer platforms (GS-FLX and GS-Junior), generating a total of 492,402 sequence reads (377,522 from GS-FLX and 114,880 from GS-Junior) with an overall average read length of 230.61 and 406.31 bp, respectively (Additional file 3: Table S2). In order to identify genes with preferential expression in either roots or buttons, an additional two sequencing runs were performed using a PGM™ sequencer (Personal Genome Machine™; Life Techonologies), for which each organ (button and roots) was distinguished by using Ion barcode adapters. A total of 5,541,550 reads was generated with the PGM sequencer (2,293,355 for buttons and 3,248,195 for roots). 454-reads were masked using the SeqClean software in order to eliminate sequence regions that could cause incorrect assembly. Targets for masking included poly A/T tails, ends rich in undetermined bases, and low complexity sequences. For the Ion-reads, we first used the FASTX tool kit (http://hannonlab.cshl.edu/fastx_toolkit) to separate reads according to the specific barcodes, whereafter the barcodes were trimmed and the remaining sequences were filtered. Only high quality Ion-reads ranging from 50 to 150 bp, for which a maximum of 5 bases with a quality phred score below 17 were allowed, were used in this study. In total, 459,106 masked 454-reads and 1,979,580 high quality Ion-reads (1,147,370 from roots and 832,210 from buttons), were considered to obtain de novo assembly of the peyote transcriptome. An overview of the sequencing dataset is presented in Additional file 3: Table S2. The sequenced reads were assembled using MIRA [13], which is an assembler that can integrate various platform data and perform true hybrid assemblies specially for de novo transcriptomes [13]. This assembly resulted in a total of 129,358 transcripts (contigs) that were used in a second assembly step using CAP3 [14] to eliminate redundant sequences and increase transcript length. This assembly resulted in 63,704 contigs with lengths ranging from 0.1 to ~4.2 Kbp. Only contigs larger than 200 bp (40,436 sequences, 63.47 % of the total), for which the mean length was 564.04 bp, were considered as unigenes for further analysis (Additional file 4).
Sequence annotation
The 40,436 L. williamsii unigenes were annotated by sequence similarity BLASTx searches against Arabidopsis thaliana (www.arabidopsis.org) and reference plant proteins (Refseq; comprising all green plants, ftp://ftp.ncbi.nlm.nih.gov/refseq/release/plant/) datasets. A total of 25,149 (62.19 %) unigenes showed significant similarity (e-value 10−3) to Arabidopsis or the RefSeq databases (Additional file 3: Table S3). The high percentage of L. williamsii unigenes obtained in this study that did not match the RefSeq protein database (37.81 %) indicates that there is potential for the discovery of as-yet undescribed and novel plant genes in peyote, although many of these unigenes may represent non-coding RNAs or untranslated regions (UTRs). In addition, the significance of a BLAST search depends on the length of the query sequence; therefore, short sequences are rarely matched to known genes [15], or these sequences may represent rapidly evolving parts of genes that have diverged substantially from their homologs [16]. Based on the Arabidopsis top hits, we obtained the gene ontology annotations (GOs) for 23,729 L. williamsii unigenes (Additional file 3: Table S4). Using the WEGO software [17] unigenes with Arabidopsis hits were assigned to gene ontology classes with 85,988 functional terms. Biological processes comprised the majority of the functional terms (39,957; 46.47 %), followed by cellular components (23,619; 27.47 %) and molecular functions (22,412; 26.06 %) (Additional file 2: Figure S2; see also Additional file 3: Table S4). In addition, using the KEGG Automatic Annotation Server (KAAS; http://www.genome.jp/tools/kaas/) from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database [18], Enzyme Commission (EC) numbers [19] and KEGG Orthology (KO) codes were also associated to each annotated L. williamsii unigene. 5,008 unigenes having KO codes were assigned to metabolic, genetic and environmental information processing pathways (Additional file 3: Tables S3 and S5). KEGG metabolic pathways that are well-represented by L. williamsii unigenes belong to carbohydrate, amino acid, energy and lipid metabolism (Fig. 1a). In the subclass secondary metabolism, the greatest number of unique sequences was mapped to phenylpropanoid biosynthetic pathways, for which tyrosine is the initial precursor (Fig. 1b). This result was expected considering that L. williamsii classes I and II alkaloids are also derived from this amino acid.
Fig. 1.
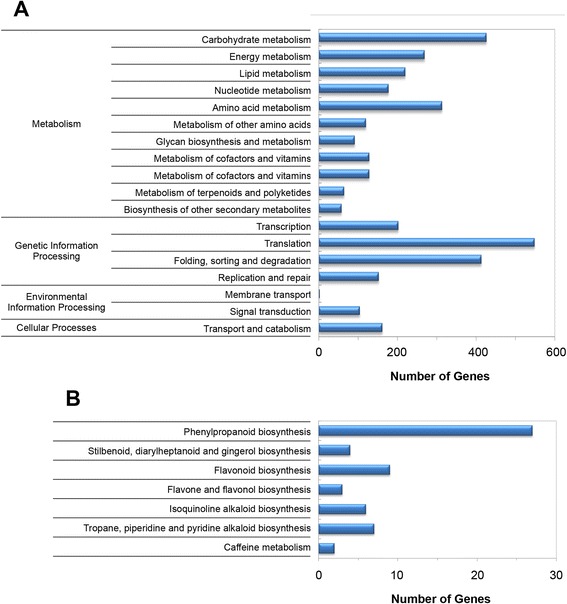
Pathway assignment based on KEGG. a Major categories based on molecular interaction and reaction networks. b Subclasses and gene distribution in the ‘Biosynthesis of other secondary metabolites’ category
In order to explore if the L. williamsii unigene database comprised a deep representation of the complex metabolic pathways that characterize plant genomes, some well-known metabolic pathways such as glycolysis/gluconeogenesis, starch and sucrose metabolism, and carbon fixation during photosynthetic process (Crassulacean acid metabolism; CAM and C4-dicarboxylic acid cycle) were reconstructed (Additional file 2: Figures S3–S5, respectively). With only a few exceptions, the proteins involved in these metabolic pathways were found in the peyote transcriptome. In most cases, more than one transcript sequence was found to encode the same enzyme. Such unique sequences may represent different fragments of a single transcript or different members of a gene family. These results indicate that the L. williamsii unigene database comprises a good representation of the peyote transcriptome, permitting its use as a source to discover candidate genes responsible for mescaline biosynthesis.
Detection of preferentially expressed unigenes
Given that mescaline accumulates only in the peyote buttons, it is tempting to think that genes involved in mescaline biosynthesis are also preferentially expressed in these organs. Considering that independent samples of mRNA from L. williamsii isolated from button and roots were used to construct the PGM libraries, an RNA-seq approach was used to analyze the expression profile of obtained unigenes. All of 1,979,580 high quality Ion-reads (1,147,370 from roots and 832,210 from buttons) were mapped independently to the L. williamsii transcriptome, and an expression profile matrix containing the unigenes (rows) and the number of mapped reads in each normalized organ-specific transcriptome (columns), was created (Additional file 3: Table S6).
It has been argued that a high number of reads (in the tens of millions) are required to perform gene expression analysis [20, 21]. Considering the number of sequences generated in the present study, we used relative frequency values (counts divided by the effective library sizes) to make reads counts comparable among samples. This method has been previously used for the efficient detection of differentially expressed genes [22, 23] (see methods for more details). The significance of differential gene expression between buttons and roots was determined using the likelihood ratios (R) method described by Stekel et al. [24]. This approach is based on a single statistical test that can be used to describe the extent to which a gene is differentially expressed between libraries. Briefly, all unigenes (and their corresponding read-counts values) were used to calculate a log likelihood ratio that trends asymptotically to a χ2 distribution in which R values ≥ 12 can be considered significantly preferentially expressed genes. Additionally, a fold change of at least 2-fold (buttons/roots) was also considered. A total of 652 unigenes was identified as preferentially expressed, 400 in buttons and 281 in roots (Additional file 3: Table S7). Interestingly, theorgan-enriched, differentially expressed genes reflect known organ-specific biological activities. Sixty unigenes (15 % of the total) selected as preferentially expressed in buttons are involved in energy metabolism (photosynthesis, oxidative phosphorylation and carbon fixation). Considering these results we suggest that the gene expression profiles found for each library can be used for the identification of genes involved in specific metabolic pathways.
Crassulacean acid metabolism
Crassulacean acid metabolism, which often operates in species subjected to low-water conditions, is a key adaptation in flowering plants. This metabolism increases the efficiency of photosynthesis by increasing CO2 and carbon capture in plant tissues, where temporal separation of the photosynthetic stages prevents water loss due to transpiration. This mechanism is mediated mainly by phosphoenolpyruvate carboxylase (PEPC, which catalyzes nocturnal CO2 fixation and producing oxaloacetate), phosphoenolpyruvate carboxylase kinase (PEPK, which controlsphosphorylation state of PEPC), malate dehydrogenase (MDH, which reduces oxaloacetate to malate), NADP-dependent malate dehydrogenase (NADP-ME, which decarboxylates malate in the cytoplasm), and pyruvate phosphate dikinase (PPDK, which converts pyruvate into PEP) [25, 26]. At the end, CO2 is available as a substrate for RUBISCO in the Calvin cycle. Besides these enzymes, a set of sugar transporters, Na+/H+ antiporters and aquaporins are needed to maintain favorable conditions for CAM [27].
Available genetic and genomic resources in CAM plants, including Ananas comosus L, Agave deserti, Agave tequilana, Opuntia ficus-indica,and Mesembryanthemum crystallinum, have revealed CAM genetic determinants [27–30], some of which have been proposed to encode homologs of candidate regulatory proteins such as TOC1, CCA1, RPP9, and ZTL [30–32]. TOC1 binds to the G-box and EE-motif promoter regions of genes involved in both the morning and evening transcription-translation feedback loops that drive the plant circadian clock; these genes include PRR9 and CCA1 in the morning feedback loop. Discrete induction of TOC1 gene expression results in reduced CCA1 and PRR9 expression, indicating that TOC1 plays a repressive rather than stimulatory role in regulating circadian gene expression [33]. The dark-dependent degradation of TOC1 protein requires expression of ZTL, and is prevented by inhibiting the proteosome pathway; therefore, the TOC1-ZTL interaction is important in the control of TOC1 and is responsible for the regulation of circadian period [34]. Furthermore, recent work in Orchidaceae demonstrated that CAM has evolved at least 10 times independently in different families including both monocots and eudicots [35]. A full understanding of CAM biology requires the identification and analysis of the genes that code for enzymes and regulators of this mechanism. Sequencing efforts in the present work have captured most of the putative orthologous genes of CAM metabolism in L. williamsii. Additional file 2: Figure S3 shows the enzymes involved in CAM whose genes are identified in our unigene dataset, including phosphoenolpyruvate carboxylase [EC:4.1.1.31], pyruvate, orthophosphate dikinase [EC:2.7.9.1], malate dehydrogenase (oxaloacetate-decarboxylating) (NADP+) [EC:1.1.1.40], malate dehydrogenase [EC:1.1.1.39]. Also, several sequences in the unigene dataset were annotated as sugar transporters, antiporters or aquaporins (Additional file 3: Table S5).
Among the annotated unigenes, UN03078 and UN24453 were highly similar to Arabidopsis ZTL (AT5G57360) and CCA1, respectively (Additional file 3: Table S3). These unigenes are unique sequences matching known circadian clock regulators related with CAM. It remains to be determined whether these sequences indeed represent the orthologs of Arabidopsis ZTL and CCA1, and whether they play a role in the circadian regulation of CAM in peyote.
Identification of Lophophora williamsii unigenes with potential relevance to the biosynthetic pathway of mescaline
The in vivo pathway for the synthesis of mescaline was first proposed by Paul et al., [36] (Fig. 2). Starting from tyrosine (1), the main intermediates are dopa (2), dopamine (3), and 3,4,5-trihydroxy-β-phenyethylamine (4), resulting in mescaline (5). These reactions include hydroxylation of (1) to (2), decarboxylation to (3), hydroxylation to (4) and methylation to (5). All enzymes required for the formation of tyrosine from glucose are represented in the L. williamsii unigene database (Additional file 2: Figure S6). Enzymes selected as candidates for involvement in mescaline biosynthesis were postulated based on the high sequence similarity of homologous genes and their expression profiles.
Fig. 2.
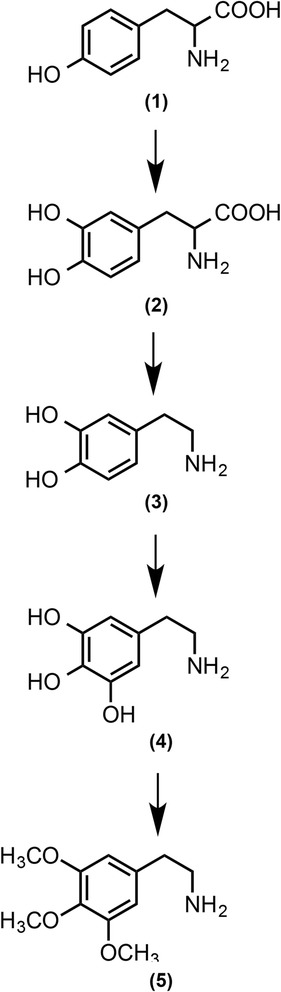
Biosynthetic pathway of mescaline in Lophophora williamsii. The main intermediates in the pathway are tyrosine (1), dopa (2) dopamine (3) 3,4,5-trihydroxy-β-phenyethylamine (4), and mescaline (5). This figure was modified from [84]
Tyrosine/DOPA decarboxylation
Opium poppy (Papaver somniferum) is the source of several pharmaceutical benzylisoquinoline alkaloids including morphine, codeine and sanguinarine. The biosynthesis of these alkaloids starts with the condensation of two tyrosine derivatives, dopamine and 4-hydroxyphenylacetaldehyde. The formation of dopamine involves the decarboxylation of tyrosine [EC:4.1.1.25] and/or dihydrophenylalanine (DOPA) by tyrosine/DOPA decarboxylase [EC:4.1.1.28] [37]. The members of the tyrosine/dopa decarboxylase (TYDC) gene family in opium poppy can be categorized into two subgroups according to their sequence homology. Representative members of each subgroup (TYDC1 and TYDC2) share 73 % amino acid identity, and both encoded enzymes exhibit L-dopa and L-tyrosine decarboxylase activities [37]. Three different L. williamsii unigenes (UN08840, UN13591 and UN15671) were annotated as homologous to Arabidopsis aromatic aldehyde synthase (ATAAS; AT2G20340). This enzyme catalyzes the conversion of phenylalanine and 3,4-dihydroxy-L-phenylalanine to phenylacetaldehyde and dopaldehyde, respectively [38]. According to the best BLAST hits annotation derived from the RefSeq-plant database, these unigenes are homologs of tyrosine decarboxylase (Medicago truncatula), a predicted protein in Populus trichocarpa,and a tyrosine/DOPA decarboxylase in Glycine max. Part of the distinctive pyridoxal-dependent decarboxylase conserved domain (PF00282) was identified in both UN13591 and UN15671 peyote unigenes by motif/domain search against the Pfam database (http://pfam.janelia.org). In contrast, the coding region represented in unigene UN08840 is homologous only to the carboxy-terminal portion of the ATAAS protein. Using the SeaView program [39], the protein-coding nucleotide sequences were aligned based on their corresponding amino acid translations (Additional file 2: Figure S7 and Additional file 5). A phylogenetic tree of peyote unigenes based on their pyridoxal-dependent decarboxylase domain sequence, including the TDYC1, TDYC2 and ATAAS proteins, was generated (Fig. 3). UN15671 was grouped with opium poppy TDYC sequences while the UN13591 unigene was grouped in the same clade with ATAAS. These data suggest that at least an ortholog to the P.somniferumTYCD (represented by the unigene UN15671) is present in L. williamsii, which could be responsible for tyrosine conversion to dopamine in this species. Interestingly, according to the relative frequency values obtained, these peyote unigenes showed higher levels of transcripts in buttons than in roots (Fig. 3).
Fig. 3.
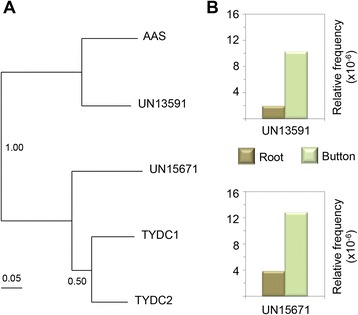
a Maximum Likelihood (ML) phylogenetic tree based on amino acid sequences of the conserved pyridoxal-dependent decarboxylase domain. The alignment includes the deduced protein sequences of the UN13591 and UN15671 unigenes of L. williamsii, A. thaliana aromatic aldehyde synthase (ATAAS), and P. somniferum tyrosine/DOPA decarboxylases (TYDC1 and TYDC2). Branch numbers represent the robustness of the three analyzed by approximate likelihood-ratio test (aLRT). b Expression patterns of L. williamsii unigenes in buttons and roots derived from RNA-seq analysis. RNA-seq data are shown as relative frequency values
Hydroxylation of aromatic compounds
Considering that during mescaline biosynthesis, dopamine is hydroxylated to form 3,4,5-trihydroxy-β-phenyethylamine, the L. williamsii transcriptome was surveyed in order to identify homologs of some biochemically characterized plant enzymes capable of introducing the hydroxyl group (-OH) into aromatic compounds (reviewed in [40]), especially those whose substrate is an aromatic amino acid that contains a single benzene ring.
Polyphenol oxidases (PPO) catalyze the O-hydroxylation of monophenols (phenol molecules in which the benzene ring contains a single hydroxyl substituent) to O-diphenols (phenol molecules containing two hydroxyl substituents). They can also further catalyze the oxidation of O-diphenols to produce O-quinones [41]. Tyrosinases are bifunctional PPOs that catalyze the O-hydroxylation of monophenols and subsequent oxidation of O-diphenols to quinines [42]. Thus, tyrosinases accept both mono- and di-phenols as substrates. The hydroxylation ability of the enzyme is also referred to as cresolase or monophenolase activity [EC:1.14.18.1], and the oxidation ability as catecholase or diphenolase activity [EC:1.10.3.1]. The monophenolase activity of tyrosinases is known to be the initial rate-determining reaction [42, 43]. In tyrosinase-catalyzed reactions, molecular oxygen is used as an electron acceptor that is reduced to water. Tyrosinases and their corresponding genes have been characterized from various sources, including bacteria, fungi, plants and mammals [41, 44]. A sequence comparison of recently published tyrosinases reveals high heterogeneity concerning their length and overall identity. However, highly conserved regions among all tyrosinases can be found in the active site domain [44, 45]. The peyote unigene UN14261 was annotated as a homolog of Glycine max PPO (RefSeq accession number XP_003522849.1). A Motif/domain search revealed that a KFDV-containg PPO1 C-terminal domain (PF122143) can be identified in the translated sequence corresponding to this peyote unigene (Additional file 2: Figure S8 and Additional file 6). This domain family is found in association with the common central domain of tyrosinase (PF00264), and is typically between 138 and 152 amino acids in length. Even though the functional significance of these domains is not known, many members of this family are plant or plastid polyphenol oxidases with the highly conserved sequence motif KFDV, from which the name derives [46].
An in-depth blast search, for which the coding sequence of the POP1 KFDV domain was used as reference (e-value 10−03), revealed that no additional PPOs could be identified in the peyote unigene dataset. Although unigene UN14261 could be considered as an interesting candidate to participate in mescaline biosynthesis, in contrast to unigene UN15671 (an ortholog of P.somniferum TYDC proteins), its transcript levels indicate that UN14261 is preferentially expressed in roots rather than buttons (Additional file 3: Table S6).
Folate- or tetrahydropterin-dependent aromatic amino acid hydroxylases (AAHs) are an additional group of enzymes localized in chloroplasts capable of catalizing the O-hydroxylation of the benzene ring of aromatic amino acids [47–49]. However, homologs of this family, which comprise phenylalanine [EC:1.14.16.1], tyrosine [EC:1.14.16.2], and tryptophan [EC:1.14.16.4] hydroxylases [48, 49], are not present in our L. williamsii unigene dataset. This was expected considering that thus far, such genes have been identified only in non-flowering plants [49].
α-keto acid dependent enzymes catalyze dealkylations, epimerizations and halogenations, hydroxylation, and a variety of oxidations [50]. Fe2+/α-keto acid-dependent oxygenases represent a group of dioxygenases [EC:1.14.11.–] that use α-ketoglutarate (α-KG) as substrate, incorporating two oxygen atoms from O2 into two different substrates where one atom is transferred to the actual substrate, and the second one to the α-KG acting as the co-substrate (reviewed in [50]). A large variety of compounds such as flavonoids and alkaloids may be synthesized by Fe2+/α-KG-dependent dioxygenases in plants [51–53]. Other types of enzymes such as 4-hydroxyphenylpyruvate dioxygenase (HPPD; EC:1.13.11.27) have no relationship in sequence but do show a similar reaction mechanism to Fe2+/α-KG hydroxylases, and these enzymes also contain ferrous iron (Fe2+) in the active site, but the α-keto acid that is decaoxylated is part of the substrate and the hydroxylation is associated with an ‘NIH’ shift (a chemical rearrangement wherein a hydrogen atom on an aromatic ring undergoes an intramolecular migration primarily during a hydroxylation reaction; [54, 55]). 4-Hydroxyphenylpyruvate dioxygenase, which catalyzes the formation of 2,5-dihydroxyphenylacetate (homogentisate), has been found in all aerobic forms of life where it is involved in tyrosine metabolism [54, 56].
Through a BLAST search based on the distinctive 2OG-FeII-Oxy domain (PF03171) of the Fe2+/α-KG oxygenases, a total of 16 peyote unigenes were identified as members of this family. These unigenes were aligned against their plant homologs, identified in the annotation process as the top BLAST hits against the RefSeq database. To identify the coding sequences in their correct open reading frames, we used the known plant homologs as references and protein-coding nucleotide sequences were aligned based on their corresponding amino acid translations using the SeaView program (Additional file 7). Among 12 peyote unigenes, complete (or almost complete) coding sequences (CDS) ranging between 510 and 1,083 bp were identified. Additionally, a highly conserved non-heme dioxygenase N-terminal domanin (DIOX-N; PF14226), commonly associated with 2-oxoglutarate/Fe(II)-dependent dioxygenase proteins, was identified in the peyote unigene set (7 in total). The phylogenetic relationships among peyote Fe2+/α-KG oxygenases, and their expression profiles, were analyzed (Fig. 4). Six of these unigenes showed highest expression levels in buttons, and four of them had two-fold or greater transcription in roots (UN07954, UN02288, UN02485 and UN03447).
Fig. 4.
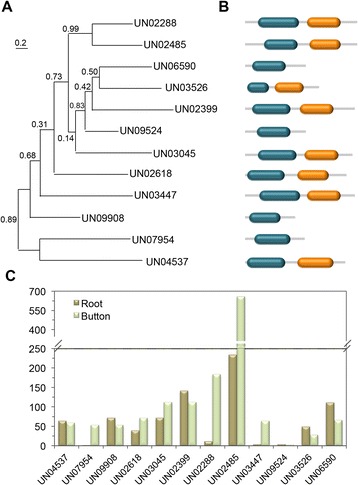
Phylogenetic relationships, primary protein structures and expression patterns of L. williamsii Fe2+/α-keto acid-dependent oxygenases. a ML un-rooted phylogenetic tree. b Schematic representation of domain structure along the length of protein. DIOX-N (PF14226) and 2OG-FeII-Oxy (PF03171) domains are represented by green and orange boxes, respectively. c Expression analysis of L. williamsii unigenes in buttons and roots performed by RNAseq
With regard to HPPD proteins [EC:1.13.11.27], a similar approach was used to identify putative homologs. Using the glyoxalase domain (PF00903), a single putative L. williamsii HPPD CDS was identified (UN03443; Additional file 8). The predicted protein showed around 77.0 % amino acid identity compared with the published Medicago truncatula HPPD. The relative abundance values indicate that the transcripts of this HPPD are almost 200-fold higher in roots than in the peyote buttons (Additional file 3: Table S6).
Considering the results described above, it is proposed that one of the Fe2+/α-KG oxygenases expressed preferentially in buttons is a prime candidate for involvement in the hydroxylation of dopamine during mescaline biosynthesis.
O-methyltransferases of L. williamsii
Enzymatic O-methylation consists of the transfer of the methyl group of a common co-substrate such as S-adenosyl-L-methionine (AdoMet) to the hydroxyl group of an acceptor molecule. This process is catalyzed by an O-methyltransferase (OMT) [EC 2.1.1.6.x], a group of enzymes that methylate a wide range of compounds with a high degree of selectivity [57, 58]. These enzymes are present in diverse organisms, including bacteria, fungi, plants and mammals. However, a few OMTs have been shown to be multifunctional enzymes that catalyze the methylation of structurally related compounds such as phenylpropanoids and flavonoids [59–63]. In plants, O-methyltransferases constitute a large family of enzymes. Novel OMT-like gene sequences have been reported using a framework phylogenetic tree encompassing 61 biochemically characterized plant OMT protein sequences for improved prediction of their putative function [64]. In order to identify OMT-like sequences, these proteins were used as reference in a BLAST similarity search (e-value 10−06) against the L. williamsii unigene dataset. A total of eleven OMT-like sequences (unigenes UN00812, UN01722, UN01870, UN02190, UN02462, UN02547, UN03053, UN03207, UN05101, UN06792 and UN28302), in which the CDS for the O-methyltransferase conserved domain (PF00891) was present, were found in the peyote transcriptome database. To identify the coding sequences in their correct open reading frames, these peyote unigenes were aligned based on their corresponding amino acid translations against their best-blast-hits homologs (Additional file 9). With only two exceptions (UN28302 and UN06792), these unigenes contain complete (or almost complete) coding sequences. A phylogenetic tree was generated in which the proteins derived from these peyote CDS and their homologs were included (Fig. 5).
Fig. 5.
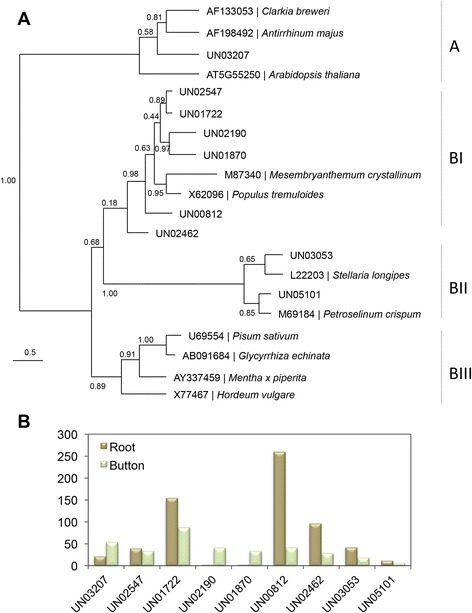
Expression patterns of OMT peyote unigenes and the phylogenetic relationships of their deduced proteins with those from various other plant species. a Phylogenetic relationship of plant OMTs. b Expression profile of OMTs genes in buttons and roots of L. williamsii plants
The phylogenetic structure of the L. williamsii OMT gene family was similar to that previously reported by Lam et al. [64]. Two distinct monophyletic lineages were recognized (A and B, respectively), one of which (clade B), includes three sister clades (BI-BIII). With only one exception (clade BIII), the remaining clades contain at least one putative OMT identified in the peyote transcriptome (Fig. 5). Clade A contains plant OMTs with S-adenosyl-L-methionine (SAM) dependent methyltransferase activity, which acts on a diverse group of metabolites including salicylic acid, benzoic acid and indole-3-acetic acid (IAA) (identified in the species Clarkia breweri, Antirrhinum majus and Arabidopsis thaliana, respectively) [65–67].
Clade BI contains six different peyote unigenes (UN00812, UN01722, UN01870, UN02190, UN02462 and UN02547) grouped with only two different plant OMTs: one lignin bispecific caffeic acid/5-hydroxy ferulic acid OMT (from Populus tremuloides) [68] and one flavonoids/caffeoyl-CoA OMT (from Mesembryanthemum crystallinum) [63]. Clade BII contains two peyote unigenes (UN03053 and UN05101) that were grouped with two plant caffeoyl-CoA 3-o-methyltransferases (from Stellaria longipes and Petroselinum crispum; respectively) [69, 70]. Finally, clade BIII, which contained no peyote unigenes, includes some flavonoid OMTs [58, 71–73].
Mescaline biosynthesis in peyote plants requires the triple-methylation of 3,4,5-trihydroxy-β-phenyethylamine (in Fig. 2 exemplified by reaction of (4) to (5)). Considering the chemical structure of substrates such caffeoyl-CoA, 5-hydroxy ferulic and/or caffeic acid, and that plant OMTs may have specific or promiscuous activities, we propose that 3,4,5-trihydroxy-β-phenyethylamine could serve as the methyl acceptor (substrate) for one or more L. williamsii OMTs classified in the BI and/or BII monophyletic lineages, especially those whose transcript levels are more abundant in buttons than in peyote roots (Fig. 5). Interestingly, only three peyote OMTs showed increased levels of transcripts in buttons compared to roots.
UN03207 was resolved as a member of clade A (SAM dependent methyltransferases), and even though the abundance of the transcript was calculated to be 2.6-fold higher in buttons than in roots, transcripts related to this OMT were detected in both organs. This was expected since metabolites such as salicylic acid, benzoic acid, and jasmonic acid play crucial roles in numerous pathways, controlling the way in which plants grow and develop [74]. With respect to unigenes UN02190 and UN01870, which were grouped in clade BI as two closely related OMTs, their transcript levels were quite low in roots and 20-fold higher in buttons. Based on these results, we propose that UN02190, UN01870, or both unigenes could be considered as primary candidates to belong to the mescaline biosynthesis pathway.
Real-time quantitative PCR verification
Transcriptional regulation revealed by RNA-seq data was confirmed by real-time quantitative reverse transcription PCR (qRT-PCT). A total of seven peyote unigenes was selected as primary candidate genes involved in mescaline biosynthesis (one TYDC, four Fe2+/α-KG oxygenases and two OMTs, respectively). A linear regression analysis showed an overall correlation coefficient of R = 0.796, which indicates a good correlation between transcript abundance assayed by real-time PCR and transcription profiles revealed by RNA-seq data (Additional file 2: Figure S9).
Conclusions
The L. williamsii transcriptome provides a detailed genomic window on metabolic processes that are carried out in a cactus plant capable of accumulating high levels of alkaloids. The transcriptome of buttons (which are photosynthetic organs) and roots of peyote vary significantly in terms of the genes that can be identified in each and their consequent expression profiles. These variations could be responsible, at least in part, for the identities of the organs but also could explain differences in terms of the metabolites that are accumulated. Most genes involved in CAM metabolism, typical of slow-growing cacti in desert regions, were identified and can be used as starting points to study the regulation of genes that allow CO2 fixation and stomatal aperture during nighttime, as opposed to C3 and C4 plants that carry out this process during the day. Genes putatively involved in the biogenesis of mescaline were identified in this work by comparing their expression levels in button and roots. This report provides a catalogue of specific candidate genes that can be further expressed in heterologous systems in order to confirm their putative roles in mescaline biosynthesis.
Methods
Plant material
Since L. williamsii is listed on CITES (the Convention on International Trade in Endangered Species of Wild Fauna and Flora; http://www.cites.org) Appendix II and it is legally protected in Mexico by the national list of species at risk of extinction, NOM-059-SEMARNAT-2010, where it is listed under the category “subject to special protection” (Pr; SEMARNAT 2010), the plants used in this study were collected in the Coahuila desert (Cuatrociénegas municipality) according to permissions granted by Mexican authorities (DE/5336/2007 official letter issued by Subsecretaría de gestión para la protección ambiental, Dirección general de protección contra riesgos sanitarios, and Dirección general de la vida silvestre).
Alkaloid extraction
15 g of both powdered samples (roots and buttons, respectively) were extracted at room temperature three times with EtOH (300 ml) for 48 h each time with stirring. The combined ethanol extracts were filtered and evaporated in vacuo to give 1915.4 and 1779.56 mg of oily residues, respectively. The residues were dissolved in H2O, made alkaline with concentrated ammonia (pH 9) and extracted twice with CHCl3 and once with CHCl3:EtOH (3:1). The combined extracts were dried over anhydrous Na2SO4, filtered and evaporated to dryness to yield 463.9 mg from roots and 431.0 mg from buttons, respectively, of total alkaloids.
Gas chromatography–mass spectrometry
A gas chromatograph (Hewlett Packard 6890) coupled to a quadrupole electron impact ionization mass spectrometer (Hewlett Packard 5973) was used. The obtained mass spectra were compared with the NIST v 2008 Mass Spectra Data Base (National Institute of Standards and Technology, U.S. Department of Commerce). The GC oven temperature program was as follows: initial temperature 150 °C, increasing at a rate of 4 °C/min. to 280 °C and maintained for 20 min. Injector temperature was set to 250 °C. At the MS the ionization source was set at 250 °C and the quadrupole at 130 °C. A 2 μl aliquot of the extract was injected in split-less mode injection into a HP-5 MS capillary column (30 m × 0.25 mm × 0.25 μm Agilent Technologies, CA, USA) using Helium, chromatographic grade as carrier gas at 1 mL/min.
RNA extraction, library construction, pyrosequencing and de novo assembly
Total RNA was isolated from roots and buttons using the Trizol reagent (Invitrogen) and re-purified with the RNeasy kit (Qiagen) following the manufacturer’s recommendations. RNA purity was checked using the Agilent 2100 Bioanalyzer RNA 6000 Nano Assay chip (Agilent Technologies, Stockport, U.K). cDNA synthesis was performed from 3.5 μg of total RNA using the Message Amp-II kit (Ambion, Foster City, CA) following the manufacturer’s protocol as described earlier [75]. cDNA derived from buttons and peyote roots was pooled (using the same amount of each) and sequencing on the GS-FLX and GS-Junior platforms (one run per equipment). Non-pooled cDNA was used to prepare independent libraries from buttons and roots (represented by Ion barcode adapters). cDNA samples were prepared with the Ion Xpress™ Template Kit (Life Technologies) according to the Ion Xpress™ Template Kit User Guide and sequenced on a Personal Genome Machine™ (PGM™) sequencer using two 3.18 semiconductor chips.
After a filtering process, high quality reads generated with both GS (454) and PGM (IonTorrent) technologies were pooled and subjected to de novo assembly using the MIRA assembler [13] and the resultant contigs were re-assembled with CAP3 [14]. As a result, 40,436 non-redundant contigs (unigenes) ranging from 200 to 4,170 bp in length were obtained. Files containing sequence reads and quality scores were deposited in the Short Read Archive of the National Center for Biotechnology Information (NCBI) [BioProject PRJNA261064].
Annotation of the Lophophora williamsii unigenes
The unigenes were compared against the Arabidopsis thaliana (http://www.arabidopisis.org/) and RefSeq (ftp://ftp.ncbi.nlm.nih.gov/refseq/release/plant/) protein databases. In both alignments, a cut-off e-value of 10−3 and bit score ≤ 30 was applied and only the top blast hit was considered. Top protein matches from Arabidopsis or additional plant proteins were assigned to each of the L. williamsii unigenes. The gene ontology (GO) functional classes and pathways for each unigene were assigned based on Arabidopsis GO SLIM and pathway annotation (ftp://ftp.arabidopsis.org/home/tair/Ontologies/). Additionally, the unigenes were also analyzed using the KEGG Automatic Annotation Server (KAAS [76]; http://www.genome.jp/tools/kaas/). The bi-directional best hit (BBH) method was used to provide annotations of KEGG Orthology (KO) codes. Enzyme Commission (EC) numbers were also assigned based on the annotations extracted from Kyoto Encyclopedia of Genes and Genomes (KEGG) [77]. Finally, conserved domains in the selected unigenes were confirmed by motif/domain search against the Pfam database (http://pfam.janelia.org).
Comparative analysis of transcript levels
The Ion-reads were aligned using Bowtie [78] to the L. williamsii assembled transcriptome. More than 95 % of total reads from each peyote organ (button and root) were mapped back to the peyote unigenes, nearly to 65 % with a single match. Read hits per unigene (reads counts) were normalized using the relative frequency of reads, i.e., the number of reads mapped for a given unigene relative to the total number of reads in a specific library. In order to identify unigenes with significant differences in their transcripts levels, the R statistical differential expression value was calculated as described by Stekel et al. [24].
Phylogenetic analysis
Phylogenetic analyses were performed in a maximum likelihood (ML) framework using SeaView v2.4 software [39], which drives the Muscle [79] (for alignment) and PhyML [80] (for phylogenetic analysis) programs. The PhyML option was used under LG (Le and Gascuel) model [81]. Equilibrium frequencies, topologies, and branch lengths were optimized, the starting tree was determined using BioNJ, and both nearest-neighbor interchange (NNI) and subtree pruning and regrafting (SPR) algorithms for tree searching were used. Branch robustness was analyzed by approximate likelihood-ratio test (aLRT) [82].
Evaluation of genes expression by qRT-PCR
A total of 1 μg of RNA was reverse transcribed for first-strand cDNA synthesis using SuperScript III reverse transcriptase (Invitrogen) according to the manufacturer’s instructions. Gene-specific primer pairs (Additional file 3: Table S8), which were designed using the Primer3 v.0.4.0 web tool (http://bioinfo.ut.ee/primer3-0.4.0/primer3/), were used for real-time PCR. Reactions were performed with the SYBR Green PCR Master Mix (Applied Biosystems) in an ABI PRISM 7500 Fast Real-time system. Actin was used as the standard to normalize the content of cDNA and the 2-ΔΔCt method [83] was employed to compare button and root samples. Ten microliters of the reaction mixture was added to each well. The thermal cycling program was set to 50 °C for 2 min, 95 °C for 1 min, and 40 cycles of 95 °C for 15 s and 60 °C for 1 min. The real-time PCR was conducted with at least three technical replicates for each sample, and data are indicated as means ± standard error (SE).
Acknowledgements
The authors would like to thank María de Jesus Ortega-Estrada for her technical support in the lab and real-time PCR services. We also thank Beatriz Jiménez-Moraila for collection permit management, and the staff of the “Laboratorio de Servicios Genómicos” at LANGEBIO for their assistant in RNAseq library preparation and sequencing. Special thanks go to Alexandro Alonso-Sánchez and Emanuel Villafan De La Torre for their valuable discussion on bioinformatics processes and phylogenetic analyses. This paper is dedicated to our colleague and freind, Hienz Wagner-Hellmann, in memory of his contributions to preserve and expand the knowledge of the American cacti.
Additional files
(FASTA 978 bytes)
GC-MS chromatograms obtained from ethanol extracts of buttons (A) and roots (B) of L. williamsii. Figure S2. Gene ontology classification of the L. williamsii transcriptome. Figure S3. The carbon fixation pathway in photosynthetic organisms. Figure S4. Glycolysis / Gluconeogenesis biosynthesis pathway reconstructed based on the de novo assembly and annotation of the L. williamsii transcriptome. Figure S5. Starch and sucrose metabolic pathway in L.williamsii. Figure S7. Alignment of pyridoxal-dependent decarboxylase conserved domains in the deduced amino acid sequences of the UN13591 and UN15671 L. williamsii unigenes. Figure S8. (A) Alignment of the PPO1-KFDV C-terminal conserved domain of the UN14261 L. williamsii unigene and Glycine max PPO1 (XP_003522849.1). (B) Expression analysis of the L. williamsii unigene by RNA-seq. Figure S9. Real-time PCR validation of RNA-seq results. (PPTX 877 kb)
Alkaloids found in the ethanol extracts of buttons and roots of L. williamsii. Table S2. Reads statistics of the L. williamsii dataset. Table S3. Annotation of Lophophora williamsii unigenes. (TAIRv10; http://www.arabidopsis.org/ and NCBI; ftp://ftp.ncbi.nih.gov/refseq/release/plant/ Table S4. Funtional classification of L williamsii unigenes based on Arabidopsis GO annotation. Table S5. Detailed information for L. williamsii unigenes mapped on KEGG pathways. Table S6. Expression profile matrix of L. williamsii unigenes. Table S7. Preferentially expressed unigenes in specific organs (buttons and roots) of L. williamsii. (XLSX 16694 kb)
L. williamsii unigenes. (ZIP 7124 kb)
(FASTA 2 kb)
(FASTA 1009 bytes)
(FASTA 33 kb)
(FASTA 2 kb)
Alignment of O -methyltransferase proteins. (FASTA 25 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
EIL performed transcriptome assembly, annotation, statistical analysis and manuscript writing. EIL, CAPT, FZH, and ERC performed the experiments. EIL, VAA, CAPT, ERC, JMT, CCV, AFC, JLOR, AHE and LHE analyzed the data. LHE conceived the project and was responsible for directing all of the research activities. All authors have read and approved the final submitted version of the manuscript.
Contributor Information
Enrique Ibarra-Laclette, Email: enrique.ibarra@inecol.mx.
Flor Zamudio-Hernández, Email: fzamudio@langebio.cinvestav.mx.
Claudia Anahí Pérez-Torres, Email: claudia.perez@inecol.mx.
Victor A. Albert, Email: vaalbert@buffalo.edu
Enrique Ramírez-Chávez, Email: eramirez@ira.cinvestav.mx.
Jorge Molina-Torres, Email: jmolina@ira.cinvestav.mx.
Araceli Fernández-Cortes, Email: araceli@langebio.cinvestav.mx.
Carlos Calderón-Vázquez, Email: ccalderon@ipn.mx.
José Luis Olivares-Romero, Email: jose.olivares@inecol.mx.
Alfredo Herrera-Estrella, Email: aherrera@langebio.cinvestav.mx.
Luis Herrera-Estrella, Email: lherrera@langebio.cinvestav.mx.
References
- 1.Anderson EF. Peyote: The Divine Cactus, 2nd ed. edn. Tucson: University of Arizona Press; 1996.
- 2.Lundström J, Agurell S. A complete biosynthetic sequence from tyrosine to mescaline in two cactus species. Tetrahedron Lett. 1969;10(39):3371–3374. doi: 10.1016/S0040-4039(00)99766-1. [DOI] [PubMed] [Google Scholar]
- 3.Kapadia GJ, Fayez MB. Peyote constituents: chemistry, biogenesis, and biological effects. J Pharm Sci. 1970;59(12):1699–1727. doi: 10.1002/jps.2600591202. [DOI] [PubMed] [Google Scholar]
- 4.Jansen MAK, Gaba V, Greenberg BM. Higher plants and UV-B radiation: balancing damage, repair and acclimation. Trends Plant Sci. 1998;3(4):131–135. doi: 10.1016/S1360-1385(98)01215-1. [DOI] [Google Scholar]
- 5.Kerr R. Natural Products: Their Chemistry and Biological Significance. Econ Bot 1995;49(3):308-308.
- 6.Kutchan TM. Alkaloid biosynthesis - the basis for metabolic engineering of medicinal plants. Plant Cell Online. 1995;7(7):1059–1070. doi: 10.1105/tpc.7.7.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wink M. Introduction: Biochemistry, physiology and ecological functions of secondary metabolites. In: Annual Plant Reviews Volume 40: Biochemistry of Plant Secondary Metabolism. Wiley-Blackwell; 2010: 1-19.
- 8.Bruhn J, Holmstedt B. Early peyote research an interdisciplinary study. Econ Bot. 1973;28(4):353–390. doi: 10.1007/BF02862854. [DOI] [Google Scholar]
- 9.McLaughlin JL. Peyote: an introduction. Lloydia. 1973;36(1):1–8. [PubMed] [Google Scholar]
- 10.Schultes RE. The appeal of peyote (Lophophora williamsii) as a medicine. Am Anthropol. 1938;40(4):698–715. doi: 10.1525/aa.1938.40.4.02a00100. [DOI] [Google Scholar]
- 11.Aragane M, Sasaki Y, Nakajima J, Fukumori N, Yoshizawa M, Suzuki Y, et al. Peyote identification on the basis of differences in morphology, mescaline content, and trnL/trnF sequence between Lophophora williamsii and L. diffusa. J Nat Med. 2011;65(1):103–110. doi: 10.1007/s11418-010-0469-7. [DOI] [PubMed] [Google Scholar]
- 12.Gottlieb A, Todd L. Peyote and Other Psychoactive Cacti: Ronin Publishing; 1997.
- 13.Chevreux B, Pfisterer T, Drescher B, Driesel AJ, Müller WEG, Wetter T, et al. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 2004;14(6):1147–1159. doi: 10.1101/gr.1917404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang X, Madan A. CAP3: A DNA sequence assembly program. Genome Res. 1999;9(9):868–877. doi: 10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Novaes E, Drost DR, Farmerie WG, Pappas GJ, Jr, Grattapaglia D, Sederoff RR, et al. High-throughput gene and SNP discovery in Eucalyptus grandis, an uncharacterized genome. BMC Genomics. 2008;9:312. doi: 10.1186/1471-2164-9-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumar S, Filipski A. Multiple sequence alignment: in pursuit of homologous DNA positions. Genome Res. 2007;17(2):127–135. doi: 10.1101/gr.5232407. [DOI] [PubMed] [Google Scholar]
- 17.Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006;34(Web Server issue):W293–297. doi: 10.1093/nar/gkl031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bairoch A. The ENZYME database in 2000. Nucleic Acids Res. 2000;28(1):304–305. doi: 10.1093/nar/28.1.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Y, Zhou J, White KP. RNA-seq differential expression studies: more sequence or more replication? Bioinformatics. 2014;30(3):301–304. doi: 10.1093/bioinformatics/btt688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tarazona S, García-Alcalde F, Dopazo J, Ferrer A, Conesa A. Differential expression in RNA-seq: a matter of depth. Genome Res. 2011;21(12):2213–23. doi: 10.1101/gr.124321.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bizuayehu T, Lanes C, Furmanek T, Karlsen B, Fernandes J, Johansen S, et al. Differential expression patterns of conserved miRNAs and isomiRs during Atlantic halibut development. BMC Genomics. 2012;13(1):11. doi: 10.1186/1471-2164-13-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gongora-Castillo E, Ibarra-Laclette E, Trejo-Saavedra D, Rivera-Bustamante R. Transcriptome analysis of symptomatic and recovered leaves of geminivirus-infected pepper (Capsicum annuum) Virol J. 2012;9(1):295. doi: 10.1186/1743-422X-9-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stekel DJ, Git Y, Falciani F. The comparison of gene expression from multiple cDNA libraries. Genome Res. 2000;10(12):2055–2061. doi: 10.1101/gr.GR-1325RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cushman JC, Bohnert HJ. Crassulacean acid metabolism: molecular genetics. Annu Rev Plant Physiol Plant Mol Biol. 1999;50:305–332. doi: 10.1146/annurev.arplant.50.1.305. [DOI] [PubMed] [Google Scholar]
- 26.Winter K, Holtum JAM. Facultative crassulacean acid metabolism (CAM) plants: powerful tools for unravelling the functional elements of CAM photosynthesis. J Exp Bot. 2014;65(13):3425–41. doi: 10.1093/jxb/eru063. [DOI] [PubMed] [Google Scholar]
- 27.Zhang J, Liu J, Ming R. Genomic analyses of the CAM plant pineapple. J Exp Bot. 2014;65(13):3395–404. doi: 10.1093/jxb/eru101. [DOI] [PubMed] [Google Scholar]
- 28.Cushman JC, Tillett RL, Wood JA, Branco JM, Schlauch KA. Large-scale mRNA expression profiling in the common ice plant, Mesembryanthemum crystallinum, performing C3 photosynthesis and crassulacean acid metabolism (CAM) J Exp Bot. 2008;59(7):1875–1894. doi: 10.1093/jxb/ern008. [DOI] [PubMed] [Google Scholar]
- 29.Gross S, Martin J, Simpson J, Abraham-Juarez M, Wang Z, Visel A. De novo transcriptome assembly of drought tolerant CAM plants, Agave deserti and Agave tequilana. BMC Genomics. 2013;14(1):563. doi: 10.1186/1471-2164-14-563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mallona I, Egea-Cortines M, Weiss J. Conserved and divergent rhythms of crassulacean acid metabolism-related and core clock gene expression in the cactus Opuntia ficus-indica. Plant Physiol. 2011;156(4):1978–1989. doi: 10.1104/pp.111.179275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suetsugu N, Wada M. Evolution of three LOV blue light receptor families in green plants and photosynthetic stramenopiles: phototropin, ZTL/FKF1/LKP2 and aureochrome. Plant Cell Physiol. 2013;54(1):8–23. doi: 10.1093/pcp/pcs165. [DOI] [PubMed] [Google Scholar]
- 32.Nagel DH, Kay SA. Complexity in the wiring and regulation of plant circadian networks. Curr Biol. 2012;22(16):R648–657. doi: 10.1016/j.cub.2012.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang W, Pérez-García P, Pokhilko A, Millar AJ, Antoshechkin I, Riechmann JL, et al. Mapping the core of the Arabidopsis circadian clock defines the network structure of the oscillator. Science. 2012;336(6077):75–79. doi: 10.1126/science.1219075. [DOI] [PubMed] [Google Scholar]
- 34.Mas P, Kim W-Y, Somers DE, Kay SA. Targeted degradation of TOC1 by ZTL modulates circadian function in Arabidopsis thaliana. Nature. 2003;426(6966):567–570. doi: 10.1038/nature02163. [DOI] [PubMed] [Google Scholar]
- 35.Silvera K, Winter K, Rodriguez BL, Albion RL, Cushman JC. Multiple isoforms of phosphoenolpyruvate carboxylase in the Orchidaceae (subtribe Oncidiinae): implications for the evolution of crassulacean acid metabolism. J Exp Bot. 2014;65(13):3623–3636. doi: 10.1093/jxb/eru234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paul AG, Khanna KL, Rosenberg H, Takido M. Biosynthesis of peyote alkaloids. J Chem Soc D. 1969;14:838–8. doi: 10.1039/c29690000838. [DOI] [Google Scholar]
- 37.Facchini PJ, De Luca V. Differential and tissue-specific expression of a gene family for tyrosine/dopa decarboxylase in opium poppy. J Biol Chem. 1994;269(43):26684–26690. [PubMed] [Google Scholar]
- 38.Gutensohn M, Klempien A, Kaminaga Y, Nagegowda DA, Negre-Zakharov F, Huh J-H, et al. Role of aromatic aldehyde synthase in wounding/herbivory response and flower scent production in different Arabidopsis ecotypes. Plant J. 2011;66(4):591–602. doi: 10.1111/j.1365-313X.2011.04515.x. [DOI] [PubMed] [Google Scholar]
- 39.Gouy M, Guindon S, Gascuel O. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;27(2):221–224. doi: 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
- 40.Ullrich R, Hofrichter M. Enzymatic hydroxylation of aromatic compounds. Cell Mol Life Sci. 2007;64(3):271–293. doi: 10.1007/s00018-007-6362-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mayer AM. Polyphenol oxidases in plants and fungi: going places? Annu Rev Plant Physiol Plant Mol Biol. 2006;67(21):2318–2331. doi: 10.1016/j.phytochem.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 42.Robb DA. Tyrosinae. In: Lontie R, editor. Copper proteins and copper enzymes. Boca Raton, Florida: CRC Press Inc; 1984. [Google Scholar]
- 43.Rodriguez-Lopez JN, Tudela J, Varon R, Garcia-Carmona F, Garcia-Canovas F. Analysis of a kinetic model for melanin biosynthesis pathway. J Biol Chem. 1992;267(6):3801–3810. [PubMed] [Google Scholar]
- 44.Marusek CM, Trobaugh NM, Flurkey WH, Inlow JK. Comparative analysis of polyphenol oxidase from plant and fungal species. J Inorg Biochem. 2006;100(1):108–123. doi: 10.1016/j.jinorgbio.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 45.Selinheimo E, NiEidhin D, Steffensen C, Nielsen J, Lomascolo A, Halaouli S, et al. Comparison of the characteristics of fungal and plant tyrosinases. J Biotechnol. 2007;130(4):471–480. doi: 10.1016/j.jbiotec.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 46.Tran L, Taylor J, Constabel C. The polyphenol oxidase gene family in land plants: Lineage-specific duplication and expansion. BMC Genomics. 2012;13(1):395. doi: 10.1186/1471-2164-13-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fitzpatrick PF. Mechanism of aromatic amino acid hydroxylation†. Biochemistry. 2003;42(48):14083–14091. doi: 10.1021/bi035656u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Farquharson KL. Introducing aromatic amino acid hydroxylases from plants. Plant Cell Online. 2010;22(10):3192. doi: 10.1105/tpc.110.221011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pribat A, Noiriel A, Morse AM, Davis JM, Fouquet R, Loizeau K, et al. Nonflowering plants possess a unique folate-dependent phenylalanine hydroxylase that is localized in chloroplasts. Plant Cell. 2010;22(10):3410–3422. doi: 10.1105/tpc.110.078824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Purpero V, Moran G. The diverse and pervasive chemistries of the α-keto acid dependent enzymes. J Biol Inorg Chem. 2007;12(5):587–601. doi: 10.1007/s00775-007-0231-0. [DOI] [PubMed] [Google Scholar]
- 51.Hausinger RP. FeII/alpha-ketoglutarate-dependent hydroxylases and related enzymes. Crit Rev Biochem Mol Biol. 2004;39(1):21–68. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 52.Anzellotti D, Ibrahim RK. Novel flavonol 2-oxoglutarate dependent dioxygenase: affinity purification, characterization, and kinetic properties. Arch Biochem Biophys. 2000;382(2):161–172. doi: 10.1006/abbi.2000.2002. [DOI] [PubMed] [Google Scholar]
- 53.Turnbull JJ, Nakajima J, Welford RW, Yamazaki M, Saito K, Schofield CJ. Mechanistic studies on three 2-oxoglutarate-dependent oxygenases of flavonoid biosynthesis: anthocyanidin synthase, flavonol synthase, and flavanone 3 beta-hydroxylase. J Biol Chem. 2004;279(2):1206–1216. doi: 10.1074/jbc.M309228200. [DOI] [PubMed] [Google Scholar]
- 54.Rundgren M. Tritium isotope effects in the reaction catalyzed by 4-hydroxyphenylpyruvate dioxygenase from Pseudomonas sp. strain P.J. 874. Biochim Biophys Acta. 1982;704(1):59–65. doi: 10.1016/0167-4838(82)90132-7. [DOI] [PubMed] [Google Scholar]
- 55.Neidig ML, Decker A, Choroba OW, Huang F, Kavana M, Moran GR, et al. Spectroscopic and electronic structure studies of aromatic electrophilic attack and hydrogen-atom abstraction by non-heme iron enzymes. Proc Natl Acad Sci U S A. 2006;103(35):12966–12973. doi: 10.1073/pnas.0605067103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moran GR. 4-Hydroxyphenylpyruvate dioxygenase. Arch Biochem Biophys. 2005;433(1):117–128. doi: 10.1016/j.abb.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 57.Ibrahim R, Bruneau A, Bantignies B. Plant O-methyltransferases: molecular analysis, common signature and classification. Plant Mol Biol. 1998;36(1):1–10. doi: 10.1023/A:1005939803300. [DOI] [PubMed] [Google Scholar]
- 58.Willits MG, Giovanni M, Prata RT, Kramer CM, De Luca V, Steffens JC, et al. Bio-fermentation of modified flavonoids: an example of in vivo diversification of secondary metabolites. Phytochemistry. 2004;65(1):31–41. doi: 10.1016/j.phytochem.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 59.Gauthier A, Gulick PJ, Ibrahim RK. Characterization of two cDNA clones which encode O-methyltransferases for the methylation of both flavonoid and phenylpropanoid compounds. Arch Biochem Biophys. 1998;351(2):243–249. doi: 10.1006/abbi.1997.0554. [DOI] [PubMed] [Google Scholar]
- 60.Frick S, Kutchan TM. Molecular cloning and functional expression of O-methyltransferases common to isoquinoline alkaloid and phenylpropanoid biosynthesis. Plant J. 1999;17(4):329–339. doi: 10.1046/j.1365-313X.1999.00379.x. [DOI] [PubMed] [Google Scholar]
- 61.Chiron H, Drouet A, Claudot AC, Eckerskorn C, Trost M, Heller W, et al. Molecular cloning and functional expression of a stress-induced multifunctional O-methyltransferase with pinosylvin methyltransferase activity from Scots pine (Pinus sylvestris L.) Plant Mol Biol. 2000;44(6):733–745. doi: 10.1023/A:1026507707186. [DOI] [PubMed] [Google Scholar]
- 62.Wein M, Lavid N, Lunkenbein S, Lewinsohn E, Schwab W, Kaldenhoff R. Isolation, cloning and expression of a multifunctional O-methyltransferase capable of forming 2,5-dimethyl-4-methoxy-3(2H)-furanone, one of the key aroma compounds in strawberry fruits. Plant J. 2002;31(6):755–765. doi: 10.1046/j.1365-313X.2002.01396.x. [DOI] [PubMed] [Google Scholar]
- 63.Ibdah M, Zhang X-H, Schmidt J, Vogt T. A Novel Mg2+-dependent O-methyltransferase in the phenylpropanoid metabolism of Mesembryanthemum crystallinum. J Biol Chem. 2003;278(45):43961–43972. doi: 10.1074/jbc.M304932200. [DOI] [PubMed] [Google Scholar]
- 64.Lam KC, Ibrahim RK, Behdad B, Dayanandan S. Structure, function, and evolution of plant O-methyltransferases. Genome. 2007;50(11):1001–1013. doi: 10.1139/G07-077. [DOI] [PubMed] [Google Scholar]
- 65.Ross JR, Nam KH, D’Auria JC, Pichersky E. S-adenosyl-l-methionine: salicylic acid carboxyl methyltransferase, an enzyme involved in floral scent production and plant defense, represents a new class of plant methyltransferases. Arch Biochem Biophys. 1999;367(1):9–16. doi: 10.1006/abbi.1999.1255. [DOI] [PubMed] [Google Scholar]
- 66.Dudareva N, Murfitt LM, Mann CJ, Gorenstein N, Kolosova N, Kish CM, et al. Developmental regulation of methyl benzoate biosynthesis and emission in snapdragon flowers. Plant Cell. 2000;12(6):949–961. doi: 10.1105/tpc.12.6.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zubieta C, Ross JR, Koscheski P, Yang Y, Pichersky E, Noel JP. Structural basis for substrate recognition in the salicylic acid carboxyl methyltransferase family. Plant Cell. 2003;15(8):1704–1716. doi: 10.1105/tpc.014548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bugos RC, Chiang VL, Campbell WH. cDNA cloning, sequence analysis and seasonal expression of lignin-bispecific caffeic acid/5-hydroxyferulic acid O-methyltransferase of aspen. Plant Mol Biol. 1991;17(6):1203–1215. doi: 10.1007/BF00028736. [DOI] [PubMed] [Google Scholar]
- 69.Zhang X-H, Chinnappa CC. Molecular characterization of a cDNA encoding caffeoyl-coenzyme A 3-O-methyltransferase of Stellaria longipes. J Biosci. 1997;22(2):161–175. doi: 10.1007/BF02704730. [DOI] [Google Scholar]
- 70.Schmitt D, Pakusch AE, Matern U. Molecular cloning, induction and taxonomic distribution of caffeoyl-CoA 3-O-methyltransferase, an enzyme involved in disease resistance. J Biol Chem. 1991;266(26):17416–17423. [PubMed] [Google Scholar]
- 71.Wu Q, Preisig CL, VanEtten HD. Isolation of the cDNAs encoding (+)6a-hydroxymaackiain 3-O-methyltransferase, the terminal step for the synthesis of the phytoalexin pisatin in Pisum sativum. Plant Mol Biol. 1997;35(5):551–560. doi: 10.1023/A:1005836508844. [DOI] [PubMed] [Google Scholar]
- 72.Christensen AB, Gregersen PL, Olsen CE, Collinge DB. A flavonoid 7-O-methyltransferase is expressed in barley leaves in response to pathogen attack. Plant Mol Biol. 1998;36(2):219–227. doi: 10.1023/A:1005985609313. [DOI] [PubMed] [Google Scholar]
- 73.Akashi T, Sawada Y, Shimada N, Sakurai N, Aoki T, Ayabe S-i. cDNA cloning and biochemical characterization of S-adenosyl-l-methionine: 2,7,4′-trihydroxyisoflavanone 4′-O-methyltransferase, a critical enzyme of the legume isoflavonoid phytoalexin pathway. Plant Cell Physiol. 2003;44(2):103–112. doi: 10.1093/pcp/pcg034. [DOI] [PubMed] [Google Scholar]
- 74.Davies PJ. Plant hormones and their role in plant growth and development. Dordrecht: Springer Netherlands; 1987. [Google Scholar]
- 75.Vega-Arreguin J, Ibarra-Laclette E, Jimenez-Moraila B, Martinez O, Vielle-Calzada J, Herrera-Estrella L, et al. Deep sampling of the palomero maize transcriptome by a high throughput strategy of pyrosequencing. BMC Genomics. 2009;10(1):299. doi: 10.1186/1471-2164-10-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35(Web Server issue):W182–185. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32(Database issue):D277–280. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52(5):696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 81.Le SQ, Gascuel O. An improved general amino acid replacement matrix. Mol Biol Evol. 2008;25(7):1307–1320. doi: 10.1093/molbev/msn067. [DOI] [PubMed] [Google Scholar]
- 82.Anisimova M, Gascuel O. Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst Biol. 2006;55(4):539–552. doi: 10.1080/10635150600755453. [DOI] [PubMed] [Google Scholar]
- 83.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods (San Diego, Calif) 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 84.Kovacic P, Somanathan R. Novel, unifying mechanism for mescaline in the central nervous system: electrochemistry, catechol redox metabolite, receptor, cell signaling and structure activity relationships. Oxid Med Cell Longev. 2009;2(4):181–190. doi: 10.4161/oxim.2.4.9380. [DOI] [PMC free article] [PubMed] [Google Scholar]