

Abstract
Staphylococcus aureus is an important pathogenic bacterium that causes various infectious diseases. Extracellular vesicles (EVs) released from S. aureus contain bacterial proteins, nucleic acids, and lipids. These EVs can induce immune responses leading to similar symptoms as during staphylococcal infection condition and have the potential as vaccination agent. Here, we show that active immunization (vaccination) with S. aureus-derived EVs induce adaptive immunity of antibody and T cell responses. In addition, these EVs have the vaccine adjuvant ability to induce protective immunity such as the up-regulation of co-stimulatory molecules and the expression of T cell polarizing cytokines in antigen-presenting cells. Moreover, vaccination with S. aureus EVs conferred protection against lethality induced by airway challenge with lethal dose of S. aureus and also pneumonia induced by the administration of sub-lethal dose of S. aureus. These protective effects were also found in mice that were adoptively transferred with splenic T cells isolated from S. aureus EV-immunized mice, but not in serum transferred mice. Furthermore, this protective effect of S. aureus EVs was significantly reduced by the absence of interferon-gamma, but not by the absence of interleukin-17. Together, the study herein suggests that S. aureus EVs are a novel vaccine candidate against S. aureus infections, mainly via Th1 cellular response.
Introduction
Staphylococcus aureus is one of the most significant bacteria in human health care. Globally, 10~20% of the population is S. aureus carrier [1]. S. aureus infection, in addition to soft tissue or skin infection and sinusitis, is the major cause of pneumonia, osteomyelitis, and sepsis [2]. In addition, S. aureus is also one of the main causative bacteria of nosocomial infections [3]. Recently, with the increasing burdens by the S. aureus strains that have resistance toward methicillin, the methicillin-resistant S. aureus (MRSA) has become prominent in hospitals, together with the community-acquired S. aureus [4].
Vaccination, or active immunization, is one of the most cost-effective methods for prevention and control of bacterial infectious diseases [5]. Multidrug resistant S. aureus strains that are ineffectively treated by the current antibiotics are emerging. Therefore, the development of a vaccine against S. aureus is in fast needs. For effective prevention of bacterial infections, immune responses should be pathogen-specific and be able to induce long-term memory [6].
Extracellular vesicles (EVs) derived from bacteria are lipid bi-layered particles of 20~200 nm in size [7]. Gram-negative bacteria including Escherichia coli, Pseudomonas aeruginosa, and Acinetobacter baumanii produce EVs [7,8,9]. These EVs have pathogenic components or virulence factors. Therefore, EVs derived from Gram-negative bacteria can be used as vaccination agents by inducing both the innate and adaptive immunity [10]. Recently, we found that Gram-positive bacteria, such as S. aureus, also have the ability to produce EVs [11]. EVs derived from S. aureus (SEVs) contain peptidoglycan, lipoteichoic acid, and many pathogenic molecules, such as enterotoxin (SEQ), IgG-binding protein (Sbi), and hemolysin [11]. We also found that the skin application of SEVs induces both the antibody and T cell responses [12]. These findings indicate the potential of SEVs as a vaccine for prevention of S. aureus infection. To test this, we evaluated the effects of SEV immunization concerning pneumonia and mortality due to S. aureus infection.
Results
In vitro innate immune response of SEVs
Both the innate immune responses and adaptive immune responses have important roles in clearing infectious bacteria. Dendritic cells (DCs) bridge innate and adaptive immunity via antigen-presentation and the production of cytokines [13]. To assess the immunogenicity of SEVs toward DCs, bone marrow-derived DCs (BMDCs) were treated with SEVs for 24 h prior to the measurement of the expression of co-stimulatory molecules and pro-inflammatory or T cell polarizing cytokines. The uptake of SEVs by BMDCs was observed (Fig 1A). The expression of co-stimulatory molecules such as CD80 and CD86 in BMDCs was enhanced by the treatment with SEVs compared to non-treated control (Fig 1B). In addition, the production of pro-inflammatory mediators, such as tumor necrosis factor-alpha (TNF-α), interleukin 6 (IL-6), and IL-12 from BMDCs were also enhanced by the treatment with SEVs (Fig 1C). These findings suggested that SEVs have the vaccine adjuvant ability to induce adaptive immunity.
Fig 1. In vitro immunogenicity of S. aureus-derived EVs (SEVs).

(A) Uptake of SEVs by bone marrow-derived dendritic cells (BMDCs). BMDCs were treated with SEVs (10 μg/ml) for 24 h. BMDCs cytoplasm were stained with CellTracker Green CMFDA (5-chloromethylfluorescein diacetate, green), nuclei with Hoechst (blue), and SEVs with DiI (red). The quantification of SEV-florescence in no-treatment and SEV-treatment group (n = 20, each group). (B) The expression of co-stimulatory molecules in BMDCs. The expression of CD80 and CD86 in BMDCs were measured 24 h after treatment with SEVs (10 μg/ml) or PBS. (C) Production of pro-inflammatory cytokines from BMDCs 24 h after SEVs treatment. BMDCs were treated with various concentrations of SEVs, and the levels of TNF-ɑ, IL-6, and IL-12 in the cell supernatants were measured by ELISA. *** indicates p< 0.001.
Effect of SEV vaccination against lethality induced by S. aureus infection
To test the efficacy of SEV vaccination against S. aureus-induced pneumonia, mouse pneumonia model was established by oropharyngeal application of different doses of S. aureus (Fig 2A). The lethal and sub-lethal doses of S. aureus in this model were 4 × 108 and 1 × 108 colony forming units (CFU), respectively. Histologic findings showed that pneumonia was elicited 24 h after the application of the sub-lethal dose. It was observed that alveolar space and airways were occupied by inflammatory exudates and immune cells (Fig 2B). Based on these results, we evaluated the efficacy of SEVs vaccination on the protection of lethality induced by S. aureus pneumonia. Mice were immunized intramuscularly with different doses of SEVs three times with an interval of 7 days and then challenged with the lethal dose of S. aureus 7 days after the last immunization, as shown in Fig 2C. All mice immunized with 5 or 10 μg of SEVs survived, whereas 40% of mice immunized with 1 μg of SEVs and none of the sham-treated mice survived (Fig 2D). Based on these data, we chose 5 μg of SEVs as the vaccination dose for further experiments. We evaluated the vaccination efficacy by the dosing frequency of SEV vaccination. All mice survived after three SEV immunizations, 70% of mice survived after two SEV immunizations, and 30% of mice survived after a single SEV immunization, and none of the sham-immunized mice survived after three sham immunizations (Fig 2E).
Fig 2. Efficacy of S. aureus EVs (SEVs) vaccination on protection against lethality induced by S. aureus lung infection.

(A) Determination of lethal and sub-lethal doses of S. aureus in mouse pneumonia model. Survival rates in mice were evaluated after oropharyngeal application with different doses (1 × 108, 2 × 108, 3 × 108 and 4 × 108 CFU) of S. aureus. Survival was monitored every 12 h for 3 days (n = 10, each group). (B) Histologic image of mouse lung after oropharyngeal application of S. aureus (1 × 108 CFU) 24 h post-infection. (C) Study protocol for SEV-immunization and challenge of the lethal dose (4 × 108 CFU) of S. aureus. SEVs and sham (PBS) were injected intramuscularly at weekly intervals for 3 weeks, and then S. aureus was applied via oropharyngeal route one week after the last immunization. (D) Efficacy of different doses (1, 5, and 10 μg) of SEV vaccination. Survival was monitored every 12 h for 3 days (n = 10, each group). (E) Efficacy of SEV vaccination according to immunization frequency. Survival rates were monitored every 12 h for 3 days in mice immunized with SEVs (5 μg) once, twice, or three times (n = 10, each group).
Effect of SEV vaccination on protection against S. aureus-induced pneumonia
To assess the efficacy of SEVs vaccine candidate on the protection against S. aureus-induced pneumonia, mice immunized three times with SEVs (5 μg) or sham injections, were oropharyngeally applied with the sub-lethal dose of S. aureus (1 × 108 CFU), and were monitored for 24 h after the challenge. The lung tissues of SEV-immunized mice had almost no bacteria burdens, but the lung tissues of sham-immunized mice showed bacterial colonies (Fig 3A). Histologically, inflammatory exudates (pneumonic consolidation) were present in alveolar spaces 24 h after S. aureus infection only in the tissues isolated from sham-immunized mice (Fig 3B). In addition, in vivo imaging system (IVIS) spectrometry analyses showed reduction in the signals for S. aureus in the whole body and in the lung of mice immunized with SEVs, whereas stronger signals for S. aureus was detected for sham-immunized mice compared to SEV-immunized mice (Fig 3C and 3D). To evaluate the degree of onset of systemic inflammation in mice with S. aureus-induced pneumonia, we measured the level of IL-1β and IL-6 in blood as the representative cytokines for systemic induction of sepsis. Significant reductions of both cytokines were detected in SEV-immunized mice compared with sham-immunized mice (Fig 3E). Taken together, these findings indicate an effective vaccination efficacy of SEV immunization against S. aureus pneumonia.
Fig 3. Efficacy of SEV vaccination on protection against pneumonia induced by sub-lethal dose of S. aureus.

For all figures, SEVs (5 μg) and sham (PBS) were injected intramuscularly to mice at weekly intervals for 3 weeks, and then sub-lethal dose (1 × 108 CFU) of S. aureus was applied via the oropharyngeal route one week after the last immunization. Normal: PBS-immunized and PBS-challenged mice; PBS: PBS-immunized and S. aureus-challenged mice; SEV: SEV-immunized and S. aureus-challenged mice. (A) Colony forming unit (CFU) counts from lung of SEV- and sham (PBS)-immunized mice 24 h after the S. aureus challenge (n = 10, each group). (B) Histology (left panel) and gross image (right panel) of lung from SEV- and sham (PBS)-immunized mice after the sub-lethal dose of S. aureus challenge. (C) Distribution of S. aureus before and after SEV-immunization. Cy7-labeled S. aureus was applied via the oropharyngeal route to SEV- and sham (PBS)-immunized mice. Cy7 fluorescence of whole mouse (upper panel) or lung (lower panel) was acquired by IVIS spectrum 24 h after the S. aureus challenge. (D) Bioluminescence signal in the lung tissue after Cy7-labeled S. aureus administration. The amount of the bioluminescence signal (photons/s) in the lung tissue was measured by IVIS spectrum 24h after S. aureus challenge (n = 5, each group). (E) The levels of IL-β and IL-6 in serum of SEV- and sham (PBS)-immunized mice 24 h after the S. aureus challenge (n = 10, each group). * indicates p< 0.05 vs. PBS.
Adaptive immunity induced by SEV vaccination
We next evaluated the adaptive immune responses induced by SEV immunization. Mice were immunized intramuscularly with SEVs (5 μg) three times. Serum SEV-reactive IgG levels were enhanced after the second immunization and further enhanced after the third immunization compared to sham-immunization (Fig 4A). To evaluate the T cell responses induced by SEV vaccination, mouse spleens were extracted 72 h after the final immunization and then T cells were isolated and re-stimulated by co-incubation with anti-CD3 and anti-CD28 antibodies or PBS. The production of IFN-γ, IL-17, and IL-4 from splenic T cells was significantly increased in SEV-immunized mice after anti-CD3/CD28 stimuli, but these responses were not observed in sham-immunized mice (Fig 4B). Adaptive immunity was evaluated according to the administration routes of SEV vaccine. SEV-reactive antibody production measured 72 h after the third immunization was similar among intraperitoneal, subcutaneous, and intramuscular vaccination routes (Fig 4C). However, the production of IFN-γ, IL-17, and IL-4 from splenic T cells was significantly enhanced by intramuscular injection compared to intraperitoneal and subcutaneous applications (Fig 4D). Intramuscular vaccination with SEV induced Th1, Th17, and Th2 cell as well as IgG antibody responses.
Fig 4. Antibody and T cell responses after SEV vaccination.

For (A) and (B), SEVs (5 μg) and sham (PBS) were injected intramuscularly to mice at weekly intervals for 3 weeks (n = 10, each group). (A) The levels of SEV-reactive IgG in serum. Sera were obtained from SEV- and sham-immunized mice 7 days after each immunization and serum levels of SEV-reactive IgG were measured by ELISA. (B) SEV-specific production of IFN-γ, IL-17, and IL-4 from splenic T cells. Splenic T cells were isolated from spleens of SEV- and sham-immunized mice, and then stimulated with anti- CD3/CD28 for 72 h. The levels of IFN-γ, IL-17, and IL-4 in the cell supernatants were measured by ELISA. For (C) and (D) SEVs (5 μg) and sham (PBS) were applied intraperitoneally (IP), subcutaneously (SC), or intramuscularly (IM) at weekly intervals for 3 weeks (n = 10, each group). (C) The levels of SEV-reactive IgG in serum. Sera were obtained from SEV- and sham (PBS)-immunized mice 7 days after the last immunization. (D) SEV-specific production of IFN-γ, IL-17, and IL-4 from splenic T cells. Splenic T cells were isolated from spleen of SEV- and sham (PBS)-immunized mice, and then stimulated with anti-CD3/CD28 for 72 h. The levels of IFN-γ, IL-17 and IL-4 in the cell supernatants were measured by ELISA. * indicates p< 0.05 vs. PBS and ** indicates p< 0.01 vs. the other groups.
Role of antibody and CD4+ T cells on the protective effect of SEV vaccination
Both the cell-mediated and humoral immune responses are important in providing protective immune responses to the host. To examine whether cell-mediated or humoral immunities are mainly involved in protective responses induced by SEV-immunization, we compared the resistance to S. aureus infection in naïve mice who received serum or CD4+ T cells isolated from the immunized mice. The protocol used for the serum transfer assay was the same as the previous protocol in a study that reported effective anti-bacterial efficacy using passive immunization by serum-transfer [14]. For CD4+ T cell transfer, naïve mice received CD4+ T cells isolated from SEV-immunized mice. The majority (70%) of the mice that received CD4+ T cells survived the lethal-dose challenge of S. aureus. None of the mice that received CD4+ T cells from sham-immunized mice survived over 48 h after the lethal-dose of S. aureus challenge (Fig 5A). These results suggested that the adoptively transferred CD4+ T cells survived in naïve mice and operated properly. Therefore, mice that received CD4+ T cells from SEV-immunized mice showed significant effective protective response against S. aureus infection compared with the mice that received CD4+ T cells from sham-immunized mice. In contrast, no protective ability were observed in both the mice that received serum from SEV-immunized and sham-immunized mice (Fig 5B). These results indicated that the efficacy of vaccination induced by SEV-immunization is mediated mainly by CD4+ T cell response, rather than B cell-mediated humoral immune response.
Fig 5. Role of antibody and T helper cells on the protective effect of SEV vaccination.
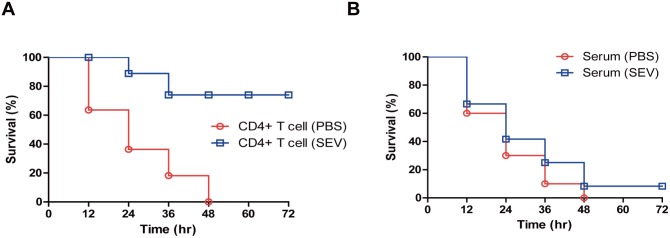
CD4+ T cells and blood sera were isolated from SEV-and sham (PBS)-immunized mice 6 days after three immunizations. Either CD4+ T cells (A) or sera (B) were applied intraperitoneally to naïve mice. After 24 h of the injection, lethal dose (4.0 × 108 CFU) of S. aureus was applied via the oropharyngeal route to the adoptive transferred mice. The survival rates were monitored every 12 h after the bacterial challenge for 3 days (n = 10, each group).
Role of TLR-mediated signaling on SEV vaccine efficacy
Both the acquired immunity and innate immunity are important in defense against bacterial infection. In innate immunity, antigen recognition is one of the main steps in the antigen-processing of presentation by antigen presenting cells, including DCs. Toll-like receptor (TLR) signaling pathways, with their down-stream signal of Myd88, are the representative signaling pathways and are divided into nine sub-types. To examine the recognition pathway of SEVs in mice, we evaluated survival rate after SEV-immunization in Myd88-/-, TLR2-/-, TLR4-/-, TLR9-/- and wild type (WT) mice after challenge with the lethal dose of S. aureus. Like WT mice, TLR4-/- and TLR9-/- mice immunized with SEVs showed 100% survival rate after the challenge. However, all of the sham-immunized and SEV-immunized Myd88-/- and TLR2-/- mice died within 48 h of the challenge (Fig 6A). These results indicate that the recognition of SEVs in the host innate immunity is dependent on the TLR2 signaling, but not on TLR4 and TLR9.
Fig 6. Role of Toll-like receptor signaling and T cell-derived cytokines on the protective effect of SEV vaccination.
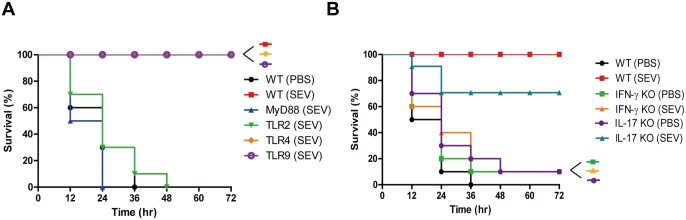
For all figures, SEVs (5 μg) and sham (PBS) were injected intramuscularly to mice at weekly intervals for 3 weeks, and then the lethal dose (4.0 × 108 CFU) of S. aureus were challenged oropharyngeally to the immunized mice one week after the last immunization. The survival was monitored every 12 h after the bacterial challenge for 3 days (n = 10 each group). (A) The survival rates of wild type (WT), MyD88-deficient, TLR2-deficient, TLR4-deficient, and TLR9-deficient (all C57BL/6 background) mice after S. aureus challenge. (B) The survival rates of WT, IFN-γ-deficient, and IL-17-deficient (all BABL/c background) mice after S. aureus challenge.
Role of Th1 and Th17 cytokines on SEV vaccine efficacy
IFN-γ and IL-17 from T cells are important in defense against bacterial infection. To investigate which T helper cells play major roles in SEV vaccination, the survival of IFN-γ-/- and IL-17-/- mice was assessed after SEV-immunization. The SEV-immunized groups of IFN-γ-/- and IL-17-/- mice showed different survival rates after challenge with the lethal dose of S. aureus (Fig 6B). All the SEV-immunized WT mice survived the bacterial challenge, whereas only 10% and 70% of the SEV-immunized mice survived for IFN-γ-/- and IL-17-/- mice, respectively. All of the sham-immunized WT and knock-out mice died. These data indicated that Th1 mediated cellular response is mainly involved in inducing vaccine efficacy by SEVs immunization, with partial involvement of Th17 mediated cellular response.
Long-term vaccination effect of SEV immunization for protection against lethality induced by S. aureus infection
Finally, we evaluated the long-term vaccination effect of SEV immunization for protection against lethality induced by S. aureus infection. As shown in Fig 7A, SEVs (5 μg) or sham (PBS) were administered intramuscularly to mice at weekly intervals for 3 weeks, then challenged with S. aureus (4 × 108 CFU) by oropharyngeal application 40 days after the last immunization. As shown in Fig 7B, all the mice that were immunized with SEVs survived the S. aureus infection, whereas all the sham-immunized mice died. This result suggests that SEV induced protective immune response is maintained in long term.
Fig 7. Long-term effect of SEV vaccination on the protection against lethality induced by S. aureus infection.
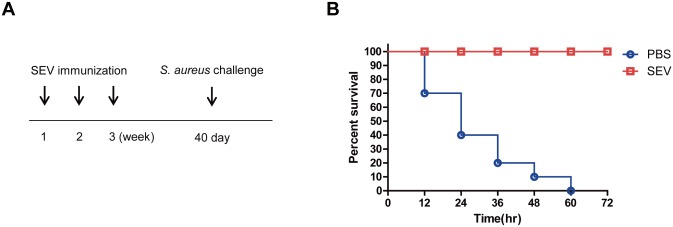
(A) Study protocol for SEV vaccination and bacterial challenge. SEVs (5 μg) and sham (PBS) were injected intramuscularly at weekly intervals for 3 weeks, and then S. aureus (4 × 108 CFU) was challenged by oropharyngeal application 40 days after the last immunization. (B) Survival rates of SEV- and sham-immunized mice challenged with S. aureus (n = 10, each group). Survival was monitored every 12 h for 40 days.
Toxicity of SEV vaccination
Safety is the one of the important requirement to be considered when developing and controlling vaccine. Therefore, in order for the SEV vaccine to be developed for use clinically, toxicity is a critical issue that must be assured. To test the toxicity of SEVs immunization, mice were injected intramuscularly with SEVs of the dose used for immunization experiments and of 10 times higher. The survival rate, body temperature, and body weight were monitored for 14 days for examination of any inflammatory responses (Fig 8A). All of the mice survived after SEV administration. Moreover, all the mice injected with SEVs did not show any phenotypical difference compared to the sham-injected mice. In addition, we measured the blood serum cytokines after the SEV administration to evaluate the evidence of systemic inflammation (Fig 8B). As a result, there was no difference in the blood serum cytokine levels between the sham and SEVs administration groups. These results indicate that the administration of SEVs does not cause notable toxic effect on mice.
Fig 8. Toxicity of SEV vaccine.

Mice were intramuscularly administered with 5 μg or 50 μg of SEV and monitored for 14 days. (n = 10) (A) The survival rate, body temperature, and body weight of mice measured at indicated times. (B) The levels of IL-1β, IL-6 and TNF-α in serum of SEV- and sham (PBS)-immunized mice 24 h after the SEV administration. n.s. indicates not significant.
Discussion
The hope for an efficacious vaccine against S. aureus infection has been blunted by the failures of the clinical trials of S. aureus vaccines [15,16,17]. Multivalent vaccines may work better in humans; previous results have shown that multiple antigen vaccines have better vaccine efficacy than single antigen vaccines in vivo [17,18,19]. In addition, as S. aureus pathogenic and defense mechanisms became better understood, both humoral and T cell-mediated immunities have been revealed to be important in protection against S. aureus infection [20]. Our present data implicate SEVs as a novel vaccine candidate. In particular, SEVs effectively protect against lethality and pneumonia induced by S. aureus infection, mainly via IFN-γ-producing Th1 cellular response rather than B cell mediated antibody response.
In our previous study with the SEVs, we have found that SEVs contain membrane proteins, cytosolic proteins, and other virulence factors. Although not fully understood, the pathogenic molecules inside the SEV and EV-associated proteins may induce host immune response differently than secreted proteins. The EV-associated proteins would be protected from antibody-mediated neutralization or protease. Therefore EV-associated proteins could retain longer in the host and travel long-distance. This characteristic, coupled with other physical properties of EVs, like having nano-sized diameters, make EVs ideal for both drug delivery vehicle and immune induction for vaccines [21]. In addition, we have also previously elucidated the vaccination mechanism of EVs using EVs derived from Gram-negative Escherichia coli to show that T cell-mediated immunity is the major inducer of protective immune response in mice [10]. Our results in the present study also showed that CD4+ T cell response is more effective as defensive mechanism than B cell-mediated humoral immunity in S. aureus infection (Fig 5A). These finding demonstrate that T cell-mediated immunity is the major immune response in SEV vaccination.
Studies have sought to develop a vaccine against S. aureus using capsular polysaccharides or surface proteins as target antigens. However, most failed when applied to human [22,23]. A combinational vaccine of capsular polysaccharides type 5 and 8 (CPS5 and CPS8) originated from Gram-positive S. aureus was combined with exoprotein A originated from Gram-negative P. aeruginosa. The combination was used as vaccine agent for human, but it failed in phase III clinical trial. Because CPS8 is expressed in only 40% of S. aureus strains, the antigen lacks sufficient capacity to prevent wide range of S. aureus infections [24]. In addition, although CPS5 has proven efficacious in animal models of staphylococcal infection, the use of CPS5 as a target antigen has been hampered by the fact that expression is limited to the stationary phase of S. aureus growth [25,26]. The other example of failure for S. aureus vaccine is a trial using a single surface staphylococcal protein, iron surface determinant B (IsdB). Although IsdB is highly conserved and is an important virulence factor, IsdB vaccination showed only partial protective effect against the lethal infection in the mouse model [27]. In addition, passive immunization using antibody specific to single antigen for example, the antibody against ClfA, CP5, CP8, and ABC transporter has also failed [26,28,29]. Combined with the fact that S. aureus expresses many toxins and immune evasion factors, previous evidence indicates that a single antigen is insufficient for inducing protective immune response. Recently, our proteomic study have demonstrated that SEVs harbors various surface proteins as well as toxins [11]. This prompted the notion that SEVs can be a good vaccine candidate against S. aureus infection. The present observations that vaccination with SEVs effectively protects lethality and pneumonia induced by S. aureus infection supports the view that a multivalent vaccine works well for S. aureus infections.
The previous failures of passive immunization strategies using antibody raise the possibility that B cell-derived humoral responses may not be as important in mediating protection against S. aureus infection. Previous preclinical results have shown that antibody-mediated responses are important in conferring protection against S. aureus. For example, passive transfer of antibodies against staphylococcal antigens in animal models conferred protection against S. aureus infection [30,31]. However, whether antibody-focused vaccine strategies provide any protective effect against S. aureus infection can be questioned based on the findings from several clinical studies [26,29,32]. Studies have shown the presence of S. aureus antibodies in most of clinical samples [33,34,35], and colonized subjects have a higher antibody titer against staphylococcal antigens [35]. Moreover, no clinical trial using passive immunization strategies has succeeded [24]. Presently, T cell-mediated immunity rather than antibody response conferred protection against lethality induced by S aureus infection. Previous results confirmed that patients with T cell deficiency were highly at risk of staphylococcal infections and B-cell deficient mice were not susceptible to S. aureus infection than wild type control [36]. These collective findings suggest that T cell-mediated immunity rather than antibody-mediated immunity is critical in the protection against S. aureus infection.
Th1 and Th17 cell responses, rather than Th2, are considered to have a key role in host defense toward staphylococci infections [19,37]. Animal experiments indicate that IFN-ɤ-deficient mice are more susceptible to S. aureus infections compared to WT mice [36,38], suggesting that the Th1 cellular response is important in providing protection against S. aureus. Moreover, IL-17-producing Th17 cells are important for fighting S. aureus infection by recruiting neutrophils to the site of infection [38]. Furthermore, superoxide-deficient mice were more susceptible to S. aureus infection [24,39]. These collectively suggest that effective elimination of S. aureus infections requires both the phagocyte recruitment and phagocytosis, mainly via Th17 cell immunity, and also that the bactericidal activity of phagocytes is mediated mainly by Th1 cell immunity. In the present study, SEV vaccination conferred protection against severe S. aureus infection, which was mainly dependent on IFN-ɤ rather than IL-17. These findings suggest that Th1 cell-mediated immunity is important in the protection against severe S. aureus infections, and imply that vaccine development focused on Th1 cell-mediated immunity could provide better vaccine efficacy.
Adjuvants play important roles for promoting antibody production or directing T cells to generate proper cytokines for cellular response [24]. Aluminum hydroxide (alum) and oil-in-water emulsions have been shown to increase antibody titers [40,41,42]. However, increasing studies have shown that T cell response can also be stimulated by the role of adjuvants [42,43,44]. Therefore, using the right adjuvant is important as much as selecting antigens in the vaccine development. SEVs do not require adjuvants since SEVs themselves already contain plethora of bacterial components including various cell wall components[11]. Indeed, the present study showed that SEVs could lead to sufficient innate and adaptive immune responses, without the aid of adjuvants. It is presumed that cell wall components in SEVs may provoke effective immune responses similar to the adjuvants. Peptidoglycan and lipoteichoic acid cell wall components of Gram-positive bacteria induce innate immune responses such as the up-regulation of co-stimulatory molecule expression and Th1/Th17-polarizing cytokine production through TLR2- and Myd88- dependent pathways [45,46,47]. Our results here also have shown that SEVs could enhance the expression of co-stimulatory molecules and the production of IL-12 and IL-6 (Th1 and Th17 polarizing cytokines, respectively) by antigen-presenting cells.
The present study reports an innovative strategy in vaccine development against S. aureus infections. The vaccine strategy using SEVs provides two main advantages. First, SEVs harbor many bacterial proteins, including cell surface proteins and toxins. Secondly, they do not require adjuvants to elicit an effective adaptive immune response since SEVs themselves can induce both cellular and antibody responses, which confer protection against S. aureus infections. Collectively, our findings indicate that vaccine development using native EVs from Gram-positive bacteria is a powerful and innovative strategy.
Material and Methods
Ethics Statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the Pohang University of Science and Technology. The experimental protocols were approved by the Institutional Animal Care and Use Committee at Pohang University of Science and Technology, Pohang, Republic of Korea (Permit Number: 2013-01-0004). All animal experiments were planned to minimize mice suffering.
Mice
Animal experiments were performed using WT (both C57BL/6 and BALB/c background), TLR2-, TLR4-, TLR9-, and MyD88-deficient (C57BL/6 background), and IFN-γ– and IL-17-deficient (BALB/c background) mice (all 6-week-old females).
Bacteria strain and culture
For mouse lung infection models, S. aureus (ATCC 14458) were grown at 37°C in nutrient broth (MERCK 1.05443.0500) until the optical density at 600 nm reached 1.5. Culture aliquots of 500 ml were centrifuged, washed in PBS, and suspended in sterile PBS for mortality studies. Bacteria cultures ranging from 1–4 × 108 CFU were administered in 25 μl of total volume.
Preparation of SEVs
Overnight bacterial cultures were pelleted at 10,000 g for 20 min, and the supernatant was filtered through a 0.45 μm vacuum filter. The filtrate was concentrated using a QuixStandBenchtop System (Amersham Biosciences), filtered through a 0.22 μm vacuum filter, and was pelleted by ultra-centrifugation in a 45 Ti rotor (Beckman Instruments) at 150,000 g for 3 h at 4°C. The isolated SEV containing pellet was resuspended in PBS and stored at -80°C until use.
Measurement of SEV-reactive antibody titer
SEV-specific IgG was evaluated in serum harvested from SEV- and sham-immunized mice. SEVs (100 ng) were coated in wells of a 96-well black plate and were blocked with 1% bovine serum albumin in PBS, and loaded with serum samples diluted 500-fold in the 1% suspension. Peroxidase-conjugated anti-mouse IgG was added as secondary antibody and SEV–specific IgG were detected after incubation the addition of a chemiluminescence substrate.
Adoptive transfer
Serum and spleen were isolated from SEV- and sham-immunized mice one week after the last immunization. For serum transfer experiment, 100 μl serum from either SEV- or sham-immunized mice were intraperitoneally injected in normal naïve mice. For CD4+ T cell transfer, spleens were ground using a cell strainer and were incubated with ammonium chloride solution to lyse red blood cells. The spleen cells were collected from the pellet after centrifugation and were resuspended in RPMI 1640 containing 10% fetal bovine serum and β-mercaptoenthanol (50 μM). Splenocyte T cells were isolated by positive selection using anti-CD3 magnetic bead (Miltenyi Biotec) and CD4+ T cells were serially separated by anti-CD4 microbead positive selection. Cell purity was assessed by flow cytometric analysis. The purified CD4+ T cells (2 × 106 cells in 100 μl PBS) were intravenously injected in naïve mice.
Immunization protocol and induction of bacterial pneumonia
For SEV immunization, mice were injected intramuscularly three times in weekly intervals in 100 μl PBS. Serum was obtained by eye bleeding 72 h after the last immunization to measure the antibody titers. For the bacteria-induced pneumonia model, mice were administered with S. aureus via the oropharynx airway. Survival was monitored every 12 h for 6 days. Blood and lung tissues from SEV- and sham-immunized mice were collected 24 h after bacterial challenge for evaluation. To establish bacterial pneumonia in the mouse, anesthetized mice were inoculate by oropharyngeal application with different doses of S. aureus.
Mice euthanasia
For humane animal experiment, mice showing severe SIRS indexes were euthanized before death to minimize suffering. After bacterial challenge, mice were monitored every 6 h for 6 days and mice having phenotypical symptoms of systemic inflammatory response syndrome (SIRS) indexes like eye-exudates and piloerection were measured for body temperatures[48,49]. If the body temperatures of the mice dropped below 34°C, mice were euthanized by cervical dislocation. There was no unexpected death in this study.
Determination of viability as colony forming units (CFU)
A day after the bacteria injection, the bacterial burden in the lung was examined. The lung tissue was homogenized in sterile PBS. The homogenized samples were serially diluted and were plated in nutrient broth agar plates. After overnight incubation at 37°C, CFU were determined.
Cy-7 labeling of S. aureus
For labeling S. aureus with Cy-7, Cy-7 dye (GE Healthcare; PA17101) was-incubated with S. aureus at 4°C overnight. S. aureus were pelleted at 10,000 g for 30 min, resuspended in PBS, aliquoted, and stored at -80°C until use. An IVIS spectrometer was used for analysis.
Measurement of cytokines
The levels of cytokines in fluids and cell-culture supernatants were measured by ELISA in accordance with the manufacturer’s instructions (R&D Systems): IL-1β and IL-6 in serum and IL-1β, IL-6, IL-12p40 and TNF-α in the culture supernatant of DCs, and IFN-γ, IL-17 and IL- 4 in supernatant of isolated splenic T cells and CD4+ T cells.
Histological analysis
Isolated lungs were fixed in paraffin, sectioned, and stained with hematoxylin and eosin (H&E). Lung tissues were analyzed at 40× and 400× magnification.
Ex vivo studies of isolated DCs
Bone marrow-derived DCs from mice were prepared using a high concentration (20 ng/ml) of granulocyte macrophage colony stimulating factor (R&D Systems). To detect polarizing cytokine from DCs stimulated by SEVs, DCs were exposed to SEVs (5 × 105 per well of a 24-well plate) and incubated for 24 h. After 24 h, the conditioned medium was collected from SEV-treated cells for evaluation of cytokines by ELISA.
Statistical analyses
For multiple comparisons, one-way analysis of variance (ANOVA) was used first. If significant differences were found, individual t-tests or Wilcoxon’s rank-sum tests were performed between pairs of groups. Differences were considered statistically significant if P < 0.05.
Acknowledgments
We thank the members of the POSTECH animal facility for their experimental expertise.
Data Availability
All relevant data are within the paper.
Funding Statement
This research was supported by Korea Drug Development Fund (KDDF) funded by MSIP, MOTIE, and MOHW (Grant No. KDDF-201212-05) and a grant of the Korea Health Technology R & D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI14C2694).
References
- 1. Kluytmans J, van Belkum A, Verbrugh H (1997) Nasal carriage of Staphylococcus aureus: epidemiology, underlying mechanisms, and associated risks. Clin Microbiol Rev 10: 505–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lowy FD (1998) Staphylococcus aureus infections. N Engl J Med 339: 520–532. [DOI] [PubMed] [Google Scholar]
- 3. Richards MJ, Edwards JR, Culver DH, Gaynes RP (2000) Nosocomial infections in combined medical-surgical intensive care units in the United States. Infect Control Hosp Epidemiol 21: 510–515. [DOI] [PubMed] [Google Scholar]
- 4. David MZ, Daum RS (2010) Community-associated methicillin-resistant Staphylococcus aureus: epidemiology and clinical consequences of an emerging epidemic. Clin Microbiol Rev 23: 616–687. 10.1128/CMR.00081-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Levine MM, Sztein MB (2004) Vaccine development strategies for improving immunization: the role of modern immunology. Nat Immunol 5: 460–464. [DOI] [PubMed] [Google Scholar]
- 6. Fearon DT, Locksley RM (1996) The instructive role of innate immunity in the acquired immune response. Science 272: 50–53. [DOI] [PubMed] [Google Scholar]
- 7. Lee EY, Bang JY, Park GW, Choi DS, Kang JS, Kim HJ, et al. (2007) Global proteomic profiling of native outer membrane vesicles derived from Escherichia coli. Proteomics 7: 3143–3153. [DOI] [PubMed] [Google Scholar]
- 8. Bomberger JM, Maceachran DP, Coutermarsh BA, Ye S, O'Toole GA, Stanton BA, et al. (2009) Long-distance delivery of bacterial virulence factors by Pseudomonas aeruginosa outer membrane vesicles. PLoS Pathog 5: e1000382 10.1371/journal.ppat.1000382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kwon SO, Gho YS, Lee JC, Kim SI (2009) Proteome analysis of outer membrane vesicles from a clinical Acinetobacter baumannii isolate. FEMS Microbiol Lett 297: 150–156. 10.1111/j.1574-6968.2009.01669.x [DOI] [PubMed] [Google Scholar]
- 10. Kim OY, Hong BS, Park KS, Yoon YJ, Choi SJ, Lee WH, et al. (2013) Immunization with Escherichia coli outer membrane vesicles protects bacteria-induced lethality via Th1 and Th17 cell responses. J Immunol 190: 4092–4102. 10.4049/jimmunol.1200742 [DOI] [PubMed] [Google Scholar]
- 11. Lee EY, Choi DY, Kim DK, Kim JW, Park JO, Kim S, et al. (2009) Gram-positive bacteria produce membrane vesicles: proteomics-based characterization of Staphylococcus aureus-derived membrane vesicles. Proteomics 9: 5425–5436. 10.1002/pmic.200900338 [DOI] [PubMed] [Google Scholar]
- 12. Hong SW, Kim MR, Lee EY, Kim JH, Kim YS, Jeon SG, et al. (2011) Extracellular vesicles derived from Staphylococcus aureus induce atopic dermatitis-like skin inflammation. Allergy 66: 351–359. 10.1111/j.1398-9995.2010.02483.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, et al. (2000) Immunobiology of dendritic cells. Annu Rev Immunol 18: 767–811. [DOI] [PubMed] [Google Scholar]
- 14. Bubeck Wardenburg J, Schneewind O (2008) Vaccine protection against Staphylococcus aureus pneumonia. J Exp Med 205: 287–294. 10.1084/jem.20072208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cook J, Hepler R, Pancari G, Kuklin N, Fan H, Wang WM, et al. (2009) Staphylococcus aureus capsule type 8 antibodies provide inconsistent efficacy in murine models of staphylococcal infection. Hum Vaccin 5: 254–263. [DOI] [PubMed] [Google Scholar]
- 16. Tuchscherr LP, Buzzola FR, Alvarez LP, Lee JC, Sordelli DO (2008) Antibodies to capsular polysaccharide and clumping factor A prevent mastitis and the emergence of unencapsulated and small-colony variants of Staphylococcus aureus in mice. Infect Immun 76: 5738–5744. 10.1128/IAI.00874-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stranger-Jones YK, Bae T, Schneewind O (2006) Vaccine assembly from surface proteins of Staphylococcus aureus. Proc Natl Acad Sci U S A 103: 16942–16947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim HK, Kim HY, Schneewind O, Missiakas D (2011) Identifying protective antigens of Staphylococcus aureus, a pathogen that suppresses host immune responses. FASEB J 25: 3605–3612. 10.1096/fj.11-187963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Arrecubieta C, Matsunaga I, Asai T, Naka Y, Deng MC, Lowy FD, et al. (2008) Vaccination with clumping factor A and fibronectin binding protein A to prevent Staphylococcus aureus infection of an aortic patch in mice. J Infect Dis 198: 571–575. 10.1086/590210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Spellberg B, Daum R (2012) Development of a vaccine against Staphylococcus aureus. Semin Immunopathol 34: 335–348. 10.1007/s00281-011-0293-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bachmann MF, Jennings GT (2010) Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat Rev Immunol 10: 787–796. 10.1038/nri2868 [DOI] [PubMed] [Google Scholar]
- 22. Robbins JB, Schneerson R, Horwith G, Naso R, Fattom A (2004) Staphylococcus aureus types 5 and 8 capsular polysaccharide-protein conjugate vaccines. Am Heart J 147: 593–598. [DOI] [PubMed] [Google Scholar]
- 23. Harro CD, Betts RF, Hartzel JS, Onorato MT, Lipka J, Smugar SS, et al. (2012) The immunogenicity and safety of different formulations of a novel Staphylococcus aureus vaccine (V710): results of two Phase I studies. Vaccine 30: 1729–1736. 10.1016/j.vaccine.2011.12.045 [DOI] [PubMed] [Google Scholar]
- 24. Bagnoli F, Bertholet S, Grandi G (2012) Inferring reasons for the failure of Staphylococcus aureus vaccines in clinical trials. Front Cell Infect Microbiol 2: 16 10.3389/fcimb.2012.00016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. O'Riordan K, Lee JC (2004) Staphylococcus aureus capsular polysaccharides. Clin Microbiol Rev 17: 218–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schaffer AC, Lee JC (2008) Vaccination and passive immunisation against Staphylococcus aureus. Int J Antimicrob Agents 32 Suppl 1: S71–78. 10.1016/j.ijantimicag.2008.06.009 [DOI] [PubMed] [Google Scholar]
- 27. Kuklin NA, Clark DJ, Secore S, Cook J, Cope LD, McNeely T, et al. (2006) A novel Staphylococcus aureus vaccine: iron surface determinant B induces rapid antibody responses in rhesus macaques and specific increased survival in a murine S. aureus sepsis model. Infect Immun 74: 2215–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ohlsen K, Lorenz U (2010) Immunotherapeutic strategies to combat staphylococcal infections. Int J Med Microbiol 300: 402–410. 10.1016/j.ijmm.2010.04.015 [DOI] [PubMed] [Google Scholar]
- 29. Otto M (2010) Novel targeted immunotherapy approaches for staphylococcal infection. Expert Opin Biol Ther 10: 1049–1059. 10.1517/14712598.2010.495115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Josefsson E, Hartford O, O'Brien L, Patti JM, Foster T (2001) Protection against experimental Staphylococcus aureus arthritis by vaccination with clumping factor A, a novel virulence determinant. J Infect Dis 184: 1572–1580. [DOI] [PubMed] [Google Scholar]
- 31. Vernachio JH, Bayer AS, Ames B, Bryant D, Prater BD, Syribeys PJ, et al. (2006) Human immunoglobulin G recognizing fibrinogen-binding surface proteins is protective against both Staphylococcus aureus and Staphylococcus epidermidis infections in vivo. Antimicrob Agents Chemother 50: 511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. DeJonge M, Burchfield D, Bloom B, Duenas M, Walker W, Kaufman D, et al. (2007) Clinical trial of safety and efficacy of INH-A21 for the prevention of nosocomial staphylococcal bloodstream infection in premature infants. J Pediatr 151: 260–265, 265 e261 [DOI] [PubMed] [Google Scholar]
- 33. Dryla A, Prustomersky S, Gelbmann D, Hanner M, Bettinger E, Kocsis B, et al. (2005) Kaufman Dand in acutely infected patients. Clin Diagn Lab Immunol 12: 387–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Clarke SR, Brummell KJ, Horsburgh MJ, McDowell PW, Mohamad SA, Stapleton MR, et al. (2006) Identification of in vivo-expressed antigens of Staphylococcus aureus and their use in vaccinations for protection against nasal carriage. J Infect Dis 193: 1098–1108. [DOI] [PubMed] [Google Scholar]
- 35. Verkaik NJ, de Vogel CP, Boelens HA, Grumann D, Hoogenboezem T, Vink C, et al. (2009) Anti-staphylococcal humoral immune response in persistent nasal carriers and noncarriers of Staphylococcus aureus. J Infect Dis 199: 625–632. 10.1086/596743 [DOI] [PubMed] [Google Scholar]
- 36. Spellberg B, Ibrahim AS, Yeaman MR, Lin L, Fu Y, Avanesian V, et al. (2008) The antifungal vaccine derived from the recombinant N terminus of Als3p protects mice against the bacterium Staphylococcus aureus. Infect Immun 76: 4574–4580. 10.1128/IAI.00700-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cho JS, Pietras EM, Garcia NC, Ramos RI, Farzam DM, Monroe HR, et al. (2010) IL-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. J Clin Invest 120: 1762–1773. 10.1172/JCI40891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lin L, Ibrahim AS, Xu X, Farber JM, Avanesian V, Baquir B, et al. (2009) Th1-Th17 cells mediate protective adaptive immunity against Staphylococcus aureus and Candida albicans infection in mice. PLoS Pathog 5: e1000703 10.1371/journal.ppat.1000703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Edwards SW, Say JE, Hart CA (1987) Oxygen-dependent killing of Staphylococcus aureus by human neutrophils. J Gen Microbiol 133: 3591–3597. [DOI] [PubMed] [Google Scholar]
- 40. Galli G, Hancock K, Hoschler K, DeVos J, Praus M, Bardelli M, et al. (2009) Fast rise of broadly cross-reactive antibodies after boosting long-lived human memory B cells primed by an MF59 adjuvanted prepandemic vaccine. Proc Natl Acad Sci U S A 106: 7962–7967. 10.1073/pnas.0903181106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jones T (2009) GSK's novel split-virus adjuvanted vaccines for the prevention of the H5N1 strain of avian influenza infection. Curr Opin Mol Ther 11: 337–345. [PubMed] [Google Scholar]
- 42. Dormitzer PR, Galli G, Castellino F, Golding H, Khurana S, Del Giudice G, et al. (2011) Influenza vaccine immunology. Immunol Rev 239: 167–177. 10.1111/j.1600-065X.2010.00974.x [DOI] [PubMed] [Google Scholar]
- 43. Baudner BC, Ronconi V, Casini D, Tortoli M, Kazzaz J, Singh M, et al. (2009) MF59 emulsion is an effective delivery system for a synthetic TLR4 agonist (E6020). Pharm Res 26: 1477–1485. 10.1007/s11095-009-9859-5 [DOI] [PubMed] [Google Scholar]
- 44. Kamath AT, Rochat AF, Valenti MP, Agger EM, Lingnau K, Andersen P, et al. (2008) Adult-like anti-mycobacterial T cell and in vivo dendritic cell responses following neonatal immunization with Ag85B-ESAT-6 in the IC31 adjuvant. PLoS One 3: e3683 10.1371/journal.pone.0003683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kim MR, Hong SW, Choi EB, Lee WH, Kim YS, Jeon SG, et al. (2012) Staphylococcus aureus-derived extracellular vesicles induce neutrophilic pulmonary inflammation via both Th1 and Th17 cell responses. Allergy 67: 1271–1281. 10.1111/all.12001 [DOI] [PubMed] [Google Scholar]
- 46. Lin HY, Tang CH, Chen YH, Wei IH, Chen JH, Lai CH, et al. (2010) Peptidoglycan enhances proinflammatory cytokine expression through the TLR2 receptor, MyD88, phosphatidylinositol 3-kinase/AKT and NF-kappaB pathways in BV-2 microglia. Int Immunopharmacol 10: 883–891. 10.1016/j.intimp.2010.04.026 [DOI] [PubMed] [Google Scholar]
- 47. Lee IT, Lee CW, Tung WH, Wang SW, Lin CC, Shu JC, et al. (2010) Cooperation of TLR2 with MyD88, PI3K, and Rac1 in lipoteichoic acid-induced cPLA2/COX-2-dependent airway inflammatory responses. Am J Pathol 176: 1671–1684. 10.2353/ajpath.2010.090714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, et al. (1992) Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest 101: 1644–1655. [DOI] [PubMed] [Google Scholar]
- 49. Park KS, Choi KH, Kim YS, Hong BS, Kim OY, Kim JH, et al. (2010) Outer membrane vesicles derived from Escherichia coli induce systemic inflammatory response syndrome. PLoS One 5: e11334 10.1371/journal.pone.0011334 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper.