

Summary
Presenilin-1 (PS1) deletion or Alzheimer’s Disease (AD)-linked mutations disrupt lysosomal acidification and proteolysis, which inhibits autophagy. Here, we establish that this phenotype stems from impaired glycosylation and instability of vATPase V0a1 subunit causing deficient lysosomal vATPase assembly and function. We further demonstrate that elevated lysosomal pH in PS1KO cells induces abnormal Ca2+ efflux from lysosomes mediated by TRPML1 and elevates cytosolic Ca2+. In WT cells, blocking vATPase activity or knockdown of either PS1 or the V0a1 subunit of vATPase reproduces all of these abnormalities. Normalizing lysosomal pH in PS1KO cells using acidic nanoparticles restores normal lysosomal proteolysis, autophagy, and Ca2+ homeostasis, but correcting lysosomal Ca2+ deficits alone neither re-acidifies lysosomes nor reverses proteolytic and autophagic deficits. Our results indicate that vATPase deficiency in PS1 loss of function states causes lysosomal/autophagy deficits and contributes to abnormal cellular Ca2+ homeostasis, thus linking two AD-related pathogenic processes through a common molecular mechanism.
Introduction
PS1 is primarily known to be a catalytic component of γ-secretase complex, which carries out cleavage of amyloid precursor protein yielding Abeta peptides, which in various forms have been implicated in AD pathogenesis (Chavez-Gutierrez et al., 2012; De Strooper and Annaert, 2010; Selkoe and Wolfe, 2007; Steiner and Haass, 2000). Loss of function mutations of PS1 that cause early onset AD alter the proportion of Abeta 42 and 40 peptides (Chavez-Gutierrez et al., 2012), which is considered critical to their neurotoxicity and a major contributor to AD pathogenesis. Increasing evidence, however, indicates that PS1 serves additional γ-secretase-independent roles in wnt signaling (Kang et al., 1999), ER Ca2+ regulation (Shilling et al., 2014; Tu et al., 2006) as well as in lysosomal function and autophagy (Coen et al., 2012; Lee et al., 2010; Wilson et al., 2004; Wolfe et al., 2013). PS1 dysfunction is therefore likely to contribute in multiple ways to AD pathogenesis by altering Abeta clearance, production, and oligomerization (Nixon, 2007) and corrupting diverse lysosomal functions via the massive build-up of incompletely degraded autophagic substrates in lysosomes, a characteristic feature of the neuritic dystrophy in AD (Nixon and Yang, 2012). Lysosomal dysfunction in neurons is closely tied to neurodegeneration and cell death mechanisms (Cesen et al., 2012; Nixon and Yang, 2012). Growing genetic and biochemical evidence implicates dysfunction of the endosomal-lysosomal and autophagic lysosomal pathways in the pathogenesis of a number of neurodegenerative disorders, including AD, Parkinson’s disease and ALS (Frakes et al., 2014; Ghavami et al., 2014; Menzies et al., 2015; Nixon, 2013). The therapeutic efficacy of autophagy/lysosome modulation in animal models of these disorders (Butler et al., 2011; Sun et al., 2008; Yang et al., 2011) further underscores the significance of lysosomal impairments to disease pathogenesis.
It has been shown that loss of PS1 function in multiple cell types disrupts lysosome acidification, leading to markedly impaired autophagy (Avrahami et al., 2013; Dobrowolski et al., 2012; Lee et al., 2010; Torres et al., 2012; Wolfe et al., 2013). Controversial reports from two groups (Coen et al., 2012; Zhang et al., 2012a), however, proposed that PS1 plays no role in lysosomal pH, lysosomal proteolysis, or vATPase subunit maturation, and that the V0a1 subunit specifically implicated in our studies, is not involved in lysosomal acidification.
Here we directly demonstrate deficiencies in lysosomal vATPase content and function in lysosomes of PS1KO cells, and establish the role of failed vATPase V0a1 subunit maturation in PS1-dependent lysosomal acidification failure, leading to defective autophagy and abnormal efflux of lysosomal Ca2+. We further show that the secondary abnormalities in lysosomal Ca2+ efflux are caused by a pH-modulated activation of the low H+ sensitive endolysosomal Ca2+ channel, transient receptor potential cation channel mucolipin subfamily member 1 (TRPML1) (Raychowdhury et al., 2004), and are responsible for substantial elevations of cytosolic Ca2+ in PS1-deficient cells. We present further evidence that the V0a1 subunit is essential for lysosome acidification in neurons and non-neural cells and that inhibiting vATPase function in WT cells recapitulates the PS1KO phenotype. Restoring normal lysosomal pH using lysosomal-targeted acidic nanoparticles reverses these abnormalities, but the correction of lysosomal calcium deficits alone does not, thus implying that lysosomal pH modulates TRPML1 activation and Ca2+ efflux as a secondary consequence of vATPase deficiency in PS1KO cells. Our studies, therefore, link two γ-secretase-independent effects of PS1, each having pathogenic significance in AD, and demonstrate that vATPase deficiency is the common underlying mechanism.
Results
PS1-dependent regulation of lysosomal pH is essential for lysosomal Ca2+ homeostasis mediated by TRPML1
To investigate the relationship between defects in lysosomal Ca2+ homeostasis and lysosomal acidification in PS1KO cells, we measured lysosomal Ca2+ levels and observed lowered lysosomal Ca2+ levels, as previously reported (Coen et al., 2012) and concomitantly elevated cytosolic Ca2+ levels (Fig 1A). We confirmed lysosomal Ca2+ reduction by assaying Ca2+ release after pretreatment with ionomycin to release all internal, non-lysosomal calcium stores (Lloyd-Evans et al., 2008), followed by glycyl-L-phenylalanine-beta-naphthylamide (GPN) to induce lysosomal rupture causing the release of lysosomal Ca2+ (Penny et al., 2014). Interestingly, GPN elevated cytosolic Ca2+ due to lysosomal Ca2+ release after ionomycin treatment in WT, but had minimal effect on PS1KO cells (Fig 1B). To confirm in PS1KO cells that reduced lysosomal Ca2+ release after GPN is due to deficient lysosomal calcium stores, we treated WT cells with U18666a, a control agent used to deplete lysosomal Ca2+ stores without affecting vATPase function (Lloyd-Evans et al., 2008). As expected, U18666A treated WT cells exhibited a marked reduction in the rise of cytosolic Ca2+ due to lysosomal Ca2+ release following addition of ionomycin and then GPN. Importantly, the magnitude of the reduction of lysosomal Ca2+ release was similar to that observed in untreated PS1KO cells (Fig 1B), again showing that levels of lysosomal Ca2+ are markedly lowered in PS1KO cells.
Figure 1. TRMPL1 mediates lysosomal Ca2+ efflux in PS1 KO cells.
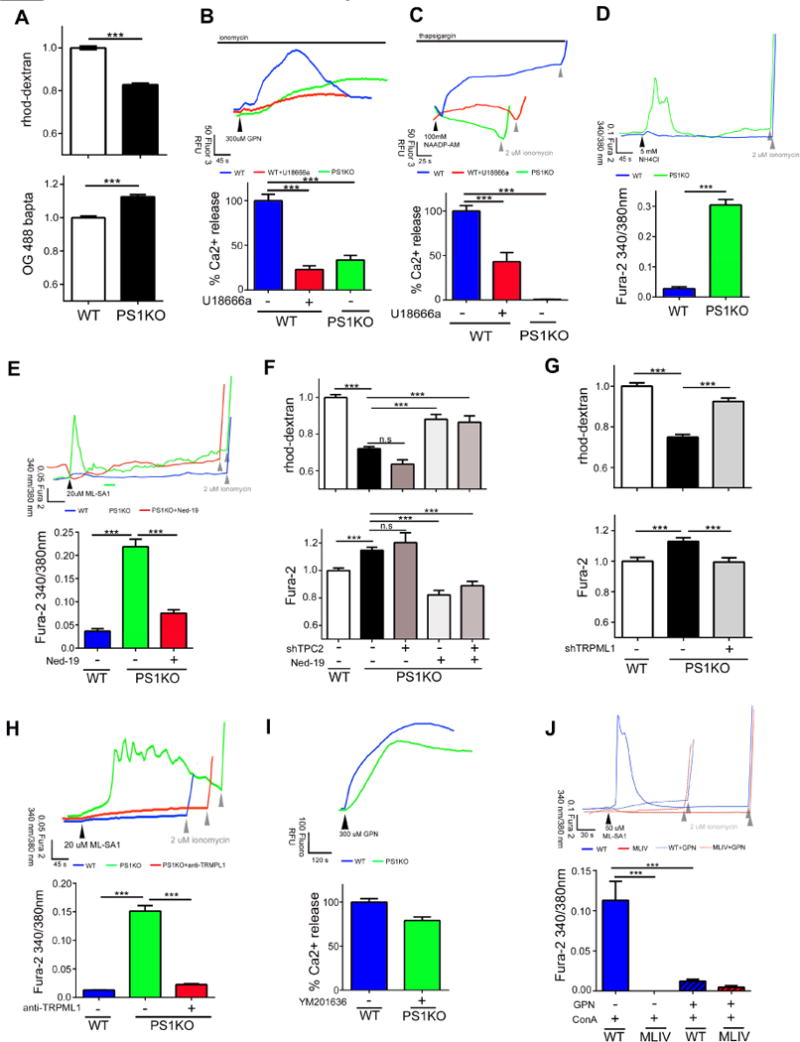
(A) Lysosomal Ca2+ is reduced (WT: n=336; PS1KO n=338) and cytosolic Ca2+ is elevated in PS1KO cells (WT: n=148; PS1KO n=150). (B) Cells were treated with 5μM ionomycin followed by GPN (300 μM) to measure lysosomal Ca2+ levels Less Ca2+ is released from PS1KO (n=18) cells WT cells treated for 24 hrs with 2μg/ml U18666a (n=33) release less lysosomal Ca2+. (C) Compared to WT (n=67), PS1KO cells (n=93) released minimal Ca2+ after 1 μM thapsigargin followed by NAADP-AM (100 nM) (arrowhead). Ca2+ release was significantly lowered in WT cells treated with U18666a (n=25). (D) Addition of 5mM NH4Cl increased cytosolic Ca2+ in PS1KO (n=64) but not WT cells (n=125). (E) 20μM ML-SA1 (black arrowhead) elevated cytosolic Ca2+ levels in PS1KO (n=151) but not WT cells (n=206) and this elevation was diminished by Ned-19 (0.5μM, 24hrs; n=167) (Gray arrowheads indicate ionomycin addition). (F) 72 hr shRNA TPC2KD had no discernible effect on lysosomal or cytosolic Ca2+ levels in PS1KO cells (n>90). 0.5μM Ned-19 (24hrs) restored lysosomal and cytosolic Ca2+ levels even when TPC2 was knocked down (G) 72hr knockdown of TRPML1 significantly elevated lysosomal Ca2+ and decreased cytosolic Ca2+ in PS1KO cells (H) Anti-TRMPL1 antibody (16h, 5μg/ml) inhibited ML-SA1 mediated lysosomal Ca2+ release in PS1KO cells (n=60). (I) Lysosomal Ca2+ levels determined using GPN are almost normalized in PS1KO cells after 10μM YM201636 for 1hr via reduced Ca2+ flux through TRPML1 (n=100). (J) ConA followed by ML-SA1 induced a rise in cytosolic Ca2+ in WT (n=33) but not MLIV fibroblasts (n=29) imaged in a Ca2+ free buffer Osmotic lysis of lysosomes with GPN prior to ML-SA1 abolished Ca2+ release in WT (n=11) and MLIV cells (n=17). ***p<0.0001. Error bars: ±S.E.M
Lysosomal Ca2+ channels, including TPC1/2, are NAADP dependent and sensitive to lysosomal pH. At normal lysosomal pH, elevated cytosolic NAADP transiently binds the channel, triggering lysosomal Ca2+ release; but at elevated lysosomal pH, TPC2 channels are blocked because NAADP cannot dissociate to re-trigger additional Ca2+ release (Pitt et al., 2010). We investigated the effects of NAADP-AM, after clamping the ER store with thapsigargin. Untreated WT cells showed an increase in cytosolic Ca2+ after NAADP-AM addition (Fig 1C) and pre-treatment with U18666A reduced the rise in cytosolic Ca2+, reflecting the loss of Ca2+ from the lysosome (Fig 1B, C). By contrast, PS1KO cells showed no NAADP-dependent Ca2+ release (Fig 1C) despite the presence of a remaining, albeit diminished, lysosomal Ca2+ pool (Fig 1A, B). Given that elevated lysosomal pH blocks TPC channels, the lack of response to NAADP-AM in PS1KO cells provided further evidence for a lysosomal pH defect and implied a different cause for Ca2+ leak. Due to a lack of NAADP mediated lysosomal Ca2+ release in PS1KO cells, we investigated the cause of the reduced lysosomal Ca2+ levels by focusing on another endolysosomal Ca2+ permeant channel reported to be activated in its endogenous state by elevated lysosomal pH, namely TRPML1 (Raychowdhury et al., 2004).
To analyze the lysosomal pH dependence of TRPML1 activity in PS1KO cells, we first altered lysosomal proton content with NH4+, which alkalinizes the lysosome only at high concentrations, thereby inducing Ca2+ leak and emptying lysosomal Ca2+ stores (Christensen et al., 2002). We hypothesized that a weak lysosomal alkalinization stimulus (5mM, < 2min), which does not affect lysosomal pH in WT cells (data not shown), would trigger Ca2+ leak more readily in the elevated lysosomal pH in PS1KO cells. As expected, this condition triggered a significant Ca2+ leak from PS1KO compared to WT cells (Fig 1D). These data indicated that PS1KO cells more readily leak Ca2+ with the addition of small amounts of an alkalinizing agent and suggested that other pH sensitive channels, like TRPML1, might become hyper-active at elevated pH. To investigate this hypothesis, we treated cells with the TRPML1 agonist ML-SA1 (Grimm et al., 2010). Although a low dose (20 μM) of ML-SA1 induced only a minimal elevation in cytosolic Ca2+ in WT cells (Fig. 1E), cytosolic Ca2+ rose markedly in PS1KO cells (Fig 1E).
While TPC2 and TRPML1 are functionally distinct (Yamaguchi et al., 2011), they are both activated by phosphoinositols (Dong et al., 2010; Jha et al., 2014) suggesting that they could both be inhibited by similar ligands. Ned-19, an NAADP analog, inhibits activation of NAADP-dependent lysosomal Ca2+ channels preventing Ca2+ efflux (Naylor et al., 2009). Surprisingly, Ned-19 pre-treatment of PS1KO cells inhibited TRPML1 and cytosolic Ca2+ elevation induced by ML-SA1 was now corrected back to WT levels in the Ned-19 treated PS1KO cells (Fig 1E).
As Ned-19 has been previously known to inhibit only lysosomal NAADP-dependent Ca2+ channels, we investigated the possibility that Ned-19 inhibits channels in addition to TPC2 by knocking down TPC2 in PS1KO cells. Knockdown of TPC2 had no effect on either lysosomal or cytosolic Ca2+ levels (Fig 1F, S1A), confirming that it is inactive at the abnormally high lysosomal pH. However, consistent with the idea that Ned-19 can target other lysosomal Ca2+ channels, Ned-19 increased lysosomal Ca2+ and decreased cytosolic Ca2+ in both PS1KO and shTPC2 PS1KO cells (Fig 1F). To verify that TRPML1 is the primary Ca2+ efflux channel in PS1KO cells, we knocked down TRPML1 in PS1KO cells, which restored both lysosomal and cytosolic Ca2+ to near WT levels (Fig 1G, S1A). Moreover, an anti-TRMPL1 antibody that inhibited the activation of the channel by ML-SA1 (Zhang et al., 2009) efficiently inhibited the rise in cytosolic Ca2+ induced by ML-SA1 (Fig 1H). As additional confirmation, we treated PS1KO cells with YM201636 to inhibit the synthesis of PI(3,5)P2, the endogenous potentiator of TRPML1 channel opening (Zhang et al., 2012b) which reduced the opening frequency of TRPML1. After GPN-induced rupture of lysosomal membranes, cytosolic Ca2+ rose in PS1KO cells to levels nearly equal to those of WT cells indicating an increase in the releasable pool of lysosomal Ca2+ (Fig 1I).
We confirmed that TRPML1 is responsible for depleting lysosomal Ca2+ at the elevated pH in PS1KO cells by demonstrating that concanamycin A (ConA) inhibition of vATPase activity in control human fibroblasts induced a PS1KO-like elevation of lysosomal Ca2+ release in response to the TRPML1 agonist ML-SA1, whereas MLIV human fibroblasts, containing inactive TRPML1, showed no response (Fig 1J) indicating an elevation of lysosomal pH can induce hyperactivation of TRPML1. The rise in cytosolic Ca2+ under ML-SA1 conditions originated at the lysosome and via TRMPL1 because cytosolic Ca2+ in control and MLIV human fibroblasts pre-treated with GPN did not rise after additional treatment with ML-SA1 (Fig 1J).
Collectively, these data indicate that the presence of an elevated Ca2+ leak out of PS1KO lysosomes is mediated by TRPML1, which is hyper-activated in PS1KO cells due to elevated lysosomal pH.
Restoring normal lysosomal pH, but not lysosomal Ca2+ homeostasis alone, rescues lysosomal deficits and autophagy
Lysosomal Ca2+ dysfunction caused by PS1 deletion was proposed to be responsible for autophagy deficits in PS1KO cells (Coen et al., 2012), although no evidence or underlying mechanism was shown. Because treatment of PS1KO cells with Ned-19 normalized TRPML1 activity (Fig 1E) and prevented abnormal lysosomal Ca2+ efflux, we investigated whether or not reducing this Ca2+ leak influences lysosomal pH or lysosome-related function. Although Ned-19 treatment of PS1KO cells restored normal lysosomal and cytosolic Ca2+ levels (Fig. 2A), its addition to PS1KO cells had no ameliorative effects on lysosomal pH (Fig. 2B), levels of LC3-II (Fig 2C), or CatD activation in lysosomes measured using Bodipy FL-pepstatin A (Fig 2D).
Figure 2. Restoring normal lysosomal pH, but not lysosomal Ca2+ homeostasis alone, rescues lysosomal deficits and autophagy.
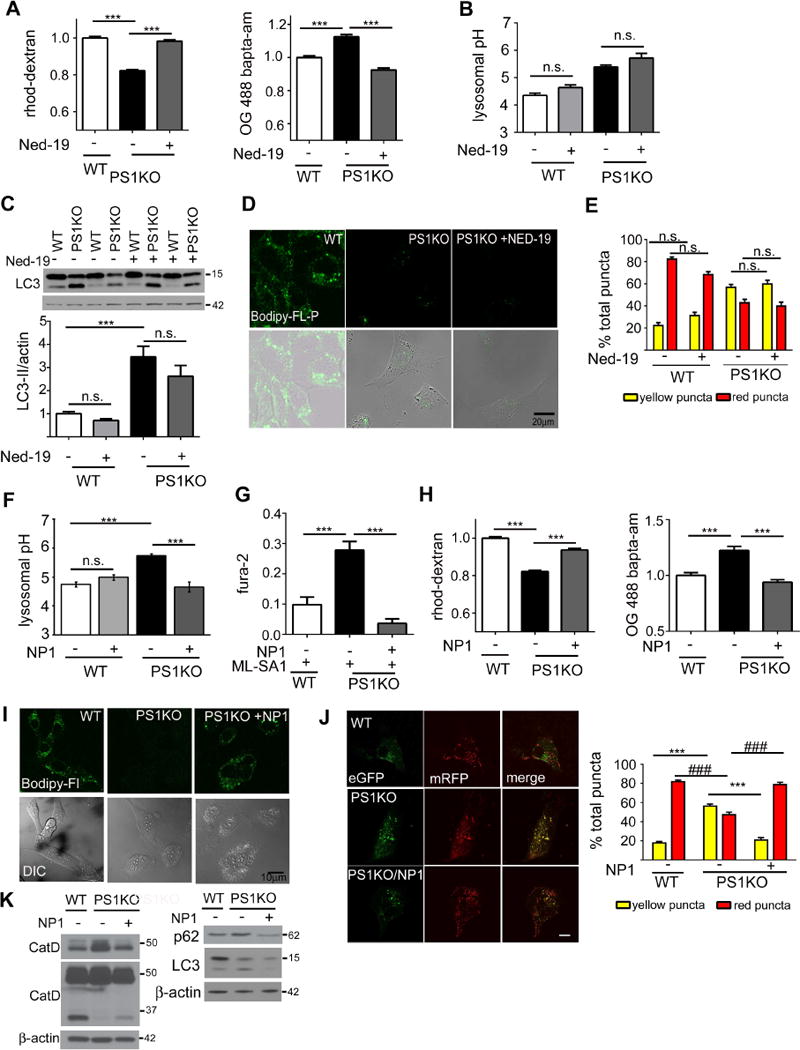
(A) Ned-19 (0.5μM, 24hrs) elevated lysosomal Ca2+ (WT n=453; PS1KO n=461; PS1KO/Ned-19 n=522) and decreased cytosolic Ca2+ levels in PS1KO cells (WT n=148; PS1KO n=150; PS1KO/Ned-19 n=150). (B) Ned-19 does not alter lysosomal pH in WT or PS1KO cells (n=9 each). (C) Ned-19 does not reduce elevated LC3-II levels in PS1KO cells (n=11) or (D) increase in situ CatD activity (n=3). Scale bars 20μm (E) PS1KO and WT cells transiently transfected for 48 hrs with EGFP-mRFP LC3, showed no change in percentages of yellow or red puncta after 0.5 μM Ned-19 for 24hrs (F) Uptake of lysosome-targeted NP-1 acidic nanoparticles (24 hrs, 1mg/ml) in PS1KO cells restored normal lysosomal pH (n=6) and (G) lowered ability of ML-SA1 to elevate cytosolic Ca2+ (WT n=180; PS1KO n=186; PS1KO/NP1 n=237). (H) NP-1 also elevated lysosomal Ca2+ levels (WT n=318; PS1KO n=305; PS1KO/NP1 n=313) and lowered cytosolic Ca2+ in PS1KO cells (WT n=148; PS1KO n=150; PS1KO/NP1 n=150). (I) NP-1 normalized levels of Cat D activity, (J) NP-1 reduced yellow puncta, and increased red puncta, in PS1KO cells, indicating restored autophagic flux Scale bars 10μM * p <0.05, ***/### p<0.0001. (K) NP-1 increased mature CatD levels and reduced elevated LC3-II/p62 levels in PS1KO cells Error bars: ±S.E.M
Cells were transiently transfected with the eGFP-mRFP-LC3 construct and treated with Ned-19 to assess any alterations in autophagic flux. Compared with WT controls, untreated PS1KO cells display a lower percentage of red puncta, and an elevated percentage of yellow puncta, indicating defective maturation of autophagosomes into acidic lysosomes. Ned-19 did not affect these percentages, indicating that lysosomal Ca2+ restoration did not restore lysosomal acidity or autophagic flux (Fig 2E). EM morphometry confirmed these results by showing that Ned-19 did not alter the abnormally lowered ratio of lysosomes to late AVs in PS1KO cells (Fig S2A). Previously, we demonstrated that autophagy induction was not altered in PS1KO cells, and that reduced autophagic flux was due specifically to delayed lysosomal degradation (Lee et al., 2010). Establishing that Ned-19 does not influence autophagy induction, we observed that Ned-19 treatment does not alter levels of either p-mTOR or p-P70S6K in PS1KO cells (Fig S2B). Thus, the key driver of autophagic dysfunction in PS1KO cells, namely markedly delayed degradation, is not due to Ca2+ dysfunction, as previously speculated (Coen et al., 2012).
Contrasting with the minimal effect of lysosomal Ca2+ correction on autophagy, reversal of lysosomal pH deficits in PS1KO cells rapidly normalized autophagy and lysosomal function, including Ca2+ efflux. We were able to lower intraluminal lysosomal pH using a poly (DL-lactide-co-glycolide) (PLGA) acidic nanoparticle which is internalized in cells by endocytosis and targeted to lysosomes where protons are released in the lysosomal lumen through hydrolysis of poly-lactic acid (Baltazar et al., 2012). We identified a nanoparticle species, designated NP-1, as having optimal uptake efficiency and ability to restore lysosomal pH as previously shown in ARPE-19 cells (Baltazar et al., 2012). Exposure of PS1KO cells to NP-1 normalized lysosomal pH, lowering it from 5.7 to 4.6 (Fig 2F). Pre-treatment with NP-1 for 24hrs normalized TRMPL1 activity, as no rise in cytosolic Ca2+ was elicited with ML-SA1 (Fig 2G), similar to the lack of effect of ML-SA1 onWT cells (Fig 1E). The normalization of lysosomal Ca2+ and decrease in cytosolic Ca2+ levels supports the hypothesis that lysosomal Ca2+ levels are dependent upon lysosomal acidification (Fig. 2H). In addition, we observed that lysosomal pH correction with NP-1 fully restored CatB and CatD activities in PS1KO lysosomes in situ measured with MR-CatB and Bodipy-pepstatin A, respectively (Fig. 2I, Fig S2C), as well as levels of mature CatD. Accumulation of the autophagy substrates, p62 and LC3-II, were also markedly decreased (Fig. 2K). Moreover, NP-1 decreased the percentage of yellow eGFF-mRFP-LC3 puncta by 50%, and increased the percentage of red puncta by ~30% in PS1KO cells, reflecting a restoration of autophagic flux at the lysosomal clearance stage of autophagy (Fig 2J) given that autophagy induction markers, p-mTOR and p-p70S6K were unaltered by NP-1(Fig S2D). Furthermore, V0a1 glycosylation was unchanged by NP-1, indicating that re-acidification was due to lactic acid release from the nanoparticle and not to amelioration of vATPase deficits (Fig S2E).
Elevated lysosomal Ca2+ efflux in PS1KO cells is secondary to defective lysosomal acidification and can be induced in WT cells by vATPase inhibition
Treatment of WT cells with the vATPase inhibitor, ConA, elevated lysosomal pH to a level similar to that in untreated PS1KO cells (Fig 3A) and did not further elevate lysosomal pH in PS1KO cells (Fig 3A). These results are consistent with previous evidence that, despite a high level of vATPase inhibition, cells can still achieve a moderate degree of lysosomal acidification (Mindell, 2012). Supporting the close relationship between pH and Ca2+ content in lysosomes, ConA decreased lysosomal Ca2+ and elevated cytosolic Ca2+ in WT cells (Fig 3B) but, as expected, did not alter the already abnormal levels of lysosomal and cytosolic Ca2+ in PS1KO cells where vATPase function is substantially compromised (Fig 3B). Importantly, ConA inhibition of vATPase activity in WT cells reproduced previously reported PS1KO lysosome and autophagy deficits, including elevated LC3-II levels (Fig 3C), lowered CatD activity (Fig 3D), lowered CatB activity (Fig 3E), increased late AV accumulation, and decreased lysosome number (Fig 3F) and autophagic flux measured using the tandem EGFP-mRFP LC3 reporter (Fig 3G). Moreover, ConA treatment had no effect on autophagic induction, as both p-mTOR and p-p70S6K levels remained unchanged (Fig S2F).
Figure 3. Inhibition of VATPase function induces a PS1KO-like defective autophagy phenotype.
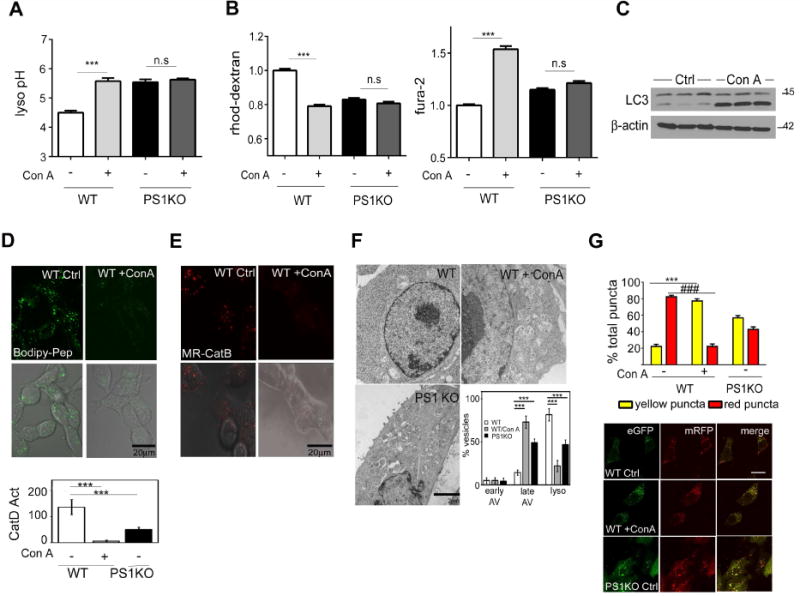
(A) ConA (50nM, 24hrs) elevated lysosomal pH in WT (n=6 each), but had no effect on PS1KO cells (B) ConA decreased lysosomal Ca2+ (WT n=155; WT/ConA n=153) and elevated cytosolic Ca2+ (WT n=154; WT/ConA n=152) in WT but had no significant effect on Ca2+ levels in PS1KO cells (PS1 n=153 and 164; PS1/ConA n=161 and 151, lyso and cyto). (C) Con A elevated LC3-II levels, decreased in vivo and in vitro CatD enzyme activities and CatB activity (D, E respectively), and (F) increased late autophagic vacuole accumulation while decreasing lysosome number (n=25). (G) ConA (50nM) elevated yellow puncta, and markedly decreased red puncta in EGFP-mRFP-LC3 cells Scale bars 10μm * p<0.05, ***/### p<0.0001. Error bars: ±S.E.M
PS1 deletion leads to loss of lysosomal vATPase activity
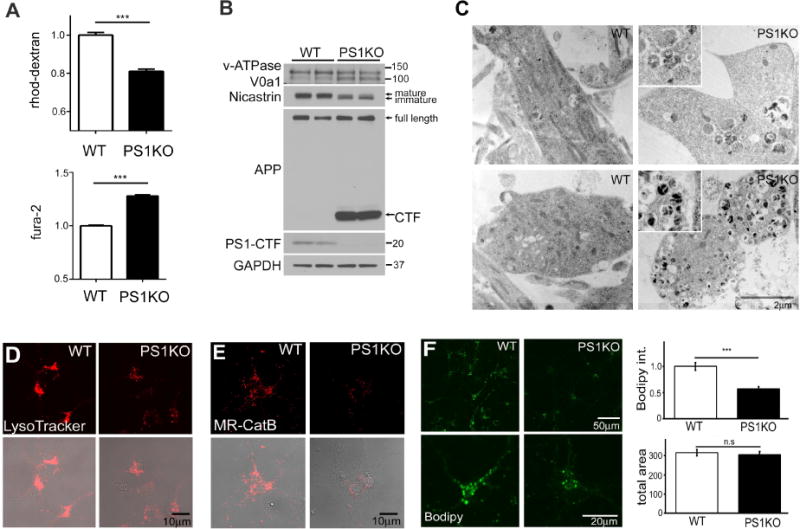
We previously reported reduced V0a1 subunit maturation and levels in PS1KO cells (Lee et al., 2010; Wolfe et al., 2013). To investigate this issue further specifically in lysosomes, we used a superparamagnetic chromatography isolation procedure (Walker and Lloyd-Evans, 2015) to isolate highly enriched lysosomal preparations from PS1KO and WT cells, comprising 3% of the total cell lysate (protein: protein) and representing an enrichment of the LAMP2 lysosomal marker. Western blot analysis of enriched lysosomes confirmed the purity of lysosome fractions by demonstrating the presence of minimal calnexin and EEA1 and strong enrichment of LC3-II and CatD (Fig 4A). The mature active form of CatD was diminished in PS1KO lysosomes, as previously seen in intact cells (Lee et al., 2010). The V0a1 subunit in purified lysosomes from PS1KO cells was present at markedly lowered levels compared to those in WT lysosomes and existed predominantly at a lower apparent molecular weight in PS1KO lysosomes, consistent with it being incompletely glycosylated, as previously reported (Lee et al., 2010). The ratio of V0a1 relative to LAMP2 used as a lysosome loading control was lowered 70% in PS1KO lysosomes (Fig 4B). We next analyzed the assembly of the vATPase complex on isolated lysosomes by measuring levels of lysosomal V1E1 subunit, which actively assembles with the membrane bound V0 sub-complex during formation of the complete vATPase. V1E1 on PS1KO lysosomes was reduced to similar levels as V0a1 (Fig 4B), substantiating earlier findings on crude membrane fractions from PS1KO cells (Lee et al., 2010).
Figure 4. PS1KO lysosomes are markedly deficient in vATPase V0al subunit and vATPase activity.
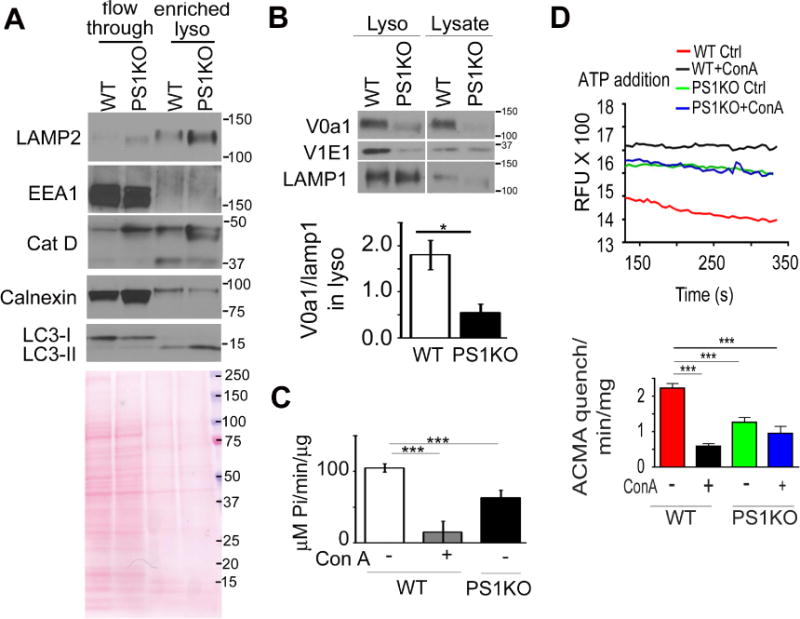
(A) In highly enriched lysosomes from PS1KO cells, (B) V0al subunit levels were decreased (n=3), (C) ATP hydrolysis activity was reduced and (D) proton translocation ability of vATPase was decreased ConA treatment of WT cells for 24 hr prior to lysosomal enrichment decreased proton translocation, but had no effect in PS1KO cells* p<0.05, *** p<0.0001. Error bars: S.E.M
To assess activity of the assembled vATPase in purified lysosomes, we next measured the ATP-hydrolytic activity of the complex and its proton pumping capacity directly. The ATP hydrolysis rate, monitored by release of inorganic phosphate (Ramirez-Montealegre and Pearce, 2005), was >40% lower in lysosomes isolated from PS1KO cells relative to WT lysosomes, while ConA treatment of WT cells reduced ATP hydrolysis by ~90%, indicating that, despite the marked loss of V0a1, there is some residual ATPase activity in PS1KO (Fig 4C). We next measured the rate of proton translocation into the lysosomal lumen using the quenching of 9-amino-6-chloro-2-methoxyacridine (ACMA), which was ~50% lower in PS1KO lysosomes than in WT lysosomes. As expected, pre-treatment of lysosomes with ConA significantly inhibited proton translocation in WT cells, but minimally reduced this rate in PS1KO lysosomes, confirming that loss of PS1 markedly impairs vATPase function (Fig 4D).
The vATPase V0a1 subunit is essential for lysosomal acidification and autophagic function
PS1 siRNA treatment of WT cells for 48 hrs only partially decreased expression of mature V0a1 and 96 hrs of PS1 siRNA was needed to decrease V0a1 levels more than 85% (Fig 5A), and significantly increase LC3-II levels (Fig 5B, C), showing diminished lysosomal clearance. LysoTracker signal decreased 50% after 96 hrs of V0a1 siRNA (Fig. 5D) but only 20% after 48hrs, again underscoring the slow turnover of preexisting vATPase. Similarly, in situ CatB enzyme activity decreased 60% only after 96 hrs of V0a1 siRNA exposure (Fig 5E). Thus, even after 4 days of siRNA exposure, residual V0a1 on lysosomes is enough to maintain a minor fraction of normal proton pumping activity. These results support the idea that V0a1 is essential for lysosomal acidification and function and that its turnover is quite long, consistent with previous findings (http://helixweb.nih.gov/ESBL/Database).
Figure 5. vATPase V0a1 knockdown inhibits lysosomal acidification.
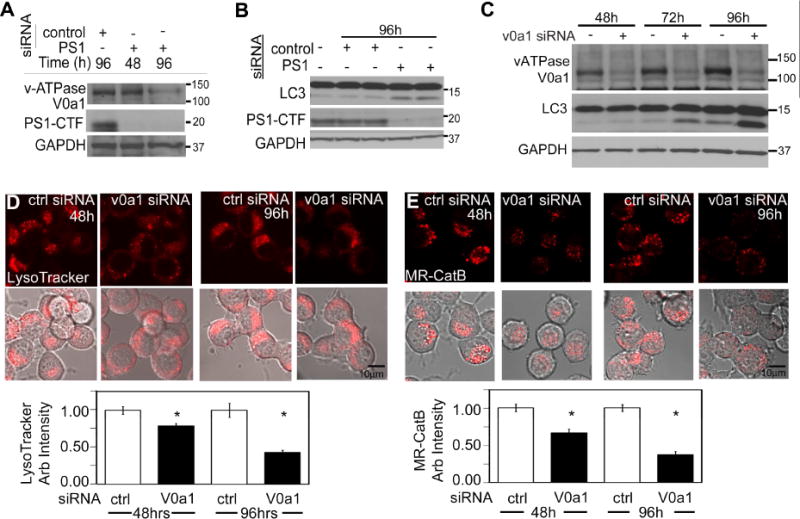
PS1 siRNA in WT cells decreased V0a1 levels (A) and increased LC3-II (B). (C) Levels of LC3-II were increased after V0a1 subunit siRNA treatment LysoTracker intensity (n=55) (D) and CatB activity (n=30) (E) decreased after V0a1 siRNA Scale bars 10 μm * p<0.05. Error bars: S.E.M
Glycosylation of V0a1 is critical for its stability and functions
Glycosylation of the V0a1 subunit has been proposed to influence its stability and function (Gillespie et al., 1991; Lee et al., 2010). It was recently demonstrated that glycosylation of residue R444 in V0a3, an osteoclast specific V0a isoform, is required for its proper delivery to lysosomes (Bhargava et al., 2012). To investigate the role of glycosylation on the V0a1 subunit, we generated an R447L mutant analogous to the R444L mutation in V0a3, using a C-terminal flag tagged V0a1 (V0a1-flag) that enabled the mutant to be differentiated from endogenous V0a1 subunit in murine neuroblastoma (N2a) cells.
To investigate the R447L effects on V0a1 maturation, we treated lysates stably expressing V0a1-flag with PNGase F. PNGase converted the mature V0a1WT-flag to its 100 kDa immature form, whereas there was no discernible shift in the mobility of V0a1R447L after PNGase F treatment. Moreover, levels of V0a1R447L in the stably transfected cells were markedly lower than levels of V0a1WT in the corresponding cells (Fig 6A). To analyze whether this decrease was due to rapid degradation of the unglycosylated protein, cells were treated for 24 hours with the proteasome inhibitor MG-132 and lysates were immunoblotted with anti-flag antibody. As predicted, the proteasome inhibitor significantly delayed V0a1R447L degradation (Fig 6B). To document premature turnover of V0a1R447L, cells were exposed to cycloheximide for various times. Immunoblot analysis of these lysates confirmed that the glycosylation mutant V0a1R447L is rapidly degraded by the proteasome (Fig 6C). Previous studies demonstrated that immature V0a1 is not correctly delivered to the lysosome but rather retained in the ER (Lee et al., 2010). To identify the localization of V0a1R447L, we fractionated cells on an Opti-prep gradient. Immunoblot analyses of these fractions revealed that V0a1R447L subunit co-sedimented with ER-enriched fractions, whereas V0a1WT-flag was observed primarily in lysosome-enriched fractions (Fig 6D). Double-immunofluorescence analysis using compartmental markers confirmed the mislocalization of V0a1R447L (Fig 6D).
Figure 6. The mutant protein construct V0a1R447L-flag is not glycosylated and is degraded in N2a cells.
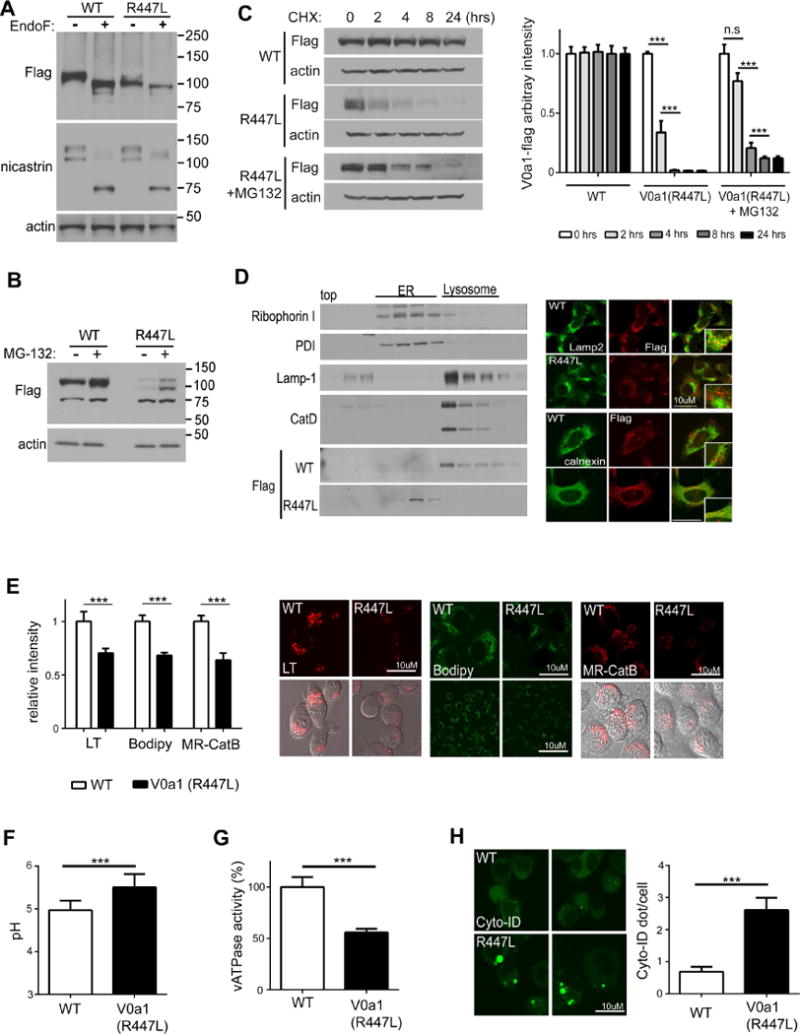
(A) Cell lysates were immunoblotted with anti-flag antibody followed by PNGase F treatment Nicastrin blots provided as a positive control for PNGase treatments (B) Cell lysates were immunoblotted with anti-flag antibody followed by proteasome inhibitor (MG-132) treatment for 24 hrs (C). Cell lysates were immunoblotted with anti-flag antibody followed by CHX treatment for the indicated time Cell lysates form V0a1R447L-flag were pre-incubated with MG-132 for 24 hrs then treated with CHX for the indicated time Levels of V0a1-flag were quantified Results were plotted as ratios normalized to the non-treated sample per each cell line (D) Immunoblot of V0a1-flag distribution in subcellular fractions of V0a1WT-flag and V0a1R447L-flag ER marker proteins (Ribophorin-I and PDI) primary localized infractions 5–11 and lysosomal marker proteins (LAMP-1 and mature CatD) mainly in fractions 13–17. Double-immunostaining showed strong colocalization of V0a1-flag and LAMP-2 in V0a1WT -flag cells, whereas V0a1R447L-flag strongly colocalized with ER marker calnexin Scale bar 10 μm (E) Cells were incubated with LysoTracker for lysosomal acidification assay To access the in vivo lysosomal enzyme activity, cells were incubated with Bodipy-FL-pepstatin A and MR-CatB for CatD and CatB activity, respectively Scale bar 10 μm Intensity of signal was quantified (LT: n=60, Bodipy: n=50, MR-CatB: n=55). Results were plotted as ratios normalized to V0a1WT-flag (F) Lysosomal pH values were measured ratiometrically using LysoSensor Yellow/Blue-dextran (n=10, at least 6 × 103 cells/n). (G) v-ATPase activity was reduced in V0a1R447Lcells (H) Autophagosomes were immunolabeled with Cyto-ID autophagy detection kit Results were plotted as mean of AV (Cyto-ID) puncta number per cells (n=80). Scale bar 10 μm * denotes p<0.05, ** denotes p<0.001, *** denotes p<0.0001. Error bars: ±S.E.M
To further analyze the functional role of V0a1 glycosylation, we incubated V0a1R447L cells with LysoTracker, Bodipy-FL-pepstatin A and MR-CatB to assay lysosomal acidification and in situ activity of CatD and CatB, respectively. Quantitative analyses demonstrated that both LysoTracker signal and cathepsin activities were significantly decreased in V0a1R447L cells (Fig 6E), commensurate with an observed rise in lysosomal pH and decline in vATPase activity measured directly (Fig 6 F, G). As expected, levels of autophagy marker protein LC3-II increased (Fig S3A) and clearance of autophagosomes were reduced in V0a1R447L cells (Fig 6H), whereas autophagy induction related signals (i.e. p-mTOR, p-P70S6K) were not altered (Fig S3B).
These results are in accord with previous findings and establish that V0a1 subunit glycosylation is critical for the stability and lysosomal targeting that enables lysosomal acidification and hydrolytic function.
PS1 is an essential mediator of lysosomal acidification and Ca2+ homeostasis in neurons
To establish that PS1 loss causes a similar lysosomal phenotype in a neuronal PS1-deficient cell model, we assayed these parameters directly in neurons. Importantly, levels of lysosomal Ca2+ were lowered and cytosolic Ca2+ was elevated significantly in PS1KO primary neurons (Fig 7A). Western blot analysis demonstrated that V0a1 maturation is also markedly decreased (Fig 7B). Further indicative of autophagy failure, the numbers of autophagic vacuoles (AVs), consisting of mainly single membrane electron-dense autolysosomes and multilamellar organelles, were significantly increased in neuronal perikarya (Fig 7C). Consistent with abnormally elevated lysosomal pH and disrupted proteolysis, LysoTracker signal and CatB/D activity were significantly decreased in PS1KO neurons compared to WT neurons (Fig 7D, E, F), confirming earlier findings in PS1KO cells (Lee et al., 2010). Contrasting with these current findings, Coen et al. reported no lysosomal acidification deficits in PS1KO neurons. A plausible explanation for their failure to detect differences in neurons lies in the atypical features of the PS1KO neurons they used. In our PS1KO neurons, nicastrin maturation is significantly impaired and APP-CTF levels are increased, as expected from the loss of PS1, and as reported by other groups (Esselens et al., 2004; Wilson et al., 2004), while these signature properties of PS1KO were not present in the neurons used by Coen et al.
Figure 7. Lysosomal Ca2+, acidification, and enzyme activities are reduced in PS1KO neurons.
(A) Lysosomal Ca2+ (WT n=180; PS1KO n=181) was reduced and cytosolic Ca2+ (n=20) was elevated in PS1KO neurons In PS1KO neurons, (B) maturation of V0a1 and nicastrin was decreased and APP-CTF levels were increased; (C) AVs accumulate in both cell bodies and axons; (D) LysoTracker signal and in vivo CatB (E) and CatD (n=28) (F) activities were decreased Scale bars 10μm *** p<0.0001. Error bars: S.E.M
Discussion
Presenilin 1 plays an important role in Ca2+ homeostasis and autophagy/lysosomal protein degradation, beyond its well-studied catalytic role as part of γ-secretase (Lee et al., 2010; Tu et al., 2006). However, the relationship between two major γ-secretase independent functions of PS1, namely maintenance of Ca2+ homeostasis and lysosomal proteolysis, is poorly understood. Based on an earlier observation that lysosomal alkalinization increased lysosomal Ca2+ efflux (Christensen et al., 2002), it was reasonable to suspect that lysosomal acidification failure resulting from loss of PS1 may also disrupt lysosomal Ca2+ homeostasis. In this study, we show that acidic lysosomal pH is indeed essential for maintaining normal lysosomal Ca2+ levels whereas restoration of lysosomal Ca2+ levels by itself does not significantly affect lysosomal acidification under these conditions. Our direct measurements reveal significantly decreased lysosomal Ca2+ in PS1 ablated cells and neurons. Additional measurements of lysosomal Ca2+ levels after GPN-mediated lysosome rupture and Ca2+ release confirm the lysosomal Ca2+ deficit. Notably, we also demonstrate that TRPML1 mediated release of lysosomal Ca2+ in PS1KO cells substantially increases cytosolic Ca2+ levels and is a major contributor to the PS1-related Ca2+ defect previously attributed only to disruption of ER Ca2+ homeostasis (Chan et al., 2000; Cheung et al., 2010).
Increasing evidence suggests that the primary Ca2+ stores targeted by NAADP are present in acidic organelles such as the lysosome, and are distinct from pools in the ER (Calcraft et al., 2009). TPC2 is the primary NAADP dependent Ca2+ efflux channel in the lysosome, and its activation via NAADP has previously been shown to be sensitive to lysosomal pH. In fully acidified lysosomes, Ca2+ stores that are lowered by elevated NAADP levels become replenished when the dissociation rate between NAADP and TPC2 increases (Pitt et al., 2010). When lysosomal pH is normal, elevated cytosolic NAADP levels induce lysosomal Ca2+ release from lysosomal stores (Churchill et al., 2002; Lloyd-Evans et al., 2008). However, increasing lysosomal pH leads to a failure of the purified TPC2 to dissociate from NAADP, rendering the channel inactive (Pitt et al., 2010)
PS1KO cells, which have elevated lysosomal pH, treated with NAADP-AM did not release lysosomal Ca2+, whereas U1866a treated WT cells, possessing a normal lysosomal pH, released lysosomal Ca2+ in response to NAADP-AM treatment (Lloyd-Evans et al., 2008). These data are consistent with the TPC2 channel being inactive in PS1KO cells. Conversely, ML-SA1, an agonist for the lysosomal TRPML1 channel, evoked a stronger release of Ca2+ in PS1KO cells than in WT cells, despite the lower lysosomal Ca2+ content in PS1KO cells.
Our data indicate that TRPML1 in PS1KO cells is more responsive to low concentrations of the agonist, ML-SA1, and likely exists in a hyper-active state. In addition, there have been reports that endogenous TRPML1, as opposed to a constitutively active mutant form of TRPML1 overexpressed in cells (Dong et al., 2009), is less active at a lower pH range and becomes more active as pH increases (Raychowdhury et al., 2004). This property of the channel appears to contribute to its hyper-activity at elevated lysosomal pH in PS1KO cells. To establish that TRPML1, the lysosomally localized member of the TRPML family, is the primary Ca2+ efflux channel and is specifically activated by elevated lysosomal pH, we showed that TRPML1 knockdown in PS1KO cells elevates lysosomal Ca2+ and decreases cytosolic Ca2+ and that an inhibitory antibody against TRPML1 abolishes ML-SA1 mediated lysosomal Ca2+ release. In addition, cytosolic Ca2+ did not rise when ML-SA1 treated control and MLIV fibroblasts were pretreated with GPN to abolish any Ca2+ signal from lysosomal stores, implying a principal lysosomal source for this cytosolic Ca2+ rise. Moreover, we observed a large increase in cytosolic Ca2+ in ConA treated WT fibroblasts indicating that inhibition of vATPase, and thus an elevation of lysosomal pH, can induce TRPML1 sensitivity to ML-SA1. Additionally, pre-treatment of PS1KO cells with NP-1 to lower lysosomal pH normalized the rise in cytosolic Ca2+ mediated by ML-SA1 as observed in untreated cells.
Ned-19 normalized the abnormal ML-SA1 induced Ca2+ release in the PS1KO cells providing additional evidence that the increased Ca2+ efflux is due to hyper-active TRPML1. By blocking lysosomal Ca2+ leak, Ned-19 restored Ca2+ homeostasis in PS1KO cells, but had no detectable effect on lysosomal pH or any aspect of the abnormal lysosomal/autophagy phenotype in PS1KO cells. If lysosomal Ca2+ fluxes were a significant influence on lysosomal acidification, we would have expected Ned-19 treatment to alter the elevated lysosomal pH in PS1KO cells. In addition, ConA recapitulated PS1KO phenotypes in WT cells but, as expected, did not worsen the Ca2+ defect in PS1KO cells. Thus, our data provide strong evidence that elevated lysosomal pH is the basis for both reduced lysosomal Ca2+ and lysosomal/autophagy defects in PS1KO cells.
In AD, ER Ca2+ homeostasis is dysregulated and multiple Ca2+ regulatory mechanisms, including inositol 1,4,5-triphosphate and ryanodine receptors, are known to be disrupted by PS1 loss of function in early onset AD (Chan et al., 2000; Cheung et al., 2010) leading to an increase in cytosolic Ca2+. Interestingly, lysosomal Ca2+ release can trigger secondary ER Ca2+ release via nanojunctions (Fameli et al., 2014; Kilpatrick et al., 2013). Possible crosstalk between ER and lysosomal Ca2+ fluxes, however, has not been fully considered in AD pathogenesis. Our current data raise the possibility that lysosomal Ca2+ release or leak driven by lysosomal acidification deficits could promote ER Ca2+ dysregulation, in addition to directly elevating cytosolic Ca2+ levels. The findings show a role of both pH and lysosomal Ca2+ leak in maintaining normal lysosomal Ca2+ levels and a capability of lysosomal Ca2+ leak to alter cytosolic Ca2+ levels substantially.
Lysosomal acidification is maintained primarily by the vATPase, a protein complex composed of a membrane-bound V0 domain and a peripherally associated V1 domain. The largest subunit of the V0 sub-complex, V0a, is an integral membrane protein with four known isoforms (a1–a4), a1 being the major one in brain (Nishi and Forgac, 2000). We demonstrated previously that PS1 deletion, PS1/2 deletion, or FAD-related mutation of PS1 reduces vATPase immunoreactivity on lysosomes, elevates lysosomal pH, depresses lysosomal protease activation, and significantly delays autophagic protein turnover. Moreover, stable transfection of PS1 into PS1/2 DKO cells reverses these features and restores V0a1 subunit glycosylation, lysosomal acidification and autophagic clearance (Lee et al., 2010). Importantly, additional studies have also demonstrated impaired vATPase V0a1 subunit maturation and endolysosomal acidification in multiple models, including PS1 ablated cells or mutant PS1 models (Avrahami et al., 2013; Coffey et al., 2014; Dobrowolski et al., 2012; Torres et al., 2012; Wolfe et al., 2013), underscoring that the acidification defect induced by PS1 loss of function is generalisable across various cell types. In one study, PS1 and vATPase V0a1 subunit were reported to have no role in lysosomal pH regulation (Coen et al., 2012) and differences between methods and cell culture conditions might explain some discrepancies between this study and our studies. In our current study, we could show that lysosomes purified from PS1KO cells contained markedly lowered levels of V0a1 subunit, which was associated with impaired vATPase assembly on lysosomes and commensurate reductions of lysosomal proton pumping activity. Using siRNA gene knockdown, we further demonstrated that lysosomal acidification and lysosomal enzyme activation require the vATPase V0a1 subunit, in agreement with other findings that V0a1 subunit is critical for vesicular acidification (Raines et al., 2013; Saw et al., 2011). Notably, V0a1 has a long half-life on lysosomes, requiring substantial duration of siRNA exposure (96hrs) to lower lysosomal levels of the subunit sufficiently to alter acidification, which is 48hrs longer than the interval used in the Coen et al. study where a pH change could not be detected upon siRNA treatment (Coen et al., 2012). We observed that the glycosylation mutant V0a1R447L stably expressed in cells exhibited increased ER retention, decreased stability promoting loss via ERAD, and deficient lysosomal targeting and assembly. Without proper maturation of the V0a1 glycosylation mutant, lysosomal acidification and functions, including lysosomal enzyme activation and autophagic cargo degradation, were impaired. These findings strongly support the conclusion that both the expression and proper glycosylation of V0a1 are critical for its lysosomal function.
Our data also establish that defective lysosomal acidification is the essential factor accounting for the abnormal autophagy/lysosomal degradation and lysosomal Ca2+ phenotypes in PS1 deficient neurons and non-neural cells. ConA inhibition of the vATPase in WT cells reproduced all of the features of the PS1KO phenotype, whereas Ned-19 corrected lysosomal Ca2+ levels without influencing lysosomal pH deficits or other aspects of autophagy dysfunction. Most importantly, we were able to reverse both autophagy and lysosomal Ca2+ deficits specifically by re-acidifying lysosomes using a form of lysosome-targeted acidic nanoparticles previously shown to restore lysosomal pH in ARPE 19 cells, a model of macular degeneration (Baltazar et al., 2012).
In conclusion, we have shown that the vATPase V0a1 subunit is essential for lysosomal acidification and that its maturation and proper functional assembly on lysosomes require PS1. Moreover, we have established that lysosomal Ca2+ homeostasis is modulated by lysosomal pH and that the abnormal TRPML1 mediated efflux of Ca2+ from lysosomes induced by PS1 deletion is a secondary consequence of impaired lysosomal acidification caused by inadequate levels of vATPase activity. Substantial evidence suggests that both the lysosomal/autophagic and calcium homeostasis abnormalities induced by PS1 loss of function are likely to contribute significantly to disease pathogenesis in this familial form of AD, as well as in late onset AD where similar pathobiology develops possibly via different initiating factors (Nixon and Yang, 2011). Restoration of autophagy/lysosomal function in PS1-deficient cells by re-acidifying lysosomes suggests additional approaches to therapy for AD and other neuropathological conditions associated with lysosomal pH elevation.
Experimental Procedures
Methods are described in detail in Supplemental experimental procedures.
Cell lines and reagents
Murine blastocysts with different PS1 genotypes (WT, BD6; PS1KO, BD15) were previously characterized (Lee et al., 2010). Control and MLIV human fibroblasts were purchased from Coriell Cell Repository and have numbers GM005399 and GM002527 respectively and maintained in 10% FBS in DMEM with 5% CO2. Murine neuroblastoma (N2A) cells were maintained in DMEM with penicillin/streptomycin and 10% FBS at 37°C and 5% CO2. Mouse PS1 (MSS208049) and scramble (12935-200) siRNA were purchased from Invitrogen and mouse vATPase V0a1 siRNA (11975) from Thermo Scientific. Cells were transfected using Lipofectamine RNAiMAX (Invitrogen). Primary cortical neuronal culture were derived from E15 stage pups from PS1 (+/−) crosses and cultured with Neurobasal medium.
A musculus v-ATPase V0a1WT-Flag construct (EX-Mn20338-M13) was purchased from GeneCopoeia. A V0a1R447L-flag mutant DNA construct was generated using a QuikChange II XL site-directed mutagenesis kit (Agilent technologies, #200521) according to manufacturer’s instructions with 5′-GCATGGTGTTCAGCGGCCTATACATTATTCTTCTGATG-3′ and 5′-CATCAGAAGAATAATGTATAGGCCGCTGAACACCATGC-3′ primer set. After establishing the stable cell line, cells were maintained with G418 (300 μg/ml) containing medium.
Either GFP-shTPCN2 or GFP-shTRPML1 (Qiagen) were transfected with Lipofectamine 2000 (Invitrogen) for 72 hours and calcium measurements for cells containing the GFP signal were calculated for either cytosolic or lysosomal Ca2+.
Gel electrophoresis, immunoblotting, and Confocal laser scanning microscopy
Experiments were performed as previously described (Lee et al., 2010).
Deglycosylation and cycloheximide treatment
To assess flag-tagged V0a1 mutant glycosylation, samples were treated for 24 hrs at 37 C with PNGase F using an enzymatic deglycosylation kit according to the manufacturer’s instruction (PROzyme). To assess the stability of the V0a1 mutant, samples were treated with CHX (100 μg/ml) for 0, 2, 4, 8, 24 hrs with or without pre-treatment with MG-132 (1 μM/ml, 24 hrs) and then analyzed by SDS-PAGE.
Subcellular Fractionation and Enzyme activity for Cathepsins
Procedures were performed as previously described (Lee et al., 2010).
Lysosomal pH measurement and Ultrastructural analyses
Procedures were performed as previously described (Wolfe et al., 2013).
Lysosomal Isolation
Cells were incubated in growth medium containing 1mM HEPES, pH 7.2 and 10% Dextran conjugated magnetite (Liquid Research LLC) for 24 hrs, then chased in normal growth media for 24 hrs. Cells were washed in PBS then harvested in 4ml of ice cold Buffer A. Cells were then homogenized with 40 strokes of a tight fitting pestle in a Dounce homogenizer then passed through a 22G needle 5 times. After homogenization, 500ul of ice cold Buffer B (220mM HEPES, 375mM KCL, 22.5mM MgAc, 1mM DTT, DNase I) was added and samples were then centrifuged at 750 × g for 10 mins. The supernatant was then decanted over a QuadroMAC LS column that had previously been equilibrated with 0.5% BSA in PBS. The pellet was subjected to re-addition of 4ml cold Buffer A, 500μl cold Buffer B and then resuspended and recentrifuged. DNase I (10μl/ml in PBS) was added and the column was then incubated for 10 min and then washed with 1ml cold PBS. Lysosomes were eluted by removing the column from the magnetic assembly, adding 500 μl of PBS and forced through the column using a plunger.
vATPase activity assay
Lysosome-enriched fractions were mixed with 3 vol of Buffer and incubated at 37°C for 5 min. After incubation, the reaction was started by addition of 2 mM ATP and incubated 20 min at 37°C. The ATPase reaction was stopped after 25 min by the addition of 2 ml of molybdate solution and 0.2 ml of ascorbic acid, then developed for 5 min at room temperature. Control samples were measured in the presence of the vATPase inhibitor ConA (1μM) and the experimental values were subtracted accordingly. Absorbance was measured at 750 nm and solutions of KH2PO4 were used to generate a standard curve.
Proton Translocation assay
Proton transport activity into the lumen of isolated lysosomes was measured by fluorescence quenching of 9-amino-6-chloro-2-methoxyacridine (ACMA) in the presence or absence of 1μM concanamycin A. Lysosomes were added to a cuvette containing 2 ml of reaction buffer. The reaction was started by the addition of 1 mM ATP in BTP, pH 7.5, a measurement (ex412/em480) taken every 5 seconds for 600 seconds on a SpectraMax M5 multimode reader(Molecular Devices).
Lysosomal Ca2+ Measurements
Cells were plated on glass bottom dishes the night before treatment. For baseline calcium measurements, cells were incubated with 25mg/mL rhod-dextran for 12 hrs before imaging. For all cytosolic measurements, cells were incubated with either 5μM Oregon-Green 488 Bapta-1 AM (Life Technologies) or 2μM Fura-2 AM (Life Technologies) for 1 hour and then chased with complete medium for 30 minutes. Cells were washed with HBSS (Invitrogen) and imaged and analyzed using ImageJ (NIH).
Release of lysosomal Ca2+ was measured using methods adapted from those previously described (Lloyd-Evans et al., 2008). Cells were plated on μ-Slide 8 well imaging dishes (ibidi), left to adhere overnight and then treated with 2μg/ml U18666a for 24 hrs as appropriate. Post incubation, cells were loaded with either 5μM Fluo3-AM or 5μM Fura 2-AM (StratechScientific LTD) in DMEM with 1% BSA and 0.0025% Pluronic acid F127 for 1 hr at 37°C then washed, left for 10 minutes to allow deesterification of the calcium dye, and imaged in 1x HBSS with 1mM HEPES, 1mM MgCl2 and 1mM CaCl2. Ca2+ free HBSS was used for all human fibroblast experiments. Intracellular Ca2+ responses were recorded using a Zeiss Colibri LED microscope system with an AxiocamMrm CCD camera and Zeiss Axiovision software version 4.7 with the additional physiology module for live cell Ca2+ imaging.
Acidic nanoparticle treatment
Poly (DL-lactide-co-glycolide) (PLGA) ResomerH RG 502 H was purchased from Boerhinger Ingelheim Inc., VA and prepared as previously described (Baltazar et al., 2012).
Analytical Procedures
Quantitative colocalization analysis was performed using ImageJ software (NIH Image) and all experiments were performed in triplicate unless otherwise indicated. Error bars represent standard error of the mean (±S.E.M).
Supplementary Material
Highlights.
PS1 is essential for V0a1 subunit glycosylation, stability, and vATPase assembly
PS1 deficiency depletes vATPase, impairing lysosomal acidification and proteolysis
Defective lysosome acidification in PS1KO cells causes lysosomal Ca2+ efflux
TRPML1 mediates lysosomal Ca2+ efflux and cytosolic Ca2+ elevation in PS1KO cells
Acknowledgments
We are very grateful to Dr. Alan Bernstein (Global HIV Vaccine Enterprise, USA) for PS BD cells. The authors would also like to thank Nicole Gogel for assisting in manuscript preparation and Cori Peterhoff for assisting in formatting figures. This work was supported by NIH P01AG017617, Takeda Pharmaceutical Company Ltd., New York Community Trust, and Litwin Foundation, Inc to RAN. The work was also supported by a RCUK Fellowship and Royal Society grant to ELE and BBRSC PhD and MRC in vivo skills award funding support to LJH. U. K is a co-founder and share holder of Ocugen, Inc. and EyeTrans Technologies, Inc.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
J.H.L performed experiments in Fig 5, 6, 7. M.K.M performed experiments in Fig 1,2,3,7. D.M.W performed experiments in Fig 2, 3, 4. L.J.H and E.L.E. performed experiments in Fig 1. A.K performed EM analysis; Y.S and P.L established primary neuron cultures; P.M performed in vitro CatD activity assays; U.K, C.H.M contributed acid nanoparticles and guidance on their use. E.E.C assisted in nanoparticle experiments. J.H.L, M.K.M, D.M.W, L.J.H, E.L.E, and R.A.N designed experiments, analyzed data and wrote the paper.
References
- Avrahami L, Farfara D, Shaham-Kol M, Vassar R, Frenkel D, Eldar-Finkelman H. Inhibition of glycogen synthase kinase-3 ameliorates beta-amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the Alzheimer disease mouse model: in vivo and in vitro studies. J Biol Chem. 2013;288:1295–1306. doi: 10.1074/jbc.M112.409250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltazar GC, Guha S, Lu W, Lim J, Boesze-Battaglia K, Laties AM, Tyagi P, Kompella UB, Mitchell CH. Acidic nanoparticles are trafficked to lysosomes and restore an acidic lysosomal pH and degradative function to compromised ARPE-19 cells. PLoS One. 2012;7:e49635. doi: 10.1371/journal.pone.0049635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhargava A, Voronov I, Wang Y, Glogauer M, Kartner N, Manolson MF. Osteopetrosis mutation R444L causes endoplasmic reticulum retention and misprocessing of vacuolar H+-ATPase a3 subunit. J Biol Chem. 2012;287:26829–26839. doi: 10.1074/jbc.M112.345702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler D, Hwang J, Estick C, Nishiyama A, Kumar SS, Baveghems C, Young-Oxendine HB, Wisniewski ML, Charalambides A, Bahr BA. Protective effects of positive lysosomal modulation in Alzheimer’s disease transgenic mouse models. PLoS One. 2011;6:e20501. doi: 10.1371/journal.pone.0020501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcraft PJ, Ruas M, Pan Z, Cheng X, Arredouani A, Hao X, Tang J, Rietdorf K, Teboul L, Chuang KT, et al. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature. 2009;459:596–600. doi: 10.1038/nature08030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesen MH, Pegan K, Spes A, Turk B. Lysosomal pathways to cell death and their therapeutic applications. ExpCell Res. 2012;318:1245–1251. doi: 10.1016/j.yexcr.2012.03.005. [DOI] [PubMed] [Google Scholar]
- Chan SL, Mayne M, Holden CP, Geiger JD, Mattson MP. Presenilin-1 mutations increase levels of ryanodine receptors and calcium release in PC12 cells and cortical neurons. J Biol Chem. 2000;275:18195–18200. doi: 10.1074/jbc.M000040200. [DOI] [PubMed] [Google Scholar]
- Chavez-Gutierrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, Lismont S, Zhou L, Van Cleynenbreugel S, Esselmann H, et al. The mechanism of gamma-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012;31:2261–2274. doi: 10.1038/emboj.2012.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung KH, Mei L, Mak DO, Hayashi I, Iwatsubo T, Kang DE, Foskett JK. Gain-of-function enhancement of IP3 receptor modal gating by familial Alzheimer’s disease-linked presenilin mutants in human cells and mouse neurons. Sci Signal. 2010;3:ra22. doi: 10.1126/scisignal.2000818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen KA, Myers JT, Swanson JA. pH-dependent regulation of lysosomal calcium in macrophages. J Cell Sci. 2002;115:599–607. doi: 10.1242/jcs.115.3.599. [DOI] [PubMed] [Google Scholar]
- Churchill GC, Okada Y, Thomas JM, Genazzani AA, Patel S, Galione A. NAADP mobilizes Ca(2+) from reserve granules, lysosome-related organelles, in sea urchin eggs. Cell. 2002;111:703–708. doi: 10.1016/s0092-8674(02)01082-6. [DOI] [PubMed] [Google Scholar]
- Coen K, Flannagan R, Baron S, Carraro-Lacroix L, Wang D, Vermeire W, Michiels C, Munck S, Baert V, Sugita S, et al. Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. J Cell Biol. 2012;198:23–35. doi: 10.1083/jcb.201201076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey EE, Beckel JM, Laties AM, Mitchell CH. Lysosomal alkalization and dysfunction in human fibroblasts with the Alzheimer’s disease-linked presenilin 1 A246E mutation can be reversed with cAMP. Neuroscience. 2014;263:111–124. doi: 10.1016/j.neuroscience.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, Annaert W. Novel research horizons for presenilins and gamma-secretases in cell biology and disease. AnnuRevCell DevBiol. 2010;26:235–260. doi: 10.1146/annurev-cellbio-100109-104117. [DOI] [PubMed] [Google Scholar]
- Dobrowolski R, Vick P, Ploper D, Gumper I, Snitkin H, Sabatini DD, De Robertis EM. Presenilin Deficiency or Lysosomal Inhibition Enhances Wnt Signaling through Relocalization of GSK3 to the Late-Endosomal Compartment. Cell Rep. 2012;2:1316–1328. doi: 10.1016/j.celrep.2012.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong XP, Shen D, Wang X, Dawson T, Li X, Zhang Q, Cheng X, Zhang Y, Weisman LS, Delling M, et al. PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin Ca(2+) release channels in the endolysosome. Nat Commun. 2010;1:38. doi: 10.1038/ncomms1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong XP, Wang X, Shen D, Chen S, Liu M, Wang Y, Mills E, Cheng X, Delling M, Xu H. Activating mutations of the TRPML1 channel revealed by proline-scanning mutagenesis. J Biol Chem. 2009;284:32040–32052. doi: 10.1074/jbc.M109.037184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esselens C, Oorschot V, Baert V, Raemaekers T, Spittaels K, Serneels L, Zheng H, Saftig P, De Strooper B, Klumperman J, et al. Presenilin 1 mediates the turnover of telencephalin in hippocampal neurons via an autophagic degradative pathway. J Cell Biol. 2004;166:1041–1054. doi: 10.1083/jcb.200406060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fameli N, Ogunbayo OA, van Breemen C, Evans AM. Cytoplasmic nanojunctions between lysosomes and sarcoplasmic reticulum are required for specific calcium signaling. F1000Res. 2014;3:93. doi: 10.12688/f1000research.3720.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frakes AE, Ferraiuolo L, Haidet-Phillips AM, Schmelzer L, Braun L, Miranda CJ, Ladner KJ, Bevan AK, Foust KD, Godbout JP, et al. Microglia induce motor neuron death via the classical NF-kappaB pathway in amyotrophic lateral sclerosis. Neuron. 2014;81:1009–1023. doi: 10.1016/j.neuron.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghavami S, Shojaei S, Yeganeh B, Ande SR, Jangamreddy JR, Mehrpour M, Christoffersson J, Chaabane W, Moghadam AR, Kashani HH, et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. ProgNeurobiol. 2014;112:24–49. doi: 10.1016/j.pneurobio.2013.10.004. [DOI] [PubMed] [Google Scholar]
- Gillespie J, Ozanne S, Tugal B, Percy J, Warren M, Haywood J, Apps D. The vacuolar H(+)-translocating ATPase of renal tubules contains a 115-kDa glycosylated subunit. FEBS Lett. 1991;282:69–72. doi: 10.1016/0014-5793(91)80446-a. [DOI] [PubMed] [Google Scholar]
- Grimm C, Jors S, Saldanha SA, Obukhov AG, Pan B, Oshima K, Cuajungco MP, Chase P, Hodder P, Heller S. Small molecule activators of TRPML3. Chem Biol. 2010;17:135–148. doi: 10.1016/j.chembiol.2009.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha A, Ahuja M, Patel S, Brailoiu E, Muallem S. Convergent regulation of the lysosomal two-pore channel-2 by Mg(2)(+), NAADP, PI(3,5)P(2) and multiple protein kinases. EMBO J. 2014;33:501–511. doi: 10.1002/embj.201387035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang DE, Soriano S, Frosch MP, Collins T, Naruse S, Sisodia SS, Leibowitz G, Levine F, Koo EH. Presenilin 1 facilitates the constitutive turnover of beta-catenin: differential activity of Alzheimer’s disease-linked PS1 mutants in the beta-catenin-signaling pathway. J Neurosci. 1999;19:4229–4237. doi: 10.1523/JNEUROSCI.19-11-04229.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpatrick BS, Eden ER, Schapira AH, Futter CE, Patel S. Direct mobilisation of lysosomal Ca2+ triggers complex Ca2+ signals. J Cell Sci. 2013;126:60–66. doi: 10.1242/jcs.118836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolf DM, Marinez-Vicente M, Massey AG, Sovak G, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146–1158. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd-Evans E, Morgan AJ, He X, Smith DA, Elliot-Smith E, Sillence DJ, Churchill GC, Schuchman EH, Galione A, Platt FM. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med. 2008;14:1247–1255. doi: 10.1038/nm.1876. [DOI] [PubMed] [Google Scholar]
- Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci. 2015;16:345–357. doi: 10.1038/nrn3961. [DOI] [PubMed] [Google Scholar]
- Mindell JA. Lysosomal acidification mechanisms. Annu Rev Physiol. 2012;74:69–86. doi: 10.1146/annurev-physiol-012110-142317. [DOI] [PubMed] [Google Scholar]
- Naylor E, Arredouani A, Vasudevan SR, Lewis AM, Parkesh R, Mizote A, Rosen D, Thomas JM, Izumi M, Ganesan A, et al. Identification of a chemical probe for NAADP by virtual screening. NatChemBiol. 2009;5:220–226. doi: 10.1038/nchembio.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi T, Forgac M. Molecular Cloning and Expression of Three Isoforms of the 100-kDa a Subunit of the Mouse Vacuolar Proton-translocating ATPase. J Biol Chem. 2000;275:6824–6830. doi: 10.1074/jbc.275.10.6824. [DOI] [PubMed] [Google Scholar]
- Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007;120:4081–4091. doi: 10.1242/jcs.019265. [DOI] [PubMed] [Google Scholar]
- Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–997. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Yang D. Autophagy and neuronal cell death in neurological disorders. Cold Spring Harb Perspect Biol. 2012;4 doi: 10.1101/cshperspect.a008839. pii: a008839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA, Yang DS. Autophagy failure in Alzheimer’s disease-locating the primary defect. NeurobiolDis. 2011;43:38–45. doi: 10.1016/j.nbd.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penny CJ, Kilpatrick BS, Han JM, Sneyd J, Patel S. A computational model of lysosome-ER Ca2+ microdomains. J Cell Sci. 2014;127:2934–2943. doi: 10.1242/jcs.149047. [DOI] [PubMed] [Google Scholar]
- Pitt SJ, Funnell TM, Sitsapesan M, Venturi E, Rietdorf K, Ruas M, Ganesan A, Gosain R, Churchill GC, Zhu MX, et al. TPC2 is a novel NAADP-sensitive Ca2+ release channel, operating as a dual sensor of luminal pH and Ca2+ J Biol Chem. 2010;285:35039–35046. doi: 10.1074/jbc.M110.156927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raines SM, Rane HS, Bernardo SM, Binder JL, Lee SA, Parra KJ. Deletion of vacuolar proton-translocating ATPase V(o)a isoforms clarifies the role of vacuolar pH as a determinant of virulence-associated traits in Candida albicans. J Biol Chem. 2013;288:6190–6201. doi: 10.1074/jbc.M112.426197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Montealegre D, Pearce DA. Defective lysosomal arginine transport in juvenile Batten disease. HumMolGenet. 2005;14:3759–3773. doi: 10.1093/hmg/ddi406. [DOI] [PubMed] [Google Scholar]
- Raychowdhury MK, Gonzalez-Perrett S, Montalbetti N, Timpanaro GA, Chasan B, Goldmann WH, Stahl S, Cooney A, Goldin E, Cantiello HF. Molecular pathophysiology of mucolipidosis type IV: pH dysregulation of the mucolipin-1 cation channel. HumMolGenet. 2004;13:617–627. doi: 10.1093/hmg/ddh067. [DOI] [PubMed] [Google Scholar]
- Saw N, Kang S, Parsaud L, Han G, Jiang T, Grzegorczyk K, Surkont M, Sun-Wada G, Wada Y, Li L, et al. Vacuolar H(+)-ATPase subunits Voa1 and Voa2 cooperatively regulate secretory vesicle acidification, transmitter uptake, and storage. Mol Biol Cell. 2011;22:3394–3409. doi: 10.1091/mbc.E11-02-0155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ, Wolfe MS. Presenilin: Running with Scissors in the Membrane. Cell. 2007;131:215–221. doi: 10.1016/j.cell.2007.10.012. [DOI] [PubMed] [Google Scholar]
- Shilling D, Muller M, Takano H, Mak DO, Abel T, Coulter DA, Foskett JK. Suppression of InsP3 receptor-mediated Ca2+ signaling alleviates mutant presenilin-linked familial Alzheimer’s disease pathogenesis. JNeurosci. 2014;34:6910–6923. doi: 10.1523/JNEUROSCI.5441-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner H, Haass C. Intramembrane proteolysis by presenilins. Nat Rev Mol Cell Biol. 2000;1:217–224. doi: 10.1038/35043065. [DOI] [PubMed] [Google Scholar]
- Sun B, Zhou Y, Halabisky B, Lo I, Cho SH, Mueller-Steiner S, Devidze N, Wang X, Grubb A, Gan L. Cystatin C-cathepsin B axis regulates amyloid beta levels and associated neuronal deficits in an animal model of Alzheimer’s disease. Neuron. 2008;60:247–257. doi: 10.1016/j.neuron.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres M, Jimenez S, Sanchez-Varo R, Navarro V, Trujillo-Estrada L, Sanchez-Mejias E, Carmona I, Davila JC, Vizuete M, Gutierrez A, et al. Defective lysosomal proteolysis and axonal transport are early pathogenic events that worsen with age leading to increased APP metabolism and synaptic Abeta in transgenic APP/PS1 hippocampus. Mol Neurodegener. 2012;7:59. doi: 10.1186/1750-1326-7-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, Serneels L, De Strooper B, Yu G, Bezprozvanny I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell. 2006;126:981–993. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker MW, Lloyd-Evans E. A rapid method for the preparation of ultrapure, functional lysosomes using functionalized superparamagnetic iron oxide nanoparticles. Methods Cell Biol. 2015;126:21–43. doi: 10.1016/bs.mcb.2014.10.019. [DOI] [PubMed] [Google Scholar]
- Wilson CA, Murphy DD, Giasson BI, Zhang B, Trojanowski JQ, Lee VM. Degradative organelles containing mislocalized a- and b-synuclein proliferate in presenilin-1 null neurons. J Cell Biol. 2004;165:335–346. doi: 10.1083/jcb.200403061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe DM, Lee JH, Kumar A, Lee S, Orenstein SJ, Nixon RA. Autophagy failure in Alzheimer’s disease and the role of defective lysosomal acidification. EurJNeurosci. 2013;37:1949–1961. doi: 10.1111/ejn.12169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi S, Jha A, Li Q, Soyombo AA, Dickinson GD, Churamani D, Brailoiu E, Patel S, Muallem S. Transient receptor potential mucolipin 1 (TRPML1) and two-pore channels are functionally independent organellar ion channels. J Biol Chem. 2011;286:22934–22942. doi: 10.1074/jbc.M110.210930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang DS, Stavrides P, Mohan PS, Kaushik S, Kumar A, Ohno M, Schmidt SD, Wesson D, Bandyopadhyay U, Jiang Y, et al. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer’s disease ameliorates amyloid pathologies and memory deficits. Brain. 2011;134:258–277. doi: 10.1093/brain/awq341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Jin S, Yi F, Li PL. TRP-ML1 functions as a lysosomal NAADP-sensitive Ca2+ release channel in coronary arterial myocytes. J Cell Mol Med. 2009;13:3174–3185. doi: 10.1111/j.1582-4934.2008.00486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Garbett K, Veeraraghavalu K, Wilburn B, Gilmore R, Mirnics K, Sisodia S. A Role for Presenilins in Autophagy Revisited: Normal Acidification of Lysosomes in Cells Lacking PSEN1 and PSEN2. JNeurosci. 2012a;32:8633–8648. doi: 10.1523/JNEUROSCI.0556-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Li X, Xu H. Phosphoinositide isoforms determine compartment-specific ion channel activity. Proc Natl Acad Sci U S A. 2012b;109:11384–11389. doi: 10.1073/pnas.1202194109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.