

Abstract
Patients with systemic mastocytosis (SM) have a wide variety of problems, including skeletal abnormalities. The disease results from a mutation of the stem cell receptor (c-kit) in mast cells and we wondered if the function of bone marrow stromal cells (BMSCs; also known as MSCs or mesenchymal stem cells) might be affected by the invasion of bone marrow by mutant mast cells. As expected, BMSCs from SM patients do not have a mutation in c-kit, but they proliferate poorly. In addition, while osteogenic differentiation of the BMSCs seems to be deficient, their adipogenic potential appears to be increased. Since the hematopoietic supportive abilities of BMSCs are also important, we also studied the engraftment in NSG mice of human CD34+ hematopoietic progenitors, after being co-cultured with BMSCs of healthy volunteers vs. BMSCs derived from patients with SM. BMSCs derived from the bone marrow of patients with SM could not support hematopoiesis to the extent that healthy BMSCs do. Finally, we performed an expression analysis and found significant differences between healthy and SM derived BMSCs in the expression of genes with a variety of functions, including the WNT signaling, ossification, and bone remodeling. We suggest that some of the symptoms associated with SM might be driven by epigenetic changes in BMSCs caused by dysfunctional mast cells in the bone marrow of the patients.
Introduction
Mast cells express c-kit, (CD117), which is the stem cell factor (SCF) receptor. When this receptor is constitutively activated due to genetic mutation(s), an increase in mast cell (MC) proliferation occurs and leads to the disease systemic mastocytosis (SM) [1]. More than 90% of patients with SM carry the somatic D816V activating mutation in the KIT gene [2]. The over proliferation of MCs in the bone marrow (BM) results in an alteration in the expression of additional genes [3] and mediators including histamine within the bone marrow and thus a change in the immediate environment of Bone Marrow Stromal Cells (BMSCs, also known as bone marrow-derived mesenchymal stem cells or MSCs) that are responsible for generating cartilage, bone and marrow adipocytes in BM during bone turnover and repair, as well as supporting hematopoiesis.
A number of pathologic features of SM suggest a possible link between specific disease findings and a role for BMSCs. These include the well-documented skeletal disease in patients with SM [4, 5], and abnormalities in the cellular composition of the BM including anemia [6]. As we have previously established that the human BMSCs bear all four histamine receptors [7], we wanted to examine if the increase in MCs might be associated with abnormalities of the BMSC compartment which might in turn impact disease pathology.
Results
BMSCs from patients with SM show abnormal morphology and growth characteristics
BMSCs from healthy volunteers (Fig. 1 A, B) and patients with SM (Fig. 1 C, D) were plated as published earlier [7, 8] and the cultures were examined 1 week later. BMSCs from the patients were growing very slowly, thus difficult to culture and exhibited signs of senescence (large, flat morphology) in early cultures. These cells showed β-gal staining indicating senescence, while normally growing cells did not (Fig. 1 E, F). We tested the proliferative ability of the patient’s BMSCs by using BrdU and adding serum or FGF2 to the medium. In all conditions the proliferation of the patients’ BMSCs was significantly (p<0.01 to p<0.001) less than those from healthy donors (Fig. 1 G).
Fig. 1. BMSC morphology and proliferation differs between healthy controls and patients with SM.

A. (low magnification) and B. (high magnification) show a 7 day culture of BMSCs from normal volunteers compared with C. (low magnification) and D. (high magnification) that are samples of SM patients. All images were photographed under the same conditions. E. normal donor and F. patient sample demonstrate β-gal staining, a marker of senescence, strongly labeling cells in the patient’s culture. G. The rate of proliferation was assessed by BrdU incorporation, also suggesting that in all conditions, the BMSCs from the patients seem to be disadvantaged. The samples were pooled from 3 patients and three volunteers to get enough cells to perform the assays. The experiments were performed in triplicate samples. Abbreviations: FGF2 = fibroblast growth factor 2; Scale: 100 μm (A–D), 50 μm (E, F). Statistical significance is labeled as ** = p<0.01 and *** = p<0.001
Next we compared primary colony forming efficiency (CFE) of BMSCs from healthy and diseased BM. We found that from primary bone marrow aspirates, the CFE from the patients with SM was significantly below (p<0.01) that of the healthy volunteers (1.5±1.8 vs. 10.1±1.7 in 100,000 plated cells respectively) (Fig. 2 A). While the CFE was very different between the donor and the patient groups, we found that the percentage of CD34+ hematopoietic progenitors were similar (Fig. 2 B). We then compared the number of population doublings over time between healthy BMSCs (n=7) vs. BMSCs from patients with SM (n=10). As can be seen, BMSCs from patients with SM exhibited fewer doublings over time compared with BMSCs from the healthy counterparts (Fig. 2 C, D). Since the patient group was older on average than the normal volunteer group, we used the available age matched samples (all 40 +/− 2 years) and noticed a similar difference (Fig. 2 E).
Fig. 2. Primary colony forming ability and population doubling of BMSCs from healthy controls and patients with SM.

A. Primary colony forming efficiency (CFE) of patient samples (green) appears significantly lower than in normal controls (red). B. The percentage of CD34+ hematopoietic progenitors was similar (in the same samples as in A) between the groups. C. The population doubling over time of BMSCs from healthy individuals was higher than D. from patient samples. E shows age-matched patients and controls
BMSCs from the BM of patients with SMC do not carry the KIT D816V mutation
SM is associated with somatic mutations in KIT that occur in hematopoietic stem cells and affect the mast cell lineage. We next determined if in addition to MCs, the BMSCs might also harbor the KIT D816V mutation. BMSCs were cultured from 5 patients and submitted to KIT D816V analysis by real-time qPCR. The KIT D816V mutation was not detected in the cultured BMSCs of patients with SM.
BMSCs from patients appear as a mixture of normal and abnormal cells
When we plated the BMSCs from the patients, we noticed that some areas on the plate that appeared to show normal growth, exhibiting the typical “swirly” pattern and easily growing. On the other hand there were some very sparsely populated areas, where the cells looked large, flat and showed no pattern of swirling (reminiscent of senescent cells) that were growing slowly or not at all and were positive for β-gal staining. (Fig. 3 A, B). The normal vs. abnormal colonies were counted from 10 patient samples and their respective numbers were: 3 vs. 16, 13 vs. 5, 5 vs. 6, 3 vs. 11, 0 vs. 3,1 vs. 8, 0 vs. 5,4 vs. 4, 0 vs. 11, 2 vs. 8, respectively. The average of these data were 3.1 (healthy) vs. 7.7 (abnormal) that reached statistical significance (p<0.017). To further explore this observation, and after plating aspirates at a clonal density, we next isolated and cultured cells from both normal (N=5–20/patient) and abnormal appearing colonies (N=2–8/patient) from the patients’ primary BM aspirate. We first cultured the cells in a flask, and once the colonies began to proliferate we picked cells from both types of colonies for study (Suppl. Fig. 1). We then separately pooled cells from both types of colonies to obtain enough cells for assay, as dictated by the slow growth of abnormal-appearing colonies. We then examined the cells for the presence of known BMSC surface markers. We found that the mean fluorescent intensity of CD 105 (endoglin) seems to be slightly higher in cells from the pooled normal colonies and greatly overexpressed in cells from the abnormal colonies of mastocytosis patients (Fig 4A; red) compared with BMSCs from normal donors (blue). In addition, the mean fluorescent intensity of CD146 appeared to be decreased in cells from the abnormal colonies compared with cells from normal clones and BMSCs from normal donors. (Fig. 4 A).
Fig. 3. Normal and abnormal colonies of BMSCs appear within the same patient sample.
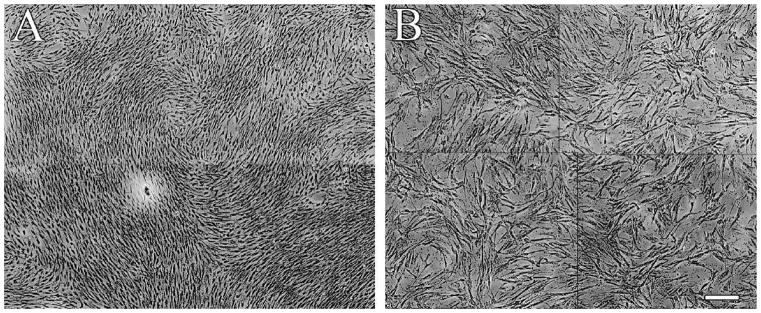
BMSCs that show normal growth and morphology (A) are shown next to scattered, abnormally looking senescent cells (B). Both cell populations were grown from single colonies from the same culture dish.
Fig. 4. Differences between normal and abnormal colonies taken from the same patient’s sample.

A. Five accepted markers of BMSCs were used in FACS analysis to compare the abnormal (left column) and normal (right column) cells (red) from patients with SM to healthy donor BMSCs (blue). Green denotes the unstained control. The mean fluorescent intensity of CD105 appeared higher in the patients’ samples, while they are lower for CD146. For details of antibodies used see Methods. B. Using osteogenic and adipogenic tissue culture media we demonstrated that the abnormal colonies are not able to form a calcified matrix (as shown by Alizarin red staining that labels calcium), but seem to produce more fat cells (as shown by Oil red staining). C. Illustration of the technique of studying engraftment of human CD34+ hematopoietic progenitors in immune-compromised NSG mice. Following co-culturing the CD34+ cells with BMSCs derived from healthy donors, or normal and abnormal colonies from patients, the CD34+ cells are injected into NSG mice previously treated with busulphan; and 4 weeks later, engraftment is determined by the percentage of human CD45+ cells in the peripheral blood. D. shows that BMSCs from healthy donors better support CD34+ progenitors to form more blood cells than those derived from normal and abnormal cells from patients. Five mice were used in each group. Statistical significance is labeled as * = p<0.05 and ** = p<0.05
Functional differences between cells from abnormal MC colonies in osteogeneic and adipogeneic differentiation compared with normal MC colonies
We next examined whether the morphologic and surface molecule differences are associated with functional differences between the normal and abnormal MC colonies. Using pooled abnormal and normal colonies we performed osteogenesis and adipogenesis assays [8]. The pooled abnormal colonies were found to produce less calcified matrix than the normal colonies. At the same time they appeared to produce more adipose tissue than their normal counterparts (Fig. 4 B).
Since human BMSCs bear all four histamine receptors (6), we wanted to examine the effect of histamine produced by mast cells on the BMSC cultures. When human BMSCs were cultured for two weeks in histamine rich media, the mRNA encoding peroxisome proliferator-activated receptor gamma (PPARγ) was significantly unregulated at a concentration of 10−7 M histamine in the medium (data not shown). This is a nuclear receptor protein that acts as transcription factor to regulate genes and its expression indicates the ability of the cells to differentiate towards adipogenic vs. osteogenic direction. In two experiments there was a 1.6 and 2.5 fold increase (p<0.002), respectivelee, of PPARγ mRNA isolated from hBMSCs cultured with 10−7 M histamine vs. control BMSCs.
Abnormal MC BMSCs are deficient in supporting hematopoiesis
Since an important and unique role of BMSCs is hematopoietic support, we next performed an experiment to see if there is any change in this function. We co-cultured healthy human CD34+ progenitors with BMSCs derived from normal donors, and BMSCs grown from both normal and abnormal colonies from patients with SM. Following a 4-day co-culture, we transplanted these hematopoietic progenitors into immune-compromised (NSG – NOD/SCID/IL2 receptor gamma chain triple KO), busulphan treated mice. Four weeks later we analyzed the peripheral blood to determine the percentage of human CD45+ cells (Fig. 4 C). We found that the mice transplanted with cells previously co-cultured with the pooled abnormal SM BMSC colonies had significantly (p<0.01) less huCD45+ cells in circulation than the mice transplanted with cells cultured with BMSCs from healthy donors (9+1.4 vs. 3.7+1.1 %). There was also a significant (p<0.05) difference in the effects on support of human blood cells between the normal and abnormal pooled clones from patients with SM; the pooled normal colonies producing lower values than healthy donors, but higher values that the abnormal clones (Fig. 4 D).
Gene Expression profiling on BMSCs from healthy donors and MS patients
Gene expression profiling was performed BMSCs from patients with SM (n=3) compared to those from normal donors (n=3). Among all of the genes evaluated, 6885 were detected to change compared to normal donors by more than 1.5-fold in at least 1 sample; and Principle Component Analysis (PCA) using these genes separated the samples into 2 clusters, one with BMSCs from donors and the other BMSCs from patients with SM (Fig. 5A), clearly indicating a disparity between these cell groups. We then identified genes that were differentially expressed between patients with SM and normal donors by using t-test (p-value <0.001). Among these genes, 410 were up-regulated in BMSCs from patients by more than 2-fold (red), while 185 were down-regulated (green) (Fig. 5B). The functions of these genes were annotated with the MetaCore database, and 16 significantly changed networks with p-value less than 0.05 were identified (Table 1). When comparing gene array data with BMSCs from normal volunteers, we found down regulation in the SM BMSCs of certain bone and cartilage and up regulation of adipose tissue differentiation related genes. BMP4 (2.3 fold p<0.009); Runx2 (2 fold p<0.001; ALPL (2 fold p<0.045) and BGN (1.25 fold p<0.19) involved in osteogenic differentiation and COL12A1 (1.69 fold p<0.045) and COL10A1 (6.1 fold p<0.001) involved in cartilaginous differentiation were all down regulated in the SM patients’ samples. PPARG (0.5 fold p<0.027) and CEBPA (0.4 fold p<0.001) on the other hand were up regulated in SM BMSCs, and are indicators of adipogenic differentiation ability.
Fig. 5. Gene expression profiling on BMSCs from mastocytosis patients and healthy donors.

Gene expression profiling was performed on BMSCs from 3 age-matched MS patients and donors. A. The genes that were detected by more than 1.5-fold change in at least 1 sample were used to run the Principle Component Analysis (PCA) [24]. The BMSCs from patients (blue spheres) were separated from those from normal donors (red spheres). B. The differentially expressed genes were identified by using a t-test, and 410 genes were up-regulated in BMSCs from MC patients (red dots, fold change >=2), while 185 were down-regulated (green dots, fold change >=2), x-axis indicates the log2 fold change of mastocytosis/donor, y-axis indicates the −log10 (p-value).
Table 1.
Metacore networks of significant genes
| MetaCore Networks | p-Value | Genes |
|---|---|---|
| Blood coagulation | 7.03E-05 | SERPINA6 (1.51), ADAMTS14 (−1.31), LMAN1 (−.24), MST1 (1.03), KRT18 (1.25), VWF (1) |
| KNG1 (1.74), F11 (1.69), TMPRSS6 (1.09) | ||
| WNT signaling | 2.63E-03 | ESR1 (1.40), TGFB3 (−1.22), WNT10B (1.47), WNT6 (1.74), FOXA2 (1.84), APC (−1.01), ADCY1 (1.29) |
| NFATC1 (−1), SFRP1 (2.47) | ||
| IL-2 signaling | 8.45E-03 | ESR1 (1.40), SYK (1.60), NFATC1 (−1), LCK (1.64), CCR2 (1.32), BCL2 (−1.02), TMPRSS6 (1.09), |
| Blood vessel morphogenesis | 1.03E-02 | PDE6B (1.56), NPR1 (1.47), TBX1 (1.43), TNNC1 (1.12), MMP19 (−1.15), VCAM1 (−1.46), |
| CEACAM6 (1.25), GRB7 (1.56), FOXF1 (2.40) | ||
| Ossification and bone remodeling | 1.73E-02 | IGFBP4 (1.12), TGFB3 (−1.22), CHRDL1 (−1.10), WNT6 (1.74), WNT10B (1.47), COL10A1 (−2.60), |
| TWIST1 (−1.06), RUNX2 (−1.03) | ||
| Connective tissue degradation | 1.93E-02 | LNPEP (−1.18), ADAMTS14 (−1.31), PCSK6 (2.56), CTSG (1.60), MMP19 (−1.15), TMPRSS6 (1.09), |
| Kallikrein-kinin system | 2.29E-02 | ADAMTS14 (−1.31), KNG1 (1.74), VWF (1.0), NOS2 (1.0), KNG1 (1.74), PTGES2 (1.09), F11 (1.69), |
| ECM remodeling | 2.46E-02 | ADAMTS14 (−1.31), PCSK6 (2.56), CSTG (1.60), MMP19 (−1.15), COL10A1 (−2.60) |
| Hedgehog signaling | 2.53E-02 | GAS1 (−1.30), TBX1 (1.43), FOXA2 (1.84), ADCY1 (1.29), JAG2 (1.60), BMI1 (−1.57), GLI1 (−1.49) |
| FOXF1 (2.40), EYA2 (−1.16), CDC37 (1.40), SOX8 (2.0), SOX3 (3.18) | ||
| Feeding and Neurohormone signaling | 2.66E-02 | TAC3 (1.25), KIT (1.64), TGFB3 (−1.22), ADCY1 (1.29), CCR2 (1.32), GLI1 (−1.49), BCL2 (−1.04) |
| Bile acids transport and its regulation | 2.70E-02 | SULT2A1 (2.25), ABCC4 (−1.02), ABCG5 (2.12), ABCB1 (1.51) |
| Muscle contraction | 3.26E-02 | THBS1 (−1.15), GUCA1B (1.25), KCNH2 (1.64), NPR1 (1.47), TNNC1 (1.12), GAL (1.64) |
| MYBPH (−1.41), GUCA1B (1.25) | ||
| Regulation of angiogenesis | 3.92E-02 | EFNA1 (1.0), IL1R1 (−1.18), THBS1 (−1.15), GLI1 (−1.49), CARD11 (1.47) |
| Neurogenesis | 4.17E-02 | CACNA1B (1.06), RIMS4 (1.36), PCP4 (1.09), SYT3 (1.32), THBS1 (−1.15), WNT6 (1.74) |
| WNT10B (1.47), POU4F1 (1.32), ERBB3 (1.03) | ||
| Visual perception | 4.45E-02 | PDE6B (1.56), GUCA1B (1.25), MYO7A (1.12), PRPH (1.15), RLBP1 (2.25) |
| Leucocyte chemotaxis | 4.53E-02 | LPAR2 (1.15), LIMK1 (−1.10), SKAP1 (2.40), VCAM1 (−1.47), CD2 (2.06), LCK (1.64) |
| CCL17 (1.22), CCR2 (1.32) |
The names of genes were shown, the values indicate the log2 (fold change) of Mastocytosis/Donor
We also looked for differences between BMSCs from normal volunteers vs. patients in expression of genes encoding factors that might affect the hematopoietic support function of the cells. The results showed a significant down regulation in SM derived BMSCs of CXCL12 (0.5 fold p<0.0003), PDGFα (0.66 fold p<0.004), VCAM1 (0.36 fold p<0.0004) and FGF7 (0.27 fold p<0.00001), which are known to be important in the hematopoietic support function of BMSCs.
Discussion
Mastocytosis is a rare disease characterized by an abnormal accumulation of mast cells in a number of tissues including the marrow, liver, spleen, lymph nodes and skin. In SM, the bone marrow is consistently affected [6] and skeletal abnormalities are well described [4, 9–11]. Since the bone marrow is home to not only the hematopoietic stem and progenitor cells, but also the BMSCs that maintain the BM environment, we were interested in whether the disease affects these cells as well and be associated with abnormal function.
In the work presented, we were found that BMSCs from patients with SM have a slower growth rate and a shorter life span than those from healthy donors. Since SM is associated with somatic activating mutations in KIT [1, 12–14], we first wanted to determine if the BMSCs also carry the KIT D816V mutation. All 5 patients’ BMSCs examined for the mutation showed no presence of a mutant KIT D816V allele. This result suggests that the slow growth rate and early senescence of the BMSCs from patients with SM is most likely due to epigenetic factors. These effects might be due to a variety of factors. For example, MCs release histamine and BMSCs bear histamine receptors. A chronically increased histamine concentration in the tissue environment might produce epigenetic changes – such as disturbances in growth properties as well as affecting their “stamens”. It is possible that as the disease progresses, increasingly more BMSC clones become abnormal, thus compounding the disease state for these patients. Histamine also induces the BMSC production of IL-6 and IL-8 [7] and high levels of these cytokines might also affect the composition of the environment surrounding the BMSCs. In addition to histamine, mast cell secretory granules contain other mediators (proteases, proteoglycans, heparin, heparan sulphates, cytokines etc.) that are released into the environment upon cell stimulation. All of these MC derived factors might have an effect on the biology of the BMSCs in the bone marrow niche and the effect of all these factors individually and in combination will also need to be examined in the future.
Our in vitro data exposing healthy BMSCs to high levels of histamine revealed an elevation of a peroxisome proliferator-activated receptor (PPAR) gamma mRNA in the cells. PPARγ is a prime inducer of adipogenesis in BMSCs and its elevation shifts the differentiation potential of BMSCs from osteogenic towards adipogenic lineage [15]. Due to the exposure of high levels of histamine over a long period of time in the BM of SM patients, this shift could partially explain the poor osteogenic differentiation of the cells and could underlie some of the skeletal problems of these patients.
It is also interesting to consider the possibility that primarily abnormal BMSCs (due to so far unknown mutations?) might actually facilitate the growth and expansion of mast cells within the marrow and that the expanded mast cell population would further impact on the misfunction of BM stroma. Additional genetic and epigenetic studies are required to elucidate the pathophysiological mechanisms for these observations.
We demonstrate that in addition to the growth deficiencies, the SM BMSCs have an altered expression of surface markers. Thus, CD 105 or endoglin is overexpressed in the abnormal SM BMSCs. The overexpression of endoglin in mouse fibroblasts resulted in a reduction in extracellular matrix components [16], and a decreased cellular migration and altered cellular morphology was observed. The expression pattern of CD146 is also of interest. CD146 (also known as melanoma cell adhesion molecule) has been reported to be the most specific marker of skeletal (self renewing) stem cells in the bone marrow [17, 18]. Thus a lower expression of this protein suggests that fewer skeletal stem cells are present in the BM, or may reflect a change in their functionality, which might explain the skeletal problems reported in patients with SM. Since the same molecule is also characteristic of the cell that supplies hematopoietic support within the BM niche, our finding that CD34+ cells that are co-cultured with the abnormal SM colonies (i.e., CD146low expression) showed a decrease in engraftment might also be due to differences in CD146 expression. CD 146 is an adhesion molecule, so the effect on CD34+ cells might thus relate to a decrease in homing ability following transplantation. Furthermore, we also observed significant down-regulation in SM derived BMSCs of CXCL12 (0.5 fold p<0.0003), PDGFα (0.66 fold p<0.004), VCAM1 (0.36 fold p<0.0004) and FGF7 (0.27 fold p<0.00001) – all of these factors are known to play a role in supporting hematopoiesis in the BM niche [19–21].
Our studies clearly demonstrate that in addition to the increased number of MCs, the bone marrow stroma is also involved in the disease and might be responsible for disease findings. It could be feasible to determine if BMSCs could serve as a new target for potential therapy in SM, a disease that at present has no cure.
Methods
Patients
Patients with SM were evaluated at the National Institutes of Health (NIH; Bethesda, MD) as part of Institutional Review Board (IRB)–approved research protocol designed to study the pathogenesis of SM (NCT00044122) and were diagnosed according to the World Health Organization (WHO) criteria. KIT D816V mutation analysis was performed described [22]. Healthy controls participated under the IRB approved protocol NCT01071577. The biopsy and aspirates were obtained from the same affected site of the patients. Neoplastic mast cell “free” aspirates were not obtained. Although of scientific interest, obtaining these samples would have been technically challenging and would have subjected the patients to repeated bone marrow aspirations.
Culturing of BMSCs
Bone marrow aspirates were diluted according to cell counts. In complete medium and plate 0.25–0.5× 106 nucleated cells/cm2 in 75-cm2 culture flasks (patient samples did not always have this number of cells available) and incubate cells in complete medium for 24 hours in a 37°C, 5% CO2 incubator. Medium was changed every 3 days and cells were passaged before reaching confluency. To isolate normal and abnormal colonies the patients’ BMSCs were grown in a flask and monitored daily. When the colonies were large enough to determine their nature, the top of the flask was sawn off and the normal and abnormal looking colonies were isolated and seeded again in separate culture plates. Since the abnormal colonies grew very slowly and stopped growing very early, we had to pool the abnormal colonies to have enough cells for experiments to perform. For more details see [8].
Proliferation studies of BMSCs
BMSCs were serum starved in basal medium for 72 hours. 5000 human serum starved BMSCs were plated in 96-well plates/200 μl each well. After an overnight incubation the serum free medium was changed to either 20% serum medium or basal medium containing 10 ng/ml FGF-2. Twenty-four hours later BrdU was added at a final cc of 10μM. After another 12 hours cells were fixed with fix-perm buffer and the next steps were done according to the Roche BrdU ELISA protocol.
Beta-galactosidase (β-gal) staining of expanded BMSC
BMSCs were stained using a Senescence Associated Beta-Gal Staining kit (Sigma Aldrich, St. Louis, MO) following the manufacturer’s protocol. Briefly, BMSCs were washed with PBS, fixed with Fixation Buffer for 7 minutes at room temperature, washed with PBS, and then incubated with staining mixture at 37°C overnight. The senescent cells under 10 randomly selected high-magnitude microscope fields (100X) were counted on the following day.
Primary Colony-forming efficiency (CFE) and measurement of CD34+ cells of bone marrow aspirate
Primary CFE of bone marrow aspirates were analyzed by methods as reported previously [23]. Briefly, 1× 105 nucleated cells were plated in T25 flask (Corning, Corning, NY) in duplicate and cultured for 13 days without changing the culture medium. The colonies were fixed with methanol for 30 minutes and stained with saturated methyl-violet water solution for 20 minutes. Colonies were observed under low magnitude light microscope field (25 X). Colonies containing 50 or more cells of fibroblastic morphology were counted [23].
The percentage of CD34+ hematopoietic progenitor cell (HPC) was determined by following standardized protocols in Cell Processing Section (CPS), Department of Transfusion Medicine, Clinical Center of NIH. Briefly, 1 × 106 nucleated cells were incubated with CD34 APC, CD3 PE-Cy7, 7AAD and CD45 APC-Cy7 (BD Biosciences, San Jose, CA) for 20 min at 4 ° and then lysed in a Lysis Washing Assistant equipment (BD Biosciences). The CD34+ percentage was measured on a FACSCanto cytometer (BD Biosciences) [23, 24].
Population Doubling (PD) curve of expanded BMSC
PD was calculated to analyze the proliferation of BMSC. The number of BMSCs at each passage was manually counted and the PD for each passage was calculated using equation: N= log2 (NH/N1) where N = population doublings, N H = cell harvest number, N I = plating cell number, and the cumulative PDs were calculated in relation to the number of cells at the first passage [23, 24].
FACS analysis of BMSCs
Cells were incubated with corresponding antibodies for 15 minutes on 4 degree in FACS buffer (1XPBS/1%BSA). FACS was performed using FACS Calibur. Data were analyzed with FlowJo. The antibodies used were as follows: Anti-CD44 PE BD Biosciences (Cat No 555479). Anti-CD45 FITC BD Biosciences (Cat No 555482) Anti-CD90-PE eBioscience (Cat No 12-0909), Anti-CD105-PE eBioscience (Cat No 12-1057), Anti-CD146-FITC Abcam (Cat No ab78451), Anti-CD166-PE BD Biosciences (Cat No 560903).
Osteogenic and adipogenic differentiation assay of BMSCs
In vitro bone and adipose stimulation assays were performed as reported earlier [8]. 0.3 million pooled MSCs were plated in 6 well plates in complete MSC medium. After 72 hours medium was changed to either osteogenic or adipogenic medium. After 14 days cells were fixed and stained with Alizarin Red or Oil Red respectively. Representative wells are shown.
Histamine stimulation of BMSCs followed by adipogenic differentiation assay
25k BMSCs were plated in 12 well plates in complete MSC medium. Histamine dihydrochloride (Abcam) was added once every day for a week to maintain a 10−7M concentration in the wells of the experimental samples, while control BMSCs received vehicle only. After one week the adipogenic differentiation assay was started in all wells. After 14 days, total RNA was isolated using the Quick-RNA Mini Prep kit (Zymo Research) according to manufacturer’s instructions. Isolated total RNA was reverse transcribed using oligo-dT primers and the Moloney-murine leukemia virus reverse transcriptase according to the instructions of the manufacturer (Promega, Madison, WI). The resultant cDNA was amplified with QuantiTect SYBR Green RT-PCR kit (Qiagen) using primers ACGAAGACATTCCATTCACAA (sense) and CTCCACAGACACGACATTC (antisense) corresponding to 166–186 and 345–363 of Genebank sequence XM_006713208.1. RT-PCR conditions were as follows: 95°C for 15 min initial activation of the Taq polymerase and denaturation of the reverse transcriptase and then 45 cycles of 94°C for 15 sec denaturation, primer-specific temperatures for 30 sec annealing (PPARG2 55°C, ACTB 60°C), and 72°C for 30 sec extension.
Human CD34+ cell engraftment experiment after BMSC co-culture
Pooled samples of human BMSCs (from healthy donors or mastocytosis patients) were seeded in 6 well plates (0.25 million cells/3 ml in each well) in complete MEM-alpha medium and were grown to confluence. Mitomycin pretreatment was performed by incubating BMSCs in complete MSC medium containing 10 ug/ml final concentration of mitomycin C for 2 hours. This was followed by vigorous washes 5 times with PBS.
Peripheral blood CD34+ cells (PB CD34+) were obtained from healthy adult volunteers. Briefly, volunteers received 5 days of 10 μg/kg recombinant human (rHu) granulocyte colony-stimulating factor (G-CSF, Amgen, Thousand Oaks, CA). On day 5 of mobilization, PB CD34+ cells were collected by one 12–15-L leukopheresis, and CD34-positive selection was performed using the Isolex 300i automated immuno-magnetic system (Nexell, Miltenyi, Auburn, CA). CD34+ cells (2 million/well) were added to each well in StemSpan medium containing 25 ng/ml of SCF, Tpo, and Flt3-ligand.
8–10 week old male NOD-SCID-Gamma (NSG) mice from JAX [25] were used as recipients (5 mice were used in each group). Busulfan (Busulfex, Otsuka Pharmaceutical, Rockville, MD) was diluted with PBS to deliver 35 mg/kg in a final volume of 400 μl and was injected into recipient mice intraperitoneally 24 h prior to the cell infusion. Cultured cells were filtered through a 40 um strainer and subsequently injected in a total volume of 200 ul PBS. After 5 days of co-culture the CD34+ cells were collected, counted, and injected intravenously via the tail vein into the busulfan pretreated mice at a concentration of 1 million cells/300 μL PBS.
Four weeks after transplantation blood was collected from the recipient animals to check for human cell chimerism. After RBC lysis, using ACK lysing buffer, FACS was performed using mouse and human CD45 specific antibodies. The percentage of human CD45+ cells was determined as a marker of engraftment.
Gene Expression profiling on BMSCs from healthy donors and MS patients
The gene expression profiling was performed by following standard protocols in CPS [24]. Briefly, the BMSCs from 3 age-matched SM patients (30.3±8.5 years) and healthy donors (34.7±6.8 years) were selected for analysis. Total RNA was extracted and labeled with Cy5-CTP and human reference RNA (was labeled with Cy3-CTP. The labeled cRNA was pooled, fragmented and then hybridized on 4 × 44K microarrays (Agilent, Santa Clara, CA) for 17 hours at 65°C. The microarrays were then washed and scanned using an Agilent Scanner and data were acquired using Agilent Feature Extraction Software. The raw data was uploaded into mAdb database (http://madb.nci.nih.gov/) and then analyzed by BRB-Array Tools [26] (http://linus.nci.nih.gov/BRB-ArrayTools.html) and Partek Genomics Suite (Partek Inc, Saint Louis, MO). Tests for differences between MS patients and healthy donors were conducted for individual genes using a t-test, considering P values of <0.001 as significant. The functional relevance of the differentially expressed genes was annotated by using MetaCore database (https://portal.genego.com/) following its instructions.
Mutational analysis of SM patients’ derived cultured BMSCs
DNA extraction
One million stromal cells cultured from mastocytosis patients were lysed and genomic DNA (gDNA) was prepared by using QIAamp DNA blood mini kit (QIAgen) and eluted in 100 μL of elution buffer. Genomic DNA from HMC1.2 cells (KIT D816V heterozygous) [27] were also prepared as was stromal cell genomic DNA and used as the KIT D816V mutation positive control. Genomic DNA from PBMCs of healthy volunteers was prepared and used as a negative control.
Allele specific qPCR for KIT D816V mutation
Two real-time qPCR assays were performed using the TaqMan Universal PCR Master Mix with AmpErase UNG on the 7500 Real Time PCR System (Applied Biosystems, Foster City, CA). The KIT D816V mutation specific assay amplifies KIT D816V alleles, and the control assay amplifies both wild-type KIT and D816V KIT alleles. The forward and reverse primer, TaqMan probe sequences and conditions for the qPCR assays were used as described [28]. A four fold dilution series of HMC1.2 cell gDNA was used as standards to perform qPCR assays on same plate for absolute quantification and for calculating PCR efficiencies of mutation-specific and control assays. Real-time qPCR was performed with 50 ng sample gDNA in a total 25μL reaction volume. All samples were performed in triplicate. Data was analyzed using SDS software version 1.3.1 (Applied Biosystems). Percent KIT D816V allele was calculated based on the formula described [28]. The mutation positive was determined as 2 of 3 or 3 of 3 reactions generating a threshold cycle (Ct) value below 41. The mutation negative was defined as none of three reactions producing a Ct value below 44.
Supplementary Material
Table 2.
Patients’ data and summary of usage of BMSCs
| Used for | Date | BSI ID | DIN | Age | Gender | Classification | Tryptase (ng/mL) | Mast cell aggregates | % invovement | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | BRDU proliferation assay | 3/31/10 | 56 | M | Smoldering | 341 | yes | 50 | ||
| 2 | clotted | 7/28/10 | 40 | M | Indolent | 20 | yes | 10 | ||
| 3 | clotted | 6/9/11 | 65 | M | Indolent | 74 | yes | 5 | ||
| 4 | clotted | 5/12/11 | 51 | F | Indolent | 188 | yes | 30 | ||
| 5 | did not grow | 9/8/10 | 49 | F | Indolent | 35 | yes | 5 | ||
| 6 | did not grow | 11/3/10 | 44 | M | Indolent | 105 | yes | 30 | ||
| 7 | colonies for morphology | 4/14/10 | 42 | F | Smoldering | 150 | yes | 40 | ||
| 8 | colonies for morphology | 11/15/07 | 48 | F | Indolent | 287 | yes | 40 | ||
| 9 | FACS analysis, repopulation assay | 5/5/10 | 63 | F | Indolent | 52 | yes | 15 | ||
| 10 | repopulation assay | 6/9/10 | 58 | F | indolent | 29 | yes | 10 | ||
| 11 | repopulation assay | 4/21/10 | 67 | M | Indolent | 123 | yes | 40 | ||
| 12 | frozen, ossicle | 7/1/10 | 37 | F | Indolent | 46 | yes | 30 | ||
| 13 | frozen, ossicle | 7/7/10 | 66 | F | No mastocytosis | 17 | no | 0 | ||
| 14 | frozen, ossicle | 8/18/10 | 51 | F | Indolent | 68 | yes | 30 | ||
| 15 | frozen, ossicle | 7/14/10 | 61 | F | Smoldering | 319 | yes | 30 | ||
| 16 | gene array | 1/20/11 | MC000299 | 11FC00007 | 23 | F | 2 minor criteria | 10 | no | <5 |
| 17 | gene array | 2/17/11 | MC000304 | 11FC00024 | 41 | F | Indolent | 13 | yes | 20 |
| 18 | gene array | 3/30/11 | MC000328 | 11FC00048 | 47 | F | Indolent | 25 | yes | 15 |
| 19 | gene array | 3/2/11 | MC000306 | 11FC00031 | 57 | M | Indolent | 68 | yes | 10 |
| 20 | gene array | 3/31/11 | MC000329 | 11FC00049 | 40 | F | Indolent | 79 | yes | 15 |
| 21 | gene array | 1/5/11 | 11FC00001 | 24 | F | Indolent | 80 | yes | 15 | |
| 22 | gene array | 1/13/11 | 11FC00003 | 27 | F | Indolent | 100 | yes | 50 | |
| 23 | gene array | 2/9/11 | MC000303 | 11FC00020 | 55 | F | Smoldering | 119 | yes | 30 |
| 24 | gene array | 3/10/11 | MC000318 | 11FC00038 | 58 | F | Aggressive | 120 | yes | 40 |
| 25 | gene array | 2/2/11 | 11FC00015 | 48 | F | Smoldering | 166 | yes | 50 | |
| 26 | gene array | 3/3/11 | MC000307 | 11FC00032 | 71 | F | indolent | 173 | yes | 30 |
| 27 | normal and abnormal colonies, adipo and ostegenic assay | 8/26/10 | 45 | F | 2 minor criteria | 6 | no | scattered mast cells | ||
| 28 | normal and abnormal colonies, adipo and ostegenic assay | 9/2/10 | 43 | F | Indolent | 17 | yes | 20 | ||
| 29 | normal and abnormal colonies, adipo and ostegenic assay | 9/9/10 | 52 | M | Indolent | 24 | yes | 15 | ||
| 30 | normal and abnormal colonies, adipo and ostegenic assay | 11/10/10 | 36 | F | Indolent | 35 | yes | 5 | ||
| 31 | normal and abnormal colonies, adipo and ostegenic assay | 10/27/10 | 32 | F | Smoldering | 215 | yes | 35 | ||
| 32 | normal and abnormal colonies, adipo and ostegenic assay | 11/18/09 | 48 | M | Indolent | 65 | yes | 5 |
| >25% Spindle shaped | CD2 | CD25 | KIT mutation | Cytopenia | Hepatosplenomegaly | Osteopenia/Osteoporosis | Cytoreductive Treatment | |
|---|---|---|---|---|---|---|---|---|
| 1 | yes | + | + | KIT D816V | no | yes | yes | |
| 2 | yes | + | + | KIT D816V | no | no | no | |
| 3 | yes | + | + | KIT D816V | thrombocytopenia | no | no | |
| 4 | yes | + | + | KIT D816V | no | no | osteoporosis | colon CA s/p R hemicolectomy with adjuvant chemotherapy |
| 5 | yes | + | + | KIT D816V | no | splenomegaly | yes | |
| 6 | yes | + | + | KIT D816V | no | no | osteoporosis | |
| 7 | yes | + | + | KIT D816V | no | yes | no | |
| 8 | yes | + | + | KIT D816V | eosinophilia | splenomegaly/hepatomegaly | osteoporosis | |
| 9 | yes | + | + | KIT D816V | no | hepatomegaly | yes | |
| 10 | yes | + | + | KIT D816V | no | no | yes | |
| 11 | yes | + | + | KIT D816V | no | no | no | |
| 12 | yes | + | + | KIT D816V | no | no | no | |
| 13 | no | − | − | - | no | no | no | |
| 14 | yes | + | + | KIT D816V | no | no | no | |
| 15 | yes | + | + | KIT D816V | thrombocytosis | no | yes | |
| 16 | yes | − | + | - | no | no | osteopenia | |
| 17 | yes | + | + | - | no | no | no | |
| 18 | yes | + | + | KIT D816V | no | no | no | imatinib |
| 19 | yes | + | + | KIT D816V | no | no | no | |
| 20 | yes | + | + | KIT D816V | no | no | no | |
| 21 | yes | + | + | KIT D816V | no | no | osteopenia | |
| 22 | no | − | − | KIT K5091 germline | no | no | no | |
| 23 | yes | + | + | KIT D816V | leukopenia | splenomegaly/hepatomegaly | no | |
| 24 | yes | + | + | KIT D816V/KRAS G12D | thrombocytopenia | splenomegaly | no | |
| 25 | yes | + | + | KIT D816V | no | hepatomegaly | no | IFN alpha |
| 26 | yes | + | + | KIT D816V | no | no | no | |
| 27 | yes | − | + | - | no | no | no | |
| 28 | yes | − | − | KIT D816V | no | no | no | |
| 29 | yes | + | + | KIT D816V | no | no | yes | |
| 30 | yes | + | + | KIT D816V | no | no | no | |
| 31 | yes | + | + | KIT D816V | no | no | no | |
| 32 | yes | + | + | KIT D816V | no | no | yes |
Highlights.
Patients with systemic mastocytosis (SM) develop anaemia and skeletal problems
Bone Marrow Stromal Cells from patients (SM-BMSCs) show abnormal growth
SM-BMSCs in vitro make less bone and more fat than normal BMSCs
SM-BMSCs do not carry mutation of the stem cell receptor (c-kit)
The gene expression profile of SM-BMSCs differ from healthy BMSCs
Acknowledgments
This work was supported by the DIR, NIDCR and NIAID of the IRP, NIH, DHHS. We thank the patients, healthy volunteers and clinical research staff for their contributions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Carter MC, Metcalfe DD, Komarow HD. Mastocytosis, Immunology and allergy clinics of North America. 2014;34:181–196. doi: 10.1016/j.iac.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nagata H, Okada T, Worobec AS, Semere T, Metcalfe DD. c-kit mutation in a population of patients with mastocytosis. International archives of allergy and immunology. 1997;113:184–186. doi: 10.1159/000237541. [DOI] [PubMed] [Google Scholar]
- 3.D’Ambrosio C, Akin C, Wu Y, Magnusson MK, Metcalfe DD. Gene expression analysis in mastocytosis reveals a highly consistent profile with candidate molecular markers. The Journal of allergy and clinical immunology. 2003;112:1162–1170. doi: 10.1016/j.jaci.2003.07.008. [DOI] [PubMed] [Google Scholar]
- 4.Delsignore JL, Dvoretsky PM, Hicks DG, O’Keefe RJ, Rosier RN. Mastocytosis presenting as a skeletal disorder. The Iowa orthopaedic journal. 1996;16:126–134. [PMC free article] [PubMed] [Google Scholar]
- 5.Rossini M, Zanotti R, Viapiana O, Tripi G, Orsolini G, Idolazzi L, Bonadonna P, Schena D, Escribano L, Adami S, Gatti D. Bone involvement and osteoporosis in mastocytosis. Immunology and allergy clinics of North America. 2014;34:383–396. doi: 10.1016/j.iac.2014.01.011. [DOI] [PubMed] [Google Scholar]
- 6.George TI, Horny HP. Systemic mastocytosis, Hematology/oncology clinics of North America. 2011;25:1067–1083. vii. doi: 10.1016/j.hoc.2011.09.012. [DOI] [PubMed] [Google Scholar]
- 7.Nemeth K, Wilson T, Rada B, Parmelee A, Mayer B, Buzas E, Falus A, Key S, Masszi T, Karpati S, Mezey E. Characterization and function of histamine receptors in human bone marrow stromal cells. Stem cells. 2012;30:222–231. doi: 10.1002/stem.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nemeth K, Mayer B, Sworder BJ, Kuznetsov SA, Mezey E. A practical guide to culturing mouse and human bone marrow stromal cells. In: Coligan John E, et al., editors. Current protocols in immunology. Unit 22F. Vol. 102. 2013. p. 12. [DOI] [PubMed] [Google Scholar]
- 9.Abramowitz JD, Weinerman SA. Osteoporosis as the sole manifestation of systemic mastocytosis in a young man. Endocrine practice: official journal of the American College of Endocrinology and the American Association of Clinical Endocrinologists. 2012;18:e158–161. doi: 10.4158/EP12062.CR. [DOI] [PubMed] [Google Scholar]
- 10.Hermine O, Lortholary O, Leventhal PS, Catteau A, Soppelsa F, Baude C, Cohen-Akenine A, Palmerini F, Hanssens K, Yang Y, Sobol H, Fraytag S, Ghez D, Suarez F, Barete S, Casassus P, Sans B, Arock M, Kinet JP, Dubreuil P, Moussy A. Case-control cohort study of patients’ perceptions of disability in mastocytosis. PloS one. 2008;3:e2266. doi: 10.1371/journal.pone.0002266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lim KH, Tefferi A, Lasho TL, Finke C, Patnaik M, Butterfield JH, McClure RF, Li CY, Pardanani A. Systemic mastocytosis in 342 consecutive adults: survival studies and prognostic factors. Blood. 2009;113:5727–5736. doi: 10.1182/blood-2009-02-205237. [DOI] [PubMed] [Google Scholar]
- 12.Horny HP, Sotlar K, Valent P. Mastocytosis: state of the art. Pathobiology: journal of immunopathology, molecular and cellular biology. 2007;74:121–132. doi: 10.1159/000101711. [DOI] [PubMed] [Google Scholar]
- 13.Longley BJ, Jr, Metcalfe DD, Tharp M, Wang X, Tyrrell L, Lu SZ, Heitjan D, Ma Y. Activating and dominant inactivating c-KIT catalytic domain mutations in distinct clinical forms of human mastocytosis. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:1609–1614. doi: 10.1073/pnas.96.4.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pardanani A. Systemic mastocytosis: disease overview, pathogenesis, and treatment. Hematology/oncology clinics of North America. 2012;26:1117–1128. doi: 10.1016/j.hoc.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 15.Takada I, Kouzmenko AP, Kato S. Molecular switching of osteoblastogenesis versus adipogenesis: implications for targeted therapies. Expert opinion on therapeutic targets. 2009;13:593–603. doi: 10.1517/14728220902915310. [DOI] [PubMed] [Google Scholar]
- 16.Guerrero-Esteo M, Lastres P, Letamendia A, Perez-Alvarez MJ, Langa C, Lopez LA, Fabra A, Garcia-Pardo A, Vera S, Letarte M, Bernabeu C. Endoglin overexpression modulates cellular morphology, migration, and adhesion of mouse fibroblasts. European journal of cell biology. 1999;78:614–623. doi: 10.1016/S0171-9335(99)80046-6. [DOI] [PubMed] [Google Scholar]
- 17.Bianco P, Robey PG, Saggio I, Riminucci M. “Mesenchymal” stem cells in human bone marrow (skeletal stem cells): a critical discussion of their nature, identity, and significance in incurable skeletal disease. Human gene therapy. 2010;21:1057–1066. doi: 10.1089/hum.2010.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sacchetti B, Funari A, Michienzi S, Di Cesare S, Piersanti S, Saggio I, Tagliafico E, Ferrari S, Robey PG, Riminucci M, Bianco P. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell. 2007;131:324–336. doi: 10.1016/j.cell.2007.08.025. [DOI] [PubMed] [Google Scholar]
- 19.Li T, Wu Y. Paracrine molecules of mesenchymal stem cells for hematopoietic stem cell niche. Bone marrow research. 2011;2011:353878. doi: 10.1155/2011/353878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagasawa T, Omatsu Y, Sugiyama T. Control of hematopoietic stem cells by the bone marrow stromal niche: the role of reticular cells. Trends in immunology. 2011;32:315–320. doi: 10.1016/j.it.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 21.Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25:977–988. doi: 10.1016/j.immuni.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 22.Maric I, Robyn J, Metcalfe DD, Fay MP, Carter M, Wilson T, Fu W, Stoddard J, Scott L, Hartsell M, Kirshenbaum A, Akin C, Nutman TB, Noel P, Klion AD. KIT D816V-associated systemic mastocytosis with eosinophilia and FIP1L1/PDGFRA-associated chronic eosinophilic leukemia are distinct entities. The Journal of allergy and clinical immunology. 2007;120:680–687. doi: 10.1016/j.jaci.2007.05.024. [DOI] [PubMed] [Google Scholar]
- 23.Sabatino M, Ren J, David-Ocampo V, England L, McGann M, Tran M, Kuznetsov SA, Khuu H, Balakumaran A, Klein HG, Robey PG, Stroncek DF. The establishment of a bank of stored clinical bone marrow stromal cell products. Journal of translational medicine. 2012;10:23. doi: 10.1186/1479-5876-10-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ren J, Stroncek DF, Zhao Y, Jin P, Castiello L, Civini S, Wang H, Feng J, Tran K, Kuznetsov SA, Robey PG, Sabatino M. Intra-subject variability in human bone marrow stromal cell (BMSC) replicative senescence: molecular changes associated with BMSC senescence. Stem cell research. 2013;11:1060–1073. doi: 10.1016/j.scr.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.King MA, Covassin L, Brehm MA, Racki W, Pearson T, Leif J, Laning J, Fodor W, Foreman O, Burzenski L, Chase TH, Gott B, Rossini AA, Bortell R, Shultz LD, Greiner DL. Human peripheral blood leucocyte non-obese diabetic-severe combined immunodeficiency interleukin-2 receptor gamma chain gene mouse model of xenogeneic graft-versus-host-like disease and the role of host major histocompatibility complex. Clinical and experimental immunology. 2009;157:104–118. doi: 10.1111/j.1365-2249.2009.03933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Simon R, Lam A, Li MC, Ngan M, Menenzes S, Zhao Y. Analysis of gene expression data using BRB-Array Tools. Cancer informatics. 2007;3:11–17. [PMC free article] [PubMed] [Google Scholar]
- 27.Sundstrom M, Vliagoftis H, Karlberg P, Butterfield JH, Nilsson K, Metcalfe DD, Nilsson G. Functional and phenotypic studies of two variants of a human mast cell line with a distinct set of mutations in the c-kit proto-oncogene. Immunology. 2003;108:89–97. doi: 10.1046/j.1365-2567.2003.01559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kristensen T, Vestergaard H, Moller MB. Improved detection of the KIT D816V mutation in patients with systemic mastocytosis using a quantitative and highly sensitive real-time qPCR assay. The Journal of molecular diagnostics: JMD. 2011;13:180–188. doi: 10.1016/j.jmoldx.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.