

Abstract
We describe an optimized system for the easy, effective, and precise modification of the Escherichia coli genome. Genome changes are introduced first through the integration of a 1.3 kbp Landing Pad consisting of a gene conferring resistance to tetracycline (tetA) or the ability to metabolize the sugar galactose (galK). The Landing Pad is then excised as a result of double-strand breaks by the homing endonuclease I-SceI, and replaced with DNA fragments bearing the desired change via λ-Red mediated homologous recombination. Repair of the double strand breaks and counterselection against the Landing Pad (using NiCl2 for tetA or 2-deoxy-galactose for galK) allows the isolation of modified bacteria without the use of additional antibiotic selection. We demonstrate the power of this method to make a variety of genome modifications: the exact integration, without any extraneous sequence, of the lac operon (~6.5 kbp) to any desired location in the genome and without the integration of antibiotic markers; the scarless deletion of ribosomal rrn operons (~6 kbp) through either intrachromosomal or oligonucleotide recombination; and the in situ fusion of native genes to fluorescent reporter genes without additional perturbation.
Introduction
The ability to introduce genes to an organism or alter their coding sequences is one of the fundamental tools of modern biology. While in many cases genes can be easily manipulated and expressed from extrachromosomal plasmids, it is frequently necessary or desirable to edit the organism’s chromosome directly; for example, in the generation of knockout mutants where the coding sequence of a gene is disrupted or eliminated from the organism entirely. As a consequence, a wide variety of tools for the molecular editing of bacterial chromosomes has been developed to generate various types of genomic modifications. These tools employ, for example, homologous recombination (e.g., recombineering, [1], KIKO [2], FRUIT [3], and PLRS [4]), the cleavage or excision of a target genomic sequence through the nuclease activity of homing endonucleases (e.g., gene gorging [5], MAGIC [6], ALFIRE [7], and earlier versions of Landing Pad technology [8, 9]), phage-derived integrases (CRIM, [10]; clonetegration [11]; ΦC31 [12–15]), and CRISPR-Cas9 based systems [16–20]. Additional tools for genome editing in eukaryotes include engineered zinc-finger nucleases (ZFNs, [20, 21]) and transcription activator-like effector nucleases (TALENs, [20, 22, 23]).
In many cases, the system used is dictated by the type of genomic modification desired. Recombineering is well suited for the integration of small DNA fragments into specific genomic locations, making it ideal for introducing point mutations, performing deletions, or integrating short coding sequences. Conversely, phage-based systems are applicable for the integration of large constructs at preexisting phage attachment sites in the genome; however, they are generally inapplicable to other tasks, such as targeting insertions to specific genomic loci or the generation of knockout mutants. Nuclease-assisted recombineering, such as CRISPR-Cas9 systems and Landing Pad technology, combine the benefits of both recombineering and phage-based editing methods: they are able to easily introduce or delete large coding sequences at any desired genomic location and in any orientation. They differ only in how the nuclease activity is targeted to the genomic location to be edited. CRISPR-Cas9 systems require the in vivo expression of a guide RNA complementary to the targeted location. The Landing Pad system, on the other hand, is guided by the recombineering of a short “landing pad” including unique recognition sites for the homing endonuclease I-SceI at the desired location.
Here, we describe an optimized Landing Pad system [8, 9] of three engineered plasmids (Fig 1) that allows for the precise modification of the E. coli genome in a wide variety of ways. A helper plasmid, pTKRED [8], allows the inducible expression of the recombinogenic λ-Red enzymes [1, 24–29], the homing endonuclease I-SceI [5, 9, 30], and RecA. The plasmid pTKLP serves as a PCR template for the amplification of a 1.3 kbp “landing pad”, which consists of a gene conferring either resistance to tetracycline (tetA) or the ability to metabolize the sugar galactose (galK, [1, 5]) flanked by I-SceI recognition sites and standardized sites for priming and homologous recombination (LP1 and LP2 in Fig 1). Because of its small size, the landing pad is easily recombineered into the desired location in the genome [8] by λ-Red mediated homologous recombination, and serves as a subsequent target for cleavage by I-SceI and recombination by λ-Red. Successful integration of the landing pad can be positively selected for through growth on medium containing tetracycline (for tetA landing pad) or galactose as the sole carbon source (for galK landing pad). Finally, the ultimately desired modification can be accomplished through excision of the landing pad and/or replacement with a DNA fragment carrying the modification; if this requires the integration of a large fragment (> ~2.5 kbp), the donor plasmid pTKDP, which harbors the integration fragment, is co-transformed into the cells and digested in vivo by I-SceI. In either case, homologous recombination results in repair or replacement of the excised landing pad with the desired integration fragment or modified sequence.
Fig 1. Plasmids of the Landing Pad System.

The helper plasmid, pTKRED, expresses I-SceI (inducible with L-arabinose) and the λ-Red enzymes (inducible with IPTG), and has a temperature sensitive pSC101 origin of replication that can be cured by growth at 42°C. The LP template plasmid, pTKLP, serves as a PCR template for amplification of either the tetA or galK landing pad, and carries a R6Kγ pir +-dependent origin of replication. The donor plasmid, pTKDP, serves as a fragment donor for the integration of large constructs that have been cloned into the purple region of the plasmid, guided by recombination with landing pad LP1 and LP2 sequences, or custom homology regions as described in this report. The sequence sizes given are for pTKLP-tetA and pTKDP-neo; tetA is exactly replaced with galK in pTKLP-galK, and neo is exactly replaced with various antibiotic resistance genes for alternate versions of pTKDP. Small green boxes are I-SceI restriction sites; Landing Pad Regions 1 and 2 are small red boxes labeled LP1 and LP2 respectively.
The efficiency of selection for successfully modified cells is enhanced through negative selection against retention of the landing pad. In the case of tetA, the bacteria are inoculated into medium containing NiCl2 after the integration step; NiCl2 is selectively lethal to E. coli expressing tetA [31], and hence those cells which are unsuccessfully modified and retain the tetA landing pad are eliminated from the population. Alternatively, the galK landing pad can be selected against by growth of the bacteria in the presence of 2-deoxy-galactose (DOG, [1, 5]). The enzyme product of galK, galactokinase, phosphorylates DOG into the non-metabolizable product 2-deoxy-galactose-1-phosphate, which builds up to lethal levels in those cells retaining the landing pad. In particular, the highly efficacious selection and counterselection for galK using galactose and DOG [1, 5], combined with the lethality of chromosomal double breaks caused by I-SceI and subsequent rescue by the repair of the break by successful modification [8, 9], allows the precise editing of the genome and integration of large constructs without the accompanying integration of any antibiotic resistance genes into the host genome at any point. Such an antibiotic-free approach can be advantageous when, for example, working with potentially pathogenic strains or species.
Furthermore, the plasmids pTKLP and pTKDP incorporate selectable countermeasures against undesired transformation or retention of the plasmids, eliminating requirements for tedious and time consuming screening [8]. pTKLP contains the R6Kγ origin of replication, and can therefore only be stably transformed and maintained in cells expressing the Pi protein required for R6Kγ replication [10]. As wildtype K-12 MG1655 and many other commonly used E. coli strains do not express Pi, this ensures that cells positively selected for tetA or galK expression are the result of successful incorporation of the landing pad rather than transformation of residual pTKLP used as PCR template. For pTKDP, the unintegrated plasmid backbone constitutively expresses levansucrase encoded by the gene sacB [32–35], which catalyzes the hydrolysis of sucrose into non-metabolizable levans. Therefore, when sucrose is added to the medium during the counterselection step, those cells which are not cured of pTKDP by I-SceI are lysed as a result of the buildup of levans within the periplasmic space [35].
We demonstrate the power of this single method to perform a variety of genome modifications that previously required the application of a variety of distinct tools and techniques. Specifically, we perform: [1] the integration of the entire lac operon (~6.5 kbp) into any desired location of the genome without the introduction of any extraneous sequence or the integration of any antibiotic resistance genes into the genome; [2] the direct, in situ fusion of native chromosomal genes to fluorescent reporter genes without any additional perturbation; and [3] the scarless deletion of ribosomal rrn operons (~6 kbp) through either intrachromosomal homologous recombination or recombination with oligonucleotides.
Materials and Methods
Bacterial strains
Strains used were wildtype E. coli K-12 MG1655 (Coli Genetic Stock Center). For exact integrations of the entire lac operon, an MG1655 strain in which the native lac operon has been deleted was used ([8], henceforth referred to as MG1655 Δlac; deletion including genomic locations 361249–367510). For antibiotic-free integrations with galK as a selection marker, an MG1655 Δlac strain where the native galK sequence was deleted using the method of Datsenko and Wanner [25] was used (referred to throughout as MG1655 Δlac ΔgalK; deletion including genomic locations 788831–789979). We have additionally generated an MG1655 ΔgalK strain for general use. Annotated sequences for pTKRED, pTKLP-tetA, pTKLP-galK, pTKDP-neo, pTKDP-cat, pTKDP-hph, and pTKDP-dhfr are available as Genbank accession numbers GU327533, KR071151, KR071150, KR071149, KR071146, KR071148, and KR071147 respectively.
Preparation of competent cells and transformation for recombineering
Cells competent for transformation by electroporation were prepared by inoculating an overnight culture into 30 ml of Super Optimal Broth (SOB medium) in a baffled 125 ml Erlenmeyer flask with appropriate antibiotics; if the cells were to be used for recombineering with pTKRED, 2 mM IPTG was also added to the medium at the time of inoculation. These cultures were grown at 30°C in a New Brunswick C76 shaking water bath until OD600 ~0.6, at which point the cultures were placed on ice. The cells were made electrocompetent by centrifugation (5 min at 5,000 rpm) and washing with sterile ice-cold 10% v/v glycerol three times. 100 μl of competent cells were then mixed with ~100 ng of DNA in a 5 ml polystyrene round bottom tube (Falcon) on ice. This mixture of DNA and competent cells was transferred to a 0.1 cm gap electroporation cuvette (USA Scientific) and shocked at 2.0 kV, 25 uF, 200 Ω in a Bio-Rad Micropulser electroporation apparatus. 1 ml of SOB medium was immediately added and transferred back to the 5 ml Falcon tube; the resulting culture was allowed to recover for four hours in a 30°C shaking water bath. At this point, 500 μl of culture was spun down in a table top microcentrifuge (5 min at 10,000 rpm), the supernatant dumped, the cells resuspended in the residual medium and spread on Lysogeny Broth (LB) plates containing the appropriate antibiotic, and then placed into a 30°C air incubator overnight. The remainder of the culture was allowed to recover overnight at room temp on the benchtop. The next day, if no colonies had grown on the plates, the remainder of the culture was spun down and plated in a similar fashion.
Construction of the donor plasmid pTKDP
To construct improved versions of the pTKIP plasmid, the donor plasmid in the previously reported integration scheme [8, 9], the sacB gene, conferring sensitivity to the sugar sucrose [32–35], and its promoter were amplified from plasmid pKO3 [36] by PCR using primers containing NdeI restriction sites. The PCR product was PCR purified (QIAquick) and digested with NdeI. The plasmid pTKIP-neo was also digested with NdeI, dephosphorylated with Antarctic phosphatase (New England Biosciences), and gel purified (QIAquick). The plasmid was ligated together with the sacB fragment, forming the plasmid pTKDP-neo.
Versions of pTKDP with alternate antibiotic resistances (pTKDP-cat chloramphenicol resistant; pTKDP-hph hygromycin B resistant; pTKDP-dhfr trimethoprim resistant) were constructed through recombineering [1, 25]. First, the alternate antibiotic resistance genes were amplified from the corresponding pTKIP plasmid using primers including 50 bp of the sequence of pTKDP flanking either side of the gene. The resulting PCR product was checked for the correct size via agarose gel electrophoresis and PCR purified. Next, pTKDP was transformed into the recombineering strain SW102 [1], and the resulting strain was transformed with the purified PCR products and plated onto LB agar containing the desired new antibiotic. The new pTKDP plasmids from colonies growing on these plates were purified (QIAprep) and verified by sequencing (ACGT Inc).
Construction of the landing pad template plasmids pTKLP-tetA/galK
To construct plasmid pTKLP-tetA, the tetA landing pad cassette was purified from plasmid pTKS/CS [8] by digestion with I-SceI (NEB) and subsequent gel purification. A plasmid backbone containing the R6Kγ replication origin was amplified from pKD3 (2) using primers including I-SceI restriction sites and the landing pad sequences LP1 and LP2. This backbone was PCR purified, digested with I-SceI, dephosphorylated with Antarctic phosphatase, and gel purified. The backbone and landing pad cassette were ligated together and transformed into strain BW23474 [10], which constitutively expresses the Pi protein required for R6Kγ replication maintenance. Plasmid pTKLP-galK was created in a similar fashion. galK was amplified from wildtype MG1655 by colony PCR using primers containing I-SceI restriction sites and the strong constitutive promoter PlacIQ1 [37]. This cassette was PCR purified and digested with I-SceI and ligated into the same R6Kγ plasmid backbone used for pTKLP-tetA.
Amplification, preparation, and integration of Landing Pad
Linear Landing Pad DNA fragment (LP) was amplified from the plasmid pTKLP by PCR using Phusion Flash High-Fidelity PCR Master Mix (Life Technologies) and locus and application specific primers; a table of all oligonucleotides used for recombineering in this study is given as S1 Table. Both pTKLP-tetA and pTKLP-galK contain standardized 25 bp priming sequences [Landing Pad Region 1 (LP1): 5’ TACGGCCCCAAGGTCCAAACGGTGA 3’; Landing Pad Region 2 (LP2): 5’ GATGGCGCCTCATCCCTGAAGCCAA 3’] that can also be used as subsequent targets for homologous recombination for general chromosomal integration of constructs [8, 9]. Optimum amplification conditions were decided for each primer set by thermal gradient PCR followed by agarose gel electrophoresis. Samples were PCR purified and digested with DpnI (New England Biolabs) for at least four hours at 37°C to eliminate template plasmid contamination, followed by PCR purification.
To integrate the landing pad, cells were first prepared by transformation with the helper plasmid pTKRED [8]. These cells were then made competent and transformed with the purified landing pad as described above (see Preparing competent cells and transformation). When plating, cells recombineered with the tetA landing pad were spread on LB plates with 10 μg/ml tetracycline and 100 μg/ml spectinomycin; for ΔgalK cells incorporating the galK landing pad, cells were plated on M63 minimal medium plates with 0.2% w/v galactose as carbon source and 100 μg/ml spectinomycin [1]. These plates were incubated at 30°C overnight, at which point the remainder of the transformed culture was plated in a similar fashion. Plates were allowed to grow until colonies were visible, requiring ~15 hours for tetA integrants and ~48 hours for galK integrants on minimal medium plates. Integrants were verified by colony PCR, gel electrophoresis, and sequencing.
Counterselection against tetA with NiCl2 and determination of growth rate
Growth rate experiments for wildtype MG1655 and MG1655 strains with the tetA landing pad incorporated at the rrnB ribosomal operon (rrnB::tetA) were performed in a 24 well Corning Costar microplate. To begin, strains were grown overnight in 5 ml of Rich Defined Medium (RDM, Teknova) + 0.5% v/v glycerol at 37°C in separate tubes with appropriate antibiotics. Then, two separate tubes for each strain containing 50 ml of RDM + 0.5% v/v glycerol were prepared by inoculation with the overnight cultures such that the calculated initial OD was 0.002. Wells of the plate were filled with 2 mL of these cultures, and appropriate dilutions of 1 M NiCl2 solution were added to the wells to cover concentrations of 0–10 mM NiCl2 in 1 mM increments. After preparation, the plate was placed into a Tecan Infinite 200 plate reader (Tecan Ltd) at 37°C with shaking, and OD values at 600 nm were recorded every 15 minutes for 48 hours.
To calculate the doubling time of each well, the data collected from the plate reader were analyzed using MATLAB (MathWorks). Each growth curve was blanked using the average OD600 reading collected from two uninoculated control wells. Regions of exponential growth for each curve were identified by visual inspection of plots of Log2(OD600) vs time and fit by linear regression to extract growth rates for each NiCl2 concentration. At high concentrations of NiCl2 (> ~8 mM) neither the wildtype nor tetA landing pad strain was able to grow and the growth curves were indistinguishable from the background; the growth rates of these cultures were set to zero in Fig 2.
Fig 2. tetA counterselection with NiCl2.
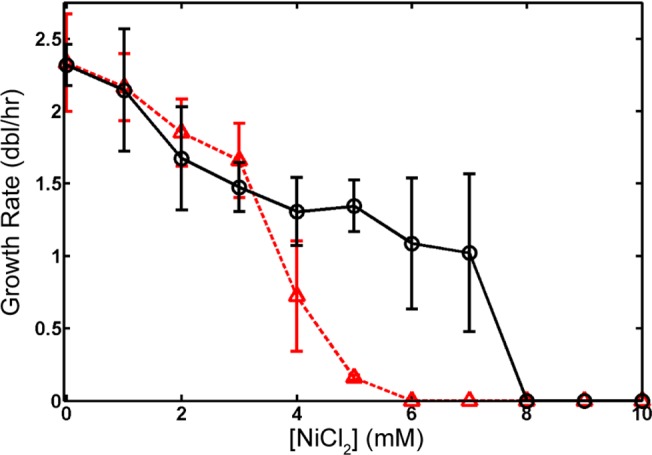
Wildtype MG1655 (black circles, solid line) and MG1655 rrnB::tetA (red triangles, dashed line) were grown in RDM + 0.5% v/v glycerol with concentrations of NiCl2 from 0–10 mM, and the effect of NiCl2 quantified by measuring the growth rate (doublings per hour) in exponential growth. Points are the average of three measurements, and error bars are the SD. The growth rate for both strains at high concentrations of NiCl2 where no observable growth was detected is set to zero. Because of the large differential in growth rate, counterselection against the tetA landing pad can be effectively performed at 5–7 mM NiCl2 in K-12 MG1655.
Construction of fragments for exact integration
To test the efficiency of the exact integration of large constructs without any extraneous sequence or antibiotic markers [9], pTKDP-neo was first digested with I-SceI and the products dephosphorylated with Antarctic phosphatase. The plasmid backbone, containing the origin of replication, bla ampicillin resistance gene, and the sacB counterselection marker, was gel purified from this reaction by gel purification (QIAquick).
The entire lac operon, including the promoter of lacI and terminators of lacA, was amplified from wildtype MG1655 by colony PCR using primers containing I-SceI restriction sites and 50 bp of homology to the targeted genomic location [either the atpI, nth, or ygcE locus [8, 9, 38]]. The PCR reactions were purified, digested with I-SceI, and gel purified. These fragments were then ligated together with the purified pTKDP backbone to form plasmids pTKDP-atpI-lacIZYA, pTKDP-nth-lacIZYA, and pTKDP-ygcE-lacIZYA.
Exact, antibiotic-free integration
Electrocompetent MG1655 Δlac ΔgalK carrying the helper plasmid pTKRED and induced to express λ-Red enzymes were prepared and transformed with ~100 ng of purified galK landing pad targeted towards either the atpI, nth, or ygcE locus. Successful integrants were obtained by selection on M63 minimal medium plates with 0.2% w/v galactose as the sole carbon source [1] and verified by colony PCR. The resulting strains were transformed with the appropriate pTKDP-xxx-lacIZYA plasmid and spread on LB + 100 μg/ml ampicillin plates, where xxx can be any of the three genomic loci tested (atpI, nth, or ygcE). Several colonies were picked and used to inoculate a 20 mm glass tube containing 5 ml of RDM + 0.5% v/v glycerol medium with 100 μg/ml spectinomycin, 2 mM IPTG to induce expression of λ-Red enzymes, and 0.4% w/v L-arabinose to induce expression of I-SceI. These tubes were allowed to grow in a shaking 30°C water bath until saturation. At this point, a small sample was taken, diluted 105 fold, and plated on LB plates with 2 mM IPTG and 20 μg/ml X-gal and allowed to grow overnight at 37°C. The number of correct integrants was quantified as the number of blue versus white colonies.
At the same time, 100 μl of the each saturated tube was used to inoculate another glass tube containing 5 ml of RDM + 0.5% glycerol with 0.2% w/v DOG for galK counterselection and 5% w/v sucrose for sacB counterselection. These tubes were allowed to grow at 37°C until saturated, usually taking 1–2 days. After saturation, a small sample was taken, diluted 105 fold, and plated on LB plates with 2 mM IPTG and 20 μg/ml X-gal and allowed to grow overnight at 37°C. The number of correct integrants was quantified as the number of blue versus white colonies. Several colonies for each integration location were picked and verified by colony PCR (Fig 3).
Fig 3. Exact, antibiotic-free integration.
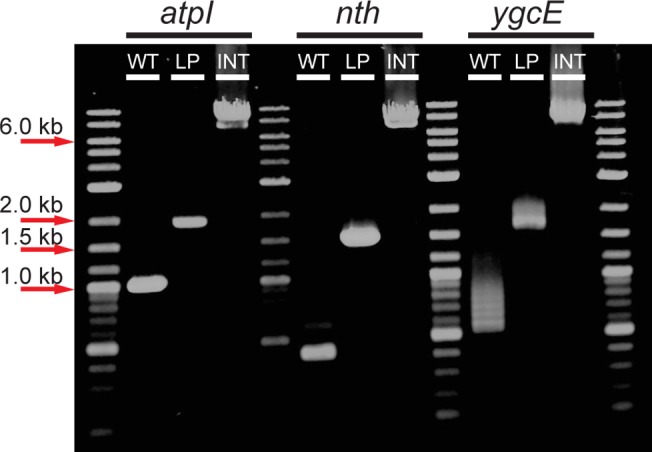
Agarose gel electrophoresis of colony PCR products using primers that bind to regions flanking each locus (atpI, nth, and ygcE). The first lanes of each show PCR products from wildtype cells (WT). The second lanes of each show the PCR product after the landing pad is inserted (LP). The last lanes of each show PCR products after integration of the lac operon at each locus (INT).
We also performed this same procedure using the tetA landing pad and counterselection with NiCl2 to gather statistics on the efficiency of integration (Table 1).
Table 1. Exact Integration Statistics.
| Without Counterselection | With Counterselection | |||||||
|---|---|---|---|---|---|---|---|---|
| tetA (Blue/White) | % Correct | galK (Blue/White) | % Correct | tetA (Blue/White) | % Correct | galK (Blue/White) | % Correct | |
| atpI | 888 / 25 | 97.3 | 54 / 829 | 6.1 | 1148 / 15 | 98.7 | 720 / 6 | 99.2 |
| nth | 1104 / 64 | 94.5 | 49 / 248 | 16.5 | 1375 / 35 | 97.5 | 601 / 9 | 98.5 |
| ygcE | 194 / 559 | 25.8 | 51 / 353 | 12.6 | 1052 / 306 | 77.5 | 232 / 200 | 53.7 |
Colony counts resulting from exact integration of the lac operon at each of three loci (atpI, nth, and ygcE) using either the tetA or galK landing pad. Counts are given both before and after counterselection against landing pad retention using 5% w/v sucrose and either 6 mM NiCl2 (for tetA landing pad) or 0.2% w/v DOG (for galK landing pad). The percent correct integrants was calculated as the percentage of blue colonies after growth on 2 mM IPTG and 20 μg/ml X-gal.
In situ gene fusion
To demonstrate in situ translational fusion of native chromosomal genes of interest to genes encoding a fluorescent reporter, we fused mCherry to rpoD (the housekeeping sigma factor σ70) and ECFP to hupA (the α subunit of the heterodimeric nucleoid associated protein HU). The strategy and results are outlined in Fig 4.
Fig 4. In situ gene fusion.

(A) Strategy for gene fusion in situ. The tetA landing pad is integrated directly between the stop codon and the next base pair by homologous recombination between regions A and B, where the last 3 bp of region A is the stop codon of the targeted gene. The landing pad is then replaced by recombineering and counterselection using NiCl2, removing the stop codon and fusing the two coding sequences together. Homologous recombination between regions A and A’ (identical to A without TAA STOP) and B and B’ (identical to B with new TAA STOP) results in translational fusion. (B) Agarose gel electrophoresis image of colony PCR products verifying fusions to rpoD and hupA, columns 1–2: wildtype; columns 3–4: tetA Landing Pad (LP) integrants; columns 5–9: rpoD::mCherry fusion; columns 10–14: hupA::ECFP fusion. (C) Sequencing results for fusions of rpoD::mCherry and hupA::ECFP. (D) 400X images of wildtype MG1655 (top) and MG1655 rpoD::mCherry hupA::ECFP (bottom) with brightfield (left column), 561 nm laser excitation for mCherry (middle column) and 457 nm laser excitation for ECFP (right column).
We first amplified by PCR the tetA landing pad from the template plasmid pTKLP-tetA. Landing pad amplification was primed from the standardized LP1 and LP2 priming sites. The primers were additionally designed to produce landing pad fragment flanked by the last 50 bp of the coding sequence of the targeted gene and the adjacent 50 bp of chromosomal sequence immediately downstream of the stop codon of each gene. The landing pad was then integrated and verified as described above (see Amplification, preparation, and integration of Landing Pad). This procedure therefore generated strains in which the tetA landing pad was successfully integrated exactly between the TAA stop codon of the targeted gene and the base pair immediately adjacent to TAA.
We next amplified the coding sequences of the fluorescent reporters mCherry from plasmid pRSET-B mCherry [39] and the cyan fluorescent reporter ECFP from plasmid pLAU53 [40]. The primers used were designed to amplify a linear fragment including the coding sequence of the fluorescent reporter flanked at the C terminal end by a TAA stop codon and the 50 bp of sequence immediately downstream of TAA for the targeted gene. At the N terminal end, the primer includes homology to the last 50 bp of the targeted gene excluding the TAA stop codon, and an additional 15 bp encoding five glycine residues as a flexible linker between the targeted gene and the fluorescent reporter gene. To complete the fusion, this PCR product was transformed into tetA landing pad integrants carrying pTKRED as described above (see Preparing competent cells and transformation for recombineering). After electroporation and recovery for four hours at 30°C, the entire culture was then added to 10 ml of RDM + 0.5% v/v glycerol with 6 mM NiCl2 and incubated at 37°C in a shaking water bath until saturation (usually ~48 hours). Finally, a sample of the saturated culture was diluted 105 fold and plated on LB agar. Successful integrants were verified by colony PCR and sequencing.
Scarless deletion by intrachromosomal recombination
The procedure for scarless deletion by intrachromosomal homologous recombination is based upon simplifications of the method of Yu et al [30]. The primers used to amplify the tetA landing pad for scarless intrachromosomal recombination were designed as shown in Fig 5A. Region A (50bp) and region C (75bp) consist of homology to two genes upstream and downstream of the target gene. In this case, region C is 75 bp of sequence immediately downstream of the 3’ end of target gene, the rRNA operon rrnB. Region B (50 bp) is the 3’ end of the targeted gene. The deletion landing pad was amplified by PCR using these primers and template pTKLP-tetA. This deletion landing pad includes homology regions A, B, C, the antibiotic marker tetA, and recognition sites for I-SceI (Fig 5A).
Fig 5. Scarless deletion by intrachromosomal homologous recombination.

(A) Strategy. The target gene is replaced by recombineering using a landing pad amplified using primers containing homology regions A, B, and C. The landing pad is then eliminated by in vivo I-SceI digestion and λ-Red mediated homologous recombination between regions C, followed by counterselection against tetA landing pad retention with 6 mM NiCl2. (B) Verification of the deletion by colony PCR using primers flanking the rrnB operon. Lane 1: wildtype rrnB from MG1655 (WT; ~6 kbp); Lane 2: MG1655 rrnB::tetA landing pad integrant (LP; ~1.6 kbp). Lane 3–10: 8 randomly picked colonies after deletion (270bp). (C) Sequence of the operon rrnB and the sequencing result after deletion. Targeted homology regions are indicated by same color scheme as in (A).
The deletion landing pad was then integrated into chromosome of the wildtype strain MG1655 as described previously (see Amplification, preparation, and integration of Landing Pad) to replace the target gene rrnB facilitated by λ-Red enzymes expressed from pTKRED. Colonies with deletion landing pad were selected for on LB plates containing 100 μg/ml spectinomycin and 10 μg/ml tetracycline. Colony PCR and sequencing was used to verify the presence of the deletion landing pad in the place of rrnB.
The scarless deletion of the landing pad was performed by growing the resulting landing pad strain with pTKRED in a 20 mm glass test tube containing 5 ml RDM + 0.5% v/v glycerol media with 100 μg/ml spectinomycin, 2 mM IPTG, and 0.4% w/v L-arabinose in a 30°C shaking water bath until saturation. 20μl of this saturated culture was then used to inoculate another 20 mm glass test tube containing 5 ml RDM + 0.5% v/v glycerol with 6 mM NiCl2 to eliminate cells retaining the landing pad before plating them on LB plate. Colonies were then screened on LB plates with and without 10 μg/ml tetracycline to verify the absence of the landing pad. Final verification of the deletion was performed by colony PCR and sequencing.
Scarless deletion with oligonucleotides
Scarless deletion can also be accomplished using two sets of small oligos rather than the single set of large primers used for scarless deletion by intrachromosomal recombination. Here, the landing pad was amplified using primers for the standardized LP1 and LP2 priming sites and including 30 bp of homology to chromosomal sequence adjacent to the desired deletion. This landing pad was then integrated into the chromosome as previously described (see Amplification, preparation, and integration of Landing Pad), and the integration was verified by colony PCR and sequencing.
Scarless deletion of the landing pad was then performed by transformation of the cells with small oligonucleotides consisting solely of the two 30 bp homology sequences included in the landing pad primers (Fig 6A). The landing pad strains with pTKRED were made competent and transformed with equimolar amounts of oligos as described above (see Preparing competent cells and transformation for recombineering). After electroporation, 1 ml of SOB with 2 mM IPTG and 0.4% w/v L-arabinose was added, and the culture was allowed to recover for four hours. The entire culture was then added to 10 ml of RDM + 0.5% v/v glycerol with 6 mM NiCl2 and incubated at 37°C in a shaking water bath until saturation (usually ~48 hours). Finally, a sample of the saturated culture was diluted 105 fold and plated on LB agar. Successful integrants were verified by screening for appropriate antibiotic resistances, followed by colony PCR and sequencing.
Fig 6. Scarless deletion by recombineering with oligonucleotides.

(A) Strategy. The target gene is replaced by recombineering with the tetA landing pad. The landing pad is then replaced by recombineering with short, synthesized oligonucleotides and counterselection with 6 mM NiCl2. (B) Agarose gel electrophoresis of colony PCR products verifying deletion of rrnB. Lane 1: Wildtype rrnB (WT); Lane 2: tetA landing pad integrant; Lanes 3–10 8 randomly sampled colonies after deletion. (C) Sequencing result after deletion compared to the original sequence. Targeted homology regions are indicated by the same color scheme as in (A).
Results
NiCl2 counterselection against tetA
To increase the efficiency of isolation of successfully modified bacteria, unsuccessfully modified bacteria which retain tetA after the attempted excision of the landing pad by I-SceI can be selected against using NiCl2 [31]. We have verified and quantified the efficacy of tetA counterselection using NiCl2 in E. coli MG1655 within the context of the Landing Pad system by growing wildtype and rrnB::tetA landing pad integrants (see Methods, Scarless Deletion) in a spectrum of NiCl2 concentrations from 0–10 mM (Fig 2). The large error bars on the data points at high nickel chloride concentrations are a consequence of fitting an exponential growth model to noisy growth curves resulting from NiCl2 severely impacting exponential growth. However, our results indicate that wildtype and landing pad integrants grow well at low NiCl2 concentrations, and are both completely arrested at high concentrations. However, wildtype MG1655 is able to grow satisfactorily in a range of 5–7 mM NiCl2, while the growth of tetA-expressing landing pad integrants is arrested at these concentrations.
Because of the higher average copy number of genes located near the origin of replication, oriC, the absolute level of tetA expression will vary as a function of the location of integration relative to oriC [38, 41–44]. However, we have verified for a variety of landing pad integration locations around the chromosome ([8, 38] and this report) that 6 mM NiCl2 is effective for tetA landing pad counterselection regardless of the integration location, as will be shown for integrations, deletions, and gene fusions in a variety of locations in the following sections.
Exact and antibiotic-free integration
In a previous version of the Landing Pad method, two 25 bp “landing pad regions” (LP1 and LP2) were used as targets for homologous recombination [8]. However, in some instances, the introduction of these extra 25 bp regions may be undesirable, such as when fusing two genes together, or integrating a new gene into an existing operon. In such cases, it is required that the integration be exact; that is, no additional sequence without direct and desired coding function can be included in the integration fragment.
Exact integration of very large constructs can be accomplished by making minor modifications to the donor plasmid pTKDP to replace the LP regions with regions of sequence homology to the desired chromosomal integration locus [9]. To demonstrate this, we generated linear DNA fragment encoding the entire lac operon by PCR using primers designed so that the fragment is flanked on either end by I-SceI sites and 50 bp homology to one of three loci in the E. coli chromosome: near the chromosomal origin of replication (atpI locus), near the terminus of replication (nth locus), or halfway between the origin and terminus on the left hand replichore (ygcE locus) [8]. These fragments were ligated into pTKDP plasmid backbone also previously digested with I-SceI and dephosphorylated. With these two simple steps, we generated three donor plasmids containing a large integration fragment (the ~6.5 kbp lac operon) without any antibiotic marker and flanked by locus-specific homology regions.
These plasmids were transformed into the corresponding strains where the lac operon had been deleted (MG1655 Δlac [8]) and where the tetA landing pad had been integrated at each of the three loci. The integration of the lac operon was performed as described in Methods, and the number of correct integrants both before and after NiCl2 + sucrose counterselection was quantified as the number of blue/white when grown on 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal). Using the tetA landing pad, the integration is extremely efficient at the atpI and nth loci, yielding ~95–97% correct integrants even without any additional selection or counterselection. Integration at the ygcE locus without counterselection was less efficient, yielding ~26% successful integrants; this efficiency was increased to ~76% when counterselection with 6 mM NiCl2 and 5% w/v sucrose was applied. PCR amplification across the integration locus and sequencing of failed ygcE integrants showed that the landing pad had been excised and the break repaired by intrachromosomal nonhomologous recombination. It is unclear what the differences are between the ygcE locus and the atpI and nth loci that render the ygcE locus, but not the atpI or nth loci, amenable to such repair. For each strain, representative blue colonies for each locus were verified by colony PCR across the integration, and sequencing of these PCR products verified that the junctions between the integrated fragment and the adjacent chromosomal sequence were correct and exact.
We repeated this same procedure using MG1655 Δlac ΔgalK strains where a galK landing pad was integrated at each of the three chromosomal loci, and we quantified the efficiency of integration as outlined above. The number of correct integrants before counterselection was substantially lower (~6% -16%) using the galK instead of the tetA landing pad for unknown reasons. However, after counterselection with 5% w/v sucrose and 0.2% w/v DOG, the efficiency of integration at each locus using the galK landing pad was comparable to that obtained with the tetA landing pad. Again, representative blue colonies for each strain were verified by colony PCR (Fig 3) and sequencing. Therefore, using the galK landing pad, we have exactly integrated a ~6.5 kbp fragment into specific chromosomal loci without the introduction of any extraneous sequence and without the integration of any antibiotic markers at any step of the procedure.
In situ gene fusion
Using recombineering and counterselection against the tetA landing pad, we used the Landing Pad system to demonstrate the in situ translational fusion of the native chromosomal genes encoding the housekeeping sigma factor σ70, rpoD, and the α subunit of the histone-like nucleoid associated protein HU, hupA, to fluorescent reporter genes without any additional disruption of surrounding sequence. The strategy is outlined in Fig 4A. Using recombineering and positive selection for the tetA landing pad with tetracycline, we first integrated the landing pad between the stop codon for the targeted gene and the next immediate base. Then, by counterselecting against landing pad retention with NiCl2, we replaced the landing pad with the fluorescent reporter gene, eliminating the target gene’s stop codon in the process and translationally fusing the two genes together.
Because both the coding sequences of mCherry and ECFP are short (711 bp and 720 bp respectively), replacement of the landing pad can be accomplished by direct transformation of the cells with linear DNA amplified by PCR as outlined in Methods—in situ gene fusion. Following counterselection in RDM medium with 0.5% v/v glycerol and 6 mM NiCl2, the culture was diluted 105 fold and plated on LB agar. For each of the gene fusions, we picked five colonies from these plates and performed colony PCR and sequencing to verify the integrations (Fig 4B and 4C); all colonies tested had successfully integrated the fluorescent reporter gene. Finally, to demonstrate repeatability, we again performed the hupA::eCFP fusion in the previously generated MG1655 rpoD::mCherry fusion strain. The success rate for verified colonies was again 100%, and example images of both wildtype MG1655 and MG1655 rpoD::mCherry hupA::eCFP in the brightfield and fluorescent channels are shown in Fig 4D.
Scarless deletion by intrachromosomal recombination
We also demonstrated the usage of Landing pad system in scarless deleting of genes. As an example, the rRNA operon rrnB was selected as a target gene of deletion. The deletion landing pad was first integrated into chromosome to replace rrnB, then the deletion of this landing pad was performed in the recombination growth with IPTG and L-arabinose (see Methods, Scarless deletion by intrachromosomal recombination)
After the NiCl2 selection step, we selected several colonies for screening; none grew on plates containing 10 μg/ml tetracycline. Eight colonies were picked randomly to verify the result of the deletion by colony PCR and sequencing (Fig 5B and 5C). Agarose gel electrophoresis of the colony PCR products showed that the original operon rrnB (size of about 6kbp) (Lane 1) was replaced by the deletion landing pad (size of 1.6 kbp) (Lane 2) after landing pad integration. After deletion and counterselection, all of eight representative colonies had the band of size 270bp demonstrating excision of the tetA landing pad (Fig 5B). Sequencing results of the PCR product (270bp) confirmed the desired scarless deletion of rrnB (Fig 5C). We have subsequently used this procedure to delete each of four other ribosomal operons (data not shown), with the same level of success.
Scarless deletion with oligonucleotides
While scarless deletion by intrachromosomal recombination is effective and efficient, it requires the synthesis of two large, expensive primers (75 bp and 150 bp) including the homology regions A, B, and C for the targeted locus. We have demonstrated another scarless deletion method requiring lower cost by using small oligonucleotides. These oligos contain the same two homology regions used to integrate the landing pad primer synthesized directly adjacent to one another. Therefore, when these oligos are transformed into the cells and replace the landing pad by homologous recombination enhanced by I-SceI- induced double strand breaks, the entire intervening sequence is eliminated (Fig 6A). The efficiency of integration was tested with the usages of single stranded oligos targeting either the leading or lagging strand, or annealed double stranded oligos.
We prepared the landing pad integrants for recombineering and transformed them with equimolar amounts of single stranded and double stranded oligos as described in Methods—Scarless deletion with oligonucleotides. After transformation and recovery for four hours, the culture was transferred to 10 ml RDM + 0.5% v/v glycerol with a final concentration of 6 mM NiCl2 and allowed to grow to saturation for tetA counterselection. A sample of this culture was diluted 105 fold and plated on LB agar, and 20 representative colonies for deletions with each type of oligo were verified by colony PCR and sequencing (Fig 6B and 6C). Colony PCR for all clones demonstrated that rrnB and the landing pad was deleted with 100% efficiency; 10 representative samples from Okazaki fragment-like lagging strand targeting single stranded oligos are shown in Fig 6B. Sequencing, however, showed that double stranded and Okazaki-like single stranded oligos deleted the operon without any error in 60% of samples, while leading stranded-targeting single stranded oligos were without error in only 2 out of 20 samples. These results are in accord with previous studies showing that recombineering is much more efficient using double or single stranded oligos that incorporate into the lagging strand during replication [45].
Discussion
We have shown here how, using NiCl2 or DOG for counterselection against landing pad retention, the Landing Pad system [8, 9] can be extended to generate a wide variety of genome modifications that would previously require the application of a wide variety of editing systems. The procedures described here take ~1–2 weeks from start to verified end product. Using this single system, we have explicitly demonstrated the translational fusion of native chromosomal genes to fluorescent reporters without additional perturbation, scarless gene deletion, and the exact, markerless integration of very large constructs into any locus in the E. coli genome. Using a galK landing pad, we have demonstrated the integration of the very large (~6.5 kbp) lac operon into specific loci without the integration of antibiotic markers into the genome at any step. As far as we are aware, such a feat was previously impossible with any other existing technologies. Using judiciously designed primers, oligonucleotides, and integration locations, it is also possible to use the Landing Pad system to generate single base pair substitutions and indels into specific genes in situ. Furthermore, we describe upgrades to the Landing Pad system: the landing pad template pTKLP and donor plasmids pTKDP [previously referred to as pTKS/CS and pTKIP, respectively [8]] have been optimized to include modifications that eliminate the need for screening against transformation or retention of these plasmids.
We have demonstrated and quantified the efficacy of nickel chloride (NiCl2) as a reagent for the effective negative selection against tetA expression within the context of the Landing Pad system [8, 31]. By employing such negative selection against the landing pad, the efficiency of selection of exact modifications without additional antibiotic markers can be significantly increased. The tetA gene in the landing pad is ideal for this purpose: its successful integration into the chromosome can be positively selected for by growth in medium containing tetracycline, while its retention after replacement can be negatively selected against using fusaric acid or nickel chloride [31, 34, 46, 47]. Previous studies have shown that counterselection against tetA using fusaric acid can be effective if simultaneously combined with additional negative selection against the marker sacB when grown in the presence of sucrose [34]. However, both fusaric acid and DOG are significantly more expensive than nickel chloride, and the necessity to combine fusaric acid counterselection with sacB for adequate selective pressure eliminates other possible simultaneous applications of sacB.
To effect the exact integration of small fragments into the chromosome without the inclusion of any additional unwanted sequence (e.g. antibiotic selection markers), previously existing methods have employed the gene encoding galactokinase, galK [1]. Integration of galK into a galK - host can be selected for by the ability to metabolize the sugar galactose. The expression of galK can also be selected against by supplying the cells with the galactose analogue 2-deoxygalactose (DOG), which galactokinase phosphorylates into nonmetabolizable and ultimately lethal products. Positive and negative selection of galK using galactose and DOG is effective, and, since it does not confer resistance to antibiotics, provides advantages when working with pathogenic strains or species. We have therefore created a version of the landing pad employing galK rather than tetA for such applications. However, use of galK also has disadvantages. One must be sure that the intended recipient strain is incapable of galactose metabolism such that positive selection on galactose can be successful. Furthermore, galactose must be the only carbon source available for positive selection to be effective, and hence growth and selection must be performed in minimal medium [1]. The preparation of, and extremely slow bacterial growth on, minimal medium plates makes this approach tedious and time consuming, while positive and negative selection of tetA can be performed rapidly and at low cost in liquid rich medium containing NiCl2.
We have demonstrated the scarless deletions of large regions of the chromosome using two methods: recombineering of a complex landing pad to replace the targeted gene, followed by its in vivo digestion by I-SceI and λ-Red mediated repair by intrachromosomal recombination; and recombineering of a simpler landing pad to replace the targeted gene, followed by excision of the landing pad by I-SceI and replacement by recombineering with small oligonucleotides. Both methods are equally efficient and effective, and the choice of method will be dictated by where it is desired to allocate time and resources. Deletion by intrachromosomal recombination has the advantage of requiring only a single recombineering step, eliminating the work required for additional recombineering, e.g. the preparation and transformation of competent bacteria. However, the large primers required are relatively expensive. Alternatively, the deletion can be completed by an additional recombineering step with short oligonucleotides. Using this method and completing the deletion with a single-stranded Okazaki-like oligonucleotide, the complete set of primers and oligos was much less expensive, but requires the time and work entailed by the additional recombineering step.
The Landing Pad system compares favorably to other E. coli genome editing technologies, including recombineering, phage-based approaches, and CRISPR-Cas9 systems. Recombineering excels at the site-specific integration of short DNA fragments into the genome to accomplish integrations, deletions, and exact replacements [1–3, 5, 13, 24–30, 34, 45, 48, 49]. However, we find that correctly integrating fragments larger than 2.5–3 kbp is prone to nonspecific integration and can be prohibitively difficult [8]. Phage based systems [10–13, 15] have the opposite problem: they are extremely efficient at integrating large constructs into the genome, but without extensive engineering these integrations can only be performed at previously existing phage attachment sites in the genome. For the same reason, phage-based systems are generally not applicable for other genome modification tasks, such as generating deletions, knockouts, or mutations.
CRISPR-Cas9 based systems show great potential for genome editing [16–20, 50–52], and are based upon the same underlying principle as the Landing Pad system, and the transcription activator-like effector nucleases (TALENs) and zinc-finger nucleases (ZFNs) employed in eukaryotes [20–23]: genome modification can be performed or enhanced by endonuclease mediated site-specific cleavage of the genome. In the case of the Landing Pad system, this is accomplished by integrating unique I-SceI recognition sites into the desired genomic location along with the landing pad by PCR amplification of the landing pad using locus-specific primers. CRISPR-Cas9 systems have the advantage that they do not require such prior modification of the genome. Cleavage by Cas9 can be directed to any sequence in the genome, guided by short CRISPR RNAs (crRNAs) that are designed to be complementary to the targeted sequence. The weakness, however, is that current existing CRISPR-Cas9 genome editing systems require the design and implementation of unique constructs to express the guiding crRNA; for example, a new plasmid must be created to express the complementary crRNA for each desired targeted location to guide Cas9 cleavage to that locus. Here, the Landing Pad system has the advantage in that targeting different genetic loci only requires new sets of locus-specific primers to amplify the landing pad.
Supporting Information
Oligos used for amplification and recombineering of the landing pad are designated with “LP”. Primers used for colony PCR verification are designated “ver”. Primers for amplifying the lac operon, mCherry, or ECFP with locus specific homology are designated as “lac”, “mCherry”, or “CFP” respectively. Finally, oligos used to complete the deletion of rrnB are designated “Pos” or “Neg”.
(DOCX)
Data Availability
Annotated sequences files for pTKRED, pTKLP-tetA, pTKLP-galK, pTKDP-neo, pTKDP-cat, pTKDP-hph, and pTKDP-dhfr are available as Genbank accession numbers GU327533, KR071151, KR071150, KR071149, KR071146, KR071148, and KR071147 respectively. All relevant data are within the paper and Genbank.
Funding Statement
This work was supported by the Center for the Physics of Living Cells NSF Physics Frontier Center (PHY 1430124) and startup funds from the Department of Physics at the University of Illinois at Urbana-Champaign.
References
- 1. Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 2005;33(4):e36. Epub 2005/02/26. doi: 33/4/e36 [pii] 10.1093/nar/gni035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sabri S, Steen JA, Bongers M, Nielsen LK, Vickers CE . Knock-in/Knock-out (KIKO) vectors for rapid integration of large DNA sequences, including whole metabolic pathways, onto the Escherichia coli chromosome at well-characterised loci. Microb Cell Fact. 2013;12:60 Epub 2013/06/27. doi: 10.1186/1475-2859-12-60 1475-2859-12-60 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stringer AM, Singh N, Yermakova A, Petrone BL, Amarasinghe JJ, Reyes-Diaz L, et al. FRUIT, a scar-free system for targeted chromosomal mutagenesis, epitope tagging, and promoter replacement in Escherichia coli and Salmonella enterica. PLoS One. 2012;7(9):e44841 10.1371/journal.pone.0044841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ryu YS, Biswas RK, Shin K, Parisutham V, Kim SM, Lee SK. A simple and effective method for construction of Escherichia coli strains proficient for genome engineering. PloS one. 2014;9(4):e94266 10.1371/journal.pone.0094266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Herring CD, Glasner JD, Blattner FR. Gene replacement without selection: regulated suppression of amber mutations in Escherichia coli. Gene. 2003;311:153–63. Epub 2003/07/11. doi: S0378111903005857 [pii]. . [DOI] [PubMed] [Google Scholar]
- 6. Li MZ, Elledge SJ. MAGIC, an in vivo genetic method for the rapid construction of recombinant DNA molecules. Nat Genet. 2005;37(3):311–9. Epub 2005/02/26. doi: ng1505 [pii] 10.1038/ng1505 . [DOI] [PubMed] [Google Scholar]
- 7. Rivero-Muller A, Lajic S, Huhtaniemi I. Assisted large fragment insertion by Red/ET-recombination (ALFIRE)—an alternative and enhanced method for large fragment recombineering. Nucleic Acids Res. 2007;35(10):e78. Epub 2007/05/23. doi: gkm250 [pii] 10.1093/nar/gkm250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kuhlman TE, Cox EC. Site-specific chromosomal integration of large synthetic constructs. Nucleic Acids Res. 2010;38(6):e92. Epub 2010/01/06. doi: gkp1193 [pii] 10.1093/nar/gkp1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kuhlman TE, Cox EC. A place for everything: chromosomal integration of large constructs. Bioeng Bugs. 2010;1(4):296–9. Epub 2011/02/18. 10.4161/bbug.1.4.12386 ; PubMed Central PMCID: PMC3026472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Haldimann A, Wanner BL. Conditional-replication, integration, excision, and retrieval plasmid-host systems for gene structure-function studies of bacteria. J Bacteriol. 2001;183(21):6384–93. Epub 2001/10/10. 10.1128/JB.183.21.6384-6393.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. St-Pierre F, Cui L, Priest DG, Endy D, Dodd IB, Shearwin KE. One-Step Cloning and Chromosomal Integration of DNA. ACS Synthetic Biology. 2013;2(9):537–41. 10.1021/sb400021j [DOI] [PubMed] [Google Scholar]
- 12. Groth AC, Fish M, Nusse R, Calos MP. Construction of transgenic Drosophila by using the site-specific integrase from phage phiC31. Genetics. 2004;166(4):1775–82. Epub 2004/05/06. doi: 166/4/1775 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Groth AC, Olivares EC, Thyagarajan B, Calos MP. A phage integrase directs efficient site-specific integration in human cells. Proc Natl Acad Sci U S A. 2000;97(11):5995–6000. Epub 2000/05/10. doi: 10.1073/pnas.090527097 090527097 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Keravala A, Lee S, Olivares EC, Thyagarajan B, Farruggio A, Gabrovsky VE, et al. 525. Mutants of phiC31 Integrase with Increased Efficiency and Specificity. Mol Ther. 2006;13(S1):S201–S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sekhavati MH, Tahmoorespur M, Ghaedi K, Dormiani K, Nassiri MR, Khazaie Y, et al. Cloning, Expression, and in vitro Functional Activity Assay of phiC31 Integrase cDNA in Escherichia coli. Cell J. 2013;14(4):264–9. Epub 2013/04/12. [PMC free article] [PubMed] [Google Scholar]
- 16. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–23. Epub 2013/01/05. doi: 10.1126/science.1231143 science.1231143 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gomaa AA, Klumpe HE, Luo ML, Selle K, Barrangou R, Beisel CL. Programmable Removal of Bacterial Strains by Use of Genome-Targeting CRISPR-Cas Systems. mBio. 2014;5(1). 10.1128/mBio.00928-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol. 2013;31(3):233–9. Epub 2013/01/31. doi: 10.1038/nbt.2508 nbt.2508 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jiang W, Zhou H, Bi H, Fromm M, Yang B, Weeks DP. Demonstration of CRISPR/Cas9/sgRNA-mediated targeted gene modification in Arabidopsis, tobacco, sorghum and rice. Nucleic Acids Res. 2013;41(20):e188 Epub 2013/09/04. doi: 10.1093/nar/gkt780 gkt780 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gaj T, Gersbach CA, Barbas CF III. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends in Biotechnology. 2013;31(7):397–405. 10.1016/j.tibtech.2013.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet. 2010;11(9):636–46. 10.1038/nrg2842 [DOI] [PubMed] [Google Scholar]
- 22. Bedell VM, Wang Y, Campbell JM, Poshusta TL, Starker CG, Krug Ii RG, et al. In vivo genome editing using a high-efficiency TALEN system. Nature. 2012;491(7422):114–8. doi: http://www.nature.com/nature/journal/v491/n7422/abs/nature11537.html#supplementary-information. 10.1038/nature11537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol. 2013;14(1):49–55. doi: http://www.nature.com/nrm/journal/v14/n1/suppinfo/nrm3486_S1.html. 10.1038/nrm3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Costantino N, Court DL. Enhanced levels of lambda Red-mediated recombinants in mismatch repair mutants. Proc Natl Acad Sci U S A. 2003;100(26):15748–53. Epub 2003/12/16. 10.1073/pnas.2434959100 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97(12):6640–5. Epub 2000/06/01. 10.1073/pnas.120163297 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu P, Jenkins NA, Copeland NG. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 2003;13(3):476–84. Epub 2003/03/06. 10.1101/gr.749203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pohl T, Uhlmann M, Kaufenstein M, Friedrich T. Lambda Red-mediated mutagenesis and efficient large scale affinity purification of the Escherichia coli NADH:ubiquinone oxidoreductase (complex I). Biochemistry. 2007;46(37):10694–702. Epub 2007/08/29. 10.1021/bi701057t . [DOI] [PubMed] [Google Scholar]
- 28. Posfai G, Kolisnychenko V, Bereczki Z, Blattner FR. Markerless gene replacement in Escherichia coli stimulated by a double-strand break in the chromosome. Nucleic Acids Res. 1999;27(22):4409–15. Epub 1999/10/28. doi: gkc658 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, Court DL. An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci U S A. 2000;97(11):5978–83. Epub 2000/05/17. 10.1073/pnas.100127597 100127597 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yu BJ, Kang KH, Lee JH, Sung BH, Kim MS, Kim SC. Rapid and efficient construction of markerless deletions in the Escherichia coli genome. Nucleic Acids Res. 2008;36(14):e84 Epub 2008/06/24. 10.1093/nar/gkn359gkn359 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Podolsky T, Fong ST, Lee BT. Direct selection of tetracycline-sensitive Escherichia coli cells using nickel salts. Plasmid. 1996;36(2):112–5. Epub 1996/09/01. doi: S0147-619X(96)90038-4 [pii] 10.1006/plas.1996.0038 . [DOI] [PubMed] [Google Scholar]
- 32. Blomfield IC, Vaughn V, Rest RF, Eisenstein BI. Allelic exchange in Escherichia coli using the Bacillus subtilis sacB gene and a temperature-sensitive pSC101 replicon. Molecular Microbiology. 1991;5(6):1447–57. 10.1111/j.1365-2958.1991.tb00791.x [DOI] [PubMed] [Google Scholar]
- 33. Gay P, Le Coq D, Steinmetz M, Ferrari E, Hoch JA. Cloning structural gene sacB, which codes for exoenzyme levansucrase of Bacillus subtilis: expression of the gene in Escherichia coli. Journal of Bacteriology. 1983;153(3):1424–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li X-t, Thomason LC, Sawitzke JA, Costantino N, Court DL. Positive and negative selection using the tetA-sacB cassette: recombineering and P1 transduction in Escherichia coli. Nucleic Acids Research. 2013;41(22):e204 10.1093/nar/gkt1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pelicic V, Reyrat JM, Gicquel B. Expression of the Bacillus subtilis sacB gene confers sucrose sensitivity on mycobacteria. J Bacteriol. 1996;178(4):1197–9. Epub 1996/02/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Link AJ, Phillips D, Church GM. Methods for generating precise deletions and insertions in the genome of wild-type Escherichia coli: application to open reading frame characterization. Journal of Bacteriology. 1997;179(20):6228–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Calos MP, Miller JH. The DNA sequence change resulting from the IQ1 mutation, which greatly increases promoter strength. Mol Gen Genet. 1981;183(3):559–60. Epub 1981/01/01. . [DOI] [PubMed] [Google Scholar]
- 38. Kuhlman TE, Cox EC. Gene location and DNA density determine transcription factor distributions in Escherichia coli. Mol Syst Biol. 2012;8:610 Epub 2012/09/13. doi: 10.1038/msb.2012.42 msb201242 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22(12):1567–72. Epub 2004/11/24. doi: nbt1037 [pii] 10.1038/nbt1037 . [DOI] [PubMed] [Google Scholar]
- 40. Lau IF, Filipe SR, Søballe B, Økstad O-A, Barre F-X, Sherratt DJ. Spatial and temporal organization of replicating Escherichia coli chromosomes. Molecular Microbiology. 2003;49(3):731–43. 10.1046/j.1365-2958.2003.03640.x [DOI] [PubMed] [Google Scholar]
- 41. Bremer H, Churchward G. An examination of the Cooper-Helmstetter theory of DNA replication in bacteria and its underlying assumptions. J Theor Biol. 1977;69(4):645–54. Epub 1977/12/21. doi: 0022-5193(77)90373-3 [pii]. PubMed PMID: 607026. [DOI] [PubMed] [Google Scholar]
- 42. Bremer H, Dennis PP. Modulation of Chemical Composition and Other Parameters of the Cell by Growth Rate In: Neidhardt FC, editor. Escherichia coli and Salmonella: Cellular and Molecular Biology. 2. 2 ed. Washington, D.C.: ASM Press; 1996. p. 1553–69. [Google Scholar]
- 43. Cooper S, Helmstetter CE. Chromosome replication and the division cycle of Escherichia coli B/r. Journal of Molecular Biology. 1968;31(3):519–40. 10.1016/0022-2836(68)90425-7 [DOI] [PubMed] [Google Scholar]
- 44. Helmstetter CE, Cooper S. DNA synthesis during the division cycle of rapidly growing Escherichia coli B/r. Journal of Molecular Biology. 1968;31(3):507–18. 10.1016/0022-2836(68)90424-5 [DOI] [PubMed] [Google Scholar]
- 45. Mosberg JA, Lajoie MJ, Church GM. Lambda Red Recombineering in Escherichia coli Occurs Through a Fully Single-Stranded Intermediate. Genetics. 2010;186(3):791–9. 10.1534/genetics.110.120782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bochner BR, Huang HC, Schieven GL, Ames BN. Positive selection for loss of tetracycline resistance. J Bacteriol. 1980;143(2):926–33. Epub 1980/08/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Maloy SR, Nunn WD. Selection for loss of tetracycline resistance by Escherichia coli. J Bacteriol. 1981;145(2):1110–1. Epub 1981/02/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Narayanan K, Warburton PE. DNA modification and functional delivery into human cells using Escherichia coli DH10B. Nucleic Acids Res. 2003;31(9):e51 Epub 2003/04/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Orford M, Nefedov M, Vadolas J, Zaibak F, Williamson R, Ioannou PA. Engineering EGFP reporter constructs into a 200 kb human beta-globin BAC clone using GET Recombination. Nucleic Acids Res. 2000;28(18):E84 Epub 2000/09/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bikard D, Jiang W, Samai P, Hochschild A, Zhang F, Marraffini LA. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 2013;41(15):7429–37. Epub 2013/06/14. doi: 10.1093/nar/gkt520 gkt520 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hatoum-Aslan A, Samai P, Maniv I, Jiang W, Marraffini LA. A ruler protein in a complex for antiviral defense determines the length of small interfering CRISPR RNAs. J Biol Chem. 2013;288(39):27888–97. Epub 2013/08/13. doi: 10.1074/jbc.M113.499244 M113.499244 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jiang W, Maniv I, Arain F, Wang Y, Levin BR, Marraffini LA. Dealing with the evolutionary downside of CRISPR immunity: bacteria and beneficial plasmids. PLoS Genet. 2013;9(9):e1003844 Epub 2013/10/03. doi: 10.1371/journal.pgen.1003844 PGENETICS-D-13-00288 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Oligos used for amplification and recombineering of the landing pad are designated with “LP”. Primers used for colony PCR verification are designated “ver”. Primers for amplifying the lac operon, mCherry, or ECFP with locus specific homology are designated as “lac”, “mCherry”, or “CFP” respectively. Finally, oligos used to complete the deletion of rrnB are designated “Pos” or “Neg”.
(DOCX)
Data Availability Statement
Annotated sequences files for pTKRED, pTKLP-tetA, pTKLP-galK, pTKDP-neo, pTKDP-cat, pTKDP-hph, and pTKDP-dhfr are available as Genbank accession numbers GU327533, KR071151, KR071150, KR071149, KR071146, KR071148, and KR071147 respectively. All relevant data are within the paper and Genbank.