

Abstract
mRNA translation into proteins is highly regulated, but the role of mRNA isoforms, noncoding RNAs (ncRNAs), and genetic variants remains poorly understood. mRNA levels on polysomes have been shown to correlate well with expressed protein levels, pointing to polysomal loading as a critical factor. To study regulation and genetic factors of protein translation we measured levels and allelic ratios of mRNAs and ncRNAs (including microRNAs) in lymphoblast cell lines (LCL) and in polysomal fractions. We first used targeted assays to measure polysomal loading of mRNA alleles, confirming reported genetic effects on translation of OPRM1 and NAT1, and detecting no effect of rs1045642 (3435C>T) in ABCB1 (MDR1) on polysomal loading while supporting previous results showing increased mRNA turnover of the 3435T allele. Use of high-throughput sequencing of complete transcript profiles (RNA-Seq) in three LCLs revealed significant differences in polysomal loading of individual RNA classes and isoforms. Correlated polysomal distribution between protein-coding and non-coding RNAs suggests interactions between them. Allele-selective polysome recruitment revealed strong genetic influence for multiple RNAs, attributable either to differential expression of RNA isoforms or to differential loading onto polysomes, the latter defining a direct genetic effect on translation. Genes identified by different allelic RNA ratios between cytosol and polysomes were enriched with published expression quantitative trait loci (eQTLs) affecting RNA functions, and associations with clinical phenotypes. Polysomal RNA-Seq combined with allelic ratio analysis provides a powerful approach to study polysomal RNA recruitment and regulatory variants affecting protein translation.
Introduction
Extensive studies have revealed molecular features of mRNAs enabling polysomal loading and translation [1, 2]. Protein expression was proposed to be regulated with near equal magnitude at the level of transcription and translation [3]. Protein levels can increase up to 20-fold without corresponding alterations in mRNA abundance, and mRNA levels can increase up to 30-fold without reflection on protein levels [4–6]. Correlations between cellular mRNA and protein levels can be drastically improved by measuring mRNAs bound to polysomes, a series of ribosomes held together with a strand of messenger RNA undergoing active translation (Fig 1) [6, 7]. Differential recruitment of mRNAs and isoforms to polysomes contribute a critical layer of regulation involving sequence specific regulatory events [8, 9]. Moreover, cell stimulation with EGF has revealed strong uncoupling of the transcriptome and the translatome, through rapid recruitment of mRNAs onto polysomes rather than new transcription [8, 9]. These studies, using sequencing (RNA-Seq) and hybridization arrays, respectively, have employed poly-dT for cDNA synthesis, thereby lacking ability to detect RNAs without poly-A tails and small RNAs.
Fig 1. Measuring allelic mRNA occupancy on polysomes.

The flowchart depicts the strategy used to fractionate cellular components for RNA analysis and to determine Allelic Expression Imbalance (AEI), defined as a ratio of reference allele (REF) to alternative allele (ALT). Chromatographic peaks obtained from SNaPshot [25] represent relative allelic abundance in gDNA and mRNA (as cDNA). Significant deviation from unity (after normalization to allelic ratios of gDNA or plasmid DNA) indicates unequal polysomal occupancy of allelic mRNA. RNPs represent cytosolic mRNA either polysome-free or bound to ribonucleotide particles.
Whereas loading of protein-coding mRNAs onto translating polysomes has been studied extensively, less is known about other classes of RNAs, including antisense and pseudogene RNAs, long (intergenic) noncoding RNAs (l(i)ncRNAs), and short RNAs such as microRNAs [10]. Therefore, one of the goals of this study was to perform an initial survey of all RNA classes associated with polysomes. To detect all RNAs and their isoforms, we have performed RNA-Seq with random hexamer-priming and separate analyses of short RNAs, encompassing the entire transcriptome, using a procedure that depletes ribosomal RNA.
A second focus of this study was to develop an approach for detecting genetic variants altering polysomal RNA loading. The human genome contains numerous variants affecting transcription [11] and RNA processing events [12, 13]. Since mRNA and protein levels are often poorly correlated [14–17], mRNA expression quantitative trait loci (eQTLs) sometimes fail to reflect corresponding changes in protein expression [18, 19] and only partially overlap with protein eQTLs [20]. Comprehensive studies of genetic factors acting specifically on polysomal loading and translation are sparse. Targeted molecular studies examining the effects of genetic variants on translation typically involve reporter gene assays; however, results obtained in cell culture system, may not reflect native tissue conditions [21]. A genome-wide mRNA analysis in cytosol and polysomes of lymphoblast cell lines (LCLs), with hybridization microarray methods combined with genome-wide single nucleotide polymorphism (SNP) assays, revealed numerous distinct eQTLs, suggesting pervasive genetic effects on translation [7]. At the post-transcriptional level, genetic factors could directly affect polysomal loading, or alter the formation of mRNA isoforms with differential access to polysomes. Both processes can be readily measured with mRNA microarray analysis.
A novel approach used here is to measure differential polysome loading of RNAs and their isoforms, coupled with allele-specific measurements, to assess the impact of genetic variation on translation. Differences in the allelic RNA ratios between cytosolic and polysomal extracts reveal the presence of regulatory factors that determine polysomal loading. The vast majority of genes present multiple RNA isoforms with different 3′ and 5′ UTRs and alternative splicing events, or undergoing RNA editing. To resolve these processes we employed full transcriptome sequencing (RNA-Seq) in LCLs to measure differential loading of all RNAs and their isoforms onto translating ribosomes. RNA-Seq further provides estimates of allelic ratios at heterozygous SNPs [22], revealing regulatory variants affecting transcription, RNA processing, and translation.
To test the utility of allelic RNA ratio analysis comparing total cellular to polysomal RNAs, we first employed a targeted gene approach investigating the effects on polysomal loading exerted by regulatory variants known to alter translation (OPRM1 (118 A>G; rs1799971) and NAT1*10 (rs1057126)) [23, 24]. ABCB1 (multidrug resistance 1 polypeptide, MDR1) was also included to test two proposed alternative mechanisms of post-transcriptional regulation. The synonymous SNP 3435C>T (rs1045642) had been shown to alter mRNA stability [25], while another study suggested that usage of the rare codon introduced by 3435T reduces translation [26], an event affecting polysome interactions, and thereby, could alter allelic ratios in the polysomes.
Here we report exploratory study analyzing the entire cellular transcriptome in comparison to the translatome (RNAs on polysomes). We also present a new approach to discover regulatory polymorphisms affecting translation, by measuring changes in allelic RNA ratios upon loading onto translating polysomes.
Results
Targeted measurements of polysomal allelic mRNA ratios in cell cultures
RNA fractions were recovered from total cytoplasmic lysate (cytosol) and from polysomal fractions (Fig 1), converted to cDNA, and analyzed by qRT-PCR. Spiking of the cytosolic and polysome fractions with luciferase mRNAs served as internal control. Target mRNAs were amplified with low cycle RT-PCR (non-saturating) from eluate fractions and the amplicons analyzed by gel electrophoresis, showing distributions of individual mRNAs across the gradient (S1A–S1C Fig). We then used qRT-PCR to determine the differences in cycle threshold (ΔCt) values relative to luciferase, yielding distinct profiles for OPRM1, NAT1, and ABCB1 mRNAs across polysomal fractions (S1D–S1F Fig), consistent with previous findings [8, 9]. We then measured allelic mRNA ratios in cytosol and polysomal fraction to determine differential loading for each allele.
A. OPRM1 118A>G (rs1799971) affects both expression of cytoplasmic RNA alleles and polysome loading
Equal amounts of plasmids carrying the entire OPRM1 coding region with either the 118A or the 118G allele were transfected into CHO cells. As the OPRM1 118G allele had been shown to reduce both the overall OPRM1 RNA expression and translational efficiency relative to the 118A allele, measured by luciferase assays [23], we expected to observe increasingly greater allelic A/G ratios from cytoplasmic lysates to the polysomal fractions, reflecting preferential loading of 118A. Accordingly, the normalized allelic mRNA ratios for 118A/G was 1.3 in cytoplasmic lysate (n = 6, p<0.01), while allelic A/G ratios increased to 1.5–1.6-fold in polysomes, significantly greater than the cytosolic allelic mRNA ratio (n = 6, p<0.01) (Fig 2A), consistent with the proposed dual mechanism by which 118G reduces OPRM1 protein expression [23].
Fig 2. Allelic mRNA expression measurement in polysomes of transfected cell lines and cells with native expression of NAT1 and ABCB1.
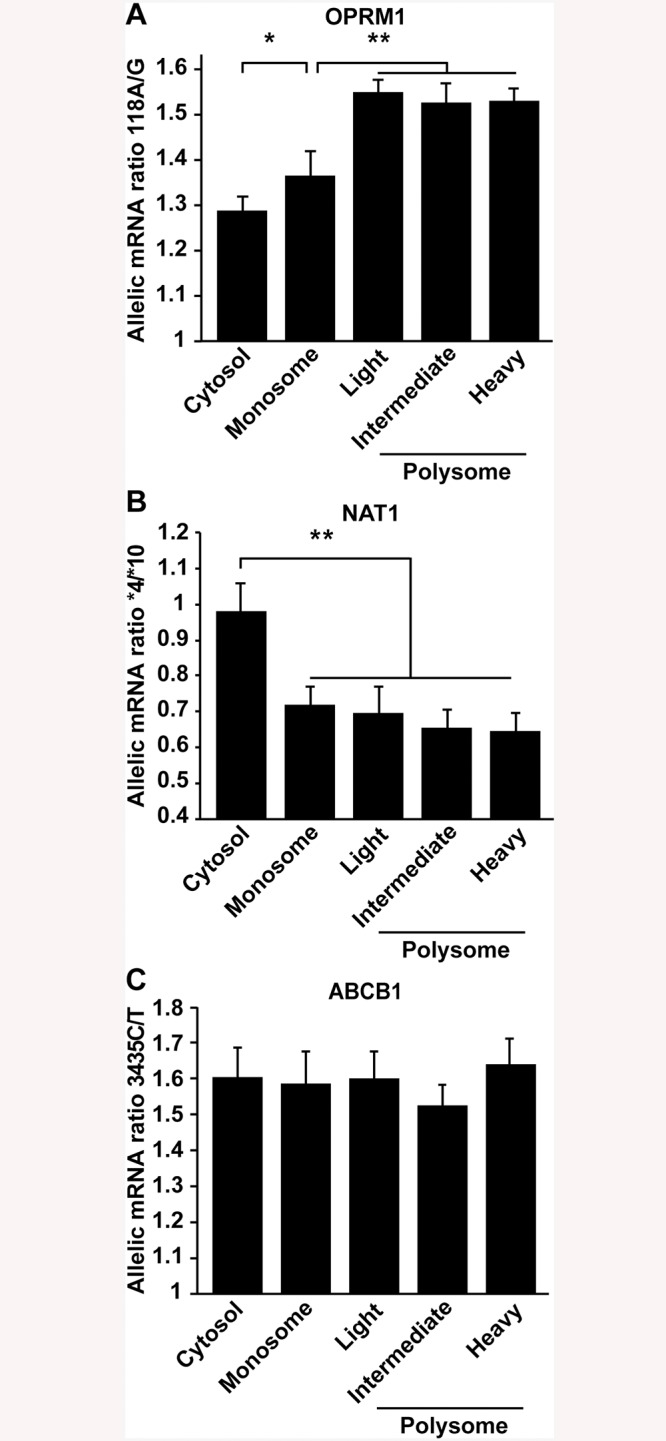
A. Effect of OPRM1 118A>G polymorphism on cytoplasmic mRNA expression and polysome loading; an A>G ratio >1 indicates reduced levels of the minor G allele relative to the A allele. Equal amounts of OPRM1 plasmids containing 118A and G were co-transfected into CHO cells. The 118A/G allelic ratios, measured with SNaPshot, represent the mean ± s.(n = 6), *p<0.049 for monosome versus cytoplasmic and **0.003 for monosome versus polysomes, two-tailed Student’s t-test. These profiles are representative of results from 3 independent cultures. B-C. AEI measurement in polysomes of lymphoblastoid cells with native expression of NAT1 and ABCB1. LCL (1 x 107 cells) heterozygous for *4/*10 were selected for polysome preparation. N-acetyltransferase 1 (NAT1) *10 allelic mRNA increased polysomal loading (lower *4/*10 ratios, p<0.01, corrected for gDNA ratios (B). Ribosomal occupancy of ABCB1 (MDR1) 3435C>T allelic mRNA. ABCB1 3435C>T allelic mRNA ratios (C). Reduced 3435C/T ratios demonstrated a significant reduction of the 3435T allele in the cytoplasmic mRNA extract, with no significant difference in any gradient fraction. These profiles are representative of gradients done with extracts from 3 independent cultures (mean ± s.d., n = 6).
B. N-acetyltransferase 1 NAT1*10 (rs1057126) increases polysomal mRNA loading
NAT1 is natively expressed in LCLs at levels sufficient for analysis. The NAT1*10 3′ UTR SNP had been shown to enhance translation in LCLs without affecting total cytoplasmic mRNA content [24], suggesting enhanced polysomal loading of the *10 allele versus the *4 wild-type allele. This hypothesis was tested in LCL sample GM07341, which is heterozygous for the NAT1*4/*10 alleles. Consistent with a lack of effect of NAT1*10 on transcription and mRNA processing, the allelic mRNA ratio NAT1*4/*10 in the cytoplasmic lysate did not differ from unity (0.98 ±0.08, p-value ≤ 0.05, n = 6) (Fig 2B). In contrast, monosomes and polysome fractions displayed allelic *4/*10 mRNA ratios below unity (0.72–0.64) (n = 6 for each category), demonstrating greater ribosome loading for the minor *10 allele, accounting for enhanced protein expression [24]. In contrast to OPRM1 (Fig 2A), the allelic *4/*10 was already strongly reduced in the monosome fraction, suggesting a main effect on first ribosomal loading, possibly via long-range interactions between the 3′ UTR *10 allele with the 5′ UTR.
C. Polysomal occupancy is not affected by the ABCB1 (MDR1) rs1045642 (3435T) allele
We tested the effect of SNP 3435C>T on mRNA expression and polysomal occupancy of ABCB1 mRNA in LCLs natively expressing ABCB1 and in HeLa cells transfected with an equal mixture of full-length ABCB1 expression plasmid with either 3435C or 3435T. Consistent with our earlier findings supporting enhanced mRNA turnover [24], the allelic mRNA ratio was 1.6 ± 0.1 (n = 6) in the cytosolic lysate, demonstrating a lower abundance of the minor allele 3435T, presumably as a result of increased turnover. The allelic mRNA ratios did not significantly differ in the polysomal fractions (allelic ratio 1.64 ± 0.05 (n = 6) (Fig 2C). In ABCB1 transfected HeLa cells, similar results were obtained, except that the allelic mRNA ratios were 1.3 ± 0.04 (n = 6) in total cytoplasm and remained the same throughout all gradient fractions (S2 Fig).
These results demonstrate the utility of detecting differences in allelic mRNA ratios between cytosol and polysomes.
Polysome RNA loading in three LCLs measured with RNA-Seq
The purpose of this exploratory study was to determine the loading of all RNA classes onto polysomes, including mRNAs and noncoding RNAs, and their isoforms and alleles. Genetic effects can be indirect, for example by altering transcription start sites, splice sites, and polyadenylation sites leading to isoforms undergoing differential loading, or by altering sequestration into other cell structures such as P-bodies.
Polysomal loading of mRNAs and other RNA classes (>200 bp long)
RNA-Seq was performed on cytosolic extracts and polysomal fractions (3 ribosomes or more) from three LCLs. Previous studies have measured mRNAs in the heavy fractions of the sucrose gradient eluate; however, this fraction does not contain smaller particles that sequester RNA (e.g., RNPs), potentially further confounding isoform and allele distributions. For better comparison with widely available RNA-Seq profiles, we measured total RNA content in the cytosol, expecting to detect significant changes in RNAs sequestered on polysomes. As each RNA fraction was amplified (NuGen kit), and relative contributions from various RNA classes differed between cytosol and polysomes, the fraction of any given RNA species loaded onto polysomes was difficult to compare. Therefore, we focused on relative recovery of the various RNA classes and rank order of single RNAs compared to total RNA. The number of RNA-Seq reads per sample averaged ~40 million with a mean length of ~120 bases, of which ~70% aligned to annotated sequences representing mRNAs and various classes of noncoding RNAs including lncRNA and pseudogenes.
The distributions of long RNA classes (>200 bases) within the cytosolic and polysomal fractions are shown in Fig 3A. While all RNA classes are represented on polysomes, individual RNAs among each class display large differences in their access to polysomes, consistent with polysomal loading as a critical step in regulation. Protein-coding mRNAs showing significant differences in relative abundance between polysomes and cytosol are listed in Table 1. Despite of low number of LCLs in this study relative to the large number of mRNAs tested, we were able to identify twenty-two mRNAs showing significantly different polysomal loading. Fourteen of these mRNAs, predominantly encoding histone proteins, showed robust enrichment on polysomes, reflecting the continuous demand for this type of protein. Among polysome-depleted genes, we observed mRNAs encoding MT-RNR2-like proteins, tumor protein translationally-controlled 1 (TPT1) and translation initiation regulators: polyadenylate-binding proteins 1 and 3 (PABPC1, 3). Possibly, these mRNAs reside mostly outside polysomes but can be rapidly recruited into polysomes when cellular conditions require rapid acceleration of protein synthesis.
Fig 3. RNA classes in cytosol and polysomes from three LCLs, measured by RNA-Seq.

After amplification of total cytosol RNA fraction and polysome fractions with NuGen, equal amounts of RNA were subjected to RNA-Seq, and sequence reads were aligned to annotated RNA to determine expression levels. A. Average expression level (normalized to FKPM) in cytoplasmic and polysome fractions from 3 different LCLs after exclusion of rRNA, tRNA, and mt-RNA. The pseudogenes and lncRNAs are reduced on polysome fractions. In cytosol lncRNA represent antisense-RNA (21.3%), lincRNA (4.7%) and lncRNA (74%). On polysomes the lncRNA distribution was drastically different: antisense-RNA (73%), lincRNA (14%) and lncRNA (13%). Error bars represent expression s.d. from 3 different LCL cells. B-D. Hierarchical clustering of the profiles of protein coding genes (B), pseudogenes (C) and lncRNA (D) performed with similarity indices (see Materials and Methods) for cytosol and polysome fractions. Observed difference of similarity indices between cytosol and polysome clusters is higher in lncRNA than in other RNA classes (note the difference on vertical scale).
Table 1. Protein coding mRNAs showing significant differences of polysome to cytosol ratios, adjusted p-value < 0.05.
| Gene ID | Gene name | Polysome/Cytosol log2(fold change) |
|---|---|---|
| ENSG00000198327.3 | HIST1H4F | 2.9 |
| ENSG00000188987.2 | HIST1H4D | 2.8 |
| ENSG00000196226.2 | HIST1H2BB | 2.5 |
| ENSG00000197409.6 | HIST1H3D | 2.3 |
| ENSG00000124529.3 | HIST1H4B | 2.3 |
| ENSG00000158406.2 | HIST1H4H | 2.1 |
| ENSG00000196331.5 | HIST1H2BO | 2.1 |
| ENSG00000197061.3 | HIST1H4C | 2.1 |
| ENSG00000256018.1 | HIST1H3G | 2.0 |
| ENSG00000187990.4 | HIST1H2BG | 2.0 |
| ENSG00000160932.6 | LY6E | 2.0 |
| ENSG00000198518.5 | HIST1H4E | 1.9 |
| ENSG00000180573.8 | HIST1H2AC | 1.9 |
| ENSG00000196532.4 | HIST1H3C | 1.8 |
| ENSG00000070756.9 | PABPC1 | -1.9 |
| ENSG00000151846.7 | PABPC3 | -2.0 |
| ENSG00000133112.12 | TPT1 | -2.2 |
| ENSG00000255823.1 | MTRNR2L8 | -2.9 |
| ENSG00000255633.3 | MTRNR2L9 | -3.2 |
| ENSG00000256045.1 | MTRNR2L10 | -3.3 |
| ENSG00000256618.1 | MTRNR2L1 | -3.4 |
| ENSG00000270672.1 | MTRNR2L6 | -3.4 |
While the polysome fractions contained high levels of protein-coding transcripts as expected, pseudogenes and long noncoding RNAs were also detected but at reduced relative levels compared to the cytosol (Fig 3A). Pseudogenes appeared to be more efficiently loaded onto polysomes compared to lncRNAs. Moreover, among the various RNA subclasses, large differences were observed. For example, lncRNA comprises antisense-RNA (21%), lincRNA (5%) and additional RNAs annotated in lncipedia as generic lncRNA (74%) in the cytosol, but on polysomes the distribution is drastically different: antisense-RNA (73%), lincRNA (14%) and lncRNA (13%). Pseudogenes and lncRNAs showing statistically significant polysome enrichment are listed in Table 2. For example, lncRNA lnc-EPHA6-1 was differentially enriched on polysomes and U47924.28, CTD-2369P2.2, and RP11-16E12.2 showed similar trends. Their role in translation should be further studied. In contrast, RNA encoded by MT-RNR2-like 11, which is annotated as a pseudogene but nevertheless protein-coding, was significantly lower in the polysome fraction (log2 fold change -3.3, adjusted p-value 0.018), possibly accounting for the discrepant annotation. Polysome to cytosol ratio distributions of mRNA and ncRNAs for all tested genes are shown in S3A Fig mRNAs were significantly enriched on polysomes compared to cytosol.
Table 2. Noncoding RNAs showing significant differences of polysome to cytosol ratios, adjusted p-value < 0.05.
| Gene ID | Gene name | Polysome/ Cytosol log2(fold change) | RNA type |
|---|---|---|---|
| lnc-EPHA6-1 | lnc-EPHA6-1 | -3.4 | lncRNA |
| ENSG00000270188.1 | MTRNR2L11 | -3.3 | Pseudogene |
| ENSG00000234782.2 | RP11-442A13.1 | -2.5 | Pseudogene |
| ENSG00000214460.3 | RP11-30P6.3 | -2.5 | Pseudogene |
| ENSG00000225471.2 | RP11-262D11.2 | -2.3 | Pseudogene |
| ENSG00000217027.1 | RP1-83M4.2 | -2.1 | Pseudogene |
| ENSG00000223361.5 | FTH1P10 | -2.0 | Pseudogene |
| ENSG00000226221.1 | AC022431.1 | -1.9 | Pseudogene |
| ENSG00000234009.1 | RPL5P34 | -1.7 | Pseudogene |
| ENSG00000226948.1 | RPS4XP2 | -1.7 | Pseudogene |
| ENSG00000220842.5 | RP11-572P18.1 | -1.7 | Pseudogene |
| ENSG00000235552.4 | RPL6P27 | -1.7 | Pseudogene |
| ENSG00000220793.4 | RPL21P119 | -1.7 | Pseudogene |
| ENSG00000226608.2 | FTLP3 | -1.6 | Pseudogene |
| ENSG00000223803.1 | RPS20P14 | -1.6 | Pseudogene |
| ENSG00000243802.2 | RP11-390K5.1 | -1.6 | Pseudogene |
| ENSG00000218175.2 | AC016739.2 | -1.6 | Pseudogene |
| ENSG00000239559.2 | RPL37P2 | -1.6 | Pseudogene |
| ENSG00000243199.1 | RP11-408P14.1 | -1.5 | Pseudogene |
To understand broadly the processes that govern the distribution of RNA classes, we determined similarity indices and constructed dendrograms of the three cytosolic and polysomal samples. Fig 3B–3D depicts the distance between branches of the dendrogram, calculated for expression profiles of various classes of RNA, revealing higher similarity of the profiles within cytosol and polysomes than between individual LCLs. lncRNAs displayed significantly greater diversity in profiles between cytosol and polysomes (Fig 3D, note different scales) compared to protein coding and pseudogene RNAs (Fig 3B and 3C), suggesting differences in the regulation of polysome loading between RNA classes, consistent with distinct overall functions.
Differential polysome loading of microRNAs
We also sequenced short noncoding RNAs and focused our analysis on microRNAs. We observed a large number of microRNAs in the polysomal fraction, consistent with previous findings [27]. Table 3 lists the microRNAs with greatest differences between cytosol and polysomes, showing large enrichment or depletion of microRNAs on polysomes, suggesting specific functions, possibly in translation. Distribution of polysome to cytosol ratios for all detected microRNAs is shown in S3B Fig. Again, we performed clustering analysis, comparing microRNAs in both fractions (Fig 4A), highlighting the microRNAs consistently over- or underrepresented. The analysis of microRNA expression profiles, performed using Renyi’s divergence followed by hierarchical clustering, revealed substantial differences of microRNA profiles between polysomes and cytosol (Fig 4B). Specifically, the microRNA profiles of polysomes displayed higher similarity as compared to cytosol (Fig 4B), suggesting tight regulation of polysomal access as described previously [27].
Table 3. Top-scoring 20 microRNAs most strongly enriched on polysomes and most strongly polysome-depleted compared to cytosol, non-adjusted p-value < 0.05.
| Polysome/Cytosol ratio | Polysome/Cytosol ratio | ||||||
|---|---|---|---|---|---|---|---|
| microRNA (enriched) | LCL14 | LCL19 | LCL31 | microRNA (depleted) | LCL14 | LCL19 | LCL31 |
| miR-340-3p | 28 | 31 | 72 | miR-21 | 0.12 | 0.10 | 0.08 |
| let-7b | 24 | 56 | 30 | miR-1246 | 0.02 | 0.09 | 0.26 |
| miR-181b | 27 | 25 | 13 | miR-155-3p | 0.11 | 0.15 | 0.15 |
| miR-423 | 13 | 18 | 15 | miR-30b | 0.15 | 0.20 | 0.24 |
| miR-1275 | 11 | 12 | 4.1 | miR-4301 | 0.12 | 0.18 | 0.32 |
| miR-574 | 13 | 7.3 | 5.9 | miR-181a-3p | 0.09 | 0.32 | 0.23 |
| miR-320a | 6.8 | 8.3 | 9.2 | miR-5100 | 0.06 | 0.45 | 0.12 |
| miR-342 | 6.8 | 8.0 | 8.9 | miR-1260b | 0.12 | 0.30 | 0.24 |
| miR-145 | 3.4 | 6.5 | 11 | miR-551b-3p | 0.14 | 0.32 | 0.28 |
| miR-423-3p | 6.7 | 5.1 | 8.3 | miR-342-3p | 0.21 | 0.27 | 0.30 |
| miR-181c | 3.5 | 6.9 | 4.8 | miR-191 | 0.27 | 0.26 | 0.28 |
| miR-181a | 6.3 | 3.4 | 4.5 | miR-106b | 0.32 | 0.32 | 0.30 |
| miR-193b-3p | 3.4 | 3.1 | 7.0 | miR-425 | 0.38 | 0.34 | 0.29 |
| miR-877 | 1.9 | 4.2 | 6.9 | miR-140-3p | 0.40 | 0.24 | 0.37 |
| let-7c | 3.3 | 3.1 | 6.1 | miR-625-3p | 0.32 | 0.29 | 0.41 |
| miR-1307-3p | 4.6 | 4.1 | 3.3 | miR-374b | 0.39 | 0.29 | 0.41 |
| miR-193b | 3.5 | 2.9 | 5.4 | miR-629-3p | 0.27 | 0.55 | 0.31 |
| miR-92a-3p | 4.3 | 3.3 | 4.2 | miR-27a-3p | 0.39 | 0.34 | 0.42 |
| let-7a | 5.4 | 3.2 | 2.3 | miR-4485 | 0.16 | 0.77 | 0.25 |
| miR-7 | 3.5 | 4.4 | 2.3 | miR-4448 | 0.19 | 0.29 | 0.76 |
Fig 4. microRNA expression in cytosol and polysomal loading in three LCLs.

A. Hierarchical clustering of microRNA expression profiles between cytosol and polysomes. microRNAs highly enriched on polysomes such as let-7a, mir-1275, and mir-145 had docking sites on mRNAs also enriched on polysomes. B. Hierarchical clustering of microRNA expression profiles measured with similarity indices (see Materials and Methods) reveal significant differences of microRNA profiles between cytosol and polysome fractions, showing reduced relative distances in comparison to cytosolic fractions.
Analysis of putative targets of the top 20 microRNAs with greatest preferential loading onto polysomes revealed 167 polysome-enriched mRNAs, with polysome/cytosol ratio ranging from 2 to 5.5 (non-adjusted p-value <0.01). Lymphocyte antigen 6 complex, locus E (LY6E), the putative target of mir-1275 (ratio >4), showed significantly higher polysomal occupancy (polysome/cytosol ratio 3.9, adjusted p-value 0.018). Polysome/cytosol mRNA ratio of PIN1, RHOG, H2AFX, and NAT14 ranged from 2.8 to 3.8, (adjusted p-value 0.06 to 0.077), while corresponding microRNAs predicted to target these genes, mir-1275, let-7a (for RHOG and NAT14), and mir-145, had ratios from 2.3 to 12. These results support the model proposed by Molotski and Soen, in which microRNA occupancy on polysomes is determined by their interaction with target mRNA [27].
Differential loading of mRNA isoforms onto polysomes
We then tested whether RNA isoforms, such as splice variants or RNAs with varying 3′ and 5′ UTRs, undergo differential loading. Determining mRNA isoforms from the RNA-Seq data yielded estimates of relative isoform distribution between cytosol and polysome fractions. RNA-Seq data on annotated isoforms were subjected to pairwise comparison between cytosol and polysomes, yielding 327 unique genes with major isoforms displaying different distributions (change in fraction of total mRNA >20% between cytosol and polysomes) (Fig 5). Select isoforms are listed in Table 4, separated into isoform RNAs enriched and depleted in polysomes. Genes generating polysome-enriched isoforms include VEGFA, STIM2, CLIP2, IMMT, SLC24RG, FMR1, ABCC1, and SERPIND8, whereas FAM195A, AURKA, CTTN, ZNF280D, PPRC1, and SLC39A8 isoforms were depleted. Differences in isoform distribution are likely the result of distinct regulatory sequences. In addition, isoforms of annotated genes that do not contain open reading frames (ORFs), e.g., ZNF280D and FAM127A, were observed predominantly in cytosol, presumably lacking domains needed for polysomal loading.
Fig 5. mRNA isoforms consistently different between cytoplasmic and polysome identified by RNA seq in three LCLs.

Among all isoforms detected for a gene, some isoforms significantly differed in abundance between cytosol and polysomes. Isoforms showing similar pattern in all 3 LCLs are shown. Differences consistent among all cell lines suggest the possibility of isoform specific polysome recruitment. The results represent mean ± s.d. (n = 3). The isoforms are identified in S3 Table.
Table 4. Genes expressing RNA isoforms with differential loading onto polysomes, and displaying different allelic RNA ratios in cytosolic and polysomal extracts.
Targets with > 2 fold difference in allelic ratio between cytosol and polysome were analyzed for isoform differences. Results show both enrichment and depletion of isoforms on polysomes.
| % Isoform* | Allelic ratio | ||||||
|---|---|---|---|---|---|---|---|
| Isoform ID | Gene | Cytosol | Polysome | Cytosol | Polysome | Transcript Type | Location |
| ENST00000226798.4 | FRG1 a | 67 | 31 | 1 | 2.7 | Protein coding | exonic |
| ENST00000253413.5 | ATP6V1E1 a | 91 | 63 | 2.8 | 1.2 | Protein coding | 5' UTR/ 3' UTR |
| ENST00000254719.5 | RPA1 a | 72 | 12 | 4.2 | 1 | Protein coding | exonic/3' UTR |
| ENST00000285243.6 | ANKRD40 b | 60 | 83 | 2.4 | 1 | Protein coding | 3' UTR |
| ENST00000292476.5 | CPSF4 a | 99 | 73 | 2.6 | 1.1 | Protein coding | intronic |
| ENST00000292538.4 | CUX1 b | 38 | 61 | 1 | 2.2 | Protein coding | exonic |
| ENST00000312419.3 | POLD4 a | 90 | 63 | 2.9 | 1.3 | Protein coding | 3' UTR |
| ENST00000319004.5 | GEMIN4 a | 91 | 75 | 1.2 | 3.8 | Protein coding | exonic |
| ENST00000338754.4 | TPST2 a | 76 | 12 | 2.8 | 1.2 | Protein coding | exonic |
| ENST00000357984.3 | TMUB2 b | 28 | 57 | 3.9 | 1.3 | Protein coding | 3' UTR |
| ENST00000361439.4 | NSMCE1 b | 57 | 91 | 2.3 | 1 | Protein coding | exonic/ 3' UTR |
| ENST00000361445.4 | MTOR a | 82 | 57 | 3 | 1.1 | Protein coding | exonic |
| ENST00000368324.4 | SYT11 a | 100 | 55 | 3.1 | 1.3 | Protein coding | 5' UTR |
| ENST00000372235.3 | TMEM53 b | 27 | 59 | 2.8 | 1.1 | Protein coding | 3' UTR |
| ENST00000373616.5 | MTCH1 a | 87 | 57 | 3 | 1.3 | Protein coding | 3' UTR |
| ENST00000398110.2 | TPST2 b | 24 | 88 | 2.8 | 1.2 | Protein coding | exonic |
| ENST00000440410.1 | DLD a | 55 | 30 | 2.5 | 1.1 | Protein coding | exonic |
| ENST00000444411.1 | MMS19 b | 39 | 62 | 2.2 | 1.7 | Protein coding | exonic |
| ENST00000447062.2 | WDTC1 a | 51 | 20 | 2.4 | 1 | Nonsense mediated decay | 3' UTR |
| ENST00000483469.1 | GET4 a | 52 | 32 | 2.3 | 1.1 | Retained intron | exonic |
| ENST00000483996.1 | SUN1 b | 51 | 70 | 2.3 | 1.1 | Retained intron | exonic |
| ENST00000491660.1 | SERP1 b | 27 | 56 | 1.1 | 2.8 | Protein coding | exonic |
| ENST00000492590.1 | FHIT a | 100 | 74 | 3.2 | 1.1 | Protein coding | exonic |
| ENST00000492955.1 | LY75 a | 55 | 30 | 4 | 1.8 | Processed transcript | exonic/ intronic |
| ENST00000518016.1 | FBXO16 b | 3 | 48 | 2.6 | 1.1 | Retained intron | exonic |
| ENST00000529725.1 | SIPA1 a | 55 | 33 | 2.8 | 1.3 | Retained intron | exonic |
| ENST00000534474.2 | MPV17L2 a | 65 | 39 | 3.4 | 1.1 | Novel Protein coding | exonic |
| ENST00000540523.1 | STAM b | 52 | 71 | 1 | 2.6 | Protein coding | exonic |
| ENST00000570487.1 | RABEP1 b | 47 | 73 | 2.7 | 1.1 | Protein coding | exonic |
| ENST00000587040.1 | SYNRG b | 38 | 58 | 2.6 | 1.5 | Processed transcript | exonic |
aRNA isoforms depleted on polysomes.
bRNA isoforms enriched on polysomes.
* Percentage of each isoform in the pool of all isoforms for given gene.
Allelic RNA ratios in cytosol and polysomes of LCLs
Allelic differences in the cytosolic fraction result from regulatory variants altering gene expression, mRNA processing, and sequestration to cellular compartments [28], whereas differences in allelic ratios between cytosol and polysomes reflect differential allelic mRNA loading of the mRNAs or of their isoforms. We calculated allelic mRNA ratios for all RNAs with sufficient expression (>20 reads across a SNP) in the cytosol and polysome fractions of three LCLs as described [22]. Where available, we used several SNPs per transcript to determine mean allelic ratios and S.D. per transcript in one or more LCLs. This approach detected 630 genes in the cytosol and 559 genes in the polysomes with allelic RNA ratio ≥2-fold, with an approximate 25% overlap. RNAs with the highest allelic ratios are provided in S2 Table, including mRNAs and ncRNAs.
To identify genetic factors altering polysomal loading, we searched for mRNAs showing ≥2-fold difference in the allelic RNA ratio (major/minor allele) between cytosolic and polysomal fractions of the same LCL sample (Fig 6). This approach yielded 60 mRNAs (11%) with the main allele enriched on polysomes compared to the cytosol, and 112 mRNAs (18%) with the main allele enriched in the cytosol (Fig 6A). To guard against false positive results, we selected mRNAs providing allelic ratio at more than one SNP (15%), shown in Fig 6A, some displaying large differences in allelic ratios between the cytosol and polysomes.
Fig 6. Allelic RNA ratios determined in whole transcriptome and translatome (polysomal RNAs) of three LCLs.

A. Allelic RNA ratio differences measured in genes with two or more heterozygous SNPs are shown. To improve accuracy of allelic ratios measured using sequencing data, the ratios were measured at multiple SNPs of same transcript. B. Validation of mRNA allelic ratios obtained with RNA-Seq using SNaPshot assay. Both methods yielded similar allelic ratio differences between cytosol and polysomes.
To validate allelic ratios calculated from RNA-Seq data, we employed a robust method (SNaPshot) [25] for accurate ratio analysis, selecting mRNAs with heterozygous states at two or more SNPs in the same LCL. Shown in Fig 6B, the results are consistent in detecting different allelic ratios between cytosol and polysomes for all test RNAs, even if the ratios in some samples were shifted to higher or lower values with SNaPshot (example FOXK2). FOXK2, GOT2, LMO2, PMS2, NSA2 and TOB2 mRNAs displayed a higher allelic ratio on polysomes, while HSP1, IFFO2, and ITPK1 presented with a lower ratio on polysomes, as observed by RNA-Seq (Fig 6B).
Among the pseudogenes, MGC70870, and GSTM2P1 had significant differences in allelic RNA ratios between cytosol and polysome fractions, suggesting similar processes regulating polysomal loading compared to mRNAs; possible translation into proteins should be evaluated. We also observed a few examples of robust allelic ratios deviating from unity in lncRNAs (LINC00665, RP11-94L15.2, lnc-CTR9-3 (ZBED5-AS1)), but the ratios did not differ between cytosol and polysome, suggesting regulation of transcription or RNA processing. Overall, our results show that subcellular RNA samples have different allelic compositions, adding to the regulatory processes arising from genetic factors.
Relationship between mRNA isoforms and measured allelic ratios in cytosol and polysomes
To distinguish effects of regulatory variants on isoform formation and on polysomal loading of isoforms, we surveyed all 327 genes with substantial differential polysomal loading of a major isoform (at least 20% difference between fractions relative to the total number of reads for transcripts at a gene locus) for allelic mRNA expression imbalance. Measured allelic RNA ratios can reflect contributions from different RNA isoforms if the marker SNP resides in an exon shared between them. For specific isoform analysis one needs to select SNPs in an exon not shared with other isoforms. Among genes with differential polysomal loading of isoforms, 82% displayed allelic ratios ≥2 fold in either cytosol or polysome in at least one LCL, but no significant difference between the two fractions, suggesting regulatory effects on transcription or RNA processing affecting all isoforms equally, for example equal changes in turnover rates. In contrast, 18% of mRNA with substantial isoforms detectable (>50% of total RNA expressed for each gene), for example MTOR, POLD4, and FHIT, displayed distinct allelic ratios between the cytosol and polysomes, suggesting a role in isoform formation or polysomal loading (Fig 7). The isoforms are identified in Table 4. Among isoforms depleted on polysomes, most have reduced allelic ratios of total RNA expressed a gene (major/minor allele) on polysomes (such as WDTC1, SIPA1, LY75, DLD, RPA1, TPST2, MTOR), except for FRG1 and GEMIN4 with higher polysomal allelic ratios (Fig 7A). A reduced ratio suggests that the minor allele isoform is enriched on polysomes. Similar results are observed with isoforms enriched on polysomes, again mostly showing reduced allelic mRNA ratios in polysomes (examples TPST2, TMEM53, TMUB2, SYNRG, MMS19), with only 3 isoforms having higher allelic ratios on polysomes (SERP1, CUX1, STAM) (Fig 7B). The interpretation of these results is hampered by allelic ratios at SNPs that could represent more than one mRNA isoform.
Fig 7. mRNA isoforms differentially loaded onto polysomes, and in addition displaying twofold different allelic RNA ratios (major/minor alleles) between cytosol and polysomes.

A. Isoforms underrepresented on polysomes. B. Isoforms enriched on polysomes. Allelic mRNA ratios reflect the sum of all isoforms where the marker SNP is located. The allelic mRNA ratios are shown above the respective bar.
Overlap between published eQTLs or GWAS associations and instances of differential allelic RNA ratios between polysomes and cytosol
A finding of allelic expression imbalance (AEI) signals the presence of regulatory variants. While distinct cytosol-polysome allelic RNA ratios likely arise from regulatory variants residing in the transcribed region of a gene locus, epigenetic effects and RNA editing are also possible [22]. As we have examined only three LCL, a single finding of AEI could implicate regulatory variants that could have a broad range of allele frequencies in the population, some showing high frequency. The instances of distinct AEI in polysomes reported here reveal a robust effect on RNA expression and presumably protein levels, with likely physiological consequences. Therefore, we searched for overlaps with GWAS associations (NHGRI GWAS Catalog) and published mRNA expression quantitative trait loci (eQTLs) from several databases, including Genotype-Tissue Expression project (GTEx), with expression data and eQTLs for multiple human tissues. In addition, Li et al. had identified polysomal eQTLs by measuring mRNA polysome recruitment with hybridization arrays in LCLs [7]. Table 5 summarizes overlaps with eQTLs for 37 genes identified here with strong allelic differences between polysomes and cytosol [29–57]. Strikingly, all but 4 of the 37 AEI genes also displayed eQTLs. Specifically, 11 genes had also been identified by Westra et al. [29]. Because such regulatory variants likely reside in the transcribed portion of the gene, we expected that in some cases the same SNP would be identified as an eQTLs as the one showing AEI, suggesting functional relevance. Indeed, in all 11 genes overlapping with the Westra study, all AEI SNPs were identical to eQTL SNPs (Table 5). Examples include CDCA2 (rs6990278; minor allele frequency (MAF) = 0.19)–the regulator of chromosome structure during mitosis, mitochondrial ribosomal protein MRPL13 (rs6650; MAF = 0.473), and RAS oncogene family member RAB38 (rs1027027, MAF = 0.279). eQTLs in transcripts of genes such as nuclear proteins involved in regulation of proliferation and cell cycle, such as NSA2 (rs6874609, MAF = 0.381) or ceramide synthase CERS5 (rs7279, MAF = 0.201), were also identified by GTEx.
Table 5. Genes implicated as having genetic variants affecting polysomal loading (either directly or via RNA isoforms) were tested for associations with clinical phenotypes (all GWAS from dbGaP) and eQTLs.
| Allelic ratio | ||||||
|---|---|---|---|---|---|---|
| Gene ID | Gene name | Cytosol | Polysomes | GWAS catalog | eQTL | References |
| ENSG00000154945.6 | ANKRD40 | 2.4 | 1 | x | [30] | |
| ENSG00000131100.8 | ATP6V1E1 | 2.8 | 1.2 | x | [30] | |
| ENSG00000122515.10 | ZMIZ2 | 2.3 | 1.1 | x * | [29] | |
| ENSG00000184661.9 | CDCA2 | 1.1 | 2.2 | x * | [29] | |
| ENSG00000170779.10 | CDCA4 | 2.2 | 1 | x * | [29] | |
| ENSG00000139624.8 | CERS5 | 2.2 | 1.3 | x * | [29, 30] | |
| ENSG00000080819.2 | CPOX | 3.1 | 1.2 | x * | [29] | |
| ENSG00000241258.2 | CRCP | 2 | 1.7 | x * | x * | [29, 31] |
| ENSG00000079459.8 | FDFT1 | 2 | 1.2 | x | x | [30, 32, 33] |
| ENSG00000109536.7 | FRG1 | 1 | 2.7 | x | x | [30, 34] |
| ENSG00000125166.8 | GOT2 | 1.3 | 2.6 | x | x | [31, 35] |
| ENSG00000107521.14 | HPS1 | 2 | 1.1 | x | [30] | |
| ENSG00000113013.8 | HSPA9 | 1.2 | 1.7 | x * | [29] | |
| ENSG00000161405.12 | IKZF3 | 1.2 | 1.7 | x * | x * | [29, 36–43] |
| ENSG00000100605.12 | ITPK1 | 2.6 | 1.3 | x | x | [30, 44–46] |
| ENSG00000226025.5 | LGALS17A | 1.7 | 3.6 | x | x | [47] |
| ENSG00000135363.7 | LMO2 | 1.2 | 1.7 | x | [48] | |
| ENSG00000054219.9 | LY75 | 4.0 | 1.8 | x | x | [30, 49] |
| ENSG00000172172.3 | MRPL13 | 2.3 | 1.6 | x * | [29] | |
| ENSG00000137409.14 | MTCH1 | 3 | 1.3 | x | [30] | |
| ENSG00000198793.8 | MTOR | 3 | 1.1 | x | x | [30, 50] |
| ENSG00000164346.5 | NSA2 | 1.2 | 2.5 | x * | [29, 30] | |
| ENSG00000078674.13 | PCM1 | 1.1 | 1.4 | x | x | [30, 51] |
| ENSG00000122512.10 | PMS2 | 1.3 | 1.8 | x * | [29, 30] | |
| ENSG00000123892.7 | RAB38 | 1 | 2.4 | x | x * | [29, 30, 52] |
| ENSG00000029725.12 | RABEP | 2.7 | 1.1 | x | [30] | |
| ENSG00000168421.7 | RHOH | 1.1 | 1.7 | x | [53] | |
| ENSG00000132383.7 | RPA1 | 4.2 | 1 | x | [30] | |
| ENSG00000163902.7 | RPN1 | 2 | 1.2 | x | x * | [29, 54, 55] |
| ENSG00000213445.4 | SIPA1 | 2.8 | 1.3 | x | [30] | |
| ENSG00000140199.7 | SLC12A6 | 1.2 | 1.4 | x * | [29, 30] | |
| ENSG00000136738.10 | STAM | 1 | 2.6 | x | [30] | |
| ENSG00000006114.11 | SYNRG | 2.6 | 1.5 | x | x | [30, 56] |
| ENSG00000136270.9 | TBRG4 | 2.7 | 1.4 | x * | [29] | |
| ENSG00000183864.4 | TOB2 | 1.3 | 2.3 | x | [57] | |
| ENSG00000105939.8 | ZC3HAV1 | 1.2 | 1.7 | x * | [29] | |
| ENSG00000183309.7 | ZNF623 | 1.5 | 1.8 | x * | [29] | |
*The same SNP implicated in allelic RNA analysis in polysomes is also identified as a candidate variant in the databases.
Overlap between polysome AEI and clinical phenotypes in the database of Genotypes and Phenotypes (dbGaP) was observed in 16 of the 37 genes (Table 5). This finding suggests a link between regulatory variants and downstream phenotypes. In two genes, CRCP and IKZF3, the implicated dbGaP SNP was identical to the SNP identified by our AEI analysis, suggesting that it is the causative SNP or in high Linkage Disequilibrium (LD) with a functional variant.
We also searched for databases with protein eQTLs (pQTLs), but these are less well developed [58]. In one case, the level of GOT2, which showed polysome-specific AEI in this study, was associated with SNP rs7194417 [59]. Also showing allele selective polysome loading of one of its isoforms, IMMT had been identified in Battle’s study as having a pQTL [60].
Discussion
Delineating the contributions of regulatory variants on protein translation is critical for understanding a substantial proportion of unresolved genetic regulation. The overall regulation of translation is considered a main factor determining protein abundance in cell lines [61], and improved correlations between polysomal mRNA and protein levels are better correlated to protein levels than total cellular mRNA levels [6]. Both findings together indicated that differential loading onto polysomes reflects a significant portion of translational regulation. By measuring allelic mRNA ratios with precision and accuracy, we demonstrate that one can detect differential loading of mRNA alleles onto monosomes and further progression into polysomes, as a means for uncovering cis-acting variants affecting translation. Here, we utilize two novel approaches for detecting the effect of genetic variants in mRNA on translational activity by measuring allelic mRNA ratios. The first approach to establish a proof of principle uses targeted expression of cloned constructs for OPRM1, NAT1, and ABCB1 alleles [23, 24, 26], while the second utilizes endogenous expression in human cells to survey the entire polysome-bound transcriptome (i.e. the translatome) in LCLs and to assess the role of genetic variants in polysomal loading.
Our targeted gene results support the use of measuring allelic ratios during polysomal loading as a sensitive means for detecting genetic effects on mRNA translation, such as initial and subsequent ribosome loading, with OPRM1 and NAT1 and ABCB1 as examples. Full sequencing of the transcriptome and translatome detects widespread differences in allelic ratios on translating mRNAs, providing evidence for cis-acting variants that influence translation. However, measuring allelic RNA ratios alone in short sequencing reads reduces the ability to attribute genetic effects to specific RNA isoforms, such as splicing or different transcription start site or poly-adenylation site usage, that occur before polysomal loading and yield RNA isoforms that then have different ability to load ribosomes. Importantly, determining allelic RNA ratios in polysomal fractions is less confounded by RNA isoforms present in other cellular fractions, while focusing on RNA alleles with likely functions in regulating translation. Therefore, differential allelic loading signals a cis-acting influence on translation, with likely biological consequences that remain obscured when measuring total cellular RNA content alone.
Molecular genetics studies of target genes
The OPRM1 non-synonymous polymorphism 118A>G (N40D) had been shown to exert a dual effect on both overall mRNA expression levels and translational activity [23], while any effect on the protein’s function [62] remains uncertain. Upon co-transfection of full-length OPRM1 118A>G cDNA alleles, the main allele A is indeed more abundant in the cytosolic fraction, while it is further significantly enriched in the heavy polysome fractions, supporting previous findings of reduced expression and translation caused by 118G. On the other hand only a small increase of the A/G ratio was observed in the monosome fractions, indicating that reduced loading of an initial ribosome onto the G allele mRNA was less robustly impaired compared to further ribosome loading. As the 118A>G SNP resides towards the 5′ region of OPRM1, we speculate that the movement of the ribosome along the mRNA strand could be impaired, resulting in lower polysome loading.
The N-acetyltransferase allele NAT1*10 had been shown to increase expressed enzyme activity, compared to the wild-type (*4) which we had attributed to enhanced translational efficiency, presumably through the increased loading onto polysomes [24]. Here we have demonstrated in LCLs natively expressing NAT1 that NAT1*10 does promote association of the mRNA allele with polysomes. Even though the *10 SNP is located in the 3′ UTR, loading of the first ribosome was already enhanced, in contrast to the observation with OPRM1 118G. 3′-untranslated regions (3′ UTR) appear to physically interact with the 5′ UTRs via protein complexes in regulating initiation of translation [63], with genetic variants modulating this process.
The synonymous SNP 3435C>T ABCB1 (rs1045642), (MDR1) has been widely shown to lower expression of the P-glycoprotein drug transporter. In LCL cells natively expressing MDR1, we confirm a lower level of expression of the 3435T allele in the cytoplasmic lysate, consistent with enhanced mRNA turnover [25]. However, the allelic C>T ratios did not change in any of the polysomal fractions, which one would have expected if rare codon usage were to affect polysomal loading [26]. This result argues against ribosome stalling caused by 3435T that leads to nonsense mediated mRNA decay or no-go-decay, and expected allelic ratio differences in polysome fractions. Further studies, such as ribosomal profiling [64, 65] will be needed to fully resolve the mechanism attributable to 3435C>T underlying reduced MDR1 expression, a topic of considerable interest in predicting therapeutic drug response.
Transcriptome analysis in polysomes
The RNA sequencing data provided here show relative distributions of different RNA categories and their occupancy on polysomes from three lymphoblast cell lines (Fig 3). Our approach provides a framework for functional studies of coding and non-coding RNAs and the impact of genetic factors on translatome dynamics. We find large differences between polysomal loading of various RNA classes and their isoforms (Table 1) as already reported. Overrepresentation of histone coding mRNA on polysomes might reflect high demand for DNA replication processes in cycling cells. In addition, our observation of pseudogenes and long noncoding RNA transcripts on polysomes is complimentary to a recent map of the human proteome [66], reporting peptides from many of these RNA categories are translated—blurring the boundaries between coding and non-coding genes. However relatively low representation of pseudogenes on polysomes suggests that members of this class predominantly function as noncoding RNA.
Differential loading of mRNA isoforms was observed for a number of genes (Fig 5, Table 4). In addition, non-coding RNAs generate isoforms that are differentially loaded on to polysomes (Table 2), suggesting different biological functions between their isoforms. Hierarchical clustering of RNA profiles in cytosol and on polysomes (Fig 3B–3D) illustrates systematic differences between cytosol and polysomes, and between individuals. The highest level of similarity was observed with lncRNA in cytosol extracts, suggesting a high level of regulation to maintain cellular equilibrium. In contrast, mRNA levels in cytosol varied most between LCLs.
Comparison of microRNAs in cytosol and polysomes showed differential association of microRNAs with polysomes suggesting distinct interaction with their target mRNAs (Fig 4A). microRNAs either accelerate degradation of mRNA or impede (or alter) translation. Several microRNAs were found to co-sediment with polysomes as seen in this study and others [27], suggesting an effect on translation. Hierarchical clustering of similarity indices suggests that microRNAs are tightly regulated in polysomes with little deviation between subjects (Fig 4B). The interactions between microRNAs and abundant mRNAs that encode microRNA docking sites has been proposed to determine polysomal loading of microRNAs [27], a finding reproduced here. Supported by the results in this study, the interplay between non-coding and protein-coding RNAs can be revealed by simultaneous RNA-Seq analysis of all RNA classes.
Allelic RNA differences detected in transcriptomes on polysomes
Any deviation of allelic RNA ratios from unity (in autosomal genes devoid of copy number variants) reveals cis-acting regulatory factors altering polysomal loading or isoform expression, having downstream effects on isoform distribution onto polysomes. In addition, genetic variants can alter cellular trafficking by multiple mechanisms, such as microRNA binding. Any of these mechanisms can result in altered allelic ratios on polysomes, requiring further study to distinguish between them.
Transcriptome sequencing [22, 67] revealed variants covering protein-coding regions UTRs, ORFs, and noncoding RNAs, showing that allelic differences between cytosolic and polysomal mRNAs are more common than has been previously suggested by analysis of total RNA levels [7]. Therefore, regulatory variants affecting translation by differential polysome loading or isoform expression could be abundant [9].
Extensive localization of long noncoding RNAs to polysomes was reported earlier [68], but measuring the allelic ratio on polysomes to identify allele specific regulation had not been reported so far. Our findings provide a foundation for functional studies of the role of genetic variants and ncRNA in modulation of translation.
Overlap with previous results and clinical relevance of genes showing differential allelic loading onto polysomes
We have surveyed GWAS databases (dbGaP) and RNA expression data (GTEx) to search for clinical associations and eQTLs in genes showing strong, differential allele-selective loading on polysomes (Table 5). This analysis revealed substantial overlap between the AEI SNPs and associated genes identified in this study, and both eQTLs and dbGaP hits (Table 5). It is remarkable that a finding of large AEI in a single sample can identify eQTLs, which require many more samples when measuring more variable mRNA levels [29]. In particular, the large Westra’s study was designed to detect trans-eQTLs specific to mRNA expression levels, but included numerous cis-eQTLs [29]. With use of only three LCLs, the majority of genes identified by our approach were also listed as polysomal eQTLs. This result strongly supports our allele-specific approach; however, in either study, an effect of variants on RNA isoform formation cannot be excluded. Application of the approach proposed in this study to a larger number of LCLs may lead to identification of novel regulatory variants of clinical relevance.
In a number of instances, the SNPs previously identified as eQTLs or GWAS hits were identical, supporting the notion that the SNP is causative or in high LD with a causative variant. Of particular interest are two overlapping AEI-GWAS SNPs. The IKZF3 synonymous SNP rs907092 (MAF = 0.30) had been associated with primary biliary cirrhosis [36], and CRCP SNP rs875971 (MAF = 0.47, located in 3′ UTR) with aortic root diameter alternations [31]. In both cases the variant either is synonymous and does not change the amino acid sequence or is located in an untranslated region, yet both have robust effect on the extent of polysomal loading. Moreover, IKZF3 is also associated with numerous other diseases including asthma and hay fever [69], rheumatoid arthritis [37], inflammatory bowel disease [39] and Crohn’s disease [42]. As an example of genes (rather than specific SNPs) implicated by allele-specific polysome enrichment, LGALS17A has been associated with obesity [47], and RAB38 in sclerosis [52].
Upon completion of this study, a similar approach was recently published by Battle et al., employing RNA-Seq for mRNAs, ribosomal fraction of mRNA profiling and protein analysis, to identify eQTLs in mRNA associated with ribosomes, termed rQTLs, and pQTLs [60]. In this study, most eQTLs overlapped with rQTLs, while a number of unique pQTLs were found. The short sequence reads in ribosomal profiling in that study prevented analysis of RNA isoforms, shown here to play a critical role in polysomal loading. In addition, the study did not exploit allelic RNA ratios as a means to establish genetic effects on polysomal loading directly or on RNA isoform formation upstream. Ribosomal loading thus yields distinct results from polysomal RNA analysis.
In conclusion, the method developed in this study provides tools to explore functional genomics of translation and examine genetic loci linked to human disease. Using different cell or tissue types with this method is likely to reveal genetic effects vital for regulatory sequences. This approach allows us to prioritize genes showing strong cis-acting influence on protein translation for further functional studies. Our study provides a proof-of-principle that allelic effects on translation are pervasive and can be sensitively detected on polysomes, deployable on a transcriptome-wide scale using RNA-Seq to identify regulatory variants affecting translation.
Materials and Methods
Cell culture and transfection
Immortalized lymphoblastoid cell lines (LCLs; GM06994, GM06991, GM10852, GM07341, GM12250, GM13045, GM06991, GM12045, GM12043, GM10852) from the Utah Residents with European ancestry (CEPH) [70], were purchased from Coriell Institute for Medical Research. Human cervical cancer cells (HeLa), and Chinese Hamster Ovary cells (CHO) were obtained from American Type Culture Collection. Cells were cultured in DMEM (HeLa), DMEM-F12 (CHO), or RPMI1640 (LCLs), supplemented with 10% fetal bovine serum, 1% penicillin, and 1% streptomycin, in a humidified incubator at 37°C with 5% CO2. For RNA-Seq experiments, cells were grown to 5x105 cells/mL density in T75 tissue culture flask and harvested. For transient transfections, 1.5×106 cells were seeded into 10 cm2 culture plates and after 24 hours transfected with 1 μg plasmid solution containing equal concentration of two expression plasmids carrying either allele of a regulatory variants in a target gene, along with 75 ng emGFP as an internal control, using lipofectamine 2000 reagent (Life Technologies, Foster City, CA).
Allelic mRNA ratio analysis of OPRM1, ABCB1, and NAT1
We used either transfection of expression plasmids (OPRM1 (CHO cells), and ABCB1 (HeLa cells) [67]), or natively LCL-expressed mRNA (NAT1 and ABCB1). Cell lines were selected to match previous experiments with the same plasmids. pcDNA3-OPRM1-118A [23] was used to generate the pcDNA3-OPRM1-118G variant allele using Quick Change site-directed mutagenesis kit (Stratagene, La Jolla, CA) with primers OPRM1-SDM-G118-F, OPRM1-SDM-G118-R (S1 Table). The sequence was confirmed using OPRM1-seq-primer-118-R. Plasmid vectors expressing full length ABCB1 (MDR1) 3435C and 3435T mRNA alleles were used as described [25]. pcDNA6.2-emGFP was used as a co-transfection control.
Cells were lysed and DNA was extracted from the nuclear pellet as described in [67]. RNA was isolated as described [67] from the total lysate solution (400 μL). An equal volume was layered onto a sucrose linear gradient (11 mL, 12 X 75 mm polystyrene, 10–50% w/v) and centrifuged at 228,000 x g in a Sorvall TH-641 rotor for 3.5 h at 4°C. Gradients were fractionated by continuous recording of UV absorbance at 254 nm into 24 fractions of approximately 0.5 mL, using a programmable density gradient fractionation system. Gradient fractions containing monosomes (80S ribosomes), light polysomes (gradient region containing 2–3 ribosomes per mRNA), intermediate (4–5 ribosomes per transcript) and heavy polysomes (6 and more ribosomes per transcript) were identified by corresponding UV peaks (Fig 1).
Eluant fractions corresponding to monosomes, light, intermediate, and heavy polysomes were collected. To each 200 μL sucrose gradient fraction, external control luciferase mRNA was added as an internal control, and RNA was isolated using trizol reagent (Life Technologies, Foster City, CA) and quantified using a Qubit 2 fluorometer (Life Technologies, Foster City, CA). RNA was isolated from every fraction followed by cDNA synthesis and qRT–PCR to study the sedimentation pattern of each target mRNA. For cDNA synthesis with oligodT priming (SSIII, Life Technologies, Foster City, CA), 0.5 μg RNA was subjected to DpnI (for plasmid transfections experiments only) and DNaseI treatment followed by real-time PCR quantification of ABCB1, OPRM1, NAT1, and luciferase mRNAs, using FAST-SYBR Green qRT-PCR supermix (Life Technologies, Foster City, CA), (for primers see S1 Table). The averaged cycle threshold (Ct) values were analyzed by a comparative Ct method [25] to obtain relative mRNA expression levels compared to luciferase control. For analysis of PCR amplicons by gel electrophoresis, extracts were amplified for 20–25 cycles to reflect the linear range during the SYBR green reaction in each experiment.
We tested allelic ratios at polymorphic sites using the primer extension method SNaPshot, (Applied Biosystems, Foster City, CA), as previously described [25]. qRT-PCR amplification of ABCB1, OPRM1, and NAT1 mRNA, and sequencing of the respective cDNA, was performed using PCR master mix (New England Biolabs, Ipswich, MA) as described [23–25, 67]. Samples were then subjected to electrophoresis on a 3730 Genetic Analyzer (Applied Biosystems, Foster City, CA) and evaluated with GeneMapper ID software V3.2 (Applied Biosystems, Foster City, CA) (Fig 1). In each sample, the allelic genomic DNA (gDNA) ratios were also measured, either of the cell’s genomic DNA isolated from the nuclear pellet, or of the transfected plasmid DNA isolated from cytosolic lysate. Allelic mRNA ratios between samples were then normalized to the DNA ratios set at 1.0. Results are from 3 independent experiments, each with 2 independent cDNA syntheses.
RNA-Seq of LCL transcriptomes
Sucrose gradients for preparation of polysomes
Three LCL lines were obtained from the Coriell Institute: LCL14, LCL19, and LCL31. Cells were treated with 0.1 mM cycloheximide for 3 min, harvested, and washed with ice-cold PBS containing 0.1 mM cycloheximide, and harvested by centrifugation at 1000 RPM. The cell pellet was resuspended in 500 μL lysis buffer containing 150 mM NaCl, 50 mM Tris-HCl pH 7.5, 10 mM KCl, 10 mM MgCl2, 0.2% NP-40, 2 mM dithiothreitol, 2 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, and 80 units/mL RNaseOUT. After 10 min incubation on ice, samples were centrifuged at 7500 x g at 4°C for 10 min to pellet the nuclei (for gDNA analysis). 100 μL lysate was stored for the isolation of total cytoplasmic RNA and plasmid DNA. Genomic DNA was extracted by lysing the nuclear pellet. Equal volumes of total cytosol lysates (400 μL) were extracted for RNA analysis or subjected to sucrose gradient centrifugation as above.
RNA was isolated with Trizol reagent from the cytoplasm of the three LCLs (total cytosol content), and from pooled polysome sucrose fractions (>3 ribosomes; 250 μL). Two-step column purification and size separation were performed on each RNA sample. First, long RNA (>200 bases) was separated using SpinSmart RNA Purification column (Denville), and the flowthrough (<200 bases) was placed on a second column for small RNA (microRNA) separation with mirPremier microRNA isolation kit (Sigma-Aldrich, St. Louis, MO). RNA quality was evaluated on a Bioanalyzer.
Cytoplasmic (total) and polysome derived long RNA (25 ng each) was then converted to cDNA using the NuGen Ovation RNA-Seq System V2 (NuGen Technologies, San Carlos, CA), which uses both random hexamers and oligo-dT to amplify all RNA sequences, while suppressing cDNA formation from ribosomal RNA by >90%. ERCC RNA (External RNA Control Consortium, Ambion) controls were spiked into RNA prior to NuGen cDNA synthesis. The NuGen Ovation RNA-Seq kit produces non-stranded cDNA (3–5 microgram, measured with Qubit (Life Technologies, Foster City, CA)). The double-stranded cDNA derived from long RNAs was sheared to 150–200 bp fragments with a Covaris focused-ultrasonicator (Covaris, Inc. Woburn, MA) and recovered by centrifuging over an YM-30 spin filter (Amicon EMD Millipore Billerica, MA). Fragments longer than 100 bp were retained and eluted from the membrane.
Library preparation and cDNA sequencing using an Ion Torrent Proton instrument
We generated barcoded sequencing libraries from 100 ng of sheared cDNA using the NEBNext Fast DNA Library Prep Set for Ion Torrent sequencing (New England Biolabs, NEB, Ipswich, MA), as described [22, 71]. In a separate step, purified small RNA fractions (containing 100 ng RNA) were used for library construction using Ion Total RNA-Seq Kit v2 for small RNAs (Life Technologies, Foster City, CA). Pooled barcoded RNA libraries were sequenced on the Ion Torrent Proton platform (Life Technologies, Foster City, CA). In the small RNA fractions, only annotated microRNAs were studied further in this work.
Sequence alignment and mapping
RNA-Seq data were aligned using a two-step alignment approach (http://ioncommunity.lifetechnologies.com/docs/DOC-8434). Reads were initially mapped using tophat v2.11 [72]. Unmapped reads are extracted and realigned with bowtie v2 [73] using the 'local' and 'very sensitive local' options to allow clipping of the ends to improve the overall alignment score. Alignment files were sorted with samtools v1.19 [74] and merged with Picard (http://picard.sourceforge.net). Small RNA reads were aligned using miRanalyzer [75] with hg19 genome assembly-based bowtie index, and bowtie2 with index files of mature microRNA sequences. The effective library size was estimated using “DESeq” R package [76].
Estimation of RNA levels
Cufflinks v2.1.1 [77, 78] was implemented to estimate annotated RNA and isoform abundances, reported as Fragments Per Kilobase per Million reads (FPKM), normalizing the number of reads within a gene to the number of fragments per kilobase of exon and million mapped reads for a given sample. Expression measurements were quantified for transcripts annotated in GENCODE v18 [79] combined with additional transcripts present only in the lncipedia v2.0 non-coding RNA database [80]. These combined annotation sets capture a broad spectrum of protein coding and non-coding transcripts. Multi-read correction was applied to improve estimates when assigning multimapped reads to transcripts. In a first analysis, the sum of all RNA isoforms expressed from a given gene locus was compared between the cytoplasm (total) and polysome fraction and expressed as a ratio (fold-change) across the three LCL samples. Differential RNA levels between cytoplasmic total and polysomal fractions were determined with the cuffdiff application of Cufflinks [77], treating the three polysomal and cytosolic RNA-Seq samples as replicates.
SNP and allelic mRNA ratio detection
To make SNP calls, the mpileup pipeline by samtools v1.19 [74] was applied to each alignment file separately to maintain allele count estimates. Annotation was provided through ANNOVAR [81] detailing location within the gene based on UCSC gene annotations, implications of the SNP on protein-coding potential, and rs number based on dbSNP 135. For initial gene-wide allelic ratio estimates, SNPs were filtered based on SNP quality and an average mapping quality. Allelic mRNA ratios were estimated as previously described [22], applying a filter of 20 reads at any SNP as the minimum number for ratio analysis. A finding of allelic expression imbalance (AEI) was defined as a fold-change between the reference and variant alleles greater than 2X (in either direction). A difference in AEI between cytosol and polysomes was determined when the ratios differed twofold or more, accounting for cases where the minor allele is less abundant in the cytosol but more abundant in the polysomes (example major/minor allele in cytosol = 0.5, and in polysomes = 2; yielding a 4-fold difference), and vice versa.
Data analysis
Statistical significance was expressed as p-values using the two-tailed student’s t-test for RNA-Seq and targeted experiments of ABCB1, OPRM1, and NAT1. The analysis was performed using Prism (GraphPad, San Diego, California, USA). Data are expressed as mean±SEM.
The non-parametric Mann-Whitney test (u-test) was used for statistical significance analysis of read counts of microRNA data. Pairwise analysis of microRNA sample similarity was performed using Renyi’s divergence calculated for each pair of samples. The pairwise similarity matrix was used for hierarchical clustering. Renyi’s divergence is a measure of similarity between two probability distributions. It is a generalization of the standard Kullblack-Leibler distance [82]. To obtain bounds on the sampling error, we applied computational methods based on the non-parametric (multinomial) bootstrap as described [82]. The analysis was performed using R package “divo”. The package is available for download in the CRAN repository (http://cran.r-project.org). Gene expression distributions of mRNA, pseudogenes, lncRNA, and microRNAs were analyzed using Chi-squared test. The analyses were performed using scripts written in R programming language [83].
Supporting Information
A-C. RNA was isolated from sucrose gradient fractions (0.5 mL). Low cycle (20–25 cycles) RT-PCR was performed for each target RNA and the amplicons resolved on an agarose gel. A. OPRM1 mRNA transfected into CHO cells showed more mRNA in light and intermediate than heavy polysomes. B-C. NAT1 and ABCB1 mRNAs natively expressed in LCLs and HeLa cells, respectively, both showing increased levels in heavy polysome fractions. mRNP represent cytosolic mRNA either free or bound to ribonucleotide particles. D-F. Sucrose gradient fractions (0.5 mL) were pooled to reflect cytosol, monosomes, light, intermediate and heavy polysomes (Fig 1). qRT-PCR results were normalized to control mRNA added to samples prior to RNA isolation and fractionation. For details see Materials and Methods. D. OPRM1 transfection into CHO cells. E-F. NAT1 and ABCB1 mRNA polysomes represent native expression in LCL cells, both showing increases towards heavy polysomes (lower Δ-Ct values).
(TIF)
ABCB1 3435C>T allelic mRNA ratios were measured in transfected HeLa cells expressing ABCB1 3435C and T alleles, demonstrating a significant reduction of the 3435T allele to the same extent in all fractions. Data are representative of gradients done with extracts from 3 independent cultures (mean ± s.d., n = 6). B. qRT-PCR of pooled polysome ABCB1 mRNA shows increased occupancy on heavy polysomes (lower Δ-Ct values).
(TIF)
A. Comparison of distributions of polysome to cytosol ratios of RNA abundance between mRNA and noncoding genes revealed significant difference between means of the ratio frequency (p-value < 0.05). B. Distribution of polysome to cytosol ratios of microRNA abundance.
(TIF)
(DOCX)
Allelic ratios were calculated with RNA-seq data of cytosol and polysome from LCLs, and 100 mRNAs and ncRNAs with highest allelic ratios in either cytosol or polysomes are listed. An allelic ratio 1.0 suggests equal expression of both alleles.
(DOCX)
For isoforms presented in Fig 5 (differences in isoforms based on 3 LCLs) and Fig 7 (based on different AEI ratios measurements in cytosol and polysomes of individual LCLs), this table provides isoform IDs.
(DOCX)
Acknowledgments
The authors wish to thank Prof. Daniel R. Schoenberg’s lab (Ohio State University) for providing polysome fractionation facility and Ms. Katherine Hartmann for critical reading of the manuscript.
This work was supported in part by an allocation of computing time from the Ohio Supercomputer Center.
Data Availability
All raw data files are available from the at the Gene Expression Omnibus (GEO) database (accession number GSE66135).
Funding Statement
This work was supported by the National Institute of General Medical Sciences (nigms.nih.gov); (Grant No. U01GM092655 to WS). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Polychronakos C. Gene expression as a quantitative trait: what about translation? Journal of medical genetics. 2012;49(9):554–7. 10.1136/jmedgenet-2012-101199 [DOI] [PubMed] [Google Scholar]
- 2. Piccirillo CA, Bjur E, Topisirovic I, Sonenberg N, Larsson O. Translational control of immune responses: from transcripts to translatomes. Nature immunology. 2014;15(6):503–11. 10.1038/ni.2891 [DOI] [PubMed] [Google Scholar]
- 3. Wu L, Candille SI, Choi Y, Xie D, Jiang L, Li-Pook-Than J, et al. Variation and genetic control of protein abundance in humans. Nature. 2013;499(7456):79–82. 10.1038/nature12223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kleijn M, Scheper GC, Voorma HO, Thomas AA. Regulation of translation initiation factors by signal transduction. European journal of biochemistry / FEBS. 1998;253(3):531–44. [DOI] [PubMed] [Google Scholar]
- 5. Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, et al. Global quantification of mammalian gene expression control. Nature. 2011;473(7347):337–42. 10.1038/nature10098 [DOI] [PubMed] [Google Scholar]
- 6. Pradet-Balade B, Boulme F, Beug H, Mullner EW, Garcia-Sanz JA. Translation control: bridging the gap between genomics and proteomics? Trends in biochemical sciences. 2001;26(4):225–9. [DOI] [PubMed] [Google Scholar]
- 7. Li Q, Makri A, Lu Y, Marchand L, Grabs R, Rousseau M, et al. Genome-wide search for exonic variants affecting translational efficiency. Nature communications. 2013;4:2260 10.1038/ncomms3260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tebaldi T, Re A, Viero G, Pegoretti I, Passerini A, Blanzieri E, et al. Widespread uncoupling between transcriptome and translatome variations after a stimulus in mammalian cells. BMC genomics. 2012;13:220 10.1186/1471-2164-13-220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sterne-Weiler T, Martinez-Nunez RT, Howard JM, Cvitovik I, Katzman S, Tariq MA, et al. Frac-seq reveals isoform-specific recruitment to polyribosomes. Genome research. 2013;23(10):1615–23. 10.1101/gr.148585.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Beilharz TH, Preiss T. Translational profiling: the genome-wide measure of the nascent proteome. Briefings in functional genomics & proteomics. 2004;3(2):103–11. [DOI] [PubMed] [Google Scholar]
- 11. Ward LD, Kellis M. Interpreting noncoding genetic variation in complex traits and human disease. Nature biotechnology. 2012;30(11):1095–106. 10.1038/nbt.2422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lappalainen T, Sammeth M, Friedlander MR, t Hoen PA, Monlong J, Rivas MA, et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501(7468):506–11. 10.1038/nature12531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sadee W, Hartmann K, Seweryn M, Pietrzak M, Handelman SK, Rempala GA. Missing heritability of common diseases and treatments outside the protein-coding exome. Human genetics. 2014;133(10):1199–215. 10.1007/s00439-014-1476-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lian Z, Wang L, Yamaga S, Bonds W, Beazer-Barclay Y, Kluger Y, et al. Genomic and proteomic analysis of the myeloid differentiation program. Blood. 2001;98(3):513–24. [DOI] [PubMed] [Google Scholar]
- 15. Cox B, Kislinger T, Emili A. Integrating gene and protein expression data: pattern analysis and profile mining. Methods. 2005;35(3):303–14. [DOI] [PubMed] [Google Scholar]
- 16. Kislinger T, Cox B, Kannan A, Chung C, Hu P, Ignatchenko A, et al. Global survey of organ and organelle protein expression in mouse: combined proteomic and transcriptomic profiling. Cell. 2006;125(1):173–86. [DOI] [PubMed] [Google Scholar]
- 17. Fu X, Fu N, Guo S, Yan Z, Xu Y, Hu H, et al. Estimating accuracy of RNA-Seq and microarrays with proteomics. BMC genomics. 2009;10:161 10.1186/1471-2164-10-161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Parts L, Liu YC, Tekkedil M, Steinmetz LM, Caudy AA, Fraser AG, et al. Heritability and genetic basis of protein level variation in an outbred population. Genome research. 2014;24(8):1363–70. 10.1101/gr.170506.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Johansson A, Enroth S, Palmblad M, Deelder AM, Bergquist J, Gyllensten U. Identification of genetic variants influencing the human plasma proteome. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(12):4673–8. 10.1073/pnas.1217238110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Garge N, Pan H, Rowland MD, Cargile BJ, Zhang X, Cooley PC, et al. Identification of quantitative trait loci underlying proteome variation in human lymphoblastoid cells. Molecular & cellular proteomics: MCP. 2010;9(7):1383–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Johnson AD, Zhang Y, Papp AC, Pinsonneault JK, Lim JE, Saffen D, et al. Polymorphisms affecting gene transcription and mRNA processing in pharmacogenetic candidate genes: detection through allelic expression imbalance in human target tissues. Pharmacogenetics and genomics. 2008;18(9):781–91. 10.1097/FPC.0b013e3283050107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Smith RM, Webb A, Papp AC, Newman LC, Handelman SK, Suhy A, et al. Whole transcriptome RNA-Seq allelic expression in human brain. BMC genomics. 2013;14:571 10.1186/1471-2164-14-571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang Y, Wang D, Johnson AD, Papp AC, Sadee W. Allelic expression imbalance of human mu opioid receptor (OPRM1) caused by variant A118G. The Journal of biological chemistry. 2005;280(38):32618–24. [DOI] [PubMed] [Google Scholar]
- 24. Wang D, Para MF, Koletar SL, Sadee W. Human N-acetyltransferase 1 *10 and *11 alleles increase protein expression through distinct mechanisms and associate with sulfamethoxazole-induced hypersensitivity. Pharmacogenetics and genomics. 2011;21(10):652–64. 10.1097/FPC.0b013e3283498ee9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang D, Johnson AD, Papp AC, Kroetz DL, Sadee W. Multidrug resistance polypeptide 1 (MDR1, ABCB1) variant 3435C>T affects mRNA stability. Pharmacogenetics and genomics. 2005;15(10):693–704. [PubMed] [Google Scholar]
- 26. Kimchi-Sarfaty C, Oh JM, Kim IW, Sauna ZE, Calcagno AM, Ambudkar SV, et al. A "silent" polymorphism in the MDR1 gene changes substrate specificity. Science. 2007;315(5811):525–8. [DOI] [PubMed] [Google Scholar]
- 27. Molotski N, Soen Y. Differential association of microRNAs with polysomes reflects distinct strengths of interactions with their mRNA targets. RNA. 2012;18(9):1612–23. 10.1261/rna.033142.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sadee W, Wang D, Papp AC, Pinsonneault JK, Smith RM, Moyer RA, et al. Pharmacogenomics of the RNA world: structural RNA polymorphisms in drug therapy. Clinical pharmacology and therapeutics. 2011;89(3):355–65. 10.1038/clpt.2010.314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Westra HJ, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nature genetics. 2013;45(10):1238–43. 10.1038/ng.2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nature genetics. 2013;45(6):580–5. 10.1038/ng.2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wineinger NE, Patki A, Meyers KJ, Broeckel U, Gu CC, Rao DC, et al. Genome-wide joint SNP and CNV analysis of aortic root diameter in African Americans: the HyperGEN study. BMC medical genomics. 2011;4:4 10.1186/1755-8794-4-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Deng X, Sabino EC, Cunha-Neto E, Ribeiro AL, Ianni B, Mady C, et al. Genome wide association study (GWAS) of Chagas cardiomyopathy in Trypanosoma cruzi seropositive subjects. PloS one. 2013;8(11):e79629 10.1371/journal.pone.0079629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chalasani N, Guo X, Loomba R, Goodarzi MO, Haritunians T, Kwon S, et al. Genome-wide association study identifies variants associated with histologic features of nonalcoholic Fatty liver disease. Gastroenterology. 2010;139(5):1567–76, 76 e1–6. 10.1053/j.gastro.2010.07.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Teumer A, Holtfreter B, Volker U, Petersmann A, Nauck M, Biffar R, et al. Genome-wide association study of chronic periodontitis in a general German population. Journal of clinical periodontology. 2013;40(11):977–85. 10.1111/jcpe.12154 [DOI] [PubMed] [Google Scholar]
- 35. Newton-Cheh C, Eijgelsheim M, Rice KM, de Bakker PI, Yin X, Estrada K, et al. Common variants at ten loci influence QT interval duration in the QTGEN Study. Nature genetics. 2009;41(4):399–406. 10.1038/ng.364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hirschfield GM, Liu X, Xu C, Lu Y, Xie G, Lu Y, et al. Primary biliary cirrhosis associated with HLA, IL12A, and IL12RB2 variants. The New England journal of medicine. 2009;360(24):2544–55. 10.1056/NEJMoa0810440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014;506(7488):376–81. 10.1038/nature12873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Martin JE, Assassi S, Diaz-Gallo LM, Broen JC, Simeon CP, Castellvi I, et al. A systemic sclerosis and systemic lupus erythematosus pan-meta-GWAS reveals new shared susceptibility loci. Human molecular genetics. 2013;22(19):4021–9. 10.1093/hmg/ddt248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491(7422):119–24. 10.1038/nature11582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nakamura M, Nishida N, Kawashima M, Aiba Y, Tanaka A, Yasunami M, et al. Genome-wide association study identifies TNFSF15 and POU2AF1 as susceptibility loci for primary biliary cirrhosis in the Japanese population. American journal of human genetics. 2012;91(4):721–8. 10.1016/j.ajhg.2012.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Anderson CA, Boucher G, Lees CW, Franke A, D'Amato M, Taylor KD, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nature genetics. 2011;43(3):246–52. 10.1038/ng.764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nature genetics. 2010;42(12):1118–25. 10.1038/ng.717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stahl EA, Raychaudhuri S, Remmers EF, Xie G, Eyre S, Thomson BP, et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nature genetics. 2010;42(6):508–14. 10.1038/ng.582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Albertsen HM, Chettier R, Farrington P, Ward K. Genome-wide association study link novel loci to endometriosis. PloS one. 2013;8(3):e58257 10.1371/journal.pone.0058257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Porcu E, Medici M, Pistis G, Volpato CB, Wilson SG, Cappola AR, et al. A meta-analysis of thyroid-related traits reveals novel loci and gender-specific differences in the regulation of thyroid function. PLoS genetics. 2013;9(2):e1003266 10.1371/journal.pgen.1003266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gieger C, Radhakrishnan A, Cvejic A, Tang W, Porcu E, Pistis G, et al. New gene functions in megakaryopoiesis and platelet formation. Nature. 2011;480(7376):201–8. 10.1038/nature10659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Comuzzie AG, Cole SA, Laston SL, Voruganti VS, Haack K, Gibbs RA, et al. Novel genetic loci identified for the pathophysiology of childhood obesity in the Hispanic population. PloS one. 2012;7(12):e51954 10.1371/journal.pone.0051954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Need AC, Attix DK, McEvoy JM, Cirulli ET, Linney KL, Hunt P, et al. A genome-wide study of common SNPs and CNVs in cognitive performance in the CANTAB. Human molecular genetics. 2009;18(23):4650–61. 10.1093/hmg/ddp413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stanescu HC, Arcos-Burgos M, Medlar A, Bockenhauer D, Kottgen A, Dragomirescu L, et al. Risk HLA-DQA1 and PLA(2)R1 alleles in idiopathic membranous nephropathy. The New England journal of medicine. 2011;364(7):616–26. 10.1056/NEJMoa1009742 [DOI] [PubMed] [Google Scholar]
- 50. Chen P, Miyake M, Fan Q, Liao J, Yamashiro K, Ikram MK, et al. CMPK1 and RBP3 are associated with corneal curvature in Asian populations. Human molecular genetics. 2014;23(22):6129–36. 10.1093/hmg/ddu322 [DOI] [PubMed] [Google Scholar]
- 51. Benjamin DJ, Cesarini D, van der Loos MJ, Dawes CT, Koellinger PD, Magnusson PK, et al. The genetic architecture of economic and political preferences. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(21):8026–31. 10.1073/pnas.1120666109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Baranzini SE, Wang J, Gibson RA, Galwey N, Naegelin Y, Barkhof F, et al. Genome-wide association analysis of susceptibility and clinical phenotype in multiple sclerosis. Human molecular genetics. 2009;18(4):767–78. 10.1093/hmg/ddn388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chu X, Pan CM, Zhao SX, Liang J, Gao GQ, Zhang XM, et al. A genome-wide association study identifies two new risk loci for Graves' disease. Nature genetics. 2011;43(9):897–901. 10.1038/ng.898 [DOI] [PubMed] [Google Scholar]
- 54. Crosslin DR, McDavid A, Weston N, Zheng X, Hart E, de Andrade M, et al. Genetic variation associated with circulating monocyte count in the eMERGE Network. Human molecular genetics. 2013;22(10):2119–27. 10.1093/hmg/ddt010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nalls MA, Couper DJ, Tanaka T, van Rooij FJ, Chen MH, Smith AV, et al. Multiple loci are associated with white blood cell phenotypes. PLoS genetics. 2011;7(6):e1002113 10.1371/journal.pgen.1002113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Turner ST, Bailey KR, Schwartz GL, Chapman AB, Chai HS, Boerwinkle E. Genomic association analysis identifies multiple loci influencing antihypertensive response to an angiotensin II receptor blocker. Hypertension. 2012;59(6):1204–11. 10.1161/HYP.0b013e31825b30f8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jin Y, Birlea SA, Fain PR, Ferrara TM, Ben S, Riccardi SL, et al. Genome-wide association analyses identify 13 new susceptibility loci for generalized vitiligo. Nature genetics. 2012;44(6):676–80. 10.1038/ng.2272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Knight JC. Approaches for establishing the function of regulatory genetic variants involved in disease. Genome medicine. 2014;6(10):92 10.1186/s13073-014-0092-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Melzer D, Perry JR, Hernandez D, Corsi AM, Stevens K, Rafferty I, et al. A genome-wide association study identifies protein quantitative trait loci (pQTLs). PLoS genetics. 2008;4(5):e1000072 10.1371/journal.pgen.1000072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Battle A, Khan Z, Wang SH, Mitrano A, Ford MJ, Pritchard JK, et al. Genomic variation. Impact of regulatory variation from RNA to protein. Science. 2015;347(6222):664–7. 10.1126/science.1260793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wilhelm M, Schlegl J, Hahne H, Moghaddas Gholami A, Lieberenz M, Savitski MM, et al. Mass-spectrometry-based draft of the human proteome. Nature. 2014;509(7502):582–7. 10.1038/nature13319 [DOI] [PubMed] [Google Scholar]
- 62. Bond C, LaForge KS, Tian M, Melia D, Zhang S, Borg L, et al. Single-nucleotide polymorphism in the human mu opioid receptor gene alters beta-endorphin binding and activity: possible implications for opiate addiction. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(16):9608–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wells SE, Hillner PE, Vale RD, Sachs AB. Circularization of mRNA by eukaryotic translation initiation factors. Molecular cell. 1998;2(1):135–40. [DOI] [PubMed] [Google Scholar]
- 64. Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS. Genome-wide annotation and quantitation of translation by ribosome profiling. Current protocols in molecular biology / edited by Ausubel Frederick M [et al. ]. 2013;Chapter 4:Unit 4 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nature protocols. 2012;7(8):1534–50. 10.1038/nprot.2012.086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, et al. A draft map of the human proteome. Nature. 2014;509(7502):575–81. 10.1038/nature13302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Smith RM, Papp AC, Webb A, Ruble CL, Munsie LM, Nisenbaum LK, et al. Multiple regulatory variants modulate expression of 5-hydroxytryptamine 2A receptors in human cortex. Biological psychiatry. 2013;73(6):546–54. 10.1016/j.biopsych.2012.09.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. van Heesch S, van Iterson M, Jacobi J, Boymans S, Essers PB, de Bruijn E, et al. Extensive localization of long noncoding RNAs to the cytosol and mono- and polyribosomal complexes. Genome biology. 2014;15(1):R6 10.1186/gb-2014-15-1-r6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ferreira MA, Matheson MC, Tang CS, Granell R, Ang W, Hui J, et al. Genome-wide association analysis identifies 11 risk variants associated with the asthma with hay fever phenotype. The Journal of allergy and clinical immunology. 2014;133(6):1564–71. 10.1016/j.jaci.2013.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. International HapMap C. A haplotype map of the human genome. Nature. 2005;437(7063):1299–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Webb A, Papp AC, Sanford JC, Huang K, Parvin JD, Sadee W. Expression of mRNA transcripts encoding membrane transporters detected with whole transcriptome sequencing of human brain and liver. Pharmacogenetics and genomics. 2013;23(5):269–78. 10.1097/FPC.0b013e32835ff536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome biology. 2013;14(4):R36 10.1186/gb-2013-14-4-r36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature methods. 2012;9(4):357–9. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hackenberg M, Rodriguez-Ezpeleta N, Aransay AM. miRanalyzer: an update on the detection and analysis of microRNAs in high-throughput sequencing experiments. Nucleic acids research. 2011;39(Web Server issue):W132–8. 10.1093/nar/gkr247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Anders S, Huber W. Differential expression analysis for sequence count data. Genome biology. 2010;11(10):R106 10.1186/gb-2010-11-10-r106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature biotechnology. 2010;28(5):511–5. 10.1038/nbt.1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Roberts A, Pimentel H, Trapnell C, Pachter L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics. 2011;27(17):2325–9. 10.1093/bioinformatics/btr355 [DOI] [PubMed] [Google Scholar]
- 79. Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome research. 2012;22(9):1760–74. 10.1101/gr.135350.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Volders PJ, Helsens K, Wang X, Menten B, Martens L, Gevaert K, et al. LNCipedia: a database for annotated human lncRNA transcript sequences and structures. Nucleic acids research. 2013;41(Database issue):D246–51. 10.1093/nar/gks915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research. 2010;38(16):e164 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Rempala GA, Seweryn M. Methods for diversity and overlap analysis in T-cell receptor populations. Journal of mathematical biology. 2013;67(6–7):1339–68. 10.1007/s00285-012-0589-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2014. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A-C. RNA was isolated from sucrose gradient fractions (0.5 mL). Low cycle (20–25 cycles) RT-PCR was performed for each target RNA and the amplicons resolved on an agarose gel. A. OPRM1 mRNA transfected into CHO cells showed more mRNA in light and intermediate than heavy polysomes. B-C. NAT1 and ABCB1 mRNAs natively expressed in LCLs and HeLa cells, respectively, both showing increased levels in heavy polysome fractions. mRNP represent cytosolic mRNA either free or bound to ribonucleotide particles. D-F. Sucrose gradient fractions (0.5 mL) were pooled to reflect cytosol, monosomes, light, intermediate and heavy polysomes (Fig 1). qRT-PCR results were normalized to control mRNA added to samples prior to RNA isolation and fractionation. For details see Materials and Methods. D. OPRM1 transfection into CHO cells. E-F. NAT1 and ABCB1 mRNA polysomes represent native expression in LCL cells, both showing increases towards heavy polysomes (lower Δ-Ct values).
(TIF)
ABCB1 3435C>T allelic mRNA ratios were measured in transfected HeLa cells expressing ABCB1 3435C and T alleles, demonstrating a significant reduction of the 3435T allele to the same extent in all fractions. Data are representative of gradients done with extracts from 3 independent cultures (mean ± s.d., n = 6). B. qRT-PCR of pooled polysome ABCB1 mRNA shows increased occupancy on heavy polysomes (lower Δ-Ct values).
(TIF)
A. Comparison of distributions of polysome to cytosol ratios of RNA abundance between mRNA and noncoding genes revealed significant difference between means of the ratio frequency (p-value < 0.05). B. Distribution of polysome to cytosol ratios of microRNA abundance.
(TIF)
(DOCX)
Allelic ratios were calculated with RNA-seq data of cytosol and polysome from LCLs, and 100 mRNAs and ncRNAs with highest allelic ratios in either cytosol or polysomes are listed. An allelic ratio 1.0 suggests equal expression of both alleles.
(DOCX)
For isoforms presented in Fig 5 (differences in isoforms based on 3 LCLs) and Fig 7 (based on different AEI ratios measurements in cytosol and polysomes of individual LCLs), this table provides isoform IDs.
(DOCX)
Data Availability Statement
All raw data files are available from the at the Gene Expression Omnibus (GEO) database (accession number GSE66135).