

Abstract
Studies from tumor cells suggest that tumor suppressor AIP1 inhibits epithelial-mesenchymal transition (EMT). However, the role of AIP1 in the tumor microenvironment has not been examined. We show that a global or vascular endothelial cell (EC)-specific deletion of the AIP1 gene in mice augments tumor growth and metastasis in melanoma and breast cancer models. AIP1-deficient vascular environment not only enhances tumor neovascularization and increases pre-metastatic niche formation, but also secrets tumor EMT-promoting factors. These effects from AIP1 loss are associated with increased VEGFR2 signaling in the vascular EC and could be abrogated by systemic administration of VEGFR2 kinase inhibitors. Mechanistically, AIP1 blocks VEGFR2-dependent signaling by directly binding to the phosphotyrosine residues within the activation loop of VEGFR2. Our data reveal that AIP1, by inhibiting VEGFR2-dependent signaling in tumor niche, suppresses tumor EMT switch, tumor angiogenesis and tumor pre-metastatic niche formation to limit tumor growth and metastasis.
Keywords: AIP1/DAB2IP, tumor microenvironment, tumor niche, pre-metastatic niche, metastasis, VEGFR2
INTRODUCTION
In the absence of metastasis cancer is largely a treatable disease. Thus, early diagnosis of patients who will develop metastasis could reduce the mortality and morbidity associated with this disease. The development of metastasis depends on genetic and/or epigenetic alterations that are intrinsic to cancer cells, and extrinsic factors provided by the tumor microenvironment (niche). Metastasis is the spread of cancer cells from a primary site to distant, secondary host organs and requires a number of distinct steps including: epithelial-mesenchymal transition (EMT) of neoplastic epithelial cells for invasive growth, invasion into adjacent tissue, entering the systemic circulation (intravasation), extravasation into surrounding tissue parenchyma, and finally proliferation from microscopic growth (micrometastasis) into macroscopic secondary tumors (1–4). Pre-metastatic niche can also be formed at distant tissues before tumor cells arrive (5). Multiple cell types have been shown to promote tumor metastasis, including vascular endothelial cells (EC), tumor-associated macrophages (TAMs), and bone marrow-derived cells (BMDCs) (1, 6, 7). These cells are firstly activated by factors secreted by tumor cells (5, 8), subsequently promoting tumor EMT, tumor angiogenesis, tumor pre-metastatic niche formation and metastasis. Therefore, the tumor cells and the host cells are interactive and form a positive feedback loop. However, the underlying molecular mechanisms regulating the tumor cell-tumor niche interactions are not completely understood.
AIP1 (ASK1-interacting protein-1; also named as DAB2-interacting protein DAB2IP) is a recently identified signaling scaffolding protein. AIP1/DAB2IP has been shown to be downregulated in a variety of human cancer tissues including prostate cancer and breast cancer (9, 10). Two genome-wide association studies of aggressive prostate cancer suggest AIP1 as a putative prostate tumor suppressor gene (11). Further studies have shown that AIP1 downregulation in cancer cells is primarily through epigenetic regulation of the AIP1 promoter. Specifically, AIP1 expression in cancer cells is suppressed by the polycomb-group protein histone-lysine N-methyltransferase EZH2 (9, 12), which is consistently elevated in invasive breast and prostate carcinoma compared with normal breast and prostate epithelia, respectively (13). Importantly, AIP1 is a major EZH2 target and silencing of AIP1 is a key mechanism by EZH2 triggers tumor metastasis in mouse prostate cancer models (12).
The function of AIP1 in tumor cells has been assessed by in vitro proliferation and EMT assays, and by in vivo tumor progression and metastasis analyses in mouse models. AIP1 contains multiple signaling domains including the N-terminal pleckstrin homology (PH) domain for membrane targeting, the PKC-conserved region 2 (C2) domain for ASK1 interaction to induce apoptosis, the Ras-GAP domain for inhibition of Ras (therefore it has been considered as a novel member of RAS-GAP family protein), the C-terminal period-like domain for inhibition of transcriptional factor NF-κB, and the proline-rich for inhibition of PI3K-Akt survival pathway (14–17). By gain-of-function and loss-of-function approaches, we and others have shown that AIP1 inhibits tumor growth, EMT and metastasis by inhibiting Ras, PI3K/Akt, GSK-3/β-catenin and NF-κB pathways (12, 16, 17). Moreover, it has been reported that the inhibitory activity of AIP1 on NF-κB, but not its Ras-GAP activity, is critical for its suppressor effect on EMT in cancer models (12). Recent data also suggest that mutant p53 in cancer cells, by binding to AIP1 in the cytoplasm, enhances NF-κB activation to increase tumor metastasis (18).
However, the role of AIP1 in tumor niche has not been explored. AIP1-KO mice exhibit enhanced inflammation and pathological angiogenesis (15, 19). Given that inflammation and angiogenesis are required for tumor growth and metastasis, in the present study we determined the role of AIP1 in tumor microenvironment in regulating tumor growth and metastasis using various mouse breast cancer models. Our data suggest that AIP1 in vascular EC represses tumor metastasis by modulating not only tumor angiogenesis, but also tumor-associated pre-metastatic niche formation and tumor cell EMT phenotype.
MATERIALS AND METHODS
Animals
All animal studies were approved by the Institutional Animal Care and Use Committee of Yale University. Littermates of WT (AIP1lox/lox) and global AIP1-KO (AIP1lox/lox:β-actin-Cre) (15), littermates of WT (AIP1lox/lox) and the AIP1-ecKO (AIP1lox/lox:VE-cad-Cre) (20, 21) were used for experiments. All mice have been backcrossed to C57BL/6 for 12th generations.
Cells and cell lines
Primary mouse lung microvessel ECs (MLECs) were isolated and were routinely grown in M199 supplemental with 20% fetal bovine serum (FBS) and endothelial cell growth supplement (ECGS) (Corning, 356006) at 37 °C and 5% CO2 as previously reported (15). The E0771 mouse breast cancer cell line was from CH3 BioSystems (catalog #940001). B16 melanoma cell lines, 168FRAN and 4T1 breast cancer cell lines, and Lewis Lung Carcinoma (LLC) cell line were sourced from the American Tissue Culture Collection (ATCC). All cell lines were validated by STR profiling.
Mouse tumor model and in vivo evaluation of lung metastases and treatments(22)
1×106 mouse cancer cells (American Type Culture Collection, Manassas, VA) were injected subcutaneously in the right rear flank of mice in a 50μl volume of Dulbecco’s Modified Essential Medium solution. Tumor dimension (length, width and depth) was measured by electronic caliper coupled to computer that converted to tumor volume. Lung metastases were established in mice by injecting 1×105 B16 melanoma cells in 100 μl of Hank’s Balanced Salt Solution (Life Technologies), without Ca++ or Mg++, into the tail vein. After 10 to 42 days, the mice were sacrificed according to procedures approved by Yale’s Institutional Animal Care and Use Committee, and the primary tumor and draining lymph nodes were analyzed by histology.
Administration of VEGFR2 kinase inhibitor and VEGFR2-neutralizing antibodies in mouse tumor models
VEGFR2 kinase Inhibitor I (Cat# 676480) was obtained from Calbiochem. It is a highly selective, cell-permeable, reversible, and ATP-competitive indolin-2-one class of receptor tyrosine kinase (RTK) inhibitor (IC50 = 70 nM) for mouse vascular endothelial growth factor receptor 2 (VEGF-R2; KDR/Flk-1). It does not inhibit PDGF, EGF, and IGF-1 RTK activities (IC50 > 100 μM). For VEGFR2 kinase inhibitor experiments, tumor-bearing mice were subjected to intravenous injection of 100 μl of either DMSO or kinase inhibitor every other day. VEGFR2-neutralizing antibody DC101 was produced from hybridoma cells that were obtained from the America Type Culture Collection (ATCC), using the BD CELLine 1000 system (BD Biosciences) following the manufacturer’s instructions. DC101 and isotype control IgG1 (40 mg/kg) was injected intravenously every other day.
Evans blue permeability assay
Evans Blue Dye (100 μl of a 1% solution in 0.9% NaCl; Sigma-Aldrich) was injected into the retro-orbital plexus of anesthetized tumor-bearing mice. 30 minutes after the injection, mice were sacrificed and perfused with PBS through the left ventricle to clear the dye from the vascular volume. Tissues (e.g., lung) were harvested, dried in 60°C overnight, and weighed before Evans blue extraction using 1 ml formamide at 55°C for 16 h. Evans blue content was quantified by reading at 630 nm in a spectrophotometer as described previously (15, 23).
Statistical Analysis
Data are represented as mean±SEM. Comparisons between two groups were by paired t-test, between more than two groups by one-way ANOVA followed by Bonferroni’s post-hoc test or by two-way ANOVA using Prism 4.0 software (GraphPad). P values were two-tailed and values <0.05 were considered to indicate statistical significance.
Other methods
Expanded methods, including Histology, Immunoblotting and Immunofluorescence analysis, Gene expression in the tissues, Cytokine assays, transwell co-culture assay, are available in the Supplement Methods.
RESULTS
AIP1 deletion in vascular endothelial cells augments tumor growth and metastasis in syngeneic and orthotopic tumor models
To determine the role of AIP1 in the tumor microenvironment on tumor growth and metastasis, we compared tumor growth and metastasis in wild type (WT) and global AIP1-deficient mice (AIP1-KO) using subcutaneous and orthotopic implantation models. In a B16 melanoma model, AIP1-KO significantly enhanced tumor growth (Fig. 1A). Pigmented B16 melanoma nodules were readily detected and quantitated microscopically. Metastasis incidences (% of mice with detectable pigmented tumor nodules) to multiple organs including lymph node, lung, liver, spleen and bone marrow at 4 weeks were significantly increased in AIP1-KO mice (Fig. 1B). Detailed examination of lung tissues indicated that lung metastasis was detected in AIP1-KO, but not in WT mice, as early as 3 weeks following implantation (Fig. 1C with quantification in Fig. 1D). Both the number of tumor nodules and % of tumor areas in the lung were higher in AIP1-KO mice (Fig. 1E with quantification in Fig. 1F). AIP1-KO mice also exhibited enhanced tumor metastasis in lymph nodes (LN) (Fig. 1G with quantification in Fig. 1H).
Figure 1. Augmented growth and metastasis of B16F10 melanoma in AIP1-KO mice.

B16 melanoma cells were injected subcutaneously (SQ) into WT and AIP1-KO mice. A. Tumor volumes are presented. n=10 mice per group. **, p<0.01. B. Tumor metastases to distant tissues were examined microscopically at week 2–4 post-implantation. Metastasis incidence (% of mice with detectable pigmented tumor nodules) was quantified. Data for week 4 are shown. n=10 mice per group. **, p<0.01. C. Representative images of tumor metastasis at week 3 and 4 in lung are shown. Arrowheads indicate the tumor nodules. D. Lung metastasis nodules at 4 weeks were quantified. n=10 mice per group. *, p<0.05 and **, p<0.01. E. Lung metastasis was examined by H&E staining. F. Metastatic tumor areas in lung were quantified from 5 random fields per lung and n=5 mice per group. *, p<0.05 and **, p<0.01. Scale bar: 50 μm. G. Regional inguinal lymph nodes were excised from WT and KO at basal or 2–4 weeks after tumor implantation; note that the pigmented melanoma metastases were detected at 3 and 4 weeks in AIP1-KO but only visible at 4 weeks in WT mice. H. LN metastases were quantified. n=10 mice per group. **, p<0.01.
AIP1 is highly abundant in vascular endothelial cells (EC) but not in myeloid cells (14, 15, 19, 24). To determined relative AIP1 expression levels in tumor ECs, tumor-associated macrophages (TAMs) and tumor cells, we implanted GFP-expressing B16 melanoma subcutaneously or E0771 breast cancer cells orthotopically into WT mice. Tumor tissues were harvested at 2 weeks post-implantation and CD31+ ECs, CD68+ TAMs and GFP+ tumor cells were collected by FACS sorting. AIP1 mRNA levels were determined by qRT-PCR. Results indicated that AIP1 was highly expressed in tumor ECs, but only weakly in tumor cells and was not expressed in TAMs (Supplementary Fig. S1).
To define the host cell type in which AIP1 regulates tumor behavior, we applied the tumor models to the EC-specific AIP1-knockout mice (AIP1-ecKO; AIP1lox/lox:VE-cad-Cre) mice. We first validated the deletion efficiency of AIP1 by qRT-PCR in freshly isolated lung ECs from AIP1lox/lox (WT), AIP1-KO and AIP1-ecKO mice. AIP1 gene was completely deleted in ECs isolated from both AIP1-KO and AIP1-ecKO, and AIP1 gene deletion was comparable between AIP1-KO and AIP1-ecKO (Fig. 2A). In an orthotopic breast cancer model with E0771 (a mouse metastatic breast cancer cell lines derived from C57BL/6 mice), AIP1-ecKO exhibited enhanced tumor growth and lung metastasis (Fig. 2B–F), comparable to the global AIP1-KO (Supplementary Fig. S2). These results suggest that AIP1 in vascular EC suppresses tumor growth and metastasis in the mouse models.
Figure 2. AIP1 in vascular endothelium inhibits tumor growth and metastasis in an orthotopic model.
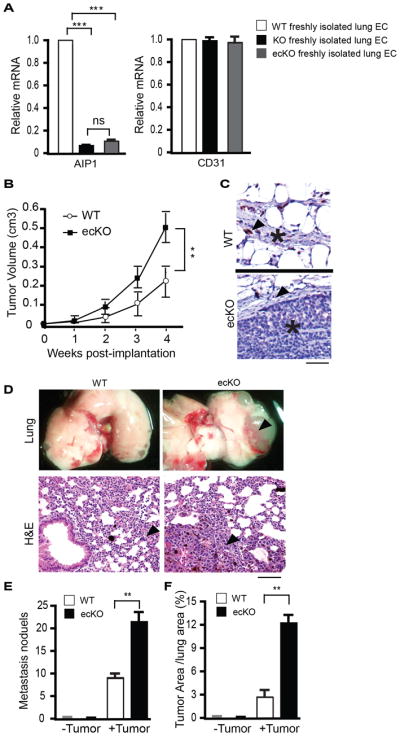
A. AIP1 deletion efficiency was verified by qRT-PCR in freshly isolated lung ECs from AIP1lox/lox (WT), AIP1-KO and AIP1-ecKO mice. n=3 mice in each group, ***, p<0.001. B–F. Mouse E0771 breast cancer cells (1×106) were injected orthotopically into the fourth mammary gland of WT and AIP1-ecKO female mice. (B) Tumor size was measured weekly. (C) A representative images of immunostaining with anti-AIP1 antibody for breast tumors from WT and AIP1-ecKO mice. Scale bar: 50 μm. Arrows indicate capillary endothelium and tumor cells are indicated (*). (D) Tumor metastasis at week 4 in lung was examined by whole tissue photography (top) and by H&E staining (bottom). Arrowheads indicate the tumor mass. Scale bar for H&E: 50 μm. (E–F) Metastasis nodules and % of tumor areas in lung were quantified. n=10 mice per group. **, p<0.01.
AIP1 deletion has no effect on tumor metastasis after a direct intravenous injection of tumor cells
We next used AIP1-ecKO mice to define the mechanism by which AIP1 in host limits tumor metastasis. Tumor metastasis involves multiple steps, including tumor cell EMT switch, intravasation into the lymphatic system or/and the bloodstream, extravasation from and survival/colonization in the host distance tissues. Primary tumor cells, by secreting soluble factors, also activate pre-metastasis niche at a distance tissue prior to their arrival to the tissue (illustrated in Supplementary Fig. S3A). To determine how AIP1 in host controls tumor metastasis, we directly injected tumor cells (1×105) into bloodstream intravenously. Tumor cells directly arrived at the lung tissue where they extravasated and colonized, therefore metastasized without processes of EMT switch, intravasation and pre-niche formation. We observed no differences between WT and AIP-ecKO mice in lung metastasis as determined by tumor nodule number and lung weight (Supplementary Fig. S3B–C). We further injected GFP+ B16 melanoma cells with fewer numbers (1×104), and examined extravasation of tumor cells to the lung. We did not observe significant differences between WT and AIP1-ecKO mice in the tumor cell retention (at 30 min post-injection) or extravasation (at 4 h post-injection) (Supplementary Fig. S3D–E). These data suggest that the AIP1 deletion in vascular EC affects tumor metastasis at a step(s) prior to extravasation, i.e., tumor cell EMT phenotypic change, tumor cell intravasation to lymphatic or/and blood systems, and the formation of pre-metastasis niche.
Tumor angiogenesis but not tumor lymphangiogenesis is increased in AIP1-ecKO mice with enhanced VEGFR2 signaling
Tumor angiogenesis and tumor lymphangiogenesis are required for tumor growth and metastasis (25). We examined tumor angiogenesis and lymphangiogenesis by immunostaining with anti-CD31 and anti-LYVE-1, respectively. Tumor vessel density and area were significantly enhanced in the AIP1-ecKO mice (Fig. 3A–B), consistent with our results in inflammatory angiogenesis models (15). Tumor implantation induced both angiogenesis and lymphangiogenesis in the sentinel lymph nodes (LN) at 2 weeks post-implantation prior to tumor cell metastasis, suggesting that LN angiogenesis and lymphangiogenesis were induced by tumor-derived angiogenic factors such as VEGFs (26). Numbers of blood vessels (Fig. 3C–D), but not lymphatic vessels (Supplementary Fig. S4), was significantly enhanced in the AIP1-ecKO mice at the peri-tumoral region and distant lymph nodes. We measured expression and activation of VEGFR2 and VEGFR3, two potent angiogenic and lymphangiogenic receptors (27). Total and phosphorylation of VEGFR2, but of VEGFR3 or total VEGFR1, were significantly increased in tumor tissues from AIP1-ecKO mice (Fig. 3E). Primary tumor induced expression and phosphorylation of VEGFR2 and VEGFR3 in distant lymph nodes at 2 weeks post-implantation. However, AIP1-ecKO exhibited enhanced expression of VEGFR2 (but not VEGFR3 or VEGFR1) (Fig. 3F–G). These results suggest that increased tumor angiogenesis, but not lymphangiogenesis, in AIP1-ecKO mice contributes to the enhanced tumor metastasis in these mice.
Figure 3. Tumor angiogenesis is increased in AIP1-ecKO mice.

Mouse breast cancer cells (1×106) were injected orthotopically into the fourth mammary gland of WT and AIP1-ecKO mice, and tumor along with surrounding mammary tissue were harvested at 2 weeks. A–B. Tumor angiogenesis was determined by immunohistochemistry and whole-mount staining with anti-CD31, and intratumoral vessel density and vessel areas were quantified from 5 sections per tumor and n=10 mice for each strain, *, p<0.05. Scale bar: 50 μm. C–D. Tumor-induced angiogenesis in lymph nodes was determined by immunostaining with anti-CD31. Scale bar: 100 μm. CD31+ areas were quantified. n=10 mice per group. **, p<0.01. E–G. Phospho-VEGFR2 (pY1054/1059) and total VEGFR2, VEGFR3, AIP1 and β-actin proteins in tumor (E) and lymph node (F–G) were determined by immunoblotting with respective antibodies. Representative blots are shown from 1 of 4 mice for each strain with fold changes compared to WT-tumor are indicated below the blots. Further quantifications of VEGFR2 and p-VEGFR2 are shown in G, n=6 mice per group. **, p<0.01.
Tumor-induced pre-metastatic niche formation is enhanced in AIP1-ecKO mice with enhanced VEGFR2 signaling
Formation of pre-metastatic niche at a distant tissue is also important for tumor metastasis which can be regulated by tumor- and bone marrow-derived cytokines(5, 8). It has been proposed that upon activation by tumor-secreted factors (e.g., VEGF, IL-6 and SDF-1), vascular endothelium at the distant tissue can be activated followed by infiltration of immune cells and bone marrow cells (5, 8). We first measured lung endothelial barrier function by “Miles assay” using Evans-blue dye. AIP1-ecKO exhibited higher permeability at day 14 post-tumor implantation in an orthotopic breast cancer model (Fig. 4A). We also observed upregulation of several pre-metastatic niche markers, including fibronectin, LOX, c-Kit, VEGFR1, CD34 and VEGFR2 in the lung as detected by qRT-PCR. These genes were more greatly upregulated in tumor-bearing AIP1-ecKO lungs (Fig. 5B). However, genes involved in lymphangiogenesis (e.g., VEGFR3) and arteriogenesis (e.g., Notch1 receptor and Dll4 ligand) were not altered. We then determined if gene expression of pre-niche markers preceded tumor cell arrival at the lung. To this end, GFP+ tumor cells were applied to orthotopic cancer model, and gene expression of tumor markers (GFP and S100A2) and pre-metastatic niche markers (fibronectin and VEGFR2) in the lung tissues were measured by qRT-PCR. Clearly, upregulation of pre-metastatic niche and VEGFR2 in the lung occurred on day 10 post-implantation before tumor-specific markers could be detected on day 17–21 (Supplementary Fig. S5). Immunoblotting confirmed that total fibronectin, phosphorylated and total VEGFR2 were upregulated in the lung tissue at day 14 post-implantation before detection of GFP+ tumor cells on day 21 (Fig. 4C). Very few activated VEGFR2+ cells were found in resting WT and AIP1-ecKO lung tissues, and tumor implantation induced a recruitment of activated VEGFR2+ cells to the lung as visualized by phospho-VEGFR2 staining. Importantly, tumor-induced activation of VEGFR2+ cells in the lung was augmented in the AIP1-ecKO mice (Fig. 4D–E). Of note, some VEGFR2+CD31− cells were detected and these cells are likely the bone marrow-derived endothelial cell progenitors (EPCs) that have not been fully differentiated into CD31+ mature EC. Interestingly, circulating VEGFR2 EPCs have been associated poor prognosis in advanced cancer patient (28). Consistently, we have observed stronger VEGFR2 signaling in bone marrow EC progenitors from AIP1-KO mice (15, 19, 24).
Figure 4. Tumor-induced pre-metastatic niche formation is augmented in AIP1-ecKO.

Mouse breast cancer cells (1×106) were injected orthotopically into the fourth mammary gland of WT and AIP1-ecKO mice. A. Lung permeability was measured on day 10 post-implantation by Miles assay with Evan’s blue dye. The dye was extracted from the lung tissue and was measured at 630 nm. Data are presented as OD/lung dry weight (g) and are mean ± SEM from n=3 for each strain. *, p<0.05. B. Lung tissues were harvested at day 10 and gene expression of pre-metastasis niche markers were analyzed by qRT-PCR with normalization by GAPDH. Fold increases in AIP1-ecKO vs WT are shown. Data are mean ± SEM, n=6 for each strain. C. GFP-expressing mouse breast cancer cells were injected orthotopically into the fourth mammary gland of WT and AIP1-ecKO mice. Lung tissues were harvested at indicated times. Fibronectin, p-VEGFR2, VEGFR2, tumor markers S100A2 and GFP, and β-actin were determined by immunoblotting with respective antibodies. n=6 mice per group at each time point. Fold changes compared to the basal level in WT are shown. D–E. Recruitment of active VEGFR2+ cells to the lung tissue were increased in AIP1-ecKO mice but was diminished by VEGFR2-specific inhibitor. Some mice of WT and AIP1-KO mice with orthotopic breast tumor were treated by intravenous injection of VEGFR2 TKI (0.4 mM) every other day. p-VEGFR2 (pY1054/1059) and CD31 in the lung at day 14 were determined by immunostaining. v: vessel. Representative images are shown from 5 sections per tumor. Scale bar: 50 μm. Arrows indicate p-VEGFR2+ cells which are quantified in E as % of total cells. Data are mean ± SEM, n=6 per group. *, p<0.05. F–G. WT and AIP1-KO mice with orthotopic breast tumor were treated by intravenous injection of VEGFR2 TKI (0.4 mM) every other day. p-VEGFR2 (pY1054/1059) and CD31 in the lung at day 14 were determined by immunostaining. p-VEGFR2+ cells were quantified in G as % of total cells. Data are mean ± SEM, n=6 per group. *, p<0.05. H. Lung tissues were harvested at day 14, and p-VEGFR2 and total VEGFR2 were determined by immunoblotting. Fold changes are shown by taking WT control as 1.0. Representative blots are shown from 1 of 3 mice for each strain. I. Tumor volumes at 3 weeks were measured. J. Lung metastasis nodules at 3 weeks were quantified. Data are mean ± SEM, n=6 mice per group. *, p<0.05; **, p<0.01.
Figure 5. AIP1 binds to the VEGFR2 kinase activation loop and inhibits VEGFR2-mediated signaling.

A. VEGFR2 mutants. Vector control (VC), VEGFR2-WT, K868M (a kinase-dead mutant), Y1175F, Y1175/Y801F or Y1054/1059F were transfected into 293T. Autophosphorylations of VEGFR2 at pY1054/1059 and pY1175 were determined by immunoblotting with phosphor-specific antibodies. B. AIP1 binds to VEGFR2 at pY1054/1059. Flag-tagged AIP1-N containing the C2 domain was transfected into 293T cells with vector control (-), VEGFR2-WT, K868M, Y1175F or Y1054/1059F. AIP1-VEGFR2 association was determined by co-immunoprecipitation with anti-VEGFR2 followed by immunoblotting with anti-Flag. C. The co-immunoprecipitation assay of AIP1 and VEGFR2-WT was performed in the presence of 0.3 mM of pY1054/1059-peptide, C2-peptide. A non-phosphor-Y1054/1059-peptide and C2KA-peptide were used as controls. D–E. Effects of the AIP1 binding on VEGFR2 signaling. AIP1-deficient mouse lung microvessel ECs (MLECs) were reconstituted by lentivirus with vector control (VC), AIP1-WT or AIP1-KA1. Cells were serum-starved for 12 h followed by treatment with VEGF (10 ng/ml for 15 min). p-VEGFR2 and p-Akt were determined by immunoblotting with phosphor-specific antibodies and ratios of p-VEGFR2/VEGFR2 and p-Akt/Akt are quantified in E. Data are mean ± SEM from three independent experiments. *, p<0.05. F. AIP1-KO MLECs were reconstituted by lentivirus with AIP1-WT or AIP1-KA1. Cells were subjected to transwell migration assays. Data are mean ± SEM from triplicates and three independent experiments. **, p<0.01.
We next determined if enhanced VEGFR2 signaling in vascular EC contributes to augmented tumor angiogenesis and pre-metastatic niche formation. To this end, a VEGFR2 tyrosine kinase inhibitor (TKI)-1 was applied to the orthotopic breast cancer model as we recently described (21). VEGFR2 TKI significantly attenuated tumor-induced recruitment of VEGFR2+ cells to the lung as detected by immunostaining (Fig. 4F–G). VEGFR2 TKI also significantly attenuated activation of VEGFR2 signaling in the lung as determined by immunoblotting (Fig. 4H). Of note, VEGFR2 TKI appeared to upregulate AIP1 expression in WT mice, suggesting a potential AIP1-VEGFR2 auto-regulatory loop. Functionally, VEGFR2 TKI blocked tumor angiogenesis (Supplementary Fig. S6A–B) and pre-metastatic niche formation (Supplementary Fig. S6C), suggesting that observed changes in tumor angiogenesis and pre-metastatic formation are dependent on vascular VEGFR2 signaling. Importantly, tumor growth and lung tumor metastasis in AIP1-ecKO mice were blunted by VEGFR2 TKI (Fig. 4I–J). VEGFR2-dependent tumor growth and metastasis in AIP1-ecKO mice were also blocked by DC101, a monoclonal antibody specifically binds and neutralizes VEGFR2 (29) (Supplementary Fig. S6D–F).
AIP1 binds to the VEGFR2 kinase activation loop and inhibits VEGFR2-mediated signaling
The results above established a critical role AIP1-regulated VEGFR2 in tumor metastasis, and we subsequently took biochemical approaches to define the mechanism by which AIP1 regulates VEGFR2 signaling. We have previously shown that AIP1 binds to an active form of VEGFR2, and a mutant AIP1 (AIP1-KA1) with mutations at the first lysine cluster within its C2 domain (K104–106 to A) loses the VEGFR2-binding activity (15). A certain type of C2, similar to the SH2 domain, associates with phosphotyrosines (30, 31). These observations prompted us to determine if AIP1 via its C2 binds to phosphotyrosine residues on VEGFR2. To this end, we mutated the tyrosine residues of VEGFR2 known to be autophosphorylated in response to VEGF-A, including Y1054/1059, Y1175 and Y1121 (32, 33). 293T cells were co-transfected with VEGFR2 mutants along with AIP1-N, the N-terminal half of AIP1 constitutively associating with VEGFR2 upon co-expression (15). Overexpression of VEGFR2-WT induces autophosphorylation at pY1054/1059 (within the kinase activation loop), pY1175 (critical for activation of PI3K and PLC-γ activation) as well as other phosphotyrosines involved in other signaling (Fig. 5A). The kinase-dead mutant (VEGFR2-K868M) and the Y1054/1059F mutant abolished VEGFR2 autophosphorylation at both pY1054/1059 and pY1157. In contrast, mutations of other Y sites (Y1175, Y1214 or Y801) had no effects on pY1054/1059. Association of VEGFR2 and AIP1 was determined by a co-immunoprecipitation assay. As shown in Fig. 5B, AIP1 associated with VEGFR2-WT and Y1175F mutant. However, the kinase-dead form and the Y1054/1059F mutant failed to associate with AIP1. These results suggest that an active form of VEGFR2 with phosphorylations at Y1054/1059 is required for the AIP1 binding. To further determine the role of VEGFR2 pY1054/1059 in the AIP1 binding, we performed a peptide competition assay using 13-mer peptides flanking the pY1054/1059 of VEGFR2 (RDIY1054KDPDY1059VRKG) in two forms: non-phosphorylated (Y1054/1059-peptide) and phosphorylated peptides (pY1054/1059-peptide). These peptides were initially designed for the generation and affinity-purification of the pY1054/1059-specific antibody (32). We performed the co-immunoprecipitation assay in the presence of 0.3 mM of either Y1054/1059-peptide or pY1054/1059-peptide. Addition of the pY1054/1059-peptide, but not the Y1054/1059-peptide, disrupted the VEGFR2-AIP1 association (Fig. 5C), suggesting that phosphorylation of VEGFR2 at Y1054/1059 is critical for the AIP1 binding. We also designed a C2-peptide derived from the lysine-cluster 1 of AIP1 C2 domain (IEAKDLPAK104KK106YLCELCL where the lysine residues are underlined; C2-KA peptide contains K to A mutations). Interestingly, the C2-peptide, but not the C2-KA peptide, also disrupted the VEGFR2-AIP1 complex, further supporting a critical role of the lysine-cluster within the AIP1 C2 domain in the VEGFR2 binding. Taken together, these data suggest that the C2 lysine-cluster on AIP1 and the phosphotyrosine residues 1054/1059 on VEGFR2 are responsible for their interactions.
We then determined effects of the AIP1 binding on VEGFR2 activation and signaling. AIP1-deficient mouse lung microvessel ECs (MLECs) (15, 34) were reconstituted by lentivirus expressing AIP1-WT or AIP1-KA1 (a mutant defective in the VEGFR2 binding). VEGF-induced phosphorylation of VEGFR2 and downstream signaling Akt were blunted by re-expression of AIP1-WT but not AIP1-KA1 (Fig. 5D–E). VEGF-stimulated migratory activities of MLECs were also inhibited by AIP1-WT but not by AIP1-KA1 in a transwell assay (Fig. 5F). Taken together, our data support that AIP1 binds to the VEGFR2 kinase activation loop and inhibits VEGFR2-mediated signaling in the vasculature.
AIP1-deficient EC enhances tumor cell EMT and invasion
When we examined tumor cell EMT and invasive markers (E-cadherin, vimentin, β-catenin, snail, NF-κB) in the orthotopic breast cancer model, we observed downregulation of E-cadherin and upregulation of vimentin, β-catenin, snail and NF-κB (p-p65) in the tumors from AIP1-ecKO mice compared to WT. These changes were more profound at the peripheral regions of the tumor (Supplementary Fig. S7). As an intrinsic EMT suppressor, AIP1 is downregulated via epigenetic regulation by EZH2 in tumor cells (12, 35). We examined if AIP1 expression in tumor cells is altered upon transplantation into AIP1-ecKO microenvironment. AIP1 expression in tumor cells was dramatically downregulated within the peripheral regions in the AIP1-ecKO host (Fig. 6A), correlating with increased EMT markers in this region. Interestingly, EZH2, a component of PRC-2 responsible for the epigenetic suppression of AIP1 expression, showed a reciprocal expression pattern of the AIP1 expression (Fig. 6A). The changes of E-cadherin, NF-κB (p-p65), AIP1 and EZH2 were also verified by immunoblotting (Fig. 6B–C). These data suggest that the host microenvironment modulates AIP1 expression and EMT phenotype of tumor cells.
Figure 6. Tumor EMT is enhanced in AIP1-ecKO mice.

Mouse breast cancer cells (1×106) were injected orthotopically into the fourth mammary gland of WT and AIP1-ecKO mice, and tumor along with surrounding mammary tissue were harvested at 2 weeks. A. AIP1 and EZH2 were detected by immunohistochemistry with respective antibodies as indicated. Tumor and surrounding fat fad are labeled. Scale bar: 50 μm. Representative images are shown from 5 sections per tumor and n=10 mice for each strain. B–C. Tumor EMT markers (E-cadherin, p-p65, AIP1 and EZH2) were determined by Immunoblotting with respective antibodies. Representative blots are shown in B from 2 of 10 mice for each strain with densitometry quantifications are shown below the blots. Quantifications are shown in C. n=10 mice per group. *, p<0.05.
AIP1-VEGFR2 signaling in vascular EC mediates tumor cell EMT switch
To determine the mechanism by which endothelial AIP1-VEGFR2 axis regulates tumor cell EMT and metastasis, we first performed a modified transwell migration assay to measure permeability of WT and AIP1-KO MLECs to tumor cells by counting tumor cells migration cross the confluent MLEC monolayer (Fig. 7A). Despite that invasive 4T1 migrated cross WT MLECs at higher rate compared to less malignant 168FRAN cells, AIP1-KO MLECs were highly permeable to both 168FRAN and 4T1 (Fig. 7B). We reasoned that AIP1-KO MLECs, by secreting factors, regulated tumor cell EMT phenotypic changes. To test this idea, we employed a transwell system to co-culture 168FARN with WT or AIP1-KO MLECs without direct cell-cell contacts (Fig. 7C). Co-culture of AIP1-KO MLECs (but not WT MLECs) strongly induced EMT responses in 168FARN tumor cells (Fig. 7D–E). However, re-expression of AIP1-WT or an ASK1-binding defective mutant KA2, but not the VEGFR2-binding defective mutant KA1, diminished the effects of AIP1-KO MLECs on tumor cell EMT. These results suggest that vascular AIP1-VEGFR2, but not AIP1-ASK1 signaling, regulates co-cultured tumor cell EMT. Moreover, treatment of MLECs with VEGFR2 kinase inhibitor (TKI) in the co-culture system blocked AIP1-KO MLEC-enhanced 168FRAN EMT responses (Fig. 7F). Of note, tumor cell EMT morphological change was accompanied by upregulation of EZH2 with a reciprocal reduction of AIP1, increase in NF-κB activity and NF-κB-dependent gene expression of metastasis-promoting factor IL-6 (Supplementary Fig. S8A–B). To test if downregulation of AIP1 is a critical step for tumor cell EMT, we examined AIP1 expression in breast cancer cell lines with various metastatic potential (168FARN, 4T07 and 4T1) (36). AIP1 expression was inversely correlated with EZH2 expression and breast cancer cell metastatic potentials in these cell lines (Supplementary Fig. S8C). Consistent with a low level of AIP1 protein and a high basal EMT in 4T1 cell, co-culture of 4T1 cell with AIP1-KO MLEC weakly enhanced its EMT (Supplementary Fig. S8D). These data suggest that the VEGFR2 signaling in vascular EC potently modulates an EZH2-AIP1-dependent EMT cascade in tumor cells.
Figure 7. AIP1-VEGFR2 signaling in vascular EC regulates tumor cell EMT program.

A–B. Endothelial permeability - tumor cell invasion assay. WT or AIP1-KO MLECs cells were cultured on the top of the transwell as monolayer for 2 days post-confluency. Tumor cells (168FARN or 4T1) were seeded at the top well of the Boyden Chamber and cells that transmigrated cross the endothelium layer were quantified at 12 h. Data presented in B are mean ± SEM from duplicates and three independent experiments. *, p<0.05; **, p<0.01. C. Diagram for the co-culture assay. MLECs were seeded at the top of the transwell. Tumor cells (e.g., breast cancer cell line 168FARN) were seeded at the bottom well of the Boyden Chamber. The two types of cells were grown without direct cell-to-cell contact and separated by a membrane with 0.4 μm pore size permeable for molecules but not for cells. D–E. Effect of MLECs on tumor cells. Co-culture of breast cancer cell line 168FARN with MLECs isolated from WT mice, AIP1-KO mice, AIP1-KO MLECs with a re-expression of AIP1, AIP1-KA1 or AIP1-KA2 by lentivirus. At 12 h, cell morphological analyses of co-cultured 168FARN cells were performed under light microscopy. Representative images are from 1 of 3 independent experiments (B). Quantifications of EMT (% of elongated spindle cells) are presented. Data are mean ± SEM from duplicates and three independent experiments. *, p<0.05. F. VEGFR2 tyrosine kinase inhibitor (TKI) diminished the effects of AIP1-KO MLECs on 168FRAN. Co-culture of breast cancer cell line 168FARN for 12 h with MLECs isolated from WT mice or AIP1-KO mice in the presence or absence of VEGFR2 TKI (20 μM). Tumor cell EMT based on morphological analyses were quantified. Data are mean ± SEM from duplicates and three independent experiments. *, p<0.05. G–H. TGFβ2 mediates MELC-enhanced tumor EMT responses. Co-culture of 168FARN with WT MLECs, AIP1-KO MLECs or AIP1-KO MLECs with TGFβ2 siRNA knockdown for 12 h. Phosphorylation of Smad2/3, expression of EZH2 and AIP1 in co-cultured 168FARN cells were determined by Western blot with respective antibodies. Fold changes are presented from 1 of 3 independent experiments, taking 168FARN alone as 1.0 (G). 168FARN cell morphological analyses were performed under light microscopy and quantifications of EMT are presented. Data are mean ± SEM from duplicates and three independent experiments. *, p<0.05. I. Models for the role of stromal AIP1-VEGFR2 signaling in regulation of tumor cell EMT and metastasis. In WT tumor niche, AIP1 inhibits VEGFR2 to repress tumor angiogenesis, pre-metastatic niche formation and tumor EMT switch. In AIP1-deficeint tumor niche, enhanced VEGFR2 signaling induces tumor angiogenesis and formation of pre-metastatic niche. Enhanced VEGFR2 activity in vascular EC also stimulates secretion of tumor EMT-promoting factors (e.g., TGFβ2), which directly augment tumor cell EMT phenotypic changes, including upregulation of EZH2 protein, reduction of AIP1 protein, increases in NF-κB activity and NF-κB-dependent IL-6 expression. Together, AIP1-deficeint niche promotes tumor invasion and metastasis.
To define the MLEC-derived EMT factors that mediate tumor cell EMT, we measured gene expression of a few known EMT factors in MLECs (37), and we found that TGFβ (TGFβ2 in particular), but not HGF, were highly upregulated in AIP1-KO MLEC (Supplementary Fig. S8E). Pre-treatment of MLECs with VEGFR2 TKI completely blunted TGFβ2 expression (Supplementary Fig. S8F), suggesting AIP1-VEGFR2 axis directly regulates TGFβ2 expression. We then determined the functional important of TGFβ2 in mediating the MLEC effect on tumor EMT responses. Co-culture of AIP1-KO MLECs induced phosphorylation of Smad2/3 and upregulation of EZH2 with a reciprocal reduction of AIP1 expression in 168FARN cells (Fig. 7G). Moreover, knockdown of TGFβ2 in AIP1-KO MLECs attenuated the EMT responses in 168FARN cells, including p-Smad2/3, upregulation of EZH2, downregulation of AIP1 and EMT morphological switch (Fig. 7G–H), supporting a role of MLEC-secreted TGFβ2 in mediating crosstalk between MELCs and tumor cells.
DISCUSSION
In the present study, by implanting tumor cells with the same genetic background into normal and AIP1-deficient (AIP1-ecKO) mice we have defined the role of AIP1 in tumor microenvironment in regulating tumor growth and metastasis. Loss of AIP1 in vascular endothelial cells is sufficient to augment tumor growth and metastasis in subcutaneous melanoma and orthotopic breast cancer models. AIP1 in the vascular EC limits tumor metastases appears to be at a step(s) prior to entrance into the bloodstream as a direct intravenously injection of tumor cells results in no differences in tumor metastasis between WT and AIP1-ecKO mice. Indeed, AIP1 deletion in the vascular EC enhances VEGFR2 signaling; this enhanced VEGFR2 not only regulates tumor angiogenesis and pre-metastatic niche formation in distant tissues, but also directly modulates tumor cell EMT phenotypic switch (Fig. 7I: Model for the role of AIP1 in tumor niche in regulating tumor EMT and metastasis). Mechanistically, we have defined a novel mechanism by which AIP1 blocks VEGFR2-dependent signaling, i.e., AIP1 through a lysine-cluster sequence within the C2 domain binds to the phosphotyrosine residues 1054/1059 within the activation loop of VEGFR2; the VEGFR2 activity in vascular EC is critical for gene expression of tumor EMT-promoting factors (e.g., TGFβ2), which directly augment tumor cell EMT phenotypic changes, including upregulation of EZH2 protein, reduction of AIP1 protein, increases in NF-κB activity and NF-κB-dependent IL-6 expression. It has been well established that EZH2 epigenetically regulates AIP1 expression in various tumor cells in vivo and in vitro, and EZH2-mediated AIP1 downregulation is critical for tumor cell EMT and metastasis (9, 12). Our data support that the upregulation of EZH2 with AIP1 reduction in tumor cells is a key mechanism by which tumor niche (vascular ECs) induce tumor EMT and metastasis. Consistently, AIP1-KO MLECs have less effect on the EMT responses of more malignant tumor cells with high EZH2/low AIP1 (e.g., 4T1 cells). Taken together, our data support that AIP1 in tumor microenvironment limits tumor metastasis by suppressing tumor cell EMT, tumor angiogenesis and tumor pre-metastatic niche formation.
It is well known that tumor-associated macrophages (TAMs) are associated with tumor EMT and metastasis (38). However, our studies indicate that AIP1 is abundantly expressed in tumor endothelial cells, but not in TAMs. Moreover, vascular EC-specific deletion of AIP1 augments tumor angiogenesis, growth and metastasis. Therefore, AIP1 in vascular endothelial cells limits tumor growth and metastasis. Our data support that the AIP1 suppresses VEGFR2 in vascular EC to limit tumor angiogenesis and formation of pre-metastatic niche formation as well as tumor cell EMT switch. VEGFR2 signaling is increased in the both primary tumor and lung tissues of AIP1-ecKO in response to subcutaneous and orthotopic tumor implantations. A VEGFR2 kinase inhibitor or a neutralizing antibody abrogates the augmented VEGFR2-dependent signaling, tumor angiogenesis and tumor metastasis in AIP1-ecKO mice. In vitro, AIP1-VEGFR2 signaling in vascular EC directly mediates tumor cell EMT switch, at least partly by regulating EMT factor TGFβ2. Taken together, we have uncovered a novel pathway - AIP1-VEGFR2-TGFβ in tumor niche and EZH2-AIP1-NF-κB in tumor cells- that mediates crosstalk between tumor niche and tumor cells (Fig. 7I).
AIP1 has been shown to mediate cytokine (e.g., TNF)-induced EC apoptosis (14–17). EC death and/or proliferation under inflammatory tumor microenvironment can have strong influence on tumor angiogenesis and hence tumor growth and on tumor metastasis (5, 8, 39). We have assessed EC apoptosis and proliferation in AIP1-ecKO mice. Specifically, we have performed immunostaining on sections of tumors and lungs from AIP1-KO animals for proliferation marker Ki-67 and apoptotic marker cleaved caspase-3. Our data indicate that AIP1-ecKO mice exhibit increased EC proliferation with decreased EC apoptosis in the primary tumor, but not in the lung tissues at two weeks post-implantation (Supplementary Fig. S9). These data suggest that increased EC proliferation with reduced EC apoptosis in AIP1-ecKO may contribute to augmented tumor angiogenesis that enhance growth and metastasis, at least in the primary tumor. We observe increased VEGFR2 signaling in both primary tumor and lung tissue, highlighting the important function of VEGFR2 signaling in primary tumor angiogenesis and pre-metastatic niche formation in distant tissues. This is further supported by the VEGFR2 inhibitor experiments presented in our study.
A significant mechanistic finding in the current study is that we have uncovered how AIP1 inhibits VEGFR2 signaling. VEGF primarily utilizes its receptor VEGFR2 (also Flk-1 or KDR) to induce angiogenic responses by activating a variety of signaling cascades including activation of phosphatidylinositol 3-kinase (PI3K)-Akt, phospholipase C-gamma (PLC-γ) and MAP kinase (40). Given the critical role of VEGFR2 signaling in angiogenesis, VEGFR2 activity/activation needs to be tightly regulated and fine-tuned. VEGFR2 activity is regulated by direct interactions with other proteins including its co-receptor neuropilins, adhesion molecule VE-cadherin and integrins. VEGFR2 could also be negatively regulated. E3 ubiquitin ligase c-Cbl and SCFβTRCP-dependent ubiquitination could induce VEGFR2 degradation (41–43). A high cell density-enhanced phosphatase (DEP-1) is associated with VEGFR2-VE-cadherin-catenin complex at cell-cell contacts and reduces VEGFR2 phosphorylation and ERK1/2 signaling (44). Therefore, increased VEGFR2 intracellular trafficking could prevent activated VEGFR2 from inactivation by phosphatases (45–47). We have previously identified several VEGFR2 modulators that directly regulate the formation, activation and stability of the VEGFR2 receptor signaling complex - two positive regulators Bmx (bone marrow tyrosine kinase located in X-chromosome, a non-receptor tyrosine kinase) and CCM3 (cerebral cavernous malformation-3 (CCM3), and two negative regulators AIP1 and endocytic adaptor protein Epsin (15, 21, 48, 49). Our current study has defined the intriguing mechanism by which AIP1 binds to active form of VEGFR2 and inhibits its signaling, i.e., AIP1 via its lysine-cluster sequence within the C2 domain directly binds to the phosphotyrosines (pY1054/1059) within the activation loop of VEGFR2. We have provided strong evidence to support our conclusion: The kinase-dead mutant (VEGFR2-K868M) and the Y1054/1059F mutant abolish both VEGFR2 autophosphorylation at pY1054/1059 and the AIP1 binding. In contrast, mutations of other tyrosine residues (Y1175, Y1214 or Y801) have no effects on VEGFR2 autophosphorylation at Y1054/1059 and the VEGFR2-AIP1 interactions. A phosphorylated pY1054/1059-peptide, but not a non-phosphorylated Y1054/1059-peptide, disrupts the VEGFR2-AIP1 association. Similarly, the C2-peptide derived from the lysine-cluster 1 of AIP1 C2 domain also disrupts the VEGFR2-AIP1 association. Based on the published crystal structure for the VEGFR2 kinase domain, the pY1054/1059 are exposed on surface and are accessible to protein interactions (50). Likewise, sequence alignment and structure-based homology modeling of AIP1 C2 domain suggests that the basic patches are surface exposed. Therefore it is possible that the VEGFR2-AIP1 interactions are mediated by electrostatic interactions. Given that AIP1 via its C2 domain directly binds to the autoactivation loop of VEGFR2, it is plausible that AIP1 directly inhibits the VEGFR2 kinase activity. We have previously shown that AIP1 can recruit PP2A phosphatase to ASK1 to dephosphorylate a negative phosphorserine-967 site, leading to ASK1 activation (51). Another possibility is that AIP1 recruits phosphatase(s) to VEGFR2 activation loop to terminate VEGFR2 signaling and this model needs to be further tested. Nevertheless, we have revealed a novel function of the AIP1-VEGFR2 complex. Besides the EC intrinsic function of AIP1-VEGFR2 signaling, AIP1-VEGFR2 in tumor niche (e.g., vascular EC) also regulates tumor cell EMT phenotype through a non-autonomous manner. Specifically, we show that the AIP1-VEGFR2 signaling in tumor niche cells, by regulating gene expression of EMT factors (e.g., TGFβ2), modulates EZH2-AIP-NF-κB-dependent EMT cascade in tumor cells (Fig. 7I).
Taken together, our results suggest that downregulation of AIP1 expression in tumor niche may be a potential biomarker for tumor malignancy, and stabilization or/and re-expression of AIP1 in tumor niche may provide a potential therapeutic strategy for the treatment of cancer metastasis.
Supplementary Material
Statement of Significance.
In this study, we demonstrate that AIP1 in tumor niche, inhibiting VEGFR2-dependent signaling, suppresses tumor epithelial-mesenchymal transition, tumor angiogenesis and tumor pre-metastatic niche formation to limit tumor growth and metastasis.
Acknowledgments
We thank Dr. Don Nguyen (Yale University School of Medicine) for insightful comments and helpful discussions. This work was supported in part by NIH grants HL109420 and HL115148 to WM; National Natural Science Foundation of China (No. 81272350 and No. 81472999), Natural Science Foundation of Guangdong (No. S2012010008908) and the University Talent Program of Guangzhou (No. 12A015G) to WJ; National Natural Science Foundation of China 81273097 and 81472998 to YH; National Natural Science Foundation of China 81371019 to YL.
Nonstandard abbreviations
- ASK1
apoptosis signal–regulating kinase 1
- AIP1
ASK1-interacting protein-1
- AIP1-KO
AIP1-knockout mice
- AIP1-ecKO
AIP1 endothelial-specific KO mice
- BMDC
bone marrow-derived cells
- BMT; BrdU
5-bromo-2′-deoxyuridine
- C2 domain
PKC-conserved region 2
- DAPI
4′,6-diamidino-2-phenylindole
- EC
endothelial cells
- EMT
epithelial-mesenchymal transition
- EZH2
enhancer of zeste homology-2
- HGF
hepatocyte growth factor
- MLEC
mouse lung microvessel EC
- PH
pleckstrin homology domain
- PRC2
polycomb-repressive complex 2
- siRNA
small interfering RNA
- SQ
subcutaneous
- TAMs
Tumor-associated macrophages
- TGFβ
transforming growth factor-β
- TUNEL
terminal deoxynucleotide transferase dUTP nick end labeling
- VEGF
vascular endothelial cell growth factor
- VEGFR2
VEGF receptor-2
- VEGFR3
VEGF receptor-3
Footnotes
Conflict of interest disclosure: None.
AUTHOR CONTRIBUTIONS
The following people performed research and analyzed data: W.J., Y.H., M.Y., Y.L., H.Z., T.B. and W.M. W.J. and W.M. designed and performed research, analyzed data. W.M. wrote the paper.
References
- 1.Fidler IJ, Poste G. The “seed and soil” hypothesis revisited. Lancet Oncol. 2008;9(8):808. doi: 10.1016/S1470-2045(08)70201-8. [DOI] [PubMed] [Google Scholar]
- 2.Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9(4):274–84. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- 3.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19(11):1423–37. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147(2):275–92. doi: 10.1016/j.cell.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438(7069):820–7. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 7.Kerbel RS, Benezra R, Lyden DC, Hattori K, Heissig B, Nolan DJ, Mittal V, Shaked Y, Dias S, Bertolini F, et al. Endothelial progenitor cells are cellular hubs essential for neoangiogenesis of certain aggressive adenocarcinomas and metastatic transition but not adenomas. Proc Natl Acad Sci U S A. 2008;105(34):E54. doi: 10.1073/pnas.0804876105. author reply E5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peinado H, Rafii S, Lyden D. Inflammation joins the “niche”. Cancer Cell. 2008;14(5):347–9. doi: 10.1016/j.ccr.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen H, Toyooka S, Gazdar AF, Hsieh JT. Epigenetic regulation of a novel tumor suppressor gene (hDAB2IP) in prostate cancer cell lines. J Biol Chem. 2003;278(5):3121–30. doi: 10.1074/jbc.M208230200. [DOI] [PubMed] [Google Scholar]
- 10.Dote H, Toyooka S, Tsukuda K, Yano M, Ouchida M, Doihara H, Suzuki M, Chen H, Hsieh JT, Gazdar AF, et al. Aberrant promoter methylation in human DAB2 interactive protein (hDAB2IP) gene in breast cancer. Clin Cancer Res. 2004;10(6):2082–9. doi: 10.1158/1078-0432.ccr-03-0236. [DOI] [PubMed] [Google Scholar]
- 11.Duggan D, Zheng SL, Knowlton M, Benitez D, Dimitrov L, Wiklund F, Robbins C, Isaacs SD, Cheng Y, Li G, et al. Two genome-wide association studies of aggressive prostate cancer implicate putative prostate tumor suppressor gene DAB2IP. J Natl Cancer Inst. 2007;99(24):1836–44. doi: 10.1093/jnci/djm250. [DOI] [PubMed] [Google Scholar]
- 12.Min J, Zaslavsky A, Fedele G, McLaughlin SK, Reczek EE, De Raedt T, Guney I, Strochlic DE, Macconaill LE, Beroukhim R, et al. An oncogene-tumor suppressor cascade drives metastatic prostate cancer by coordinately activating Ras and nuclear factor-kappaB. Nat Med. 2010;16(3):286–94. doi: 10.1038/nm.2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, Ghosh D, Sewalt RG, Otte AP, Hayes DF, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(20):11606–11. doi: 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang R, He X, Liu W, Lu M, Hsieh JT, Min W. AIP1 mediates TNF-alpha-induced ASK1 activation by facilitating dissociation of ASK1 from its inhibitor 14–3–3. J Clin Invest. 2003;111(12):1933–43. doi: 10.1172/JCI17790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang H, He Y, Dai S, Xu Z, Luo Y, Wan T, Luo D, Jones D, Tang S, Chen H, et al. AIP1 functions as an endogenous inhibitor of VEGFR2-mediated signaling and inflammatory angiogenesis in mice. J Clin Invest. 2008;118(12):3904–16. doi: 10.1172/JCI36168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xie D, Gore C, Liu J, Pong RC, Mason R, Hao G, Long M, Kabbani W, Yu L, Zhang H, et al. Role of DAB2IP in modulating epithelial-to-mesenchymal transition and prostate cancer metastasis. Proc Natl Acad Sci U S A. 2010;107(6):2485–90. doi: 10.1073/pnas.0908133107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xie D, Gore C, Zhou J, Pong RC, Zhang H, Yu L, Vessella RL, Min W, Hsieh JT. DAB2IP coordinates both PI3K-Akt and ASK1 pathways for cell survival and apoptosis. Proc Natl Acad Sci U S A. 2009;106(47):19878–83. doi: 10.1073/pnas.0908458106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Di Minin G, Bellazzo A, Dal Ferro M, Chiaruttini G, Nuzzo S, Bicciato S, Piazza S, Rami D, Bulla R, Sommaggio R, et al. Mutant p53 Reprograms TNF Signaling in Cancer Cells through Interaction with the Tumor Suppressor DAB2IP. Mol Cell. 2014;56(5):617–29. doi: 10.1016/j.molcel.2014.10.013. [DOI] [PubMed] [Google Scholar]
- 19.Zhou HJ, Chen X, Huang Q, Liu R, Zhang H, Wang Y, Jin Y, Liang X, Lu L, Xu Z, et al. AIP1 Mediates Vascular Endothelial Cell Growth Factor Receptor-3-Dependent Angiogenic and Lymphangiogenic Responses. Arterioscler Thromb Vasc Biol. 2014;34(3):603–15. doi: 10.1161/ATVBAHA.113.303053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alva JA, Zovein AC, Monvoisin A, Murphy T, Salazar A, Harvey NL, Carmeliet P, Iruela-Arispe ML. VE-Cadherin-Cre-recombinase transgenic mouse: a tool for lineage analysis and gene deletion in endothelial cells. Dev Dyn. 2006;235(3):759–67. doi: 10.1002/dvdy.20643. [DOI] [PubMed] [Google Scholar]
- 21.Pasula S, Cai X, Dong Y, Messa M, McManus J, Chang B, Liu X, Zhu H, Mansat RS, Yoon SJ, et al. Endothelial epsin deficiency decreases tumor growth by enhancing VEGF signaling. J Clin Invest. 2012;122(12):4424–38. doi: 10.1172/JCI64537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blezinger P, Wang J, Gondo M, Quezada A, Mehrens D, French M, Singhal A, Sullivan S, Rolland A, Ralston R, et al. Systemic inhibition of tumor growth and tumor metastases by intramuscular administration of the endostatin gene. Nat Biotechnol. 1999;17(499224288):343–8. doi: 10.1038/7895. [DOI] [PubMed] [Google Scholar]
- 23.Miles AA, Miles EM. Vascular reactions to histamine, histamine-liberator and leukotaxine in the skin of guinea-pigs. J Physiol. 1952;118(2):228–57. doi: 10.1113/jphysiol.1952.sp004789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang Q, Qin L, Dai S, Zhang H, Pasula S, Zhou H, Chen H, Min W. AIP1 suppresses atherosclerosis by limiting hyperlipidemia-induced inflammation and vascular endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2013;33(4):795–804. doi: 10.1161/ATVBAHA.113.301220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kerbel RS. Tumor angiogenesis. N Engl J Med. 2008;358(19):2039–49. doi: 10.1056/NEJMra0706596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hirakawa S, Kodama S, Kunstfeld R, Kajiya K, Brown LF, Detmar M. VEGF-A induces tumor and sentinel lymph node lymphangiogenesis and promotes lymphatic metastasis. J Exp Med. 2005;201(7):1089–99. doi: 10.1084/jem.20041896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nature Medicine. 2003;9(6):669–76. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 28.Massard C, Borget I, Le Deley MC, Taylor M, Gomez-Roca C, Soria JC, Farace F. Prognostic value of circulating VEGFR2+ bone marrow-derived progenitor cells in patients with advanced cancer. Eur J Cancer. 2012;48(9):1354–62. doi: 10.1016/j.ejca.2012.01.021. [DOI] [PubMed] [Google Scholar]
- 29.Lee YJ, Karl DL, Maduekwe UN, Rothrock C, Ryeom S, D’Amore PA, Yoon SS. Differential effects of VEGFR-1 and VEGFR-2 inhibition on tumor metastases based on host organ environment. Cancer Res. 2010;70(21):8357–67. doi: 10.1158/0008-5472.CAN-10-1138. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Benes CH, Wu N, Elia AE, Dharia T, Cantley LC, Soltoff SP. The C2 domain of PKCdelta is a phosphotyrosine binding domain. Cell. 2005;121(2):271–80. doi: 10.1016/j.cell.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 31.Sondermann H, Kuriyan J. C2 can do it, too. Cell. 2005;121(2):158–60. doi: 10.1016/j.cell.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 32.Zhang R, Xu Y, Ekman N, Wu Z, Wu J, Alitalo K, Min W. Etk/Bmx transactivates vascular endothelial growth factor 2 and recruits phosphatidylinositol 3-kinase to mediate the tumor necrosis factor-induced angiogenic pathway. J Biol Chem. 2003;278(51):51267–76. doi: 10.1074/jbc.M310678200. [DOI] [PubMed] [Google Scholar]
- 33.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling - in control of vascular function. Nat Rev Mol Cell Biol. 2006;7(5):359–71. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 34.Luo D, He Y, Zhang H, Yu L, Chen H, Xu Z, Tang S, Urano F, Min W. AIP1 is critical in transducing IRE1-mediated endoplasmic reticulum stress response. J Biol Chem. 2008 doi: 10.1074/jbc.M710557200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen H, Tu SW, Hsieh JT. Down-regulation of human DAB2IP gene expression mediated by polycomb Ezh2 complex and histone deacetylase in prostate cancer. J Biol Chem. 2005;280(23):22437–44. doi: 10.1074/jbc.M501379200. [DOI] [PubMed] [Google Scholar]
- 36.Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449(7163):682–8. doi: 10.1038/nature06174. [DOI] [PubMed] [Google Scholar]
- 37.Wang J, Xiao L, Luo CH, Zhou H, Zeng L, Zhong J, Tang Y, Zhao XH, Zhao M, Zhang Y. CD44v6 promotes beta-catenin and TGF-beta expression, inducing aggression in ovarian cancer cells. Molecular medicine reports. 2015;11(5):3505–10. doi: 10.3892/mmr.2015.3145. [DOI] [PubMed] [Google Scholar]
- 38.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Franses JW, Drosu NC, Gibson WJ, Chitalia VC, Edelman ER. Dysfunctional endothelial cells directly stimulate cancer inflammation and metastasis. Int J Cancer. 2013;133(6):1334–44. doi: 10.1002/ijc.28146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koch S, Tugues S, Li X, Gualandi L, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Biochem J. 2011;437(2):169–83. doi: 10.1042/BJ20110301. [DOI] [PubMed] [Google Scholar]
- 41.Duval M, Bedard-Goulet S, Delisle C, Gratton JP. Vascular endothelial growth factor-dependent down-regulation of Flk-1/KDR involves Cbl-mediated ubiquitination. Consequences on nitric oxide production from endothelial cells. J Biol Chem. 2003;278(22):20091–7. doi: 10.1074/jbc.M301410200. [DOI] [PubMed] [Google Scholar]
- 42.Singh AJ, Meyer RD, Navruzbekov G, Shelke R, Duan L, Band H, Leeman SE, Rahimi N. A critical role for the E3-ligase activity of c-Cbl in VEGFR-2-mediated PLCgamma1 activation and angiogenesis. Proc Natl Acad Sci U S A. 2007;104(13):5413–8. doi: 10.1073/pnas.0700809104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shaik S, Nucera C, Inuzuka H, Gao D, Garnaas M, Frechette G, Harris L, Wan L, Fukushima H, Husain A, et al. SCF(beta-TRCP) suppresses angiogenesis and thyroid cancer cell migration by promoting ubiquitination and destruction of VEGF receptor 2. J Exp Med. 2012;209(7):1289–307. doi: 10.1084/jem.20112446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lampugnani MG, Orsenigo F, Gagliani MC, Tacchetti C, Dejana E. Vascular endothelial cadherin controls VEGFR-2 internalization and signaling from intracellular compartments. J Cell Biol. 2006;174(4):593–604. doi: 10.1083/jcb.200602080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lanahan AA, Hermans K, Claes F, Kerley-Hamilton JS, Zhuang ZW, Giordano FJ, Carmeliet P, Simons M. VEGF receptor 2 endocytic trafficking regulates arterial morphogenesis. Dev Cell. 2010;18(5):713–24. doi: 10.1016/j.devcel.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sawamiphak S, Seidel S, Essmann CL, Wilkinson GA, Pitulescu ME, Acker T, Acker-Palmer A. Ephrin-B2 regulates VEGFR2 function in developmental and tumour angiogenesis. Nature. 2010;465(7297):487–91. doi: 10.1038/nature08995. [DOI] [PubMed] [Google Scholar]
- 47.Simons M. An inside view: VEGF receptor trafficking and signaling. Physiology (Bethesda) 2012;27(4):213–22. doi: 10.1152/physiol.00016.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.He Y, Luo Y, Tang S, Rajantie I, Salven P, Heil M, Zhang R, Luo D, Li X, Chi H, et al. Critical function of Bmx/Etk in ischemia-mediated arteriogenesis and angiogenesis. J Clin Invest. 2006;116(9):2344–55. doi: 10.1172/JCI28123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.He Y, Zhang H, Yu L, Gunel M, Boggon TJ, Chen H, Min W. Stabilization of VEGFR2 signaling by cerebral cavernous malformation 3 is critical for vascular development. Sci Signal. 2010;3(116):ra26. doi: 10.1126/scisignal.2000722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hilberg F, Roth GJ, Krssak M, Kautschitsch S, Sommergruber W, Tontsch-Grunt U, Garin-Chesa P, Bader G, Zoephel A, Quant J, et al. BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008;68(12):4774–82. doi: 10.1158/0008-5472.CAN-07-6307. [DOI] [PubMed] [Google Scholar]
- 51.Min W, Lin Y, Tang S, Yu L, Zhang H, Wan T, Luhn T, Fu H, Chen H. AIP1 recruits phosphatase PP2A to ASK1 in tumor necrosis factor-induced ASK1-JNK activation. Circ Res. 2008;102(7):840–8. doi: 10.1161/CIRCRESAHA.107.168153. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.