

Abstract
Temperate bacteriophages express transcription repressors that maintain lysogeny by down-regulating lytic promoters and confer superinfection immunity. Repressor regulation is critical to the outcome of infection—lysogenic or lytic growth—as well as prophage induction into lytic replication. Mycobacteriophage BPs and its relatives use an unusual integration-dependent immunity system in which the phage attachment site (attP) is located within the repressor gene (33) such that site-specific integration leads to synthesis of a prophage-encoded product (gp33103) that is 33 residues shorter at its C-terminus than the virally-encoded protein (gp33136). However, the shorter form of the repressor (gp33103) is stable and active in repression of the early lytic promoter PR, whereas the longer virally-encoded form (gp33136) is inactive due to targeted degradation via a C-terminal ssrA-like tag. We show here that both forms of the repressor bind similarly to the 33–34 intergenic regulatory region, and that BPs gp33103 is a tetramer in solution. The BPs gp33103 repressor binds to five regulatory regions spanning the BPs genome, and regulates four promoters including the early lytic promoter, PR. BPs gp33103 has a complex pattern of DNA recognition in which a full operator binding site contains two half sites separated by a variable spacer, and BPs gp33103 induces a DNA bend at the full operator site but not a half site. The operator site structure is unusual in that one half site corresponds to a 12 bp palindrome identified previously, but the other half site is a highly variable variant of the palindrome.
Introduction
Following adsorption and DNA injection, temperate phages must choose between two alternative outcomes: lytic growth in which the phage replicates and the cell lyses to release progeny phage particles, or lysogeny in which the lytic genes are switched off and a prophage genome is maintained either by site-specific chromosomal integration, or stable extrachromosomal replication [1]. In the well-studied example of phage lambda, lysogenic maintenance is achieved by expression of a repressor (cI) that binds to tripartite operators (OL and OR) at the early lytic promoters PL and PR [1]. Lambda cI autoregulates its synthesis by activation of its own transcription from the promoter for lysogenic maintenance (PRM) at moderate cI concentrations, and represses it when the cI concentration is high. During infection, establishment of lambda lysogeny occurs by expression of cI from the promoter for lysogenic establishment (PRE), which is independent of cI, but requires the activator, cII [2]. The decision as to the outcome of infection is determined by the overall level of cII, which is subject to degradation by host proteases including FtsH, and is modulated by lambda cIII protein [3]. Lambda cI binds as a dimer and can form DNA loops when bound at both OL and OR [4].
The temperate life style is common among bacteriophages, although the genetic diversity of the phage population is considerable [5]. Repressors have been identified in many phage genomes, although relatively few have been genetically and biochemically characterized [5]. The organization of two divergently transcribed DNA-binding proteins—typified by the cI and cro genes in lambda—separated by a control region is common but not universal. For example, the repressor of Streptomyces phage ϕC31 is located downstream of the virion structural genes and is similarly transcribed rightwards [6, 7], and in mycobacteriophage L5 (and its relatives) the repressor is located within the right arm of the genome and transcribed leftwards along with other right arm genes [8, 9]. These two systems are also unusual in that the phage genomes contain multiple (18–30) repressor binding sites dispersed across the genomes, and the ϕC31 system is further complicated by the expression of three isoforms of the repressor [10]. The L5 repressor binds as a monomer at the asymmetric non-operator binding sites (referred to as ‘stoperators’) to block transcription elongation [11]. There are few examples other than phage lambda and its relatives where the molecular basis of the decision between lytic and lysogenic outcomes is well understood [12, 13].
Comparative genomic analysis of a large number of completely sequenced mycobacteriophages shows them to be highly diverse, and they can be grouped into ‘clusters’ according to their nucleotide and gene content relationships [14, 15]. A substantial portion of these phages (~40%), including L5, are grouped in Cluster A, and share the unusual stoperator system of immunity [11, 16]. The repressor genes have been identified in phages of Clusters G, I, N, and P [12, 13], Cluster K [17], and the singleton Giles [18], but in each case they are components of pairs of divergently transcribed genes separated by a putative control region.
Mycobacteriophage BPs and its relatives in Cluster G—along with members of Clusters I, N, and P—use an unusual integration-dependent immunity system for the establishment and maintenance of lysogeny [13]. In these systems the repressor and integrase genes (BPs 32 and 33 respectively) are transcribed leftwards, but the phage attachment site (attP) is oddly located within the repressor open reading frame (see Fig 1A). As a consequence, chromosomal integration in lysogenic establishment results in separation of the 3’ end of the repressor gene and expression of a truncated version of the repressor. However, it is this short form of the repressor (e.g. BPs gp33103) that is active in conferring immunity, whereas the virally-encoded longer form (e.g. BPs gp33136) is not. Inactivation of the virally-encoded form expressed during lytic growth occurs as a result of proteolytic degradation targeted at an ssrA-like tag at the extreme C-terminus [13]. Proteolysis of the virally-encoded form is a direct determinant of lysogenization frequency, as a mutant expressing a stabilized form of BPs gp33136 establishes lysogeny at a considerably higher frequency than wild-type BPs. However, lysogenization frequency is also determined by the frequency of integration, and the integrase protein also contains a C-terminal signal for proteolysis [13].
Fig 1. The BPs Immunity Cassette.

(A) Phage BPs has three genes involved in the lytic/lysogenic growth decision; gene 32 encodes an integrase (gp32), gene 33 encodes the repressor (gp33), and gene 34 encodes a putative Cro-like protein (gp34). BPs gp33 and gp32 are expressed leftwards from the Prep promoter and gp34 is expressed rightward from the PR promoter. The attP site for BPs integration is within the 33 open reading frame, such that the prophage repressor gene is truncated the gene by 99bp. The virally-encoded repressor is 136 residues long (gp33136) and has a C-terminal tag for proteolytic degradation [13]. This prophage form of the repressor (gp33103) is 33 residues shorter, is stably expressed, and is required to maintain lysogeny. (B) The 33–34 intergenic region contains two divergent promoters, Prep and PR. The full intergenic region sequence is shown (left part above, right part below) and promoter -10 and -35 elements are in green. The operator (OR; highlighted in yellow) to which gp33103 binds to regulate PR is located between the -10 and -35 elements of PR (green). A second operator, ORep, is located proximal to 33 (highlighted in yellow) downstream of the Prep promoter (green). (C) DNA included in substrates used for binding assays are shown as horizontal lines above or below the 33–34 intergenic sequence (Mt series and TIR series of substrates). Regions of DNase I protection are indicated by black lines between the top and bottom strand bases.
The BPs 33–34 intergenic region contains two divergent promoters, PR and Prep, responsible for early lytic expression and repressor synthesis respectively [13, 19] (Fig 1B). The gp33103 active form of the repressor was shown previously to bind to a DNA substrate that includes a 12 bp palindromic sequence, which was presumed to be the operator (OR) regulating transcription from the early lytic promoter, PR [13]. This was supported by the mapping of two point mutations within this 12 bp sequence (5’-CGACATATGTCG) that give rise to a repressor-insensitive phage phenotype (i.e. they can form plaques on a repressor-expressing strain) [13]. However, the requirements for DNA binding at OR and at related sequences elsewhere in the phage genome and the nature of the protein-DNA interactions are not understood. Here we show that the two forms of the repressor bind similarly to DNA, that the short active form of the repressor is a tetramer in solution, and that the previously reported 12 bp operator sequence represents one half of a full binding site.
Results
gp33136 and gp33103 bind similarly to BPs 33–34 intergenic DNA
Electrophoretic mobility shift assays (EMSA) show that gp33103 binds to DNA substrates containing the 33–34 intergenic control region to form several distinct complexes as protein concentration increases (Fig 2A). The combined affinity for gp33103 binding is relatively weak (2.6 μM; Table 1) and the prominent complex (C4) forms at protein concentrations of 16 μM and above (Fig 2A); three faster migrating complexes are observed at lower protein concentrations and are likely binding intermediates (Fig 2A). Although DNA binding is relatively weak compared to other repressors, it is specific as little or no binding is observed with a control substrate (Fig 2A), and the binding reactions all contain 1 μg calf thymus DNA.
Fig 2. Both forms of gp33 bind similarly to the BPs 33–34 intergenic region.

(A) EMSA analysis shows that gp33103 binds to the 33–34 intergenic region with a complicated pattern of binding with four distinct complexes, labeled C1, C2, C3, and C4. No binding was seen between gp33103 and a non-speciic DNA substrate. Protein concentrations are as follows: 1) none 2) 0.16 μM 3) 0.54 μM, 4) 1.6 μM, 5) 5.4 μM, 6) 16 μM, 7) 54 μM. (B) DNA binding assays show that both gp33103 and gp33136 bind similarly to a DNA substrate containing PCR amplified DNA from the BPs gene 33–34 intergenic region (S1 Table). Protein concentrations are as shown in A. (C) Size-exclusion chromatograms for gp33103 (orange) and the molecular weight markers (green; 66kDa, 29kDa, 12.4kDa, 6.5kDa) are overlayed. (D) The inset graph shows a standard plot for the molecular mass for the protein standards against the ration of their elution volume (Ve) and void volume (Vo) (blue diamonds); the molecular mass of gp33103 is predicted to be 40.4kDa (orange diamond).
Table 1. Binding affinities of gp33103 to various DNA substrates.
| Mutant Name | Mutation in 33–34 region | Average Kd (Molar) |
|---|---|---|
| Wild type 33–34 region | None | 2.61 x 10−6 |
| 102a | T29489C | 7.05 x 10−6 |
| 102e | A29486C | 7.80 x 10−6 |
| 102j | 151bp Inversion (29282–29432) | 4.23 x 10−6 |
| 102k | 24bp Duplication (29472–29495) | 6.67 x 10−6 |
| 127b | 151bp Deletion (29323–29473) | 9.50 x 10−6 |
| 127c | 33bp Duplication (29495–29527) | 3.50 x 10−6 |
| 127d | 12bp Duplication (29474–29485) | 2.99 x 10−6 |
| 127e | 6bp Duplication (29483–29488) | 3.81 x 10−6 |
| Δ32 Clr1 | T29336C | 1.74 x 10−6 |
| Δ32 Clr4 | G29500A | 1.45 x 10−6 |
| 33A129E Clr5 | C29372T | 2.90 x 10−6 |
| 33A129E Clr6 | T29475C | 2.65 x 10−6 |
| Δ32 Clr8 | T29501C | 2.80 x 10−6 |
| 5–6 intergenic region | None | 2.75 x 10−6 |
| 26–27 intergenic region | None | 9.00 x 10−7 |
| 54–55 intergenic region | None | 2.60 x 10−6 |
| 60–61 intergenic region | None | 3.40 x 10−6 |
| O6-L | None | 1.05 x 10−5 |
| O27-R | None | 1.10 x 10−5 |
| O27-L | None | 3.85 x 10−5 |
| O55-L | None | 8.50 x 10−6 |
| O61-R | None | 1.95 x 10−5 |
| TIR-5 | None | 1.25 x 10−5 |
| TIR-6 | None | 9.50 x 10−6 |
BPs gp33136 is longer than gp33103 because of an additional 33 residues at its C-terminus. The C-terminal extension includes the ssrA tag that targets the protein for proteolysis, and stabilization of the protein by an A135E substitution gives higher levels of lysogeny. However, we noted previously that the gp33136 A135E mutant appears to give a modest increase in activity of the Prep promoter in a reporter fusion assay [13], perhaps providing transcriptional activation that is dependent on the C-terminal 33 residues. We therefore compared the binding profiles of gp33103 and gp33136 for any differences in binding to the 33–34 intergenic region that contains both Prep and PR (Figs 1 and 2B). Both proteins give similar profiles and the additional 33 C-terminal residues in gp33136 do not appear to substantially influence DNA binding. It is likely that any functional activation of Prep results from protein-protein interactions, perhaps reflecting direct contacts between BPs gp33136 and RNA Polymerase.
The 185 bp DNA segment contains only a single copy of the 12 bp sequence 5’-CGACATATGTCG that was previously predicted to be recognized by gp33103, and yet the complexes formed are more varied than would be expected for a single protein-DNA interaction. Presumably, the protein binds to additional sites within this region, binds with varying protein-DNA stoichiometries, or imposes significant DNA distortions. We note that there is a related sequence (5’-CGACATACCGGC) at the left end (33-proximal) of the intergenic region that overlaps the Prep transcription start site (Fig 1B), although the similarity is restricted to the left half of the sequence motif. We therefore sought to dissect the various determinants of gp33103 binding to this region, and to compare this to the binding of gp33103 to the other sites located elsewhere in the BPs genome.
Solution multimeric state of BPs gp33103
First, we determined the oligomeric state of gp33103 in solution. A single protein peak was observed using size-exclusion chromatography, and when compared with protein markers, has an apparent molecular mass of 40.4 kDa (Fig 2C). The monomeric mass of gp33103 is 11.2 kDa, and the simple interpretation is that the major peak corresponds to a gp33103 tetramer, although we cannot rule out the possibility that it is an alternative multimer shaped to give altered elution in the chromatography. We note that the 12 bp operator described previously [13] has dyad symmetry (Fig 1B) consistent with recognition by either a dimer or tetramer of gp33103. The gp33103 elution profile did not show any other prevalent forms and the oligomeric state is quite homogenous.
DNase I footprinting of the 33–34 intergenic region
DNase I footprinting provides further insights into the binding of gp33103 to the 33–34 intergenic region (Fig 3A). Depending on the gp33103 concentration, two different patterns of altered DNase I sensitivity are observed. At intermediate concentrations (5.4 μM– 54 μM) we see prominent protection from DNase I cutting at positions in and around the 12 bp palindromic sequence although some cut sites remain sensitive to DNase I cleavage (Fig 3A). However, there are notable enhancements of DNase I cutting to the left of the 12 bp palindrome situated approximately 20 bp and 30 bp away respectively (Fig 3A, S1 Fig). At the highest concentrations of protein used (160 μM), DNase I protection is more extensive within this region and extends to two regions (designated regions 3 and 4; Figs 1C and 3A) flanking the DNase I enhancement located about 20 bp to the left of the 12 bp palindrome. There is also protection in the gene 33 proximal end of the substrate in regions designated 1 and 2, with apparent DNase I enhancement between them (Figs 1C and 3A). As the conditions for DNase I footprinting and native gel electrophoresis of complexes are somewhat different it is not possible to draw a direct comparison between the two, although it is likely that the slowest moving of the protein-DNA complexes corresponds to the more extensive DNase I protections seen at the highest protein concentration, and that the faster migrating complexes correspond to those seen at intermediate concentrations (Figs 2A and 3A).
Fig 3. BPs gp33103 binds to several regions within the 33–34 intergenic region.

(A) DNase I footprinting assays (panels A and B) were done using 5’ end-labeled probes of the 33–34 intergenic region, which were incubated with increasing concentrations of gp33103, subjected to DNase I cleavage, and run on a denaturing gel alongside a sequencing ladder. Black arrows show the direction of the sequence from 5’ to 3’ for each DNA strand. Regions of protection from DNase I cleavage are indicated by black bars and labeled; the sequence of OR is also shown. Protein concentrations are as follows: 1) none 2) 0.16 μM 3) 0.54 μM, 4) 1.6 μM, 5) 5.4 μM, 6) 16 μM, 7) 54 μM, 8) 160 μM. Two regions of protection observed near Prep, one of which contains ORep (Regions 1 and 2). Three regions of protection are observed near PR (Regions 3 and 4, Region 5), one of which includes OR-R (Region 5). The sequences of the protected regions are shown in Fig 1C. See also S1 Fig (B) EMSAs showing gp33103 binding to 40 bp substrates spanning the 33–34 intergenic region (see Fig 1C). Binding occurs to the TIR-6 fragment containing OR-R and to substrate TIR-5, which contains Region 3 and Region 4 (OR-L) and only one base pair of OR-R. (A). Protein concentrations are as follows: 1) none 2) 0.16 μM 3) 0.54 μM, 4) 1.6 μM, 5) 5.4 μM, 6) 16 μM, 7) 54 μM.
Because of these DNase I protection patterns together with the experiments described below, we propose that the gp33103 binding site at PR spans a larger region than just the 12 bp palindrome, and that this palindrome is equivalent to a half site, of which region 4 (and perhaps part of region 3) constitute the other half site, not withstanding the sequence dissimilarities (see Fig 1C). To simplify the presentation and discussion of data below, we will refer to the 12 bp palindrome as OR-R and the region to the left that includes region 4 as OR-L (Fig 1B) reflecting the right and left half sites of OR respectively. The regions 1 and 2 of protection at Prep will be referred to as ORep.
Binding of gp33103 to subsites in the 33–34 intergenic region
The binding of gp33103 at ORep seen by DNase I footprinting was somewhat unexpected as this region lacks a site equivalent to OR or an obvious OR half site (OR-L or OR-R), although it has a distantly related sequence (Fig 1C). However, occupancy of the ORep site is evident at the highest protein concentration in DNase I footprinting (Fig 3) and may depend on concomitant binding of gp33103 elsewhere in the DNA fragment, such as at OR.
To further explore these interactions we synthesized a series of small (40 bp) dsDNA substrates, containing segments of the 33–34 intergenic region (Fig 1C) and asked whether gp33103 binds and forms protein-DNA complexes (Fig 3C). The protein binds to the TIR-6 substrate (Figs 1C and 3C) containing OR-R to form a single complex, but binds similarly to the TIR-5 substrate to form complexes with similar mobilities (Fig 3C), which was unexpected as the TIR-5 substrate lacks the 12 bp palindromic sequence previously identified as OR [13]. The TIR-5 substrate contains OR-L but does not contain OR-R (see Fig 1C). A plausible explanation is that gp33103 binds as a dimer or tetramer to OR-L and OR-R independently and at reduced affinity, notwithstanding the sequence differences. In this model, binding of a tetramer would involve two unoccupied helix-turn-helix DNA binding domains, which would be available for binding either to another site within the same substrate (perhaps ORep) with introduction of a DNA loop, or by forming intramolecular bridges between two different DNA molecules.
Binding of gp33103 to deletion substrates of the 33–34 intergenic region
To further examine the parts of the 33–34 intergenic region required for binding we generated a series of substrates containing progressive deletions from each end and tested them for binding of gp33103 (Fig 4). One notable observation is that the binding patterns with the Mt12 and Mt13 substrates (Figs 1C and 4) are similar, even though OR-R is present in Mt13 but absent from Mt12. The affinity for Mt12 is slightly reduced relative to that for Mt13, but we assume that the slowest migrating complexes (C4) have similar stoichiometries, with each containing a dimer or tetramer of gp33103 bound at OR in addition to binding elsewhere in the substrate. One of the Mt13 complexes (C3) appears to be absent from the Mt12 substrate, and presumably requires OR-R (Figs 1 and 4). Complete removal of both OR-L and OR-R (substrates Mt10 and Mt11) does not completely eliminate binding and complexes are observed albeit with reduced binding affinity (Fig 4; Table 1), reflecting weaker binding to the left part of the substrate.
Fig 4. BPs gp33103 binds with variable affinities to a series of end-deletion substrates.

Substrates were PCR amplified from the BPs 33–34 intergenic region using primers located at regular intervals from each end to sequentially shorten the 33–34 intergenic region from each end (S1 Table). The sequences included in each substrate are shown as horizontal black lines either above or below a schematic representation of the 33–34 intergenic region. Genes 33 and 34 are represented by blue and orange arrows respectively, and OR and ORep are shown in yellow. Protein concentrations are same as shown in Fig 2A. Protein affinities are shown in Table 1.
Deletion substrates lacking Prep-proximal regions but retaining OR (i.e. Mt6 and Mt7) form similar complexes, although the major complexes have somewhat different relative mobilities (0.69, 0.55, and 0.50 for Mt3, Mt4, and Mt5 respectively), perhaps reflecting DNA distortions (Fig 4). Inclusion of either part (i.e. Mt6) or all (i.e. Mt7) of the putative ORep site imposes little overall change to the pattern of complex formation (Fig 4), even though DNase I footprinting (Fig 3A) shows that gp33103 binds at the higher protein concentration to substrate similar to Mt7; furthermore, gp33103 forms complexes with substrates Mt10 and Mt 11 that lack OR (Fig 4). It is unclear whether gp33103 binds separately at OR and ORep or if one or more tetramers of gp33103 bind simultaneously at ORep and OR to form a DNA loop.
BPs gp33103 introduces a DNA bend when bound to the 33–34 intergenic region.
To further explore the binding interactions between gp33103 and this 33–34 control region, we constructed two series of substrates in plasmid pBEND2 [20], one containing only OR-R and one containing the full 33–34 intergenic region. We then determined the relative mobilities of complexes formed with gp33103 as a function of the position of the sites relative to the ends of the DNA molecules; substrates with a protein-induced bend migrate slowest if the bend is in the center of the substrate (Fig 5). We saw no evidence of DNA bending when only OR-R was present (Fig 5A), but a clear indication of a protein-induced DNA bend with the larger substrate (Fig 5B). No intrinsic bending of 33–34 intergenic region was observed, but bending was indicated in at least two of the protein-DNA complexes (Fig 5B). The magnitude of the overall bend is estimated to be about 40°, although we note that this could arise from multiple protein-DNA interactions. This is consistent with introduction of a bend when gp33103 is bound as a tetramer to OR-L and OR-R, but it is difficult to exclude the possibility of a hairpin-like DNA loop even though the overall bend is less than might be expected. These observations are also consistent with the interpretation that the relative mobilities of the complexes observed with other substrates are influenced by DNA distortions (Fig 4).
Fig 5. BPs gp33103 bends DNA but does not facilitate loop formation.

(A) The 12bp of OR was cloned into plasmid pBend2 [20] using unique restriction enzyme sites SalI and XbaI to create pVMV22. Equal-sized fragments of DNA were excised from pVMV22 with OR located at different positions relative to the edge of the probe by cleaving with appropriate enzymes. BPs gp33103 (0.54μM) was incubated with each radiolabeled probe and run on a native polyacrylamide gel. The only gp33103-DNA complex (C1) does not show any differences in relative mobility with different substrates. (B) A second plasmid containing the entire BPs 33–34 intergenic region (pVMV21) was analyzed similarly, suggesting that a bend of about 40° is introduced into the major complex (equivalent to C-4, Fig 2A) by gp33103 binding. (C) To determine if intermolecular DNA bridges are formed by gp33103, two different sized fragments of DNA containing the 33–34 intergenic region (366 bp and 256 bp) were PCR amplified and radiolabeled (S1 Table). The leftmost six lanes contain only the 366 bp substrate, the middle six lanes contain only the 256 bp substrate, and the rightmost six lanes contain both substrates. Complexes formed with each substrate are indicated by long and short arrows respectively. The concentration of protein in each series of substrates is 1) none, 2) 0.54μM, 3) 1.6μM, 4) 5.4μM, 5) 16μM, 6) 54 μM. Protein affinities are shown in Table 1.
BPs gp33103 does not promote intermolecular bridges
An alternative explanation for the observed gp33103 complexes is that gp33103 tetramers bind simultaneously to two different DNA molecules to promote intermolecular protein-bridges. To test this, we performed DNA binding assays with two different sized DNA fragments, separately and together (Fig 5C). Both DNA fragments give a similar series of complexes, and when mixed together, we observe only a combination of the complexes formed with the individual substrates, and no new complexes with mobilities suggesting that they contain more than one DNA fragment. We conclude that under these conditions although the complexes may have differing numbers of gp33103 protomers, they each contain only a single DNA molecule. We cannot exclude the possibility that bridging interactions are observed under other conditions including high substrate concentration.
Additional gp33103 binding sites in the BPs genome
BPs gp33103 is known to bind to several additional sites within the BPs genome [13] and we investigated whether there are similar complexities to the binding of gp33103 at these sites. Three other instances of sequences identical to OR-R are located in small intergenic regions, between genes 5 and 6, between genes 26 and 27, and between genes 54 and 55 (Fig 6A and 6B). A site with two base pair differences is located within the 60–61 intergenic region (Fig 6). In view of the proposition that the 12 bp palindrome at OR-R corresponds to a half-site for gp33103 binding, we examined the sequences for additional sites related to OR-R. Interestingly, in all four regions we identified, different but related sequences are positioned either 5 bp (O6, O27, O61), or 8 bp (O55) away from the other half site, consistent with the 12 bp palindrome constituting just one of two half sites. We therefore refer to the left and right halves of each of these as O6-L and O6-R, O27-L and O27-R, O55-L and O55-R, and O61-L and O61-R for O6, O27, O55 and O61 respectively (see Fig 6B).
Fig 6. Complete and partial operator sites found throughout the BPs genome.

(A) Map of the BPs genome showing open reading frames (colored boxes) with the positions of each of the operator sites indicated. (B) Sequences of the intergenic regions containing operators. Promoter sequences (green text), start codons of open reading frames (blue text), and the stop codons of open reading frames (red text) are shown. Operator half sites corresponding to the 12 bp sequence 5’-CGACATATGTCG are highlighted in yellow. Operator half sites containing sequence departures within these 12 bp sites are highlighted in orange or pink (reflecting gp33103 binding or no observed binding respectively; see Fig 7). ORep-R and OR-L are highlighted in gray. Confirmed transcription start sites are marked as +1 [[13] L. Oldfield and GFH, manuscript in preparation]. (D) Promoter fusions to a mCherry reporter gene were constructed in an integration-proficient vector, transformed into both M. smegmatis (blue bars) and a BPs lysogen (red bars) and fluorescence levels measured.
To test whether these regions play a role in the phage transcriptional program and are subject to repressor control, each was inserted upstream of a mcherry reporter gene and transformed into M. smegmatis mc2155. The 5–6, 26–27, 54–55, and 60–61 regions all have promoter activity (designated as promoters P6, P27, P55, and P61), although the strengths vary considerably, with P55 being by far the most active, and P27 the weakest (Fig 6C). Putative promoter elements containing -10 and -35 hexameric motifs are predicted for P6, P55, and P61, but not confidently for the weaker P27 (Fig 6B). The promoter-reporter plasmids were also transformed into a BPs lysogen and the promoter activities determined (Fig 6C). The P6, P27, and P55 promoters are clearly down regulated in a lysogen, presumably by gp33103 [few other phage-encoded proteins are expressed in a lysogen as indicated by RNAseq (LMO and GFH, unpublished observations)] as for the PR control [Fig 6C, [13]]. No regulation for either Prep or P61 was observed (Fig 6C).
Using native gel electrophoresis, gp33103 was shown to bind to all four substrates (O6, O27, O55 and O61) to generate a single prominent complex and a minor complex with intermediate mobility, with the exception of O6 and O55 for which additional complexes were observed (Fig 7D, 7G, 7J and 7M). Presumably, the relative simplicity of the binding patterns compared with the 33–34 DNA is because of the lack of additional sites equivalent to ORep. The overall affinities are similar to gp33103 binding to the 33–34 intergenic region (Fig 7A, Table 1), although binding is about 3-4-fold tighter to O27. To dissect out the contribution of the separate potential half sites, substrates were generated in which one half site was specifically ablated so as to leave just the 12 bp palindrome (O6-L, O27-R, O55-L, and for O61, O61-R, S1 Table), and tested for gp33103 binding (Fig 7E, 7H, 7K and 7N). BPs gp33103 binds to each of these to form a single complex, but with substantially reduced affinity (Table 1). We then inserted the other half sites (O6-R, O27-L, O55-R, O61-L) individually into a common sequence context (S1 Table) and tested gp33103 binding (Fig 7F, 7I, 7L and 7O). All of these were bound only very weakly, although complexes were detected with the O27-L substrate (Figs 6B and 7I). These binding data suggest that gp33103 binds to DNA containing two half sites, presumably as a tetramer, and the reduced mobility of the complexes formed by gp33103 and each full site is slower than for complexes with individual half sites. Although we cannot rule out the possibility that these complexes have different protein-DNA stoichiometries, it is plausible that these differences reflect a protein-induced bend when the full site is bound, as seen at OR.
Fig 7. BPs gp33103 binding to full and partial operator sites.
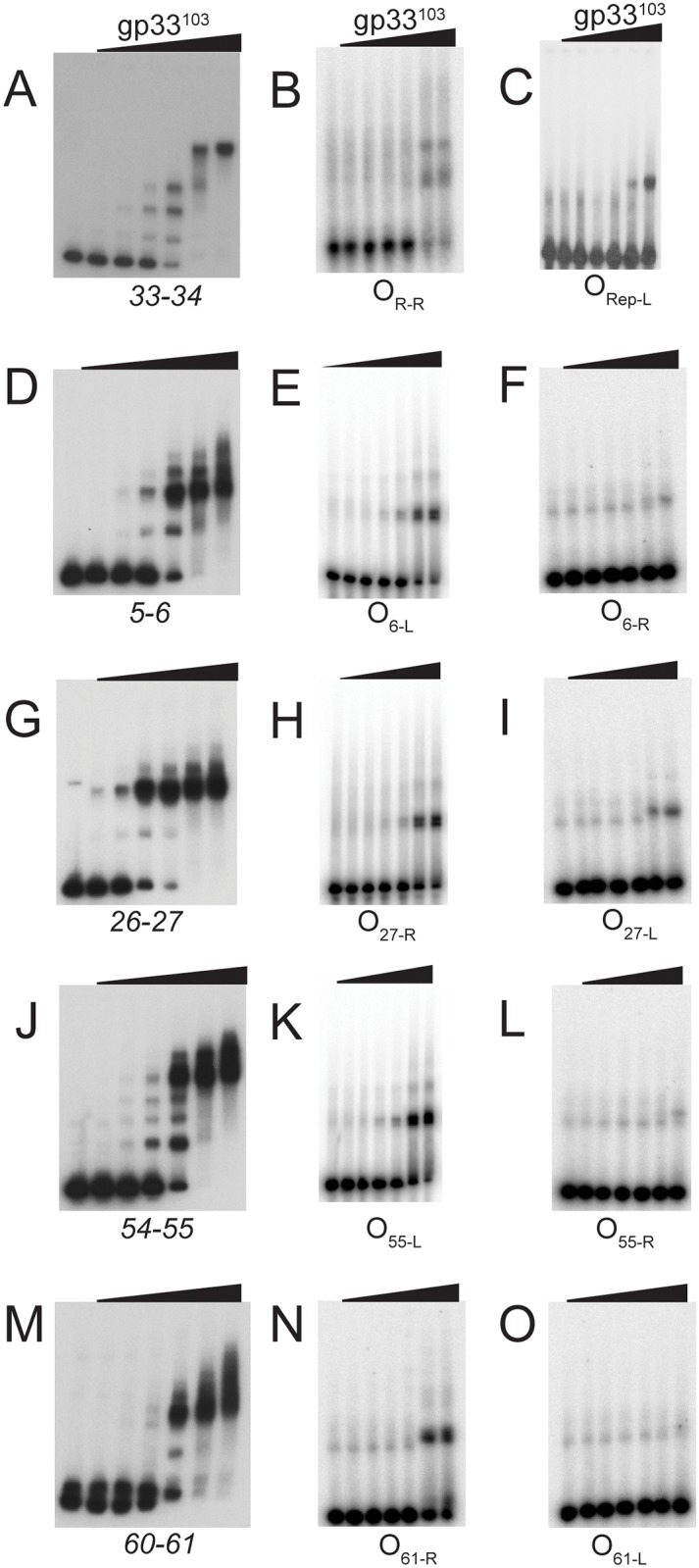
Binding of gp33103 to each of the intergenic regions where full and partial sites are found (see Fig 6; Table 1, S1 Table) are shown in gels A, D, G, J, and M. To examine binding to the O6-L, O27-R, O55-L, and O61-R sites independent from their potential partner sites, 52bp synthetic oligonucleotide substrates were generated with these sites centrally located (S1 Table), and the partner partial sites (O6-R, O27-L, O55-R, O61-L) were mutationally ablated to have no sequence similarity to the 12bp palindrome (gels E, H, K and N). Each of the partially conserved partner sites (O6-R, O27-L, O55-R, O61-L) were placed in a non-native sequence context (42bp substrates; S1 Table) to test for gp33103 binding (F, I, L, and O). Binding profiles of gp33103 to OR-R and ORep-L are shown for comparison (B and C). The concentrations of protein are as follows: 1) none, 2) 0.16μM, 3) 0.54μM, 4) 1.6μM, 5) 5.4μM, 6) 16μM, 7) 54 μM. Protein affinities are shown in Table 1.
Binding of gp33103 to adjacent 12 bp palindromes
To further test the binding of gp33103 to two half sites, we generated a series of 42 bp substrates each containing two 12 bp palindromic sequences spaced either 5 bp or 8 bp apart (Fig 8). We also made derivatives of these in which either the left or right half site was mutationally ablated, and examined gp33103 binding. With a 5 bp inter-site spacing, BPs gp33103 binds to form a single complex which has a slower mobility than those formed when each of the half sites is ablated (Fig 8B). Although the affinities for the three substrates are similar, there is suggestion of cooperative binding to two adjacent sites, as no complex corresponding to single site occupancy is present. A plausible explanation is that the binding energy gained from cooperative binding is balanced by an investment of binding energy into either DNA bending or conformational distortion of the protein. When two 12 bp palindromes are separated by 8 bp (Fig 8A), both the faster and the slower migrating complexes are formed, suggesting lack of cooperative interactions, although the overall affinities are similar to the 5bp spaced substrate suggesting that DNA bending may also be different.
Fig 8. BPs gp33103 binds to two adjacent 12 bp palindromes.
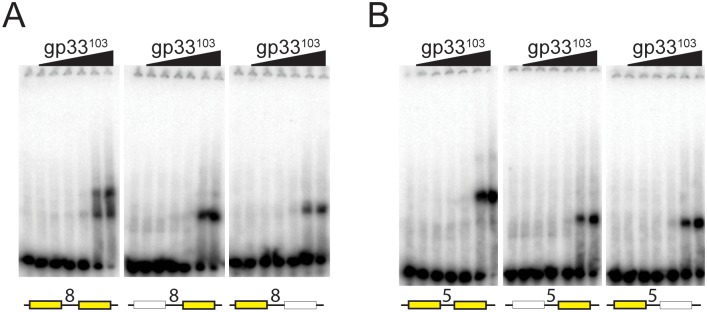
(A and B) EMSAs showing gp33103 binding to substrates containing two 12bp palindromic half sites (yellow boxes) spaced either 8 bp (panel A) or 5 bp apart (panel B). Substrates in which one half site is mutationally ablated is shown as a white box.
BPs gp33103 binding to mutant DNAs conferring a repressor-insensitive phenotype
We previously described the isolation of BPs mutants that are capable of infecting a repressor-expressing strain, and behave as though they are repressor-insensitive. Two such mutants (102a and 102e, Fig 9A) were shown to have point mutations in OR-R that reduce the affinity of gp33103 by about 3-10-fold (depending on specific DNA substrate used), apparently sufficient for the commitment to lytic growth to outcompete the resident repressor [13]. Three additional mutants (Clr4, Clr6 and Clr8, Fig 9) contain single substitutions in the regions immediately flanking OR (Fig 9A; S1 Table). Two of these (Clr4, Clr8) are to the right of OR-R in the promoter -10 region such as to influence PR promoter activity, and to give elevated gp34 synthesis which is predicted to promote commitment to lytic growth [13]. One mutant (Clr6) is in the -35 region of PR but is not predicted to influence promoter activity [19]. However, the mutation lies within the putative OR-L site and could therefore influence gp33103 binding. We examined gp33103 binding to these three mutants (Fig 8) and observed that all three form complexes with similar overall affinity as to the wild-type substrate. One interpretation is that repression of transcription requires a specific tertiary structure, and that point mutations within the binding site influence this structure, although binding affinity may be little different.
Fig 9. BPs gp33103 binding to substrate variants of the 33–34 intergenic region derived from repressor-insensitive BPs mutants.

(A) Locations of the mutations in each substrate (see Table 1). (B) Binding profiles of gp33103 to each 33–34 substrate. The concentrations of protein are as follows: 1) none, 2) 0.16μM, 3) 0.54μM, 4) 1.6μM, 5) 5.4μM, 6) 16μM, 7) 54μM. Protein affinities are shown in Table 1.
We tested the binding of gp33103 to other mutants with repressor-insensitive phenotypes, including other point mutations as well as insertions, deletions, and inversions (Fig 9B). Two mutants (Clr1 and Clr5) have mutations distal to OR and we observe that both form complexes with similar relative mobility and affinity as the parent substrate, and the basis for their phenotypes is not clear (Fig 9, Table 1).
Six other repressor-insensitive mutants have more substantial DNA rearrangements in the 33–34 region (Fig 9A; Table 1). Four of these (102k, 127c, 127d, 127e) have small duplications near the PR/OR region, and gp33103 binds to all of them with similar affinity to the wild-type substrate (Fig 9B). However, the profiles of the complexes differ to that of the wild-type, consistent again with the hypothesis that formation of a protein-DNA complex with a specific configuration is important for regulation. A mutant (127b) containing a large deletion and missing much of the intergenic region including ORep and OR-L but retaining OR-R forms complexes with much faster relative mobility that those seen with other substrates, indicating that perhaps either a monomer or dimer of gp33103 is binding, although the basis for such an unusual property is unclear.
Discussion
We have described here the unusual binding properties of the repressor encoded by mycobacteriophage BPs. The repressor is non-canonical in that it is encoded in two forms, a virally-encoded product 136-residues long (gp33136), and prophage-encoded version (gp33103) that is 33 residues shorter as a consequence of integrative recombination at the attP site situated within the gene 33 open reading frame. The two proteins bind similarly to a DNA substrate containing the regulatory region between genes 33 and 34, but the binding pattern is complex with multiple protein-DNA complexes observed (Fig 2). The binding affinity is surprisingly weak relative to other phage repressors, although the gp33103 and gp33136 binding affinities are similar to each other, and other preparations have similar affinities. The gp33 preparations retain five non-native residues at the N-terminus that we have not been able to remove without encountering insolubility, and these could influence the binding affinity. However, we also note that BPs lysogens have high levels of spontaneous lytic induction, which could reflect relatively weak binding of the repressor in vivo.
BPs gp33103 binds to a total of five loci within the BPs genome and three promoters (P6, P27, and P55) in addition to PR. Alignment of the six putative DNA binding sites (O6, O27, ORep, OR, O55, and O61) shows the juxtaposition of the half sites within each operator (Fig 10A). In three of the sites (O6, O27, and O55), one half site contains the 12 bp palindrome (O6-L, O27-R, and O55-L) and is easily recognizable, and the other half sites (O6-R, O27-L, and O55-R) contain sequence departures at two or three positions (Fig 10A). At O61, both half sites have sequence departures from the consensus (two in O61-R and and four in O61-L) although gp33103 binds with similar affinity as it does to O6 and O55 (Fig 7). OR is peculiar in that it contains the easily recognizable OR-R, but the other half site is only distantly related, although gp33103 clearly binds to it, even in the absence of OR-R (Fig 3). We propose the sequence 5’-GCGCATTTTCCA for OR-L which has only five bases of the 12 bp palindrome (Fig 10A). However, this is spaced five bases away from OR-R, which is similar to the geometries of O6, O27, and O61 and which appears to permit cooperative binding (Fig 8). We also note that position eight of this proposed OR-L is part of the PR -35 motif, and substitution with a G base both increases promoter activity, but also reduces the efficiency of repression [19], consistent with the role of OR-L in binding and regulation. The sequences of the ORep half sites are the most divergent, and the best alignment suggests that ORep-L and ORep-R have eight and seven positions conserved respectively, spaced eight bases apart with a geometry similar to that of O55. DNase I footprinting is consistent with binding to these sites, and complexes are observed with the small substrate tested that contains the complete sequence (Mt10, Fig 4). At least at higher protein concentrations, binding of gp33103 to the 33–34 intergenic regions is expected to involve occupancy of both OR and ORep. Although we cannot exclude the possibility that gp33103 forms a DNA loop by simultaneous occupancy of OR and ORep, the 113 bp OR-ORep intersite spacing is substantially below the persistence length of DNA, and DNA looping would likely require either phased A/T tracts to promote DNA bending or an accessory DNA bending protein such as HU.
Fig 10. Site alignments and models of BPs gp33103 binding.

(A) Alignment of all operators found in the BPs genome. Bases in black correspond to the consensus 12bp half site sequences. Bases in gray indicate positions that are present at that position in four or more sites. No shading indicates bases that are present in three or fewer sites. The consensus sequence is shown below the alignment. (B-I) Various plausible configurations of gp33103 binding to OR (and related sites) are shown. See text for details.
The nature of the specific protein-DNA interactions suggests there are several plausible models to consider (Fig 10). The previously identified 12 bp palindromic sequence 5’-CGACATATGTCG is typically associated with a related sequence spaced 5–8 bp away, and surprisingly even though the OR-L site is a highly redundant version with only five of the 12 positions conserved (Fig 10A), there is good evidence that gp33103 binds to it, even when OR-R is removed (Fig 3C). One set of models includes binding of two protomers to each of the 12 bp sequences within an overall site (e.g. OR or O27) (Fig 10B–10E) with the stoichiometry reflecting either a dimer bound to each 12 bp half-site (Fig 10B and 10C), or a tetramer bound to each 12 bp half site (Fig 10D and 10E). The DNA could be relatively straight (Fig 10B and 10D), or could include a modest DNA distortion (Fig 10C and 10E), although we favor the bent DNA models (Fig 10C and 10E) both because of the observed bends seen in Fig 5 and the DNase I enhancement observed at the centers of both OR-L and OR-R (Fig 3A). A second set of models proposes that a single protomer recognizes each 12 bp motif (Fig 10F–10I), with corresponding stoichiometries and bending considerations (Fig 10F–10I) as described for the two-protomer models (Fig 10B–10E). Although 12bp is a somewhat larger segment of DNA than usually recognized by a helix-turn-helix DNA binding motif, it is not unprecedented, and we note that a protomer of γδ resolvase recognizes a similarly-sized half site with contacts spanning both major and a minor grooves of the DNA [21, 22]. The palindromic nature of many of the 12 bp half sites supports two-protomer/half-site models, although we note that the symmetry with the 12 bp sequences is not conserved in all sites.
There is evidence of cooperativity in the occupancy of two 12 bp half sites, but spacing between the sites plays an important role. Cooperative binding appears to be supported by a 5 bp inter-site spacing (Fig 8) as in OR, but could occur between single protomers, dimers, or tetramers bound at each half site (Fig 10). However, investment of binding energy into DNA bending and the DNase I hypersensitivity seen within OR and ORep supports models that include an inter-site bend (Fig 10C, 10E, 10G, and 10I). We note that although models in which a protein dimer (as in Fig 10G) binds to two differently spaced half sites (e.g. 5 and 8 bp) is somewhat unusual, it is observed in the binding of γδ resolvase, where the two half sites within each of three binding sites are separated by 4, 10, and 1 bp respectively [22].
Despite the complexity of the binding profile of gp33103 to the 33–34 intergenic region, formation of properly configured complexes is necessary for normal repression in a lysogen. BPs mutants with repressor-insensitive phenotypes that have mutations mapping to the 33–34 intergenic region demonstrate the importance of this sequence and the ability of gp33103 to bind to it (Fig 9). Loss of normal repression does not closely correlate with large changes in binding affinity, and it is likely that relatively subtle sequence changes give rise to altered configurations that interfere with repression. This notwithstanding, some DNA substrates of the repressor-insensitive mutants have binding patterns that are more consistent with binding of monomers or dimers (e.g. 127b, Fig 9B), and it is unclear what determines this behavior.
The system of integration-dependent immunity seen in BPs and other phages offers a quite different perspective on phage life style decision making than seen in phage lambda and its relatives, and may represent an ancestral state for temperate phages [12]. It is perhaps not surprising that the repressor has non-canonical binding properties including tetramerization and binding to dispersed sites in the phage genome (Fig 10). Because the repressor can be expressed in two forms that differ in their C-termini, this raises the possibility that the virally-encoded product gp33136 forms mixed tetramers with the shorter protein (gp33103) and influences functionality even if not binding per se. Thus the observed tetramerization and DNA binding profiles may play roles in modulating the overall genetic switch in these phages.
Materials and Methods
Expression and Purification of gp33103 and gp33136
The gp33103 and gp33136 genes were PCR amplified from a BPs lysate using primers 5’-CAA TCG CCC ATA TGT CGC AAG CAT TCG -3’ / 5’- GAC TAC AAG CTT TCA GAA GGT TGG GGG TTC GA 3’and 5’-CAA TCG CCC ATA TGT CGC AAG CAT TCG -3’/ 5’- TGC CGG AAG AAG CTT TCA CGA CGC TTT ATC C -3’ respectively, which amplified the genes with NdeI recognition sites at the 5’ end of the gene and HindIII recognition sites at the 3’ end. Each gene was cloned into a maltose-binding fusion vector (pLC3) that was linearized with NdeI and HindIII sites for directional cloning, creating two plasmids pVMV20 and pVMV27 for gp33103 and gp33136 respectively. pVMV20 and pVMV27 were transformed into BL21(DE3)star chemically competent cells (Invitrogen) and grown until cultures reached an OD600 of 0.4–0.6. Protein expression was induced with 1 mM IPTG at 17°C overnight. Cells were pelleted and frozen at -80°C. Thawed cell pellets were resuspended in 5mL per gram of Lysis Buffer (50 mM Tris pH 8.0, 500 mM NaCl, 8% glycerol, 1 mM EDTA and 1 mM β-mercaptoethanol) and lysed in 200 mL fractions by sonicating 10 times for 10 sec at 30% output with 30 sec of cooling on ice in between bursts. Pooled cell lysates were cleared by centrifugation at 30,000 x g for 40 min at 4°C. Fusion proteins were extracted from soluble cell lysates using amylose resin affinity chromatography (Invitrogen) and the MBP tag was cleaved from the proteins of interest with TEV protease during overnight dialysis at 4°C. MBP and TEV protease contain C-terminal His tags and were removed from the gp33 proteins using nickel affinity chromatography. The flow through containing pure gp33 proteins was dialyzed into a storage buffer (50 mM Tris pH 8.0, 500 mM NaCl, 50% glycerol, 1 mM EDTA, 1 mM BME) and stored at -20°C.
DNA Binding Assays
DNA binding assays were carried out according to standard protocols [23]. Briefly, DNA substrates (either PCR substrates or annealed complimentary synthetic oligonucleotides; S1 Table) were 5’ radiolabeled using ATP, [γ-32P] with T4 polynucleotide kinase (Roche). Binding reactions contained 5–20 cps radiolabeled DNA probe, 1 μg non-specific calf thymus DNA, and varying concentrations of protein in a binding buffer containing 20 mM Tris pH 7.5, 10 mM EDTA, 25 mM NaCl, 10 mM spermidine, and 1 mM DTT for a total volume of 10 μl. Reactions were incubated at room temperature for 30 min and the resulting protein DNA complexes were resolved on a 5% native gel and detected using autoradiography and a phosphorimaging plate.
DNase I Footprinting
Footprinting assays were carried out as previously described [23]. Briefly, binding reactions were carried out in a final volume of 50 μl containing various concentrations of protein, 20cps radio labeled probe, 25 mM Tris-HCL pH 8.0, 50 mM KCl, 6.25 mM MgCl2, 0.5 mM EDTA, 10% Glycerol, 0.5 mM DTT. Binding reactions were incubated at room temperatures for 30min. After incubation, 50 μl of a solution containing 5 mM CaCl2 and 10 mM MgCl2 was added to the samples and incubated for 1 minute. Samples were treated with 1.5U DNaseI for exactly one minute, then the digestion reaction was stopped by addition of 90μl of a pre-warmed (37°C) Stop solution (200 mM NaCl, 30 mM EDTA, 1% SDS, 100 μg/mL yeast RNA). Samples were PCI (Invitrogen) extracted and ethanol precipitated. Samples were resuspended in 2–3 μl formamide loading buffer and heated to 95°C for 2 minutes before loading onto a 6% polyacrylamide 7 M urea denaturing gel for resolution. Bands were detected using autoradiography or exposed on a phosphorimageing plate and detected on a FLA-5100; FujiFilm imaging system.
Size Exclusion Chromatography
Purified gp33103 was dialyzed into a buffer containing 10 mM Tris, pH 8.0, 1 mM BME, 500 mM NaCl, and 4.5% glycerol. 1.2 mL of protein was run over a G-120 column using FPLC, 0.5 ml elution fractions were collected and peaks was identified with UV 280 absorbance measurements. Gel filtration molecular weight markers (Sigma-Aldrich) were resuspended in the same buffer at manufacturer recommended concentrations and run over the same column. The molecular mass for the gel filtration standards were plotted against elution volume (Ve) over void volume (Vo) to determine the molecular mass of gp33103 based on its elution volume.
Fluorescent Reporter Assays for Promoter Strength
The vectors were constructed by creating transcriptional fusions of regions of the BPs genome with predicted promoter elements to a codon-optimized mCherry fluorescence gene. The promoter-mCherry vectors were transformed into electrocompetent M. smegmatis mc2155 and a BPs lysogen of M. smegmatis mc2155, as previously described [24]. Fluorescence assays were performed as previously described [19]. Briefly, transformants were grown in biological triplicates under selection shaking at 37°C for 48 hours. From these cultures, 50 μl was aliquoted into 96-well plates (Falcon). Fluorescence was detected at 532 nm (FLA-5100; FujiFilm) and normalized to the optical density at 595 nm (EL800 Universal Microplate Reader; Bio-Tek Instruments) of the aliquot to account for cell density. Fluorescence units were reported as (LAU)/mm2)/OD595nm. Graphs display the mean fluorescence units ± 95% confidence interval.
Supporting Information
DNase I footprint used the same substrate and conditions as shown in Fig 3A, except that this is a separate experiment with a clearer DNA ladder.
(PDF)
(DOCX)
Acknowledgments
We thank Andrea Berman and colleagues for help with size exclusion chromatography and Andrew VanDemark and colleagues with assistance in purification of gp33103 and gp33136, as well as their kind gift of the pBEND2 vector. We also thank Travis Mavrich for comments on the manuscript.
Data Availability
The data are all contained within the paper and its Supporting Information files.
Funding Statement
This work was supported by NIH GM093901. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Ptashne M. A Genetic Switch. Oxford/Cambridge: Blackwel Science Ltd and Cell Press; 1987. [Google Scholar]
- 2. Little JW. Evolution of complex gene regulatory circuits by addition of refinements. Curr Biol. 2010;20(17):R724–34. Epub 2010/09/14. S0960-9822(10)00773-6 [pii] 10.1016/j.cub.2010.06.028 . [DOI] [PubMed] [Google Scholar]
- 3. Ptashne M, Johnson AD, Pabo CO. A genetic switch in a bacterial virus. Sci Am. 1982;247(5):128–30, 32,, 34–40. [DOI] [PubMed] [Google Scholar]
- 4. Lewis D, Le P, Zurla C, Finzi L, Adhya S. Multilevel autoregulation of lambda repressor protein CI by DNA looping in vitro. Proc Natl Acad Sci U S A. 2011;108(36):14807–12. Epub 2011/08/30. 1111221108 [pii] 10.1073/pnas.1111221108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hatfull GF, Hendrix RW. Bacteriophages and their Genomes. Current Opinions in Virology. 2011;1, 298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Smith MC, Burns RN, Wilson SE, Gregory MA. The complete genome sequence of the Streptomyces temperate phage phiC31: evolutionary relationships to other viruses. Nucleic Acids Res. 1999;27(10):2145–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sinclair RB, Bibb MJ. The repressor gene (c) of the Streptomyces temperate phage phi c31: nucleotide sequence, analysis and functional cloning. Mol Gen Genet. 1988;213(2–3):269–77. . [DOI] [PubMed] [Google Scholar]
- 8. Hatfull GF, Sarkis GJ. DNA sequence, structure and gene expression of mycobacteriophage L5: a phage system for mycobacterial genetics. Mol Microbiol. 1993;7(3):395–405. Epub 1993/02/01. . [DOI] [PubMed] [Google Scholar]
- 9. Donnelly-Wu MK, Jacobs WR Jr, Hatfull GF. Superinfection immunity of mycobacteriophage L5: applications for genetic transformation of mycobacteria. Mol Microbiol. 1993;7(3):407–17. . [DOI] [PubMed] [Google Scholar]
- 10. Smith MC, Owen CE. Three in-frame N-terminally different proteins are produced from the repressor locus of the Streptomyces bacteriophage phi C31. Mol Microbiol. 1991;5(11):2833–44. . [DOI] [PubMed] [Google Scholar]
- 11. Brown KL, Sarkis GJ, Wadsworth C, Hatfull GF. Transcriptional silencing by the mycobacteriophage L5 repressor. Embo J. 1997;16(19):5914–21. Epub 1997/10/06. 10.1093/emboj/16.19.5914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Broussard GW, Hatfull GF. Evolution of genetic switch complexity. Bacteriophage. 2013;3(1):e24186 10.4161/bact.24186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Broussard GW, Oldfield LM, Villanueva VM, Lunt BL, Shine EE, Hatfull GF. Integration-dependent bacteriophage immunity provides insights into the evolution of genetic switches. Mol Cell. 2013;49(2):237–48. 10.1016/j.molcel.2012.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hatfull GF. The secret lives of mycobacteriophages. Adv Virus Res. 2012;82:179–288. 10.1016/B978-0-12-394621-8.00015-7 . [DOI] [PubMed] [Google Scholar]
- 15. Pope WH, Bowman CA, Russell DA, Jacobs-Sera D, Asai DJ, SEA-PHAGES, et al. Whole genome comparison of a large collection of mycobacteriophages reveals a continuum of phage genetic diversity eLIFE. 2015;In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pope WH, Jacobs-Sera D, Russell DA, Peebles CL, Al-Atrache Z, Alcoser TA, et al. Expanding the Diversity of Mycobacteriophages: Insights into Genome Architecture and Evolution. PLoS ONE. 2011;6(1):e16329 10.1371/journal.pone.0016329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Petrova ZO, Broussard GW, Hatfull GF. Mycobacteriophage-repressor mediated immunity as selectable genetic markers: Adephagia and BPs repressor-selection. Manuscript submitted. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dedrick RM, Marinelli LJ, Newton GL, Pogliano K, Pogliano J, Hatfull GF. Functional requirements for bacteriophage growth: gene essentiality and expression in mycobacteriophage Giles. Mol Microbiol. 2013;88(3):577–89. 10.1111/mmi.12210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Oldfield LM, Hatfull GF. Mutational Analysis of the Mycobacteriophage BPs Promoter PR Reveals Context-Dependent Sequences for Mycobacterial Gene Expression. J Bacteriol. 2014;196(20):3589–97. 10.1128/JB.01801-14 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zwieb C, Adhya S. Plasmid vectors for the analysis of protein-induced DNA bending. Methods Mol Biol. 2009;543:547–62. Epub 2009/04/21. 10.1007/978-1-60327-015-1_32 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rimphanitchayakit V, Grindley ND. Saturation mutagenesis of the DNA site bound by the small carboxy-terminal domain of gamma delta resolvase. Embo J. 1990;9(3):719–25. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grindley ND, Whiteson KL, Rice PA. Mechanisms of site-specific recombination. Annu Rev Biochem. 2006;75:567–605. . [DOI] [PubMed] [Google Scholar]
- 23. Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, et al. Current Protocols in Molecular Biology. New York: Wiley Intersciences; 1996. [Google Scholar]
- 24. Bibb LA, Hatfull GF. Integration and excision of the Mycobacterium tuberculosis prophage-like element, phiRv1. Mol Microbiol. 2002;45(6):1515–26. . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
DNase I footprint used the same substrate and conditions as shown in Fig 3A, except that this is a separate experiment with a clearer DNA ladder.
(PDF)
(DOCX)
Data Availability Statement
The data are all contained within the paper and its Supporting Information files.