

Abstract
Biglycan (BGN), a small leucine-rich proteoglycan, binds the pro-fibrotic cytokine TGFβ and inhibits its bioactivity in vitro. Nevertheless, it is controversial whether BGN plays an inhibitory role in vivo. Therefore, the purpose of this study was to evaluate the effect of BGN deficiency on TGFβ activity in vivo by studying one-year-old Bgn null and wildtype mice on an Ldlr-null background. Phenotypic and metabolic characterization showed that the Bgn null mice had lower body weight, shorter body length and shorter femur length (all p<0.05). Surprisingly, the Bgn null mice also exhibited a striking reduction in percent body fat compared to wildtype mice (p=0.006), but no changes were observed in plasma triglycerides, total cholesterol, or glycohemoglobin. Both total and bioactive TGFβ1 concentrations in plasma were markedly elevated in Bgn null mice compared to wildtype mice (4-fold and 11-fold increase, respectively, both p<0.001), but no changes were found in hepatic levels of mRNA for Tgfβ1 or its receptors. Bgn null mice exhibited elevated expression of hepatic fibronectin protein (p=0.034) without changes in hepatic or renal histology, and Bgn null mice had decreased urinary albumin/creatinine ratio (p=0.01). Two key downstream targets of bone morphorgenic protein (BMP) 4 signaling, SMAD1/3/5 phosphorylation and Id2 gene expression, were found dramatically reduced in Bgn null livers (p=0.034). Thus, BGN deficiency decreases body fat in this hyperlipidemic mouse model without changing liver or kidney histology. Overall, we propose that this unexpected phenotype arises from effects of BGN deficiency in vivo to elevate TGFβ levels while decreasing BMP4-like signaling.
Keywords: proteoglycans, Ldlr knockout mice, TGFβ1, adiposity, liver histology, kidney histology, BMP4-like signaling
Introduction
BGN is a member of the small leucine-rich proteoglycan (SLRP) family, and consists of a distinct protein core covalently linked to 2 chondroitin/dermatan sulfate chains (1). Although BGN is abundant in the extracellular matrix (ECM) in various tissues, its biological functions in vivo are still not fully understood, despite several functions that have been found in vitro. BGN is thought to organize matrix assembly by interacting with ECM components such as collagens (2, 3). BGN is also implicated in the regulation of cell adhesion (4), migration (5), differentiation (6, 7) and apoptosis (8). BGN was found to be released from the ECM during tissue injury and in its soluble form can bind toll-like receptor (TLR) 2/4 on macrophages, resulting in their activation (9, 10).
Both BGN and decorin (DCN), a closely related class I SLRP, have been shown to bind TGFβ1, and inhibit its activity in vitro (11, 12). The TGFβ family comprises powerful and widely distributed regulators of cell division, differentiation, adhesion, migration and death, participating in ECM production, tissue homeostasis and embryogenesis (13). The TGFβ1 isoform is the most implicated in tissue fibrogenesis (14). Numerous reports have demonstrated that overexpression of TGFβ1 in mice leads to fibrosis in liver and kidney (15, 16). It is not clear, however, whether endogenous BGN serves a physiological role in the regulation of TGFβ in vivo. Endogenous decorin has been shown to inhibit TGFβ activity in vivo by reducing mouse lung fibrosis (17) and protecting against diabetic nephropathy (18) and decorin deficiency has been shown to enhance renal injury, in part due to increased TGFβ levels (18, 19). Yet, in the only head-to-head comparison of the two SLRPs in vivo, adenovirally mediated overexpression of DCN, but not BGN, protected against lung fibrosis that had been induced by concomitant TGFβ overexpression (17). Thus, the role of endogenous BGN in regulating TGFβ activity in vivo remains unclear.
The gene for the BGN core protein resides on the X-chromosome, and Bgn knockout (KO) mice were developed by Xu et al. in 1998 to study musculoskeletal diseases (20). Their phenotyping of these mice revealed osteoarthritis (21) and diminished bone mass, including shortened femur length (20). In addition, BGN has been shown to be involved in cardiovascular functions: male Bgn −/0 mice on the C57BL/6 background exhibit decreased survival after experimental myocardial infarction as a result of frequent left ventricular ruptures (22), and 50% of male BGN-deficient mice on the BALB/c background die within the first 3 months of life from spontaneous aortic dissection and rupture (23). BGN-deficiency has been shown to reduce sensitivity to exogenous TGFβ stimulation in vitro (7, 24).
The aim of this study was to determine if endogenous BGN has a physiological role in the regulation of TGFβ activity in vivo by comparing Bgn null mice and wild-type (WT) mice. We selected the Ldlr−/− background on regular chow as a model of mild-to-moderate hyperlipidemia without atherosclerosis, because hyperlipidemia has previously been shown to increase endogenous TGF-β (25), although not to the extreme extent achieved with Tgfβ transgenes.
Materials and Methods
Animals
All animal care and experimental procedures were approved by and performed in accordance with University of Kentucky, Animal Care Committee guidelines and conformed to Public Health Service policy on the humane care and use of laboratory animals. Bgn KO and littermate control Bgn WT mice, 5 × backcrossed to C57BL6J were then crossed with Ldlr−/− mice. Because Bgn is located on the X chromosome, the experimental mice for this study were generated by crossing Bgn+/− females either with Bgn+/0 littermate males (which gave Bgn +/+ and Bgn +/− females as well as Bgn+/0 and Bgn−/0 males) or with Bgn −/0 littermate males (which gave Bgn +/− and Bgn −/− females as well as Bgn +/0 and Bgn −/0 males) and then discarding heterozygous female offspring. Thus, WT and Bgn KO males were true littermates. Because of the location of the Bgn gene on the X-chromosome, WT and KO females could not be littermates, but we designed the breeding so they came from closely related litters within our colony. In all cases, the breeders were true littermates.
All mice were housed in a temperature-controlled facility with 12 hour light/dark cycles, and fed on normal rodent chow ad libitum with free access to water. Mouse urine was collected by housing mice individually for 24 h in metabolic cages at 46 weeks of age. One week later, body composition was assessed by dual energy X-ray absorptiometry (Lunar PIXImus). At 12 months of age, mice were anethetized by isoflurane inhalation, at which time body weight was measured. Body length was determined in anesthetized mice by measuring the distance from the tip of the snout to the base of the tail. Mice were then perfused through the left ventricle at constant, near-physiological pressure with 10 ml phosphate buffered saline, with exsanguination via vena caval puncture, which ensured euthanasia. Blood and major organs were collected, and the right femur was also harvested and its length measured.
Metabolic characterization
Urinary albumin and creatinine were measured using commercially available kits (Exocell, Inc., Philadelphia, PA, and R&D Systems, Minneapolis, MN, for albumin and creatinine, respectively), and data are expressed as mg albumin per g creatinine. Serum triglyceride and total cholesterol concentrations were determined using enzymatic cholesterol assay kits (Wako Chemical Co., Richmond, VA). Glycated hemoglobin levels were determined using the Glycohemoglobin Reagent Set (Pointe Scientific, Canton, MI). Plasma TGFβ1 was measured with the TGFβ1 Emax® ImmunoAssay system (Promega, Madison, WI).
Histology
Livers and kidneys were removed and divided. One piece of each tissue was fixed in 4% paraformaldehyde for liver and 6:3:1 methanol, chloroform, acetic acid for kidney and then embedded in paraffin, and the other pieces were snap-frozen in liquid nitrogen then stored at −80 °C. Histological analyses were performed on 4 μm thick tissue sections. Both liver and kidney sections were stained with Masson’s trichrome (Accustain kit, Sigma, St. Louis, MO). For measurements of mesangial matrix accumulation, kidney sections were stained with periodic acid Schiff reagent (Sigma). Using the ImageJ software (http://rsbweb.nih.gov/ij/download.html), the mesangial area and total areas were measured on at least 20 glomeruli, sectioned through the tuft, per mouse.
Immunohistochemistry
Immunohistochemical staining was performed as described previously (26). Following deparaffinization, kidney slides were incubated in Proteinase K solution (IHCWORLD, Woodstock, MD) at 37°C for antigen retrieval. Anti-fibronectin antibody (Abcam, Cambridge, MA; ab2413, 1:300) was applied overnight at 4 °C. Negative controls were performed using isotype-matched irrelevant antibodies, no primary antibody, or no secondary antibody.
RNA Extraction and Real-Time Quantitative PCR (qPCR)
Extraction of RNA from frozen liver samples was performed using TRIZOL reagent (Invitrogen, Carlsbad, CA). To remove genomic DNA contamination, RNA was further purified with DNase I (QIAGEN, Valencia, CA) and RNeasy Mini Kit (QIAGEN). 1μg of RNA was reverse transcribed into cDNA using Iscript TM cDNA synthesis kit (Bio-Rad, Hercules, CA). After 10-fold dilution, 2.5 μl cDNA was used as a template for qPCR. Amplification was done for 40 cycles using IQ™ Supermix kit (Bio-Rad, Hercules, CA) and Bio-Rad My iQ™ Single Color Real-Time PCR Detection System. Individual mRNA levels were assessed by way of qPCR, normalized to 18s RNA levels (ΔCt), and then expressed relative to the mean value from samples of male Bgn WT mice (2−ΔΔCt). Primer sequences are shown in Supplementary Table 1.
Immunoblots
Immunoblot analysis was performed as described previously (27). Mouse liver samples were homogenized with RIPA buffer (Teknova, Hollister, CA) containing protease inhibitor cocktail III (RPI, Mount Prospect, IL) and phosphatase inhibitor cocktails II (RPI), and then immunoprobed with antibodies as recommended by the manufacturer (Fibronectin antibody: Millipore, Billerica, MA; AB1954; P-SMAD1/5/8 and total SMAD1 antibodies: Cell Signaling Technology, Danvers, MA; 9511S and 9473S, respectively). For renal decorin content, total protein of mouse kidneys was extracted by Trizol followed by immunoprobing with an anti-decorin antibody (R&D systems, Minneapolis, MN). Blotting for β-actin (Sigma, St. Louis, MO; AF2066) served as a loading control.
Statistical analyses
All data are presented as means ± SEM. Statistical differences were assessed by two-way ANOVA: to assess effects of Bgn genotype and sex, followed by pairwise comparisons by Holm-Sidak method using Sigma Stat software (Jandel Scientifc). P<0.05 was considered statistically significant.
Results
Bgn KO mice were smaller, shorter, and leaner
Bgn KO mice crossed to Ldlr−/− on the C56BL6 background were born without apparent patterning defects. At twelve months of age, the body weight (p=0.02) and body length (p=0.004) of KO mice were significantly lower than that of WT littermates (Table 1). Because BGN is found within the cartilage growth plate of long bones, we also examined femur length and we found that femur length was significantly shorter in Bgn KO mice (p=0.002, Table 1). No differences were found in lean body mass or liver weight adjusted for total body weight between Bgn KO and WT mice (Table 1). Surprisingly, KO mice exhibited significantly lower percent adiposity compared to WT mice (p=0.006, Table 1). Although adiposity was never above 18% in WT animals, the reduction in adiposity in BGN-deficient mice accounted for ~60% of their lower total body weight (Table 1). There was no effect of gender on the reduction in body length, femur length, or body fat percentage in Bgn KO mice, but females had lower body weight than males for both Bgn genotypes.
Table 1.
Physical characteristics of Bgn KO mice compared to Bgn WT mice
| Physical Parameters (n) |
WT: Male (11) |
KO: Male (5) |
WT: Female (12) |
KO: Female (7) |
WT Vs KO |
M Vs F |
|---|---|---|---|---|---|---|
| Body Weight (g) | 36.2 ± 2.7 | 30.2 ± 0.9 | 26.8 ±1.2 | 22.7 ± 0.6 | p=0.022 | p<0.001 |
| Body Length (cm) | 9.8 ± 0.2 | 9.3 ± 0.2 | 9.5 ± 0.2 | 8.7 ± 0.08 | p=0.004 | N. S. |
|
Femur Length
(cm) |
1.58 ± 0.02 | 1.52 ± 0.04 | 1.60 ± 0.02 | 1.46 ± 0.04 | p=0.002 | N. S. |
|
% adiposity (100
x fat/ body weight) |
17.6 ± 2.4 | 10.5 ± 1.6 | 15.0 ± 1.2 | 10.4 ± 1.4 | p=0.006 | N. S. |
|
% lean mass (100
x lean mass/ body weight |
64.5 ± 2.5 | 64.7 ± 1.6 | 65.1 ± 2.7 | 67.5 ± 1.3 | N. S. | N. S. |
|
% Liver
weight/body weight |
4.9 ± 0.3 | 4.8 ± 0.2 | 4.6 ± 0.4 | 4.5 ± 0.2 | N. S. | N. S. |
Data are shown as means ± SEMs for N = 5-12 per group as indicated, measured after 12 months of normal rodent chow feeding. All analyses were done by two-way ANOVA.
Bgn KO mice exhibited unchanged plasma lipid profiles and glucose metabolism
After 12 months on a normal chow diet, Bgn KO mice did not differ in either plasma triglyceride or total cholesterol levels compared to wildtype mice (Suppl. Table 2). In both genotypes female mice had lower triglyceride and total cholesterol levels in plasma than male mice (p=0.002 and p=0.02 respectively, Suppl. Table 2). Neither genotype nor gender affected glycated hemoglobin levels (Suppl. Table 2), implying that BGN deficiency does not alter glucose metabolism in mice, despite increased TGFβ1 levels and decreased adiposity.
Bgn KO mice exhibited elevated TGFβ1 levels in plasma
To determine if BGN deficiency affects TGFβ in vivo, we examined plasma TGFβ1 levels in Bgn KO and WT mice. In two-month-old mice, there was no difference in TGFβ1 levels between genotypes (data not shown). However, total plasma TGFβ1 levels in Bgn KO mice were over 4-fold higher than those in Bgn WT mice by age 12 months (Figure 1A). More strikingly, the biologically active TGFβ1 levels in control mice were approximately 100 pg/ml, but those in Bgn KO mice were increased by 11 fold at 12 months of age (Figure 1B), indicating that BGN-deficiency causes TGFβ1 elevation in older mice in vivo. Hepatic mRNA levels of Tgfβ1 were not significantly increased in Bgn KO mice compared to Bgn WT mice (Figure 1C). Bgn KO mice had a trend towards increased expression of Tgfβ1 receptors (I, II, III), but this did not reach significance.
Figure 1.

Effect of BGN deficiency on TGFβ1 expression in plasma and in liver. Total TGFβ1 levels and endogenous TGFβ1 levels in plasma were determined by ELISA assay (A and B). C-F: Real-time quantitative PCR analysis was performed to determine the mRNA levels of hepatic Tgfβ1 as well as Tgfβ1receptor I, II, and III. Open bars are Bgn WT mice, and closed bars are Bgn KO mice. ***, p<0.001; WT Vs KO, by two-way ANOVA.
Bgn KO mice exhibited increased hepatic fibronectin but no changes in hepatic histology
TGFβ1, a key regulator of ECM assembly and remodeling, induces bio-synthesis of major ECM proteins such as fibronectin (28). Transgenic mice overexpressing mature (active) TGFβ1 at high levels have been shown to develop fibrosis of the liver and kidney with marked interstitial deposition of fibronectin (15). Thus, we examined the effect of BGN deficiency on fibronectin expression and the development of liver fibrosis. Bgn KO mice had a significant increase (2 fold) in hepatic fibronectin protein levels (Figure 2A) but not mRNA levels (Figure 2B) compared to Bgn WT mice. Immunohistochemical analysis showed that hepatic fibronectin in control mice was localized mainly in the peri-vascular regions, whereas Bgn KO mouse livers demonstrated increased fibronectin not only in perivascular areas but also in interstitial areas (Figure 2C). However, no apparent histological lesions (fibrosis or inflammation) were observed in Bgn KO mouse livers compared to control livers on Masson’s Trichrome staining (Figure 2D). Bgn KO mouse livers did not have significant differences in mRNA levels for the cytokines Tnfα, Il-1β or Il-6 compared to WT livers (data not shown).
Figure 2.

Effect of BGN deficiency on hepatic fibronectin expression and liver histology. A: Western blots of fibronectin were analyzed by densitometry (corrected for actin). Shown is mean±SEM from N = 5-12/group. B: real-time PCR analysis was performed to determine the mRNA levels of hepatic fibronectin. C: representative light micrographs of fibronectin immunohistochemistry staining in liver sections from 4 groups of mice. Scale bar: 20 μm. Original magnification X1000. D: Hepatic histology was evaluated with Masson’s Trichrome stain. Shown are representative sections (from N=5-12/ group) from WT or KO mice. Scale bar: 200 μm. Original magnification: X200.
Bgn KO mice exhibited decreased albuminuria but no changes in renal histology
TGFβ1 has been shown to play a critical role in contributing to the development and progression of renal injury (29, 30). However, despite the elevated plasma TGFβ1 levels, Bgn KO mice demonstrated a 50% decrease in urinary albumin excretion compared to Bgn WT mice (Figure 3A), suggesting improved kidney barrier function. Furthermore, no pathological changes such as fibrosis or inflammation were found in Bgn KO mouse kidneys (Figure 3B). Mesangial matrix area did not differ between Bgn KO mice compared to WT mice (Figure 3C and D). We found no difference in renal or aortic decorin content between Bgn KO mice and Bgn WT mice (data not shown).
Figure 3.

Effect of BGN deficiency on kidney function and structure in mice. A: Urinary albumin excretion is expressed as mg albumin per g creatinine, and was measured in 24 h urine samples obtained from individual mice. Shown is mean±SEM from N = 5-12/group. *p<0.05; WT Vs KO. B: representative light micrographs of Masson’s Trichrome stain from kidney sections. Scale bar: 20 μm. Original magnification: X200. C: Mesangial matrix was evaluated on renal sections stained with periodic acid Schiff (PAS). Shown are representative sections from 4 groups. Scale bar: 10 μm. Original magnification: X400. C: Mesangial matrix accumulation was quantified using ImageJ software.
Bgn KO livers exhibited substantially reduced activity in BMP4-like signaling
To understand the mechanisms underlying this surprising phenotype of Bgn KO mice, we measured BMP4-like signal transduction in KO vs. WT mice. BGN was previously reported to enhance BMP4 signaling in murine C2C12 cells (6) and osteoblasts (7), but the effect has not been tested in mammals in vivo. We examined key downstream targets of BMP4 – SMAD1/5/8 phosphorylation and inhibitor of differentiation/DNA binding (Id) genes (31, 32). Compared to WT mice, Bgn KO mice exhibited striking reduction in phosphorylated levels of SMAD1/5/8, whereas total SMAD1 protein did not differ between genotypes (Figure 4A). Moreover, Id2 mRNA levels were significantly decreased (p=0.013) in KO livers compared to WT livers (Figure 4B), further suggesting reduced BMP4 signaling in Bgn KO mice. Female mice had higher hepatic Id1 mRNA expression than male mice (p=0.034) but no genotype effect was observed (Figure 4C).
Figure 4.

Effect of BGN deficiency on BMP4-like signaling in liver. A. Total liver protein was analyzed by Western blot for P-SMAD1/5/8 and total SMAD1 content. Actin was used as the loading control. B and C: Real-time PCR analysis was performed to determine the mRNA levels of hepatic Id2 (B) and Id1 (C). Shown is mean±SEM from N = 5-12/group. *p<0.05; WT vs KO.
Discussion
The purpose of the present study was to determine whether endogenous BGN has a physiologic role in regulating TGFβ1 levels and biological-activity in vivo. Consistent with the findings by Xu et al. (20), one-year old hyperlipidemic Bgn KO mice (Ldlr−/− background) exhibited smaller size, lower body weight and shorter body and femur length. Surprisingly, we found that the decreased body weight is mostly the result of a decrease in body fat compared to Bgn WT mice.
Bgn KO mice had elevated total and bioactive TGFβ1 levels in plasma compared to WT mice. However, hepatic Tgfβ1 mRNA expression did not differ between genotypes, suggesting that the elevated TGFβ1 levels may be due to either lack of sequestration of TGFβ in the ECM rather than increased synthesis, or else positive feedback, given that TGFβ itself is positively regulated by active TGFβ at the protein level (18). Although Bgn KO mice had a modest increase in hepatic fibronectin expression and content, they did not have overt changes in liver histology. Despite increased TGFβ1 levels in plasma, Bgn KO mice had no changes in renal histology, and surprisingly had lower urinary albumin excretion ratio compared with Bgn WT mice. Despite decreased body weight, lower body fat, and increased bioactive TGFβ1, Bgn KO mice had no differences in plasma lipid profile or glycohemoglobin compared with WT mice. Thus, we now demonstrate that in one-year-old hyperlipidemic mice, BGN deficiency does not alter lipid metabolism, but decreases adiposity and increases plasma TGFβ1 levels without causing liver or kidney fibrosis.
BGN and DCN are the closest members of the Class I SLRPs, based on their high degree of homology (55% amino acid identity) (33). Studies in vitro have shown that both the BGN and DCN core proteins can competitively bind TGFβ1 (11), resulting in the inhibition of TGFβ1activity and downstream signaling (12, 34). DCN has been successfully employed in rodents to decrease fibrosis in kidney (35), lung (36) and vasculature (37). Kolb et al. reported that DCN, but not BGN, significantly reduced lung fibrosis induced by TGFβ1overexpression in mice (17). Our findings that BGN deficiency induced higher plasma levels of TGFβ1 in mice support the hypothesis that a physiological role of BGN in vivo is to sequester TGFβ1. Similar to the observations in skeletal tissue by Xu et al. (20), our finding of lack of any compensatory increase in renal or aortic decorin content in the Bgn KO vs WT mice suggests that these SLRPs play distinct physiological roles in the regulation of TGFβ activity in vivo.
Although the Bgn KO mice had elevated plasma TGFβ1concentrations, there was no evidence of histological changes in liver or kidney. Chen et al. demonstrated that BGN deficiency reduced the sensitivity of bone marrow stromal cells to TGFβ (7, 24). Thus, the absence of liver or kidney fibrosis and the suggestion of improved kidney function in Bgn KO mice might be the result of reduced sensitivity of tissues to the chronic elevations in TGFβ1. In our study, increased expression and altered distribution of hepatic fibronectin were observed in Bgn KO mice, implying that TGFβ1 activity was elevated in vivo, but this modest elevation was not adequate to cause pathological changes in tissues. Indeed, Ueberham et al. reported that plasma TGFβ1 levels in TGFβ1 transgenic mice ranged from 250 to 1, 200 ng/ml, which was 10-30 times the control levels(16). However, total TGFβ1 levels the plasma of our Bgn KO mice were around 2.5 ng/ml, which is only 3 times of the levels in our control mice. Thus, the TGFß1 transgenic mice in Ueberham’s study had 100-500 times more plasma TGFβ1 than the KO mice in our study, which may explain, at least in part, the more prominent fibrotic phenotype observed in their transgenic mice. A recent study demonstrated that BGN deficiency promoted a pro-proliferative and myofibroblast phenotype through increased TGFβ1/SMAD2 signaling both in primary cardiac fibroblasts and in mice after experimental myocardial infarction (38), which supports the hypothesis that BGN deficiency increases TGFβ1 activity in vivo. The present study, however, reports on one-year-old old hyperlipidemic (Ldlr−/−) Bgn KO mice on normal chow without other manipulations. In the setting of stresses, such as models of infarction, atherosclerosis or diabetes, the consequences of elevated TGFβ may be different.
A novel finding in our study is that BGN deficiency led to a striking reduction in adiposity in vivo. It is known that TGFβ1 serves as a potent negative factor in adipogenesis (39), and transgenic overexpression of active TGFβ1 caused lipodystrophy in mice (15). Thus, reduced adiposity in our Bgn KO mice may result from elevation in TGFβ1 levels. In addition, normal adipocytes have been shown to express BGN in vitro (40-43) and in vivo (44, 45), and the expression increases with high-fat diet feeding (44) or acute inflammation (45). However, BGN was found to reduce the proliferation of pre-adipocytes in vitro (8), suggesting a negative role of BGN itself in adipogenesis. Therefore, our findings seem to support that BGN deficiency caused reduced adiposity more likely through TGFβ1 elevation.
Another striking observation is that Bgn KO mice had less urinary albumin excretion compared to Bgn WT mice. This was surprising given prior literature demonstrating a pathological role for TGFβ in renal diseases. Nevertheless, our findings are consistent with previous observations in kidney disease models where BGN deficiency was shown to attenuate activation of the inflammasome and thus tubular injury in mice following unilateral ureteral obstruction (10). In a murine lupus nephritis model, BGN deficiency was also found to improve renal outcome by reducing albuminuria, pro-inflammatory cytokine expression and histological lesions, possibly through regulating the B-cell chemoattractant CXCL13 (46). Furthermore, we recently demonstrated that renal BGN showed a striking co-localization with apoB in a model of diabetic nephropathy, suggesting that one function of renal BGN may be to increase renal lipid retention, which has detrimental effects (47-50). Thus, the absence of BGN in this hyperlipidemic model could protect against renal lipid accumulation, thereby contributing to the healthy renal phenotype observed.
Previous work in cultured murine myoblasts and osteoblasts indicated a role for BGN in facilitating BMP4 signal transduction in vitro (6, 7). BMP4 belongs to the TGFβ superfamily but exerts distinct functions. BMP4 activity was shown to be associated with liver fibrosis induced by bile duct ligation (51). Tamoxifen-inducible BMP4 transgenic mice developed glomerulosclerosis with severe albuminuria (52), also suggesting that BMP4 can promote tissue fibrosis. However, in contrast to TGFβ1 which inhibits adipogenesis, BMP4 was found to be required for commitment of C3H10T1/2 pluripotent stem cells into adipocytes (53), indicating a positive role of BMP4 in adipogenesis. Our results support that BGN deficiency leads to decreased BMP4 activity in vivo, as demonstrated by reduction in phosphorylation of hepatic SMAD1/5/8 and in mRNA expression of the target gene Id2. Therefore, our results indicate that BGN deficiency causes not only an elevation in TGFβ1 levels but also a reduction in BMP4-like signaling in vivo. Decreased BMP4-like activity may antagonize the role of TGFβ1 in promoting tissue fibrosis, accounting for the lack of pathological changes in liver and kidney and improved albuminuria that we found in Bgn KO mice. However, both increased TGFβ1 and decreased BMP4-like signaling would inhibit adipogenesis, which could account for reduction in adiposity we observed in Bgn KO mice (Table 1; see the pathway schematic in Figure 5).
Figure 5.
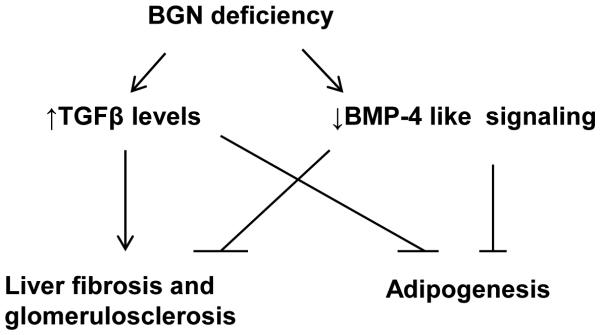
Proposed model of mechanisms involving TGFβ1 and BMP4 signaling to explain the physiological changes in Bgn null mice. In brief, BGN deficiency causes elevated TGFβ1 levels but decreased BMP4 sensitivity in this hyperlipidemic mouse model. Both TGFβ1 and BMP4 are pro-fibrotic. The lack of pathological changes such as liver fibrosis or glomerulosclerosis in Bgn null mice could be due to opposing actions from increased TGFβ1 versus decreased BMP4 signaling. Instead, reduced BMP4 activity may improve albuminuria. However, TGFβ1 and BMP4 exert opposite functions in adipogenesis, so both TGFβ1 elevation and BMP4 deactivation play negative roles in adipogenesis, potentially contributing to the striking reduction in adiposity.
One surprising phenomenon in our study is that Id1, another target gene of BMP4 signaling, showed no change between genotypes, despite a striking reduction in SMAD1/5/8 phorsphorylation as well as Id2. Indeed, the mRNA level of Id1 was found to be 8 times lower than that of Id2 according to our realtime PCR results (data not shown). In addition, liver is one of the tissues where ID1 is least expressed in mice (54). Thus, it is possible that Id1 gene is less sensitive to upstream BMP4 signaling than Id2 due to the lower abundance in gene expression. Noticeably, Id2 null mice exhibited a dramatic decrease in white adipose tissue density (55). However, the role of Id1 gene in adipogenesis is controversial, i.e., Id1 null mice demonstrated reduction in body weight and fat mass in Satyanarayana’s report (54), but this phenotype was not observed in Akerfeldt’s study (56). Therefore, our results support that BGN deficiency led to reduced adiposity through Id2 instead of Id1.
There are some limitations to this study. First, following prior studies of obesity, atherosclerosis, and other metabolic conditions (57-59), the Bgn KO mice were backcrossed 5X to C57BL6, but the small residual percentage of mixed genetic background could influence our results. As Bgn is X-linked, the male mice were true littermates; whereas WT and KO females could not be produced from single crosses (we designed the breeding so that they came from closely related litters within our single colony). We found similar effects of BGN deficiency in both male and female mice, which we analyzed separately. Because the WT and KO males were true littermates, we believe this validates our results in females. We selected the hyperlipidemic Ldlr−/− model on normal chow as a model of increased TGFβ. Further research will be required to determine the effect of BGN deficiency in other pathophysilogically realistic settings of elevated TGFβ, such as diabetes.
In summary, Bgn KO mice exhibited decreased adiposity and elevated plasma TGFβ1 levels, but no evidence of fibrogenesis in liver or kidney. Indeed, Bgn KO mice showed reduced urinary albumin output, implying improved kidney function, which agrees with the findings in several mouse models of renal damage. This paradox may be explained by decreased sensitivity to BMP4 protein in Bgn KO mice. Further studies are needed to clarify the roles of BGN in regulating adiposity and TGFβ sensitivity as well as downstream signaling.
Supplementary Material
Acknowledgments
This study was supported by grants from the National Institutes of Health, USA (RO1 HL82772 to LRT and DK043396 to KJW). TT, JCT, PGW and CN performed research; TT and LRT analyzed the data; TT, KJW, and LRT designed the research study and wrote the paper.
Footnotes
Conflict of Interest Statement
The authors confirm that there are no conflicts of interest.
References
- 1.Young MF, Bi Y, Ameye L, Chen XD. Biglycan knockout mice: new models for musculoskeletal diseases. Glycoconj J. 2002;19(4-5):257–62. doi: 10.1023/A:1025336114352. [DOI] [PubMed] [Google Scholar]
- 2.Schonherr E, Witsch-Prehm P, Harrach B, Robenek H, Rauterberg J, Kresse H. Interaction of biglycan with type I collagen. J Biol Chem. 1995;270(6):2776–83. doi: 10.1074/jbc.270.6.2776. [DOI] [PubMed] [Google Scholar]
- 3.Seidler DG, Faiyaz-Ul-Haque M, Hansen U, Yip GW, Zaidi SH, Teebi AS, et al. Defective glycosylation of decorin and biglycan, altered collagen structure, and abnormal phenotype of the skin fibroblasts of an Ehlers-Danlos syndrome patient carrying the novel Arg270Cys substitution in galactosyltransferase I (beta4GalT-7) J Mol Med. 2006;84(7):583–94. doi: 10.1007/s00109-006-0046-4. [DOI] [PubMed] [Google Scholar]
- 4.Nelimarkka L, Kainulainen V, Schonherr E, Moisander S, Jortikka M, Lammi M, et al. Expression of small extracellular chondroitin/dermatan sulfate proteoglycans is differentially regulated in human endothelial cells. J Biol Chem. 1997;272(19):12730–7. doi: 10.1074/jbc.272.19.12730. [DOI] [PubMed] [Google Scholar]
- 5.Kinsella MG, Tsoi CK, Jarvelainen HT, Wight TN. Selective expression and processing of biglycan during migration of bovine aortic endothelial cells. The role of endogenous basic fibroblast growth factor. J Biol Chem. 1997;272(1):318–25. doi: 10.1074/jbc.272.1.318. Epub 1997/01/03. PubMed PMID: 8995264. [DOI] [PubMed] [Google Scholar]
- 6.Moreno M, Munoz R, Aroca F, Labarca M, Brandan E, Larrain J. Biglycan is a new extracellular component of the Chordin-BMP4 signaling pathway. EMBO J. 2005;24(7):1397–405. doi: 10.1038/sj.emboj.7600615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen XD, Fisher LW, Robey PG, Young MF. The small leucine-rich proteoglycan biglycan modulates BMP-4-induced osteoblast differentiation. FASEB J. 2004;18(9):948–58. doi: 10.1096/fj.03-0899com. [DOI] [PubMed] [Google Scholar]
- 8.Ward M, Ajuwon KM. Regulation of pre-adipocyte proliferation and apoptosis by the small leucine-rich proteoglycans, biglycan and decorin. Cell Prolif. 2011;44(4):343–51. doi: 10.1111/j.1365-2184.2011.00763.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schaefer L, Babelova A, Kiss E, Hausser HJ, Baliova M, Krzyzankova M, et al. The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J Clin Invest. 2005;115(8):2223–33. doi: 10.1172/JCI23755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Babelova A, Moreth K, Tsalastra-Greul W, Zeng-Brouwers J, Eickelberg O, Young MF, et al. Biglycan, a danger signal that activates the NLRP3 inflammasome via toll-like and P2X receptors. J Biol Chem. 2009;284(36):24035–48. doi: 10.1074/jbc.M109.014266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hildebrand A, Romaris M, Rasmussen LM, Heinegard D, Twardzik DR, Border WA, et al. Interaction of the small interstitial proteoglycans biglycan, decorin and fibromodulin with transforming growth factor beta. Biochem J. 1994;302:527–34. doi: 10.1042/bj3020527. Pt 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Droguett R, Cabello-Verrugio C, Riquelme C, Brandan E. Extracellular proteoglycans modify TGF-beta bio-availability attenuating its signaling during skeletal muscle differentiation. Matrix Biol. 2006;25(6):332–41. doi: 10.1016/j.matbio.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 13.Ruiz-Ortega M, Rodriguez-Vita J, Sanchez-Lopez E, Carvajal G, Egido J. TGF-beta signaling in vascular fibrosis. Cardiovasc Res. 2007;74(2):196–206. doi: 10.1016/j.cardiores.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 14.Border WA, Noble NA. Transforming growth factor beta in tissue fibrosis. N Engl J Med. 1994;331(19):1286–92. doi: 10.1056/NEJM199411103311907. [DOI] [PubMed] [Google Scholar]
- 15.Clouthier DE, Comerford SA, Hammer RE. Hepatic fibrosis, glomerulosclerosis, and a lipodystrophy-like syndrome in PEPCK-TGF-beta1 transgenic mice. J Clin Invest. 1997;100(11):2697–713. doi: 10.1172/JCI119815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ueberham E, Low R, Ueberham U, Schonig K, Bujard H, Gebhardt R. Conditional tetracycline-regulated expression of TGF-beta1 in liver of transgenic mice leads to reversible intermediary fibrosis. Hepatology. doi: 10.1053/jhep.2003.50196. [DOI] [PubMed] [Google Scholar]
- 17.Kolb M, Margetts PJ, Sime PJ, Gauldie J. Proteoglycans decorin and biglycan differentially modulate TGF-beta-mediated fibrotic responses in the lung. Am J Physiol Lung Cell Mol Physiol. 2001;280(6):L1327–34. doi: 10.1152/ajplung.2001.280.6.L1327. [DOI] [PubMed] [Google Scholar]
- 18.Williams KJ, Qiu G, Usui HK, Dunn SR, McCue P, Bottinger E, et al. Decorin deficiency enhances progressive nephropathy in diabetic mice. Am J Pathol. 2007;171(5):1441–50. doi: 10.2353/ajpath.2007.070079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Merline R, Lazaroski S, Babelova A, Tsalastra-Greul W, Pfeilschifter J, Schluter KD, et al. Decorin deficiency in diabetic mice: aggravation of nephropathy due to overexpression of profibrotic factors, enhanced apoptosis and mononuclear cell infiltration. J Physiol Pharmacol. 2009;60(Suppl 4):5–13. [PMC free article] [PubMed] [Google Scholar]
- 20.Xu T, Bianco P, Fisher LW, Longenecker G, Smith E, Goldstein S, et al. Targeted disruption of the biglycan gene leads to an osteoporosis-like phenotype in mice. Nat Genet. 1998;20(1):78–82. doi: 10.1038/1746. [DOI] [PubMed] [Google Scholar]
- 21.Ameye L, Aria D, Jepsen K, Oldberg A, Xu T, Young MF. Abnormal collagen fibrils in tendons of biglycan/fibromodulin-deficient mice lead to gait impairment, ectopic ossification, and osteoarthritis. FASEB J. 2002;16(7):673–80. doi: 10.1096/fj.01-0848com. [DOI] [PubMed] [Google Scholar]
- 22.Westermann D, Mersmann J, Melchior A, Freudenberger T, Petrik C, Schaefer L, et al. Biglycan is required for adaptive remodeling after myocardial infarction. Circulation. 2008;117(10):1269–76. doi: 10.1161/CIRCULATIONAHA.107.714147. [DOI] [PubMed] [Google Scholar]
- 23.Heegaard AM, Corsi A, Danielsen CC, Nielsen KL, Jorgensen HL, Riminucci M, et al. Biglycan deficiency causes spontaneous aortic dissection and rupture in mice. Circulation. 2007;115(21):2731–8. doi: 10.1161/CIRCULATIONAHA.106.653980. [DOI] [PubMed] [Google Scholar]
- 24.Chen XD, Shi S, Xu T, Robey PG, Young MF. Age-related osteoporosis in biglycan-deficient mice is related to defects in bone marrow stromal cells. J Bone Miner Res. 2002;17(2):331–40. doi: 10.1359/jbmr.2002.17.2.331. [DOI] [PubMed] [Google Scholar]
- 25.Zhou X, Johnston TP, Johansson D, Parini P, Funa K, Svensson J, et al. Hypercholesterolemia leads to elevated TGF-beta1 activity and T helper 3-dependent autoimmune responses in atherosclerotic mice. Atherosclerosis. 2009;204(2):381–7. doi: 10.1016/j.atherosclerosis.2008.10.017. [DOI] [PubMed] [Google Scholar]
- 26.Yen TH, Chen Y, Fu JF, Weng CH, Tian YC, Hung CC, et al. Proliferation of myofibroblasts in the stroma of renal oncocytoma. Cell Prolif. 2010;43(3):287–96. doi: 10.1111/j.1365-2184.2010.00681.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang F, Thompson JC, Wilson PG, Aung HH, Rutledge JC, Tannock LR. Angiotensin II increases vascular proteoglycan content preceding and contributing to atherosclerosis development. J Lipid Res. 2008;49(3):521–30. doi: 10.1194/jlr.M700329-JLR200. [DOI] [PubMed] [Google Scholar]
- 28.Ignotz RA, Massague J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J Biol Chem. 1986;261(9):4337–45. [PubMed] [Google Scholar]
- 29.Cheng J, Grande JP. Transforming growth factor-beta signal transduction and progressive renal disease. Exp Biol Med (Maywood) 2002;227(11):943–56. doi: 10.1177/153537020222701102. [DOI] [PubMed] [Google Scholar]
- 30.Ziyadeh FN, Hoffman BB, Han DC, Iglesias-De La Cruz MC, Hong SW, Isono M, et al. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proc Natl Acad Sci U S A. 2000;97(14):8015–20. doi: 10.1073/pnas.120055097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hollnagel A, Oehlmann V, Heymer J, Ruther U, Nordheim A. Id genes are direct targets of bone morphogenetic protein induction in embryonic stem cells. J Biol Chem. 1999;274(28):19838–45. doi: 10.1074/jbc.274.28.19838. [DOI] [PubMed] [Google Scholar]
- 32.Attisano L, Wrana JL. Signal transduction by the TGF-beta superfamily. Science. 2002;296(5573):1646–7. doi: 10.1126/science.1071809. [DOI] [PubMed] [Google Scholar]
- 33.Fisher LW, Termine JD, Young MF. Deduced protein sequence of bone small proteoglycan I (biglycan) shows homology with proteoglycan II (decorin) and several nonconnective tissue proteins in a variety of species. J Biol Chem. 1989;264(8):4571–6. [PubMed] [Google Scholar]
- 34.Yamaguchi Y, Mann DM, Ruoslahti E. Negative regulation of transforming growth factor-beta by the proteoglycan decorin. Nature. 1990;346(6281):281–4. doi: 10.1038/346281a0. [DOI] [PubMed] [Google Scholar]
- 35.Border WA, Noble NA, Yamamoto T, Harper JR, Yamaguchi Y, Pierschbacher MD, et al. Natural inhibitor of transforming growth factor-beta protects against scarring in experimental kidney disease. Nature. 1992;360(6402):361–4. doi: 10.1038/360361a0. [DOI] [PubMed] [Google Scholar]
- 36.Giri SN, Hyde DM, Braun RK, Gaarde W, Harper JR, Pierschbacher MD. Antifibrotic effect of decorin in a bleomycin hamster model of lung fibrosis. Biochem Pharmacol. 1997;54(11):1205–16. doi: 10.1016/s0006-2952(97)00343-2. [DOI] [PubMed] [Google Scholar]
- 37.Fischer JW, Kinsella MG, Clowes MM, Lara S, Clowes AW, Wight TN. Local expression of bovine decorin by cell-mediated gene transfer reduces neointimal formation after balloon injury in rats. Circ Res. 2000;86(6):676–83. doi: 10.1161/01.res.86.6.676. [DOI] [PubMed] [Google Scholar]
- 38.Melchior-Becker A, Dai G, Ding Z, Schafer L, Schrader J, Young MF, et al. Deficiency of biglycan causes cardiac fibroblasts to differentiate into a myofibroblast phenotype. J Biol Chem. 2011;286(19):17365–75. doi: 10.1074/jbc.M110.192682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choy L, Derynck R. Transforming growth factor-beta inhibits adipocyte differentiation by Smad3 interacting with CCAAT/enhancer-binding protein (C/EBP) and repressing C/EBP transactivation function. J Biol Chem. 2003;278(11):9609–19. doi: 10.1074/jbc.M212259200. [DOI] [PubMed] [Google Scholar]
- 40.Calvo JC, Rodbard D, Katki A, Chernick S, Yanagishita M. Differentiation of 3T3-L1 preadipocytes with 3-isobutyl-1-methylxanthine and dexamethasone stimulates cell-associated and soluble chondroitin 4-sulfate proteoglycans. J Biol Chem. 1991;266(17):11237–44. [PubMed] [Google Scholar]
- 41.Bachem MG, Meyer D, Schafer W, Riess U, Melchior R, Sell KM, et al. The response of rat liver perisinusoidal lipocytes to polypeptide growth regulator changes with their transdifferentiation into myofibroblast-like cells in culture. J Hepatol. 1993;18(1):40–52. doi: 10.1016/s0168-8278(05)80008-x. [DOI] [PubMed] [Google Scholar]
- 42.Knippenberg M, Helder MN, Zandieh Doulabi B, Wuisman PI, Klein-Nulend J. Osteogenesis versus chondrogenesis by BMP-2 and BMP-7 in adipose stem cells. Biochem Biophys Res Commun. 2006;342(3):902–8. doi: 10.1016/j.bbrc.2006.02.052. [DOI] [PubMed] [Google Scholar]
- 43.Kim BS, Kang KS, Kang SK. Soluble factors from ASCs effectively direct control of chondrogenic fate. Cell Prolif. 2010;43(3):249–61. doi: 10.1111/j.1365-2184.2010.00680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huber J, Loffler M, Bilban M, Reimers M, Kadl A, Todoric J, et al. Prevention of high-fat diet-induced adipose tissue remodeling in obese diabetic mice by n-3 polyunsaturated fatty acids. Int J Obes (Lond) 2007;31(6):1004–13. doi: 10.1038/sj.ijo.0803511. [DOI] [PubMed] [Google Scholar]
- 45.Adapala VJ, Adedokun SA, Considine RV, Ajuwon KM. Acute inflammation plays a limited role in the regulation of adipose tissue COL1A1 protein abundance. J Nutr Biochem. 2012;23(6):567–72. doi: 10.1016/j.jnutbio.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 46.Moreth K, Brodbeck R, Babelova A, Gretz N, Spieker T, Zeng-Brouwers J, et al. The proteoglycan biglycan regulates expression of the B cell chemoattractant CXCL13 and aggravates murine lupus nephritis. J Clin Invest. 2010;120(12):4251–72. doi: 10.1172/JCI42213. 10.1172/JCI42213. PubMed PMID: 21084753; PubMed Central PMCID: PMC2993585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thompson J, Wilson P, Brandewie K, Taneja D, Schaefer L, Mitchell B, et al. Renal accumulation of biglycan and lipid retention accelerates diabetic nephropathy. Am J Pathol. 2011;179(3):1179–87. doi: 10.1016/j.ajpath.2011.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kamanna VS. Low density lipoproteins and mitogenic signal transduction processes: role in the pathogenesis of renal disease. Histol Histopathol. 2002;17(2):497–505. doi: 10.14670/HH-17.497. [DOI] [PubMed] [Google Scholar]
- 49.Kamanna VS, Roh DD, Kirschenbaum MA. Hyperlipidemia and kidney disease: concepts derived from histopathology and cell biology of the glomerulus. Histol Histopathol. 1998;13(1):169–79. doi: 10.14670/HH-13.169. [DOI] [PubMed] [Google Scholar]
- 50.Abrass CK. Cellular lipid metabolism and the role of lipids in progressive renal disease. Am J Nephrol. 2004;24(1):46–53. doi: 10.1159/000075925. [DOI] [PubMed] [Google Scholar]
- 51.Fan J, Shen H, Sun Y, Li P, Burczynski F, Namaka M, et al. Bone morphogenetic protein 4 mediates bile duct ligation induced liver fibrosis through activation of Smad1 and ERK1/2 in rat hepatic stellate cells. Journal of cellular physiology. 2006;207(2):499–505. doi: 10.1002/jcp.20593. [DOI] [PubMed] [Google Scholar]
- 52.Tominaga T, Abe H, Ueda O, Goto C, Nakahara K, Murakami T, et al. Activation of bone morphogenetic protein 4 signaling leads to glomerulosclerosis that mimics diabetic nephropathy. J Biol Chem. 2011;286(22):20109–16. doi: 10.1074/jbc.M110.179382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang H, Song TJ, Li X, Hu L, He Q, Liu M, et al. BMP signaling pathway is required for commitment of C3H10T1/2 pluripotent stem cells to the adipocyte lineage. Proc Natl Acad Sci U S A. 2009;106(31):12670–5. doi: 10.1073/pnas.0906266106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Satyanarayana A, Klarmann KD, Gavrilova O, Keller JR. Ablation of the transcriptional regulator Id1 enhances energy expenditure, increases insulin sensitivity, and protects against age and diet induced insulin resistance, and hepatosteatosis. FASEB J. 2012;26(1):309–23. doi: 10.1096/fj.11-190892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hou TY, Ward SM, Murad JM, Watson NP, Israel MA, Duffield GE. ID2 (inhibitor of DNA binding 2) is a rhythmically expressed transcriptional repressor required for circadian clock output in mouse liver. J Biol Chem. 2009;284(46):31735–45. doi: 10.1074/jbc.M109.013961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Akerfeldt MC, Laybutt DR. Inhibition of Id1 augments insulin secretion and protects against high-fat diet-induced glucose intolerance. Diabetes. 2011;60(10):2506–14. doi: 10.2337/db11-0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nawrocki AR, Hofmann SM, Teupser D, Basford JE, Durand JL, Jelicks LA, et al. Lack of association between adiponectin levels and atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2010;30(6):1159–65. doi: 10.1161/ATVBAHA.109.195826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, Doherty TM, et al. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci U S A. 2004;101(29):10679–84. doi: 10.1073/pnas.0403249101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Methia N, Andre P, Denis CV, Economopoulos M, Wagner DD. Localized reduction of atherosclerosis in von Willebrand factor-deficient mice. Blood. 2001;98(5):1424–8. doi: 10.1182/blood.v98.5.1424. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.