

Summary
T follicular helper (Tfh) cells provide essential help to B cells for the generation of high-affinity antibodies. These mechanisms provide the basis for the success of modern vaccines, but dysregulated Tfh cell responses are also linked to autoimmune diseases. In addition to their established role in driving humoral immunity, Tfh cells are also gaining attention for their role in other processes of the adaptive immune system. For example Tfh cells may serve as transitional differentiation intermediates during effector and memory T helper cell differentiation, and as a reservoir of HIV-infected cells. While B cells are required for the full maturation and maintenance of Tfh cell responses, they are dispensable for the initial induction of the Tfh cell phenotype, which in fact occurs at the priming stage through interaction with dendritic cells. Nevertheless, the precise mechanisms of these early events during Tfh cell differentiation remain relatively unknown. Here we describe a method for tracking early Tfh cell differentiation by following cell division kinetics and phenotypic changes of recently activated antigen-specific CD4+ T cells in vivo. As an example, we use this method to visualize the requirements for T cell-expressed CD28 for the differentiation of Bcl6+CXCR5+ Tfh cells.
Keywords: T follicular helper cells, follicular helper T cells, Tfh cells, flow cytometry, FACS, CFSE, CellTrace Violet, CTV, T-dependent antibody response, Bcl6, CXCR5
1. Introduction
T follicular helper (Tfh) cell differentiation begins at the priming stage when naïve CD4+ T helper cells interact with antigen-presenting dendritic cells (DCs) in the T zone of secondary lymphoid organs (Ma et al., 2012; Qi et al., 2014). Activated CD4+ T cells undergo rapid changes in their expression of co-stimulatory molecules and chemokine receptors. Downregulation of CCR7 expression, which is highly expressed on naïve CD4+ T cells, and concomitant upregulation of the chemokine receptor CXCR5 subsequently allow these activated T cells to migrate to the T-B zone border and interfollicular regions of secondary lymphoid organs, where they interact with antigen-specific B cells (Crotty, 2011; Vinuesa and Cyster, 2011). Some of these early Tfh cells, together with a few antigen-specific B cells, enter the follicle to establish full-fledged germinal centers in which somatic hypermutation and selection of high-affinity B cells results in the generation of memory B cells and plasma cells that produce high-affinity antibodies (Victora and Nussenzweig, 2012). Even though it was initially believed that B cells were essential for the differentiation of Tfh cells, more recent studies have clarified that DCs are able to induce a Tfh cell phenotype in recently activated CD4+ T cells, independent of cognate interactions with B cells (Baumjohann et al., 2011; Choi et al., 2011; Goenka et al., 2011; Kerfoot et al., 2011; Kitano et al., 2011). Nevertheless, B cells become the major antigen-presenting cell type for Tfh cells at later stages of the immune response, thus being important for the full differentiation and maintenance of germinal center Tfh cells (Baumjohann et al., 2013b; Deenick et al., 2010).
The introduction of fluorescent dyes for tracking cell divisions of labeled cells has provided important insights into various aspects of T helper cell biology. Carboxyfluorescein diacetate succinimidyl ester (CFSE) was introduced to immunology labs in the early 1990’s (Lyons and Parish, 1994) and is to date the most widely used of these dyes. More recently, several alternatives to CFSE have provided improved features and additional flexibility in the design of experiments (Quah and Parish, 2012). We have used the division status as a means to track Tfh cell development in adoptively transferred TCR-tg T cells after immunization in wild-type recipient mice. For example, we showed that those T cells in draining lymph nodes that proliferated the most became enriched for CXCR5+Bcl6+ Tfh cells (Baumjohann et al., 2011). In another study, we used this method to show that global microRNA expression in CD4+ T cells was required for the differentiation of these cells into Tfh cells, which was due to an intrinsic defect to induce the Tfh gene expression program, independent of any changes in their proliferative capacity (Baumjohann et al., 2013a). In this protocol we describe the methodologic details of these approaches.
2. Materials
2.1. Cell preparation, immunization, and antibody staining
T cell receptor-transgenic (TCR-tg) donor mice, e.g. OT-II mice (Barnden et al., 1998) in which T cells carry a transgenic TCR recognizing ovalbumin (OVA)323-339 in the context of MHC class II (I-Ab).
Recipient mice: Wildtype C57BL/6 mice or CD45.1+ congenic mice, e.g. B6.SJL-Ptprca Pepcb/BoyJ mice (The Jackson Laboratory) or B6-LY5.2/Cr mice (National Cancer Institute).
Glass slides with frosted ends.
5cm cell culture dishes.
5ml polystyrene tubes and 14ml polypropylene tubes.
Nylon mesh (70μm pore size).
Mouse CD4+ T cell isolation kit (e.g. EasySep negative selection kit by Stemcell Technologies) and a magnet for pre-enrichment.
CellTrace Violet (CTV, Life Technologies): Prepare 5 mM aliquots in PBS and store at −20°C.
NP-OVA (Biosearch Technologies): Prepare 1 mg/ml aliquots in PBS and store at −20°C. Note: Different conjugation ratios of NP and OVA are available. Here we used NP14-OVA.
Imject Alum (Thermo Scientific).
96-well round-bottom plates.
Antibodies: Fluorophore-conjugated antibodies against CD4 (clone RM4-5), CD25 (clone PC61.5), CD62L (clone MEL-14), CD44 (clone IM7), Bcl6 (clone K112-91), CD19 (clone 1D3) or B220 (clone RA3-6B2), TCR Vα2 (clone B20.1), CD45.2 (clone 104), CD45.1 (clone A20), and biotinylated anti-CXCR5 (clone 2G8).
APC-conjugated streptavidin.
Fixable Viability Dye eFluor® 780 (eBioscience), which can be detected in the APC-Cy7 channel of a flow cytometer.
Insulin syringes for the injection of cells.
2.2. Buffers and media
PBS (Mg2+/Ca2+-free).
Sorting/enrichment buffer: 2% fetal bovine serum (FBS) and 1mM EDTA in phosphate-buffered saline (PBS).
Complete medium: RPMI 1640 (with L-glutamine) supplemented with 10% FBS, 1 mM sodium pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin, 0.05 mM beta-marcaptoethanol, 10 mM HEPES.
Staining buffer: 2% FBS, 1mM EDTA, and 0.1% sodium azide (NaN3) in PBS.
Fc-block: 2% mouse serum, 2% rat serum, 5μg/ml anti-CD16/32 in staining buffer.
Foxp3/Transcription Factor Staining Buffer Set (eBioscience).
3. Methods
3.1. Adoptive Transfers and Immunizations
Dissect and pool peripheral lymph nodes (inguinal, axillary, and brachial) and spleen of TCR-tg donor mice of desired genotypes. Prepare single-cell suspensions in PBS by disrupting the tissues between the frosted ends of glass slides in 5cm plastic dishes. Filter cells through fine mesh into 14ml tubes that contain at least 1ml of sorting buffer. Fill up tubes to 14ml and spin down. Discard supernatant and resuspend cells in appropriate volumes of sorting buffer (compare next step). Keep aside a small aliquot of cells for phenotyping: Stain cells with antibodies against CD4, the specific TCR of the TCR-transgene, and congenic markers as well as other relevant markers (compare Figure 1A).
Prior to sorting by flow cytometry, CD4+ T cells are pre-enriched by negative selection using magnetic beads. Several commercial kits are available for these purposes, such as column-based products (e.g. MACS by Miltenyi Biotech) or column-free products (e.g. Dynabeads by Life Technologies). Here we use StemCell Technologies’ rapid and column-free EasySep negative selection mouse CD4+ T cell isolation kit according to the manufacturer’s instructions. In brief, cells are resuspended at a concentration of 2×108 cells/ml sorting buffer, transferred to a 5ml polystyrene tube, followed by consecutive addition of normal rat serum, primary biotinylated antibody mix, and streptavidin-conjugated microbeads. Tubes are filled up to 2.5ml with sorting buffer and placed into an EasySep magnet. After 2.5 minutes, the unbound fraction is poured off in one move into new 15ml tubes that contain 10 ml sorting buffer.
After enrichment, naïve CD4+ T cells are sorted to high purity using a flow cytometer. To this end, pre-enriched CD4+ T cells (from step 3) are spun down and resuspended in 400μl antibody mix, which includes anti-CD4, anti-CD25, anti-CD62L, and anti-CD44. Cells are stained for 10min on ice, washed once, and resuspended again in sorting buffer. Right before acquisition on a sorting machine, a viability dye (e.g. 7-AAD) should be added to exclude dead cells (compare Figure 1B for the gating strategy).
In the next step, sorted naïve TCR-tg CD4+ T cells are labeled with CTV according to the manufacturer’s instructions: Dilute the CTV stock solution 1:500 in PBS (10μm) and prewarm the tube in a 37°C waterbath. Spin down sorted cells and resuspend them at a concentration of 2×106 cells per ml of PBS. Mix equal volumes of cells with the diluted CTV (5μm final concentration). Incubate cells for 20min in a 37°C waterbath. Invert the tubes to mix at least 2-3 times during the incubation.
Fill up each tube with complete medium and incubate for an additional 5min at 37°C to quench any surplus CTV. Spin down cells and wash twice with PBS. Count cells and adjust cell numbers to 5×105 cells per 200μl PBS for injection into each recipient mouse. Ideally, injected cells are allowed to equilibrate in the host for several hours before immunization.
Prepare antigen mixture by combining 1 part of the NP-OVA stock with 1 part PBS and 2 parts of alum and slowly rotate/invert for 30-45min at room temperature.
Immunize mice with the cognate antigen: Carefully inject 20μl of antigen mixture (= 5μg antigen) s.c. into each foot pad (see notes for alternative immunization routes).
Figure 1.
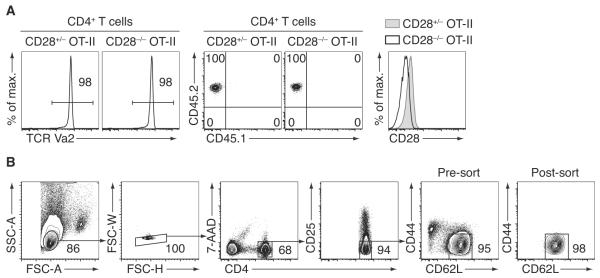
Isolation of naïve TCR-tg CD4+ T cells for adoptive transfer. (A) Single-cell suspensions were prepared from spleen and lymph nodes of CD28+/− or CD28−/− (Shahinian et al., 1993) OT-II TCR-tg mice. An aliquot was stained with PE-conjugated anti-CD4, APC-conjugated TCR Vα2, PerCP-Cy5.5-conjugated anti-CD45.2, eFluor450-conjugated anti-CD45.1, and FITC-conjugated anti-CD28 (clone 37.51) for verification of the phenotype of the donor cells. (B) The rest of the cells were pre-enriched for untouched CD4+ T cells by magnetic cell separation, followed by staining with antibodies against CD4 (conjugated to eFluor450), CD25 (APC), CD44 (Alexa Fluor 700), and CD62L (PE-Cy7). To exclude dead cells, 7-AAD was added to the stained cell suspension right before acquisition/sorting on a FACSAria II cell sorter (BD Biosciences). Small lymphocytes were gated by FSC-A vs. SSC-A characteristics. Singlet cells are identified by FSC-H vs. SSC-W gating (note that the use of EDTA in the sorting buffer significantly reduced doublet formation). Treg cells were then excluded by gating out CD25+ cells. Finally, naïve CD4+ T cells were sorted as CD62Lhigh CD44low/int cells. An optional re-sort on a small aliquot of the sorted cells was performed to verify the high purity of the obtained cell population.
3.2. Analysis by Flow Cytometry
Sacrifice recipient mice according to institutional guidelines and dissect popliteal LNs (pool 2 LNs for each mouse to increase total cell numbers). If desired, dissect non-draining LNs far away from the injection site (e.g. brachial LNs) or from non-immunized control recipients. Note that popliteal LNs from non-immunized mice are very small and will not yield many CTV+ cells.
Prepare single-cell suspensions in PBS by disrupting LNs between the frosted ends of glass slides. Filter through fine mesh (70μm). Count cells. Pipet 3×106 cells into each well of a 96-well round-bottom plate.
If cells are going to be fixed and permeabilized at a later stage, e.g. for transcription factor staining, perform staining with a fixable viability dye at this point. To this end, wash plate once with PBS and add 200μl of 1:2000 diluted eF780 viability dye to each well and incubate for 15min on ice.
Wash plate twice with staining buffer.
Add 20μl of Fc-block to each well and incubate for 5min.
Without washing, add 20μl of 2x 1° antibody mixture to the wells (40μl final staining volume). Incubate for 30min at 4°C.
Fill up with 160μl of staining buffer, spin down, and wash once more with 200μl of staining buffer.
Add 40μl of streptavidin-APC (0.2μg/ml) to each well and incubate for 15min at 4°C.
Fill up with 160μl of staining buffer, spin down, and wash once more with 200μl of staining buffer.
For immediate cell analysis or sorting with a flow cytometer, resuspend cells in 100μl flow buffer including 7AAD.
For transcription factor staining, resuspend washed cells in 100μl of freshly prepared fixation buffer and incubate for 15min ate RT, carefully shaking/tilting the plate from time to time.
Add 100μl of the freshly prepared permeabilization buffer and spin down. Resuspend in 200μl permeabilization buffer and spin down again.
Resuspend cells in 20μl Fc-block prepared in permeabilization buffer. Incubate for 5min at RT.
Without washing, add 20μl of 2x transcription factor antibody mixture to the wells (40μl final staining volume). Incubate for 30min at RT.
Fill up with 160μl of permeabilization buffer, spin down, and wash once more with 200μl of permeabilization buffer.
Resuspend cells in 100μl staining buffer and acquire cells on a flow cytometer. Include single fluorophore stains or fluorescence-minus-one control stains for compensation.
3.3. Analysis of flow cytometry data
Load data into your preferred flow cytometry analysis software (e.g. Flowjo).
Gate on singlet cells using forward/sideward scatter characteristics. Then gate on live cells, which are negative for the eF780 viability dye. In the following gates, plot CTV against B220 or CD19 to exclude B cells, against CD4 to identify CD4+ T cells, and against the congenic marker to ultimately identify a pure population of transferred TCR-tg CD4+ T cells that can then be analyzed for surface marker or transcription factor expression (compare Figure 2).
If desired, individual gates can be drawn for each cell division according to CTV dilution. Usually, a maximum number of up to 8 divisions can be reliably detected. These gates can then be used to analyze Tfh marker expression within each division (Baumjohann et al., 2013a; Baumjohann et al., 2011).
Using the CTV profile of the transferred cells together with the Cell Division function in FlowJo, further parameters can be determined, including the proliferation index.
Figure 2.

Example of the described method to dissect early steps during Tfh cell differentiation. The costimulatory molecule CD28 is known to be required for Tfh cell differentiation (Linterman et al., 2009; Walker et al., 1999). Here, we compared CD28-sufficient (heterozygous) control versus CD28-deficient OT-II cells to gain insights into the kinetics of early Tfh cell differentiation. (A) Gating strategy for the identification of adoptively transferred OT-II cells. Naïve CD45.2+ OT-II cells (purified as described in Figure 1) were labeled with CTV and injected into recipient mice. Hosts were immunized with NP14-OVA/alum s.c. in the foot pads at different time points. Draining popliteal lymph nodes from all mice were then analyzed together on the same day. Single-cell suspensions were stained with antibodies as described in the methods section and acquired on a LSR-II cytometer (BD Biosciences). First, 5×105 total cells were acquired followed by appended acquisition of CTV+ OT-II cells (see Note 13). (B) Histograms show the differences in the proliferative capacity of control vs. CD28−/− OT-II cells (gated as in A). (C) CTV profiles of activated OT-II cells at different time points after immunization. Activated CD28-sufficient OT-II cells (gated as in A) upregulate Bcl6 within the first few cell divisions. In contrast, induction of the Tfh-defining chemokine receptor CXCR5 occurs at later cell divisions and time points, preferentially in those cells that proliferated the most. CD28 is important for strong Bcl6 upregulation in activated OT-II cells and is absolutely required for the induction of CXCR5, These defects are cell intrinsic and independent of any differences in the proliferative capacity of the dividing cells.
4. Notes
Adoptively transferred TCR-tg cells can be easily identified within recipient mice if they carry distinct congenic or fluorescent marker genes. For example, different isoforms of the pan-lymphocyte marker CD45 (Ly5) are widely used. Other popular congenic markers include different isoforms of CD90 (Thy1). Crossing TCR-tg strains onto a CD45.1 or CD90.1 (Thy1.1) background provides the advantage that normal C57BL/6 mice, which are CD45.2+ and CD90.2+, can be used as hosts. Usually, heterozygosity in these alleles provides sufficient expression levels for the differentiation between transferred and recipient cells. Choosing heterozygous over homozygous cells might also reduce the risk of rejection of the injected cells, which is an important concern especially in longer-term adoptive transfer experiments. Fluorescent reporter strains (e.g. ubiquitously GFP-expressing mouse lines) can also be used to track transferred cells. Given the relatively short duration of the described protocol, we have not noticed dramatic changes in cell numbers that might have been caused by potential rejection issues.
Pre-enrichment of CD4+ T cells by magnetic bead isolation can significantly help to reduce sorting time on the flow cytometer. Since purity during the enrichment step is not a critical paramater, the volumes of antibodies and beads can be titrated down to spare reagents. We prefer to use a magnetic bead negative depletion approach that yields “untouched” CD4+ T cells for downstream applications.
It is good practice to verify the correct genotype/phenotype of the cells before injection. This can be accomplished by a quick staining of a small aliquot of LN/spleen cells and can be analyzed while at the sorter. Stain for congenic markers and the specific transgenic TCR (TCR Vα2 in the case of OT-II cells) to verify the correct congenic marker before injection of the cells into recipients.
Naïve cells are preferred over bulk T cell populations as the frequency of activated (CD44highCD62Llow) T cells might be considerably different between two different genotypes to be analyzed. These memory cells might possess different activation kinetics after adoptive transfer and immunization and could therefore mask the activation characteristics of the naïve cells.
We have optimized our protocol for the use of CellTrace Violet (CTV), as it frees the FL-1 channel (e.g. FITC, Alexa Fluor 488, GFP) on the flow cytometer. This has the advantage that GFP/YFP-expressing cells can be easily incorporated into the protocol, and CTV also requires much less compensation than CFSE. In our hands, CTV also provides a better resolution than CFSE.
One major advantage of foot pad immunization with OVA/alum is that the popliteal LN is the only direct draining LN. Furthermore, contralateral injections potentially allow for comparison within the same mouse. One disadvantage is that popliteal LNs of non-immunized mice are very small and thus harbor few cells. If no comparison is needed, both sides can be used for immunization and both popliteal LNs can be pooled for analysis. In any case, immunization routes need to be performed in accordance with institutional guidelines. In this regard, hock injections, which also drain to the popliteal LNs, represent an ethical alternative to foot pad injections (Kamala, 2007).
Dead cells may exhibit significant autofluorescence and may bind antibodies nonspecifically. Thus it is good practice to exclude dead cells during analysis by incorporating a viability dye in the staining procedure. Common dyes such as PI, 7-AAD or DAPI are usually added to samples right before acquisition and thus don’t require significant additional hands-on time. It should be noted that these dyes are not fixable. However, staining for intracellular transcription factors or cytokines requires fixation and permeabilization of cells. For these applications, several companies offer special dyes that are fixable and thus allow for the separation of live from dead cells during the acquisition on the flow cytometer and in subsequent analyses of the data.
Besides dead cells, other cell populations not necessarily of interest to the question of the experiment might interfere with the analysis of the population of interest, either by high autofluorescence, distinct forward/side scatter characteristics, or due to non-specific antibody binding. Thus, it is good practice to include Fc block (unconjugated anti-CD16/32 antibody) and mouse/rat serum incubation steps in each staining protocol. In addition, cells can be excluded in a “dump channel” by staining for multiple lineage-specific markers not expressed on the cells of interest using antibodies conjugated to the same fluorophore. For Tfh cell research, it is highly advisable to at least gate out B cells, as they are CXCR5hi cells, they potentially form T-B cell conjugates, and they are especially “sticky” because of their Fc receptor expression. Addition of EDTA to the sorting and staining buffers reduces cell doublet formation and thereby increases the yield of acquired events as well.
Activated cells become blastic, especially during the first few cell divisions. This goes hand in hand with increased FSC/SSC and increased background staining. This should be taken into account when analyzing and drawing conclusions from the data obtained. Antibodies should be carefully titrated by each investigator for optimal staining results. In addition, isotype controls, or better, cells with genetic deletion of the gene encoding the protein of interest, should be included as negative controls. It should be noted that activated CD4+ T cells from different TCR-tg mouse lines may differ significantly in their FSC/SSC characteristics and fluorescent background, so results for each TCR-tg cell type should be considered separately. For example, we have noticed that OT-II cells remain relatively small after immunization with OVA/alum, while SMARTA cells rapidly divide and become highly activated with increased FSC/SSC and autofluorescence.
The optimal amount of dye for CTV staining should be tailored to the respective TCR-tg cell line as well. OT-II cells tolerate CTV very well (Baumjohann et al., 2013a; Baumjohann et al., 2011), whereas SMARTA cells might require lower concentrations of fluorescent dyes to avoid toxicity (Choi et al., 2011).
One other issue that might contribute to different results between TCR-tg cell lines might result from the amounts of available antigen and the context of antigenic challenge. Protein immunogens decline in abundance over time, whereas viral infections with replicating virus such as LCMV typically increase rapidly after infection, providing stronger and prolonged stimuli.
GFP and other fluorescent proteins are incompatible with the fixation/permeabilization buffers used in the Foxp3 transcription factor staining set. However, they are preserved with PFA/saponin-based fixation/permeabilization procedures and may be compatible with other commercially available buffer combinations as well. This should be kept in mind when using fluorescent reporter mice together with intracellular staining. Investigators are encouraged to determine fixation/permeabilization conditions that fit their own needs.
Reducing the size of data files not only saves data storage capacity, but also results in much faster software/computer speed during analyses. We usually first acquire 5×104 to 1×105 events of all cells (to get an overview of background staining and cell types for gating purposes) and then append only gated TCR-tg cells. In this case, gates set up for acquisition should not be as narrow as those used during final analyses of the data.
Acknowledgement
This work was supported by the National Multiple Sclerosis Society (D.B.), the UCSF Program for Breakthrough Biomedical Research, funded in part by the Sandler Foundation (D.B.), and the NIH (P01 HL107202, R01 HL109102).
References
- Barnden MJ, Allison J, Heath WR, Carbone FR. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol. 1998;76:34–40. doi: 10.1046/j.1440-1711.1998.00709.x. [DOI] [PubMed] [Google Scholar]
- Baumjohann D, Kageyama R, Clingan JM, Morar MM, Patel S, de Kouchkovsky D, Bannard O, Bluestone JA, Matloubian M, Ansel KM, Jeker LT. The microRNA cluster miR-17 approximately 92 promotes TFH cell differentiation and represses subset-inappropriate gene expression. Nat Immunol. 2013a;14:840–848. doi: 10.1038/ni.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumjohann D, Okada T, Ansel KM. Cutting Edge: Distinct Waves of BCL6 Expression during T Follicular Helper Cell Development. J Immunol. 2011;187:2089–2092. doi: 10.4049/jimmunol.1101393. [DOI] [PubMed] [Google Scholar]
- Baumjohann D, Preite S, Reboldi A, Ronchi F, Ansel KM, Lanzavecchia A, Sallusto F. Persistent antigen and germinal center B cells sustain T follicular helper cell responses and phenotype. Immunity. 2013b;38:596–605. doi: 10.1016/j.immuni.2012.11.020. [DOI] [PubMed] [Google Scholar]
- Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, Lao C, Crotty S. ICOS Receptor Instructs T Follicular Helper Cell versus Effector Cell Differentiation via Induction of the Transcriptional Repressor Bcl6. Immunity. 2011;34:932–946. doi: 10.1016/j.immuni.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotty S. Follicular Helper CD4 T Cells (T(FH)) Annu Rev Immunol. 2011;29:621–663. doi: 10.1146/annurev-immunol-031210-101400. [DOI] [PubMed] [Google Scholar]
- Deenick EK, Chan A, Ma CS, Gatto D, Schwartzberg PL, Brink R, Tangye SG. Follicular helper T cell differentiation requires continuous antigen presentation that is independent of unique B cell signaling. Immunity. 2010;33:241–253. doi: 10.1016/j.immuni.2010.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goenka R, Barnett LG, Silver JS, O’Neill PJ, Hunter CA, Cancro MP, Laufer TM. Cutting edge: dendritic cell-restricted antigen presentation initiates the follicular helper T cell program but cannot complete ultimate effector differentiation. J Immunol. 2011;187:1091–1095. doi: 10.4049/jimmunol.1100853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamala T. Hock immunization: a humane alternative to mouse footpad injections. J Immunol Methods. 2007;328:204–214. doi: 10.1016/j.jim.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerfoot SM, Yaari G, Patel JR, Johnson KL, Gonzalez DG, Kleinstein SH, Haberman AM. Germinal center B cell and T follicular helper cell development initiates in the interfollicular zone. Immunity. 2011;34:947–960. doi: 10.1016/j.immuni.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitano M, Moriyama S, Ando Y, Hikida M, Mori Y, Kurosaki T, Okada T. Bcl6 protein expression shapes pre-germinal center B cell dynamics and follicular helper T cell heterogeneity. Immunity. 2011;34:961–972. doi: 10.1016/j.immuni.2011.03.025. [DOI] [PubMed] [Google Scholar]
- Linterman MA, Rigby RJ, Wong R, Silva D, Withers D, Anderson G, Verma NK, Brink R, Hutloff A, Goodnow CC, Vinuesa CG. Roquin differentiates the specialized functions of duplicated T cell costimulatory receptor genes CD28 and ICOS. Immunity. 2009;30:228–241. doi: 10.1016/j.immuni.2008.12.015. [DOI] [PubMed] [Google Scholar]
- Lyons AB, Parish CR. Determination of lymphocyte division by flow cytometry. J Immunol Methods. 1994;171:131–137. doi: 10.1016/0022-1759(94)90236-4. [DOI] [PubMed] [Google Scholar]
- Ma CS, Deenick EK, Batten M, Tangye SG. The origins, function, and regulation of T follicular helper cells. J Exp Med. 2012;209:1241–1253. doi: 10.1084/jem.20120994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi H, Liu D, Ma W, Wang Y, Yan H. Bcl-6 controlled T polarization and memory: the known unknowns. Curr Opin Immunol. 2014;28C:34–41. doi: 10.1016/j.coi.2014.01.016. [DOI] [PubMed] [Google Scholar]
- Quah BJ, Parish CR. New and improved methods for measuring lymphocyte proliferation in vitro and in vivo using CFSE-like fluorescent dyes. J Immunol Methods. 2012;379:1–14. doi: 10.1016/j.jim.2012.02.012. [DOI] [PubMed] [Google Scholar]
- Shahinian A, Pfeffer K, Lee KP, Kundig TM, Kishihara K, Wakeham A, Kawai K, Ohashi PS, Thompson CB, Mak TW. Differential T cell costimulatory requirements in CD28-deficient mice. Science. 1993;261:609–612. doi: 10.1126/science.7688139. [DOI] [PubMed] [Google Scholar]
- Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol. 2012;30:429–457. doi: 10.1146/annurev-immunol-020711-075032. [DOI] [PubMed] [Google Scholar]
- Vinuesa CG, Cyster JG. How T cells earn the follicular rite of passage. Immunity. 2011;35:671–680. doi: 10.1016/j.immuni.2011.11.001. [DOI] [PubMed] [Google Scholar]
- Walker LS, Gulbranson-Judge A, Flynn S, Brocker T, Raykundalia C, Goodall M, Forster R, Lipp M, Lane P. Compromised OX40 function in CD28-deficient mice is linked with failure to develop CXC chemokine receptor 5-positive CD4 cells and germinal centers. J Exp Med. 1999;190:1115–1122. doi: 10.1084/jem.190.8.1115. [DOI] [PMC free article] [PubMed] [Google Scholar]